
Abstract
Long noncoding RNAs (lncRNAs) are frequently dysregulated in malignancies and serve as significant regulators of tumorigenesis. The role of the lncRNA DNAH17-AS1 in gastric cancer (GC) remains incompletely understood. In this study, we explored the biological function and underlying mechanism of DNAH17-AS1 in GC. Differences in DNAH17-AS1 expression between GC and normal tissues were evaluated via The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and qRT-PCR validation. CCK-8, colony formation, animal, and flow cytometry assays were performed to detect the effects of DNAH17-AS1 on GC cell proliferation. Further biological experiments combined with bioinformatics analyses were performed to reveal the molecular mechanism involved. The results indicated that DNAH17-AS1 was strongly overexpressed in GC tissues and cells and that high expression of DNAH17-AS1 was correlated with lager tumour size, poor differentiation, and shorter survival. Silencing DNAH17-AS1 inhibited proliferation, induced G1 arrest and apoptosis in GC cells in vitro, and repressed tumorigenesis in vivo. Mechanistically, DNAH17-AS1 acted as a competitive endogenous RNA (ceRNA) for the tumour suppressor miR-202-3p and consequently prevented the degradation of ONECUT2. In addition, the DNAH17-AS1/miR-202-3p/ONECUT2 axis promoted the radioresistance of GC. In summary, DNAH17-AS1 plays crucial roles in GC progression and may be a novel promising target for therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Gastric cancer (GC), as one of the most prevalent malignant diseases, ranks fifth in incidence and fourth in mortality worldwide, with an estimated one million newly diagnosed cases and 769,000 deaths in 2020 [1]. Due to advancements in systemic treatments, including surgery, chemotherapy, molecular targeted therapy, and radiotherapy, the overall survival of patients with GC has improved. Nonetheless, several major issues, such as recurrence and distant metastasis, remain poorly addressed in the clinic [2, 3]. Therefore, exploring the underlying mechanism of GC progression is crucial for the development of alternative molecular and genetic targets.
Long noncoding RNAs (lncRNAs) are defined as transcripts longer than 200 nucleotides with no or limited protein-coding ability that are involved in tumour occurrence and development via chromosome remodelling and transcriptional or posttranscriptional modulation [4, 5]. Many studies have indicated that lncRNAs regulate various biological behaviours, such as cell growth, the Warburg effect [6], stem cell stemness properties [7], and autophagy [8]. Recently, several lncRNAs have been shown to participate in the modulation of GC progression. For example, the lncRNA CCAT1 inhibits the ubiquitin-mediated degradation of PTBP1 to increase its stability, thereby increasing the glycolytic activity of GC cells [9]. LINC00501 recruits hnRNPR to the SLUG promoter region and activates its transcription, which promotes epithelial‒mesenchymal transition and angiogenesis in GC [10]. In addition, the lncRNA CCAT2 binds to ESRP1, subsequently regulates alternative splicing of CD44 to upregulate CD44v6 expression, and ultimately facilitates GC progression. The overexpression of CCAT2 is also closely related to the unfavourable survival of patients [11]. The lncRNA DNAH17-AS1 is located on human chromosome 17q25.3 and has been reported to act as an oncogene in non-small cell lung cancer and pancreatic carcinoma by serving as a miRNA sponge [12, 13]. In GC, the specific biological functions and mechanisms of DNAH17-AS1 are not well understood.
In the present study, we aimed to explore the potential role and relevant molecular mechanism by which DNAH17-AS1 regulates cell proliferation, the cell cycle, apoptosis, and radioresistance in GC.
2 Materials and methods
2.1 Clinical subjects and specimens
We collected 65 paired GC tissues and adjacent normal tissues from the Department of General Surgery, the First Affiliated Hospital of Nanjing Medical University. No patients were treated with chemotherapy or radiotherapy before surgery. Tissues were immediately frozen after surgical resection and stored in liquid nitrogen. Every subject provided written informed consent.
2.2 RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
TRIzol reagent (Invitrogen, USA) was used to isolate total RNA from GC tissues and cells. A NanoDrop 2000 spectrophotometer (Thermo Scientific, USA) was used to determine the RNA concentration. A PrimeScript RT-PCR kit (Takara, Japan) was used to reverse transcribe the mRNAs, which produced complementary DNAs (cDNAs). The New Poly(A) Tailing 17 Kit (Thermo Scientific, USA) was used for reverse transcription of the miRNAs. QRT-PCR was performed with SYBR Green Master Mix (Roche, USA) on an ABI StepOne Plus system, after which we calculated the relative gene expression levels according to the 2−ΔΔCt method. GAPDH and U6 served as endogenous controls. The sequences of primers used were as follows: DNAH17-AS1 (forward: 5′-GGAGGGTAGAGCACATTCGG-3′, reverse: 5′-GGCACACATGGCTGTATTGG-3′); ONECUT2 (forward: 5′-CCGAACACTCTTCGCCATCT-3′, reverse: 5′-GCTCAGATCGTCTTGCCACT-3′). GAPDH (forward: 5′-CACCCACTCCTCCACCTTTG-3′, reverse: 5′-CCACCACCCTGTTGCTGTAG-3′). miR-202-3p (forward: 5′-AGAGGTATAGGGCATGGGAA-3′). Universal (5′-GCGAGCACAGAATTAATACGAC-3′); U6 (forward: 5′-CTCGCTTCGGCAGCACA-3′, reverse: 5′-AACGCTTCACGAATTTGCGT′).
2.3 Western blot
RIPA lysis buffer (Beyotime, China) was used to extract total protein from GC cells and tissues. The protein concentration was assessed with a BCA protein assay kit. After SDS-PAGE, the proteins were transferred to PVDF membranes. The membranes were subsequently blocked in TBST buffer supplemented with 5% skim milk, followed by incubation with primary antibodies at 4 °C overnight. The next day, the membranes were rinsed three times with TBST and then incubated with the appropriate secondary antibodies for 2 h. An enhanced chemiluminescence (ECL) kit (Servicebio, China) was used to visualize the protein bands after the membranes were washed three times with TBST for 10 min. Finally, the grey values of the proteins were calculated using ImageJ software. The antibodies information was listed in Supplementary Table 1.
2.4 Cell culture and transfection
The five human GC cell lines AGS, HGC-27, MKN-45, MKN-28, and NCI-N87 as well as the normal gastric mucosa epithelial cell line GES-1 were obtained from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China). We cultured these cells (with the exception of HGC-27 cells, which were cultured in F12K medium) in RPMI 1640 medium (Wisent, China) supplemented with 10% foetal bovine serum (FBS), streptomycin (100 µg/ml), and penicillin (100 U/ml) in a humidified incubator with 5% CO2 at 37 °C. Small interfering RNAs (siRNAs) targeting DNAH17-AS1 (si-DNAH17-AS1, containing si-DNAH17-AS1#1 and DNAH17-AS1#2) and a negative control (NC), a miR-202-3p mimic, a miR-202-3p inhibitor, and their NCs, as well as siRNAs targeting ONECUT2 and the corresponding NC, were designed and obtained from GenePharma (Shanghai, China). The DNAH17-AS1 sequence was subcloned and inserted into the pcDNA3.1 vector (Invitrogen, USA) (pcDNA3.1-DNAH17-AS1, shortened to DNAH17-AS1) to upregulate DNAH17-AS1 expression, while the empty vector was used as the control. DNAH17-AS1-targeting short hairpin RNA (shRNA, shDNAH17-AS1) and the NC were synthesized by Genechem (Shanghai, China) and cloned and inserted into the pLKO.1 lentiviral vector for in vivo experiments; subsequently, the lentiviruses were injected into MKN-45 cells. Lipofectamine 3000 (Invitrogen, USA) was used for all cell transfection experiments according to the manufacturer’s instructions. We performed qRT-PCR and western blot to determine the transfection efficiency.
2.5 Cell counting kit-8 (CCK-8) assay
We seeded the treated GC cells (density: 3000 cells per well) into a 96-well plate (Corning, USA) and added 100 µl of DMEM containing 10% FBS. After they were cultured for 1, 2, 3, 4, or 5 days, the cells were incubated for 2 h with 10 µl of CCK-8 reagent (Dojindo, Japan). A microplate reader (Bio-Rad, USA) was then used to measure the absorbance of each well at a wavelength of 450 nm.
2.6 Colony formation assay
The transfected GC cells (density: 500 cells/well) were seeded into a six-well plate. After 14 days of culture, the cells were washed three times with phosphate-buffered saline (PBS). Then, the cells were fixed in 4% paraformaldehyde and stained with 0.1% crystal violet (Kaigen, China) for 30 min. After removal of the dye, the number of colonies was counted.
2.7 Flow cytometry assay
For the cell cycle analysis, we collected the treated GC cells and fixed them in 70% ethanol overnight, followed by centrifugation at 1000 r/min. Then, we added 100 µl of RNAse to the cells and incubated them for 30 min at 37 °C. After that, 100 µl of propidium iodide (PI) was added and the cells were incubated for 30 min at 4 °C in the dark. Finally, we used a flow cytometer to analyse the cell cycle. For the apoptosis analysis, we seeded GC cells in a six-well plate at a density of 2 × 105 cells per well. The cells were then harvested after the incubation period and were washed with PBS. After the cells were centrifuged and resuspended in 100 µl of Annexin-binding buffer, 5 µl of Annexin V-fluorescein isothiocyanate (FITC) and 5 µl of PI were added to the sample tube and the cells were incubated for 15 min in the dark. Next, the cells were centrifuged and resuspended in 400 µl of annexin-binding buffer. Finally, flow cytometry was conducted to analyse the percentage of apoptotic cells.
2.8 Fluorescence in situ hybridization (FISH)
RiboBio Biotech (Guangzhou, China) was commissioned to design and synthesize the specific DNAH17-AS1 cDNA probes. Briefly, we seeded the cells into 24-well plates at a density of 6 × 104 cells per well, after which the cells were fixed in 4% paraformaldehyde once a confluence of 80% was reached. After permeabilization, 250 µl of DNAH17-AS1 hybridization solution containing the probe (0.5 µg/ml) was added to the cells, which were incubated in a humidified chamber overnight. The cells were stained in diluted 4,6-diamidino-2-phenylindole for 5 min. We selected signals and acquired all images with a Leica SP5 confocal microscope (Leica Microsystems, Germany).
2.9 Nuclear and cytoplasmic RNA isolation
In accordance with the manufacturer’s instructions, we used a Cytoplasmic and Nuclear RNA Purification kit (Norgenbiotek Corporation, Canada) to separate the cytoplasmic and nuclear RNA. Briefly, after the cells were lysed in cell fractionation buffer, they were then centrifuged at 500×g for 5 min. We collected the supernatant as the cytoplasmic fraction. Next, cell disruption buffer was used to lyse the remaining nuclear pellets, and the nuclear fraction was obtained. Finally, qRT-PCR was conducted to determine DNAH17-AS1 expression in both fractions. U6 and GAPDH were selected as the nuclear and cytoplasmic controls, respectively.
2.10 Luciferase reporter assay
A Mut Express II Fast Mutagenesis Kit (Vazyme, China) was used to generate mutations in the miRNA binding sites in DNAH17-AS1 and ONECUT2. The 3′-UTR wild-type and mutant sequences of DNAH17-AS1 or ONECUT2 were inserted into the pGL-3 luciferase reporter vector (Realgene, China). After the cells were cultured in 48-well plates overnight, the miR-202-3p mimic and the above reporter plasmids were cotransfected into HEK293T cells. The Dual-Luciferase Reporter Assay System (Promega, USA) was used to detect Firefly and Renilla luciferase activities.
2.11 RNA immunoprecipitation (RIP) assay
A Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Germany) was used to perform the RIP assay, detecting the interaction of DNAH17-AS1 and miR-202-3p. Briefly, anti-Ago2 antibody, the control anti-IgG antibody, and magnetic beads were coupled. Then the corresponding cell lysate was incubated with the beads at 4 °C all night. Following digestion by proteinase K, the coprecipitated RNA was extracted and measured by qRT-PCR.
2.12 Animal models and immunohistochemistry analysis
For the tumorigenesis assays, after they were suspended in 100 µl of PBS, 2 × 106 stably transfected MKN-45 cells were subcutaneously injected into the flank region of 4-week-old female nude mice. We measured the short diameter (a) and the long diameter (b) of the tumours every 3 days and calculated the volume according to the following formula: volume = 0.5 × a2 × b. Fifteen days later, the mice were sacrificed, and the subcutaneous tumours were excised, photographed, and weighed. Xenografts from nude mice were fixed in a 4% formaldehyde solution and then embedded in paraffin. The sections were incubated with anti-ki67 antibody overnight at four degrees Celsius, followed by incubation with HRP-conjugated secondary antibodies for 30 min. After they were stained with 3,3′-diaminobenzidine solution and counterstained with haematoxylin, the slides were examined under a microscope. The Ethics Committee of Nanjing Medical University requires that the maximum tumour volume should not exceed 2000 mm3. And the maximal tumour size from nude mice in our study was not exceeded.
2.13 Clonogenic survival assay
GC cells were seeded into 6-well plates at densities of 300, 500, 1000, 2000, and 4000 cells per well. After 24 h, the cells were subjected to 0, 2, 4, 6, or 8 Gy X-ray irradiation via a medical linear accelerator (Precise Accelerator, Elekta, Sweden). After incubation for 2 weeks, the cells were fixed in methanol and stained with 1% crystal violet. We counted the colonies that contained more than 50 cells. The plating efficiency (PE) was equal to the number of colonies observed/number of cells plated in the control group. The following formula was used to calculate the surviving fraction (SF): number of colonies formed/(number of cells seeded × PE). The survival curves were fitted by the single-hit multiple-target model: SF = 1 − (1 − EXP(− D/D0))N. Dq = D0 * ln(N). The mean lethal dose (D0) represents the dose required to reduce cell survival to 37%. The quasithreshold dose (Dq) represents the ability to accumulate sublethal damage. A higher D0 or Dq indicates enhanced radioresistance.
2.14 Bioinformatics analysis and statistical analysis
The RNA sequencing data of DNAH17-AS1 and miR-202-3p in GC and normal tissues were downloaded from UCSC (https://xenabrowser.net/datapages/; dataset ID: TCGA-STAD.htseq_fpkm-uq.tsv and TCGA-STAD.mirna.tsv) and the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The expression of ONECUT2 was evaluated via GEPIA online software (http://gepia.cancer-pku.cn/) and the GEO database. Survival data related to DNAH17-AS1 and ONECUT2 for gastric cancer patients were extracted from GEPIA. Sangerbox (http://www.sangerbox.com/login.html) was used to download the RNA sequencing data of lncRNAs in GC and normal tissues from the TCGA and to conduct hierarchical clustering analysis. MiRDB (https://mirdb.org/custom.html) and lncRNASNP2 (http://bioinfo.life.hust.edu.cn/lncRNASNP/#!/) were used to predict potential lncRNA‒miRNA interactions. TargetScan (http://www.targetscan.org/vert_72/), miRDB,
miRWalk (http://mirwalk.umm.uni-heidelberg.de/), miRTarBase (https://mirtarbase.cuhk.edu.cn/~miRTarBase/miRTarBase_2022/php/search.php), and DIANA (https://dianalab.e-ce.uth.gr/html/dianauniverse/index.php?r=microT_CDS) were employed to search for mRNAs that bind to miRNAs. The cytoscape software (version 3.7.1) was used to construct and visualize the lncRNA-miRNA-mRNA regulatory network. SPSS 25.0 was used to conduct all the statistical analyses. We generated the relevant diagrams with GraphPad Prism 6. The results are shown as the mean value ± standard deviation (SD) or the mean value ± standard error (SE). Student’s t test was used to assess significant differences between two independent groups. For CCK-8 assays, considering that the ability of cell proliferation was affected by two factors, namely gene expression levels and time, so the two-away ANOVA was used to compare the results of different treatment groups. The associations of DNAH17-AS1 expression with the clinical features of GC patients were evaluated via Pearson’s χ2 test. The correlations between gene expression levels were determined via linear regression. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) were considered statistically significant.
3 Results
3.1 Identification of DNAH17-AS1 upregulation in GC tissues
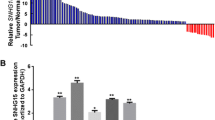
We downloaded and processed the RNA sequencing data of GC tissues and normal tissues from the TCGA. The differential expression of lncRNAs in GC and normal tissues was subsequently shown via hierarchical clustering (Fig. 1A). The volcano plot revealed 2744 differentially expressed lncRNAs, 1931 of which were upregulated and 813 of which were downregulated (Fig. 1B). DNAH17-AS1 ranked 452 in these upregulated lncRNAs. We selected DNAH17-AS1 for further study from the upregulated lncRNAs. We conducted a single heatmap analysis by extracting the RNA sequencing data of 375 GC tissues and 32 normal tissues from the TCGA and found that the expression of DNAH17-AS1 was upregulated in most cancer samples (Fig. 1C). We subsequently performed a t test and observed that DNAH17-AS1 levels were higher in GC tissues than in normal tissues in the TCGA cohort (Fig. 1D). Furthermore, we sorted 27 paired GC and normal tissues from the TCGA to perform another t test and observed the same trend (Fig. 1E). According to the data provided by the GEO database (GSE54129), DNAH17-AS1 expression was also markedly upregulated in GC tissues compared with normal tissues (Fig. 1F). In addition, data from GEPIA revealed that GC patients with high expression of DNAH17-AS1 were inclined to have worse overall survival and disease-free survival (Fig. 1G, H). UALCAN (http://ualcan.path.uab.edu/) was used to evaluate the correlation between DNAH17-AS1 expression and GC stage in the TCGA cohort, and the results indicated that DNAH17-AS1 upregulation was associated with advanced overall disease stage (Fig. 1I). Quantitative real-time PCR was used to detect the expression levels of DNAH17-AS1 in 65 paired GC and adjacent normal samples. A significant upregulation of DNAH17-AS1 was observed in GC tissues (Fig. 1J). Moreover, the expression levels of DNAH17-AS1 were greater in the GC cell lines than in the normal gastric cell line GES-1 (Fig. 1K). Clinicopathological information for the 65 GC patients revealed that high DNAH17-AS1 expression was associated with increased tumour size and poor differentiation (Table 1).

Identification of DNAH17-AS1 upregulation in GC tissues. A Hierarchical clustering of differentially expressed lncRNA between GC tissues and normal tissues from TCGA database using Sangerbox. B Volcano plot displayed the expression profile in two groups. The red points represented remarkably upregulated lncRNAs, and the green points represented remarkably downregulated lncRNAs. C Heat map analysis of DNAH17-AS1 expression in GC tissues and normal tissues from TCGA. D, E Data from TCGA were extracted to predict the relative expression of DNAH17-AS1 in unpaired and paired GC tissues. F The expression values of DNAH17-AS1 in GC samples and normal samples were validated by using GEO database (GSE54129). G, H The prognostic value of DNAH17-AS1 in GC was analyzed by GEPIA database. I The association of DNAH17-AS1 with GC TNM stage was analyzed by UALCAN website (https://ualcan.path.uab.edu/analysis.html). J Relative expression of DNAH17-AS1 in 65 GC tissues and adjacent normal tissues via qRT-PCR. K Relative expression of DNAH17-AS1 in GC cell lines by qRT-PCR. *P < 0.05, **P < 0.01, ***P < 0.001
3.2 Knockdown of DNAH17-AS1 inhibits the proliferation of gastric cancer cells
To explore the impact of DNAH17-AS1 on cell growth in GC, we transfected two specific small interfering RNAs (siRNAs) targeting DNAH17-AS1 into HGC-27 and MKN-45 cells. Next, the pcDNA3.1-DNAH17-AS1 expression plasmid was introduced into the above two cell lines and AGS cell line to generate cells overexpressing DNAH17-AS1. qRT-PCR was performed to confirm that the transfection efficiency was satisfactory (Fig. 2A). The results of the CCK-8 and colony formation assays suggested that silencing DNAH17-AS1 suppressed GC cell proliferation, whereas ectopic DNAH17-AS1 markedly accelerated cell growth (Fig. 2B–E).

Knockdown of DNAH17-AS1 inhibits proliferation of gastric cancer cells. A qRT-PCR was used to confirm the silence of DNAH17-AS1 mediated by siRNAs and its overexpression mediated by pcDNA3.1-DNAH17-AS1. B, C Cell proliferation analysis of HGC-27, MKN-45, and AGS cells after inhibiting or overexpressing DNAH17-AS1 by CCK-8 assay. D, E Influences of DNAH17-AS1 expression alteration on colony formation of GC cells. *P < 0.05, **P < 0.01, ***P < 0.001
3.3 Depletion of DNAH17-AS1 leads to cell cycle arrest in G1 phase, induces apoptosis, and impairs tumorigenesis in vivo
To further investigate whether the effect of DNAH17-AS1 inhibition on GC cell growth was caused by cell cycle arrest, flow cytometric analysis was used to detect cell cycle progression. The results indicated that HGC-27 and MKN-45 cells treated with DNAH17-AS1 siRNA1 or siRNA2 exhibited remarkable cell cycle arrest in G0/G1 phase and that the proportion of cells in S phase was reduced (Fig. 3A). Similarly, we also conducted flow cytometric analysis to determine whether GC cell growth was impacted by apoptosis and found that depletion of DNAH17-AS1 dramatically increased the number of apoptotic cells compared with that in control cells (Fig. 3B). Subsequently, western blotting was performed to examine the levels of proteins related to the cell cycle and apoptosis. We observed that the expression of cyclin D3, CDK4, and Bcl-2 decreased, whereas that of Bak and Cleaved PARP increased (Fig. 3C, Supplementary Fig. 1A). The opposite trend was found when we overexpressed DNAH17-AS1 in HGC-27 and AGS cells (Fig. 3D, Supplementary Fig. 1B), which confirms that DNAH17-AS1 is involved in both the cell cycle and apoptosis. Next, we constructed stable DNAH17-AS1-silenced MKN-45 cells and control cells and then used a mouse subcutaneous xenograft model to evaluate the effects of DNAH17-AS1 downregulation on tumour growth. Compared with those in control mice, tumour xenografts in mice treated with MKN-45 sh-DNAH17-AS1 cells had markedly decreased volumes and weights (Fig. 3E–G). We conducted ki-67 staining and detected lower proliferation in the DNAH17-AS1-inhibited group than in the control group (Fig. 3H).

Depletion of DNAH17-AS1 leads to cell cycle arrest at G1 phase, induces apoptosis, and impairs tumorigeneses in vivo. A Examination of cell cycle distribution in GC cells by flow cytometry when silencing DNAH17-AS1. B Flow cytometric analysis of the apoptosis of HGC-27 and MKN-45 cells after transfected with DNAH17-AS1 siRNAs. C, D Effects of DNAH17-AS1 on cell cycle and apoptosis-related proteins were measured by western blot. E Representative subcutaneous xenografts from the shDNAH17-AS1 group and control group. F Xenograft volumes were measured in nude mice every 3 days and growth curves are plotted. G Fifteen days after the tumor was implanted, the tumors were resected and weighed. H Immunohistochemistry staining of Ki-67 of GC xenografts in nude mice. *P < 0.05, **P < 0.01, ***P < 0.001
3.4 DNAH17-AS1 acts as a putative miR-202-3p sponge
Since the function of lncRNAs is determined by their subcellular location, we firstly used the iLoc-LncRNA tool to predict the location of DNAH17-AS1. The results indicated that DNAH17-AS1 was located mainly in the cytoplasm of cells (Fig. 4A). We subsequently analysed DNAH17-AS1 localization in HGC-27 and MKN-45 cells via nucleocytoplasmic separation and fluorescence in hybridization (FISH) and obtained the same results (Fig. 4B, C), which reveals that DNAH17-AS1 has potential for posttranscriptional regulation. Therefore, these findings indicated that DNAH17-AS1 may act as a sponge of miRNAs and can therefore prevent the regulation of target genes by miRNAs. The online databases lncRNASNP2 and miRDB were used to predict the potential target miRNAs of DNAH17-AS1. We then screened 3 candidate miRNAs (miR-765, miR-3619-5p, and miR-202-3p) from the above overlapping results and combined these miRNAs with another standard; we found that miRNA expression was significantly different between cancer and normal tissues from the TCGA (Fig. 4D). Next, we used qRT-PCR to examine the expression of candidate miRNAs after DNAH17-AS1 levels were reduced in HGC-27 cells and after DNAH17-AS1 levels were increased in MKN-45 cells. As indicated in Fig. 4E, the change in miR-202-3p expression was the most obvious, which suggested an interaction between DNAH17-AS1 and miR-202-3p. A luciferase reporter assay was then performed, the results of which revealed that the luciferase activity of the wild-type LUC-DNAH17-AS1 reporter gene was markedly reduced when it was cotransfected with the miR-202-3p mimic but not when it was cotransfected with the mutant LUC-DNAH17-AS1 reporter gene (Fig. 4F). Ago2 is the core component of the RNA-induced silencing complex (RISC), which plays a vital role in miRNA-mediated functional signals. RIP assay verified that DNAH17-AS1 was enriched by the Ago2 antibody than IgG antibody. Furthermore, we observed that miR-202-3p overexpression caused DNAH17-AS1 preferentially accumulation in Ago2-containing beads in HGC-27 cells, suggesting DNAH17-AS1 may participate in miR-202-3p-RISC complex (Fig. 4G). MiR-202-3p was clearly downregulated in GC tissues and cell lines when the TCGA dataset was analysed, and qRT-PCR was conducted in GC samples and cells collected by our group (Fig. 4H, J, and K). We also sorted 41 paired GC and normal tissues from the TCGA to perform another t test and observed the same trend (Fig. 4I). In addition, miR-202-3p expression was negatively associated with DNAH17-AS1 expression (Fig. 4L). The above results revealed that DNAH17-AS1 functions as a sponge of miR-202-3p in GC.

DNAH17-AS1 acts as a putative miR-202-3p sponge. A DNAH17-AS1 intracellular distribution was predicted using the online tool (iLoc-LncRNA). B The location of DNAH17-AS1 in GC cells was analyzed by FISH assay (scale bar: 50 μm). C The subcellular fractionation assay was conducted to validate the localization of DNAH17-AS1. D MiRNAs potentially binding with DNAH17-AS1 were screened by two bioinformatics tools (lncRNASNP2 and miRDB), which simultaneously existed differential expression in TCGA. E The impacts of DNAH17-AS1 on miRNAs expression predicted to potentially binding with DNAH17-AS1 were determined by qRT-PCR. F The predicted sequence alignment was shown, and dual luciferase reporter gene assay was performed to confirm the binding relation between DNAH17-AS1 and miR-202-3p. G Ago2-RIP assays were conducted, and DNAH17-AS1 RNA levels in the immunoprecipitates were detected by qRT-PCR. IgG was used as a negative control. H, I The relative expression of miR-202-3p in unpaired and paired GC tissues and normal tissues were analyzed via TCGA database. J Detection of the relative levels of miR-202-3p in GC tissues and matched normal tissues via qRT-PCR. K qRT-PCR was used to measure miR-202-3p expression value in normal gastric mucosa epithelial cell (GES-1) and GC cells. L The association of DNAH17-AS1 and miR-202-3p expression in GC specimens was evaluated by Spearman. *P < 0.05, **P < 0.01, ***P < 0.001
3.5 MiR-202-3p silencing counteracts the tumour-suppressing effects of si-DNAH17-AS1
To further investigate the role of miR-202-3p, we transfected a miR-202-3p mimic, inhibitor, or negative control into HGC-27 and MKN-45 cells. As shown in Fig. 5A, the miR-202-3p mimic significantly increased the expression of miR-202-3p, whereas the miR-202-3p inhibitor markedly reduced the levels of miR-202-3p. As expected, CCK-8 and colony formation assays showed an obvious suppression of proliferation when miR-202-3p was upregulated (Fig. 5B, C). Further experiments indicated that the decrease in cell proliferation caused by si-DNAH17-AS1 could be reversed when the cells were cotransfected with the miR-202-3p inhibitor (Fig. 5D, E).

MiR-202-3p silencing counteracted the tumor suppressing effects of si-DNAH17-AS1. A The miR-202-3p expression level in HGC-27 and MKN-45 cells transfected with miR-202-3p inhibitor or mimic measured by qRT-PCR. B The viability of GC cells transfected with miR-202-3p mimic and NC was detected by CCK-8 assay. C Colony formation assay on GC cells transfected with miR-202-3p mimic. D, E Growth ability of GC cells after cotransfection with miR-202-3p inhibitor, si-DNAH17-AS1 2#, or control siRNA were determined by CCK-8 and colony formation assays. *P < 0.05, **P < 0.01, ***P < 0.001
3.6 ONECUT2 is a target of miR-202-3p and is regulated by DNAH17-AS1
We used 5 target-predicting programs (TargetScan, miRDB, miRWalk, miRTarBase, and DIANA) and identified 28 candidate genes that possibly bind to miR-202-3p (Fig. 6A). Using the data predicted by the above bioinformatics tools, we constructed a competitive endogenous RNA (ceRNA) network through the cytoscape software (Supplementary Fig. 2). Three target genes (STK4, ONECUT2, and ZNF644) were subsequently selected through analysis of the RNA expression data from GEPIA according to the following criterion: gene expression in tumour tissues was more than twice as high as that in normal tissues, with a p value less than 0.05 (Fig. 6B). Next, western blotting was performed to examine whether the protein levels of the above 3 genes changed when miR-202-3p was overexpressed in HGC-27 cells. The results revealed that the ONECUT2 protein level was significantly reduced but that the proteins levels of STK4 and ZNF644 were not (Fig. 6C, Supplementary Fig. 1C), which suggested that ONECUT2 is a potential target gene of miR-202-3p. A dual-luciferase reporter assay was performed to verify whether miR-202-3p directly targets ONECUT2. As shown in Fig. 6D, the miR-202-3p mimic dramatically repressed the luciferase activity of wild-type ONECUT2 but not that of the mutated ONECUT2 3′ UTR, which revealed that miR-202-3p could specifically bind to ONECUT2. In MKN-45 cells, we performed western blotting and found that miR-202-3p overexpression downregulated ONECUT2 protein levels (Fig. 6E, Supplementary Fig. 1D). In addition, the miR-202-3p mimic decreased ONECUT2 mRNA levels in HGC-27 and MKN-45 cells, as shown by qRT-PCR (Fig. 6F). The mRNA and protein levels of ONECUT2 were clearly reduced when DNAH17-AS1 was knocked down in HGC-27 and MKN-45 cells, as shown by qRT-PCR and western blotting (Fig. 6G, H, Supplementary Fig. 1E). When cells were cotransfected with si-DNAH17-AS1 and the miR-202-3p inhibitor, silencing of miR-202-3p abolished the ability of si-DNAH17-AS1 to suppress ONECUT2, which was evident at both the mRNA and protein levels (Fig. 6I, J, Supplementary Fig. 1F). We analysed the expression levels of ONECUT2 in various tumours in the TIMER database and found that ONECUT2 was highly expressed in most malignancies, including GC (Fig. 6K). According to the data provided by GEPIA and GEO (GSE51575, GSE54129, and GSE65801), ONECUT2 expression was markedly increased in GC tissues compared with normal tissues (Fig. 6L, N). Furthermore, we selected 27 paired GC and normal tissues from the TCGA to perform another t test and observed the same trend (Fig. 6M). We validated this result in 65 paired GC tissues and adjacent normal tissues collected by our group (Fig. 6O). To evaluate the prognostic value of ONECUT2 in GC, we used the GEPIA website to construct Kaplan‒Meier plots. Patients with higher ONECUT2 expression had markedly worse overall survival and disease-free survival than those with lower ONECUT2 expression (Fig. 6P, Q). A correlation analysis revealed that ONECUT2 expression was positively associated with DNAH17-AS1 expression in GC tissues and negatively associated with miR-202-3p expression (Fig. 6R, S).

ONECUT2 is a target of miR-202-3p and regulated by DNAH17-AS1. A Venn diagram of target genes of miR-202-3p predicted by Targetscan, miRDB, miRWalk, miRTarBase, and DIANA. B Three potential target genes were further selected out whose expression levels were markedly higher in GC tissues than in normal tissues (fold change ≥ 2 and p < 0.05) by using the GEPIA database. C Western blot analyses of the relative levels of the 3 candidate genes in HGC-27 cells treated by miR-202-3p mimic. D Dual-luciferase reporter assay confirmed the direct binding between miR-202-3p and ONECUT2. E Western blot measuring ONECUT2 protein level in MKN-45 cells transfected with miR-202-3p mimic. F, G qRT-PCR detecting ONECUT2 mRNA expression in HGC-27 and MKN-45 cells treated by miR-202-3p mimic or DNAH17-AS1 siRNAs. H ONECUT2 protein expression was examined in after silencing DNAH17-AS1. I, J Rescue assays: mRNA and protein levels of ONECUT2 were determined in MKN-45 cells cotransfected with si-DNAH17-AS1 2# and miR-202-3p inhibitor. K ONECUT2 expression in all tumors was analyzed by the TIMER tool (https://cistrome.shinyapps.io/timer/). L Online tool GEPIA was employed to evaluate the expression levels of ONECUT2 in GC tissues and normal tissues. M Data from TCGA were extracted to predict the relative expression of ONECUT2 in paired GC and normal tissues. N The expression values of ONECUT2 in GC samples and normal samples were validated by using GEO database (series: GSE51575, GSE54129, and GSE65801). O Relative mRNA expression of ONECUT2 in 65 GC tissues and corresponding normal tissues. P, Q Overall survival and disease free survival rate was analyzed by using an online bioinformatics tool (GEPIA). R, S Expression correlations between ONECUT2 and DNAH17-AS1, ONECUT2 and miR-202-3p in 65 GC tissues were assessed. *P < 0.05, **P < 0.01, ***P < 0.001
3.7 ONECUT2 accelerates GC cell proliferation
We transfected ONECUT2 siRNA and siNC into HGC-27 and MKN-45 cells to silence ONECUT2 expression and examined the efficiency of this siRNA by assessing ONECUT2 expression at the mRNA and protein levels via qRT-PCR and western blotting (Fig. 7A, B, Supplementary Fig. 1G). CCK-8 and colony formation assays revealed that silencing ONECUT2 impaired the proliferation rate of HGC-27 and MKN-45 cells (Fig. 7C, D). Further rescue experiments followed by CCK-8 and colony formation assays revealed that depletion of miR-202-3p reversed the suppressive effects on GC cells caused by si-ONECUT2 (Fig. 7E, F).

ONECUT2 accelerates GC cell proliferation. A, B qRT-PCR and western blot were used to measure mRNA and protein expression levels of ONECUT2 in HGC-27 and MKN-45 cells transfected with ONECUT2 siRNA. C After inhibiting ONECUT2 expression, cell growth ability of GC cells was analyzed by the CCK-8 assay. D Colony formation assay of HGC-27 and MKN-45 cells treated with ONECUT2 siRNA or control. E, F Proliferation ability of GC cells was evaluated via CCK-8 and colony formation assays when cotransfecting miR-202-3p inhibitor, si-ONECUT2, or control siRNA. *P < 0.05, **P < 0.01, ***P < 0.001
3.8 DNAH17-AS1 promotes radioresistance in GC through miR-202-3p/ONECUT2
To further explore whether DNAH17-AS1 participates in the radioresistance of GC, we conducted a clonogenic survival assay and observed that DNAH17-AS1-depleted GC cells exhibited a remarkable reduction in colony-forming capacity after they were subjected to increasing doses of X-ray irradiation (SER = 1.61 for HGC-27 cells, SER = 1.73 for MKN-45 cells) (Fig. 8A, B). DNA double-strand breaks (DSBs) caused by irradiation can manifest as H2AX phosphorylation at Ser139 (γH2AX). Compared with that in the control cells, the expression of the DNA damage marker γH2AX in DNAH17-AS1-silenced GC cells was increased after 4 Gy dose of irradiation (Fig. 8E, Supplementary Fig. 1H). Moreover, miR-202-3p overexpression led to decreased colony formation and increased expression of γH2AX when the cells were irradiated (Fig. 8C–E, Supplementary Fig. 1I). Further rescue experiments revealed that the increase in radiosensitivity caused by si-DNAH17-AS1 could be counteracted when the cells were cotransfected with the miR-202-3p inhibitor. The D0 values were 1.36 and 1.66, and the Dq values were 0.55 and 1.29 for DNAH17-AS1-silenced HGC-27 cells and HGC-27 cells cotransfected with the miR-202-3p inhibitor, respectively (Fig. 8F, G). In addition, cells treated with si-ONECUT2 presented an obvious decrease in colony formation capacity after irradiation, and this effect was reversed by cotransfection with the miR-202-3p inhibitor (Fig. 8H, I). These data revealed that DNAH17-AS1/miR-202-3p/ONECUT2 could increase GC radioresistance.

DNAH17-AS1 promotes GC radioresistance through miR-202-3p/ONECUT2. A, B Clonogenic survival curves were generated for HGC-27 and MKN-45 cells that were treated with si-DNAH17-AS1 or negative control and were then exposed to 0, 2, 4, 6–8 Gy X-ray irradiation. Radiosensitivity parameters (D0, Dq, SF2, and SER) of the corresponding groups were shown. D0, dose to reduce survival to 37%; Dq, the repair of nonlethal injury; SF2, surviving fraction at 2 Gy; SER, sensitization enhancement ratio. The survival curve was derived from a single-hit multiple-target model: 1−(1 − EXP(− D/D0))N. C, D Clonogenic survival curves and radiosensitivity parameters of GC cells which were treated by miR-202-3p mimic or control. E γH2AX expression of GC cells transfected by si-DNAH17-AS1 or negative control, and transfected by miR-202-3p mimic or control was detected by western blot after exposed to 4 Gy dose of radiation. F, G Clonogenic survival curves and radiosensitivity parameters of GC cells after cotransfection with miR-202-3p inhibitor, si-DNAH17-AS1 2#, or control siRNA. H, I Clonogenic survival curves and radiosensitivity parameters were generated when cotransfecting miR-202-3p inhibitor, si-ONECUT2, or control siRNA. *P < 0.05, **P < 0.01, ***P < 0.001
4 Discussion
Recently, accumulating studies have shown that lncRNAs act as significant regulators of human cancer progression [14]. LncRNA LYPLAL1-DT destabilizes and decreases β-catenin protein via interacting with hnRNPK and preventing the formation of the hnRNPK/β-catenin complex, which exerts anti-oncogenic effects in triple-negative breast cancer [15]. Yu et al. reported that lncRNA LOC85009 inhibits ATG5 transcription through sequestering USP5 to reduce USF1 protein stability, ultimately, increasing docetaxel sensitivity in lung adenocarcinoma [16]. Similarly, there were also many lncRNAs exhibiting abnormal expression in GC. For example, the lncRNA DIAPH2-AS1 enhances the m5C modification of NTN1 by interacting with NSUN2, which accelerates neural invasion in GC [17]. Yang et al. demonstrated that the lncRNA CBSLR, which is induced by hypoxia, could protect GC cells from ferroptosis, resulting in chemoresistance through binding with YTHDF2 to reduce CBS mRNA stability [18]. In 2020, DNAH17-AS1 was shown to be upregulated in pancreatic carcinoma and to promote cell progression, and its high expression was related to poor prognosis [13]. DNAH17-AS1 can act as a miR-877-5p molecular sponge to upregulate CCNA2 expression, thereby promoting cell proliferation, migration and invasion in non-small cell lung cancer [12]. Nonetheless, the biological function of DNAH17-AS1 in GC has not been fully elucidated. In the present study, we first confirmed that DNAH17-AS1 was overexpressed in GC tissues and cell lines. When we explored the clinical significance of DNAH17-AS1 in 65 GC tissues, we found that high DNAH17-AS1 expression was related to larger tumour size and poor differentiation. Ectopic DNAH17-AS1 expression was closely related to poor overall survival and disease-free survival in GC patients according to the GEPIA database. Taken together, the above results revealed that DNAH17-AS1 may be a promising biomarker in GC.
In terms of cellular function, the possible effects of DNAH17-AS1 on GC cells were determined by CCK-8 assay, colony formation assay, flow cytometry, and other methods. We observed that silencing DNAH17-AS1 could significantly inhibit the proliferation of GC cells in vitro. Furthermore, the suppression of the cell cycle and induction of apoptosis were also observed when DNAH17-AS1 was knocked down. Animal experiments revealed that the downregulation of DNAH17-AS1 decreased the growth of GC cells in vivo, which was consistent with previous research. Therefore, the next logical step was to investigate the specific mechanism by which DNAH17-AS1 exerts its effects on GC.
To determine the underlying mechanism of DNAH17-AS1, FISH and subcellular fractionation were performed; these assays verified that DNAH17-AS1 was located mainly in the cytoplasm of GC cells. Salmena et al. proposed the ceRNA hypothesis, which has been subsequently accepted by most scholars [19]. Increasing evidence suggests that lncRNAs can act as miRNA decoys. For example, LINC00941 sequesters miR-335-5p, which upregulates ROCK1 and promotes pancreatic cancer progression [20]. HIF1A-AS2 enhances malignant phenotypes by binding to miR-30a-5p, which prevents this miRNA from degrading SOX4 in renal carcinoma [21]. LCAT1 knockdown caused cell proliferation and invasion arrest via miR-4715-5p in lung cancer [22]. In our study, three potential miRNAs were identified via the use of two bioinformatics tools (lncRNASNP2 and miRDB) combined with the TCGA database. The subsequent qRT-PCR results indicated that silencing DNAH17-AS1 markedly upregulated miR-202-3p expression in HGC-27 cells. Next, the luciferase and RIP assays confirmed the direct binding between DNAH17-AS1 and miR-202-3p. MiR-202-3p has been reported to act as a tumour suppressor, as this miRNA influences various key biological behaviours, including the cell cycle [23], apoptosis [24], and EMT [25], and regulates drug resistance [26], whereas the effects of miR-202-3p in GC cells are limited [24, 27]. Our study verified that upregulating miR-202-3p expression inhibited GC cell proliferation. Additionally, rescue assays further indicated that the miR-202-3p inhibitor partially counteracted the effects of DNAH17-AS1 silencing on GC cell growth, which revealed that DNAH17-AS1 could as a decoy of miR-202-3p to facilitate GC tumorigenesis.
Finally, it was essential to identify potential targets of miR-202-3p in GC cells; thus, we first screened 28 target genes using the TargetScan, miRDB, miRWalk, miRTarBase, and DIANA databases. Three targets (STK4, ONECUT2, and ZNF644) were subsequently selected based on their higher expression in GC tissues than in normal tissues according to an analysis of data from GEPIA. Next, western blotting was used to identify the target gene ONECUT2, whose protein level was markedly reduced by the miR-202-3p mimic. Combined with the results of the luciferase assay, we concluded that ONECUT2 was targeted by miR-202-3p. Moreover, the expression of ONECUT2 was downregulated by si-DNAH17-AS1, which could be abolished by a miR-202-3p inhibitor, suggesting that ONECUT2 may be directly regulated by miR-202-3p and indirectly modulated by DNAH17-AS1. ONECUT2, a member of the highly conserved family of transcription factors, is also known as OC2, is located at 18q21.31 and mediates many cellular functions, including apoptosis and differentiation [28, 29]. Previous studies revealed that ONECUT2 is involved in multiple types of human tumours. Jiang et al. reported that ONECUT2 could be regulated by the circDennd1b/miR-145-5p axis, which activates the MAPK signalling pathway, and in turn, contributes to pituitary adenoma progression [30]. ONECUT2 was also reported to influence angiogenesis in ovarian cancer [31]. Moreover, previous research has revealed a correlation between high levels of ONECUT2 and poor clinical outcomes in cancer patients, such as those with lung adenocarcinoma [32]. Our study also confirmed that ONECUT2 was overexpressed in GC tissues through analysis of data from the GEPIA and GEO databases and from our own internal samples. Further loss-of-function experiments followed by CCK-8 and colony formation assays demonstrated the suppressive effects of ONECUT2 silencing on GC cell proliferation. Rescue assays indicated that the oncogenic effects of the miR-202-3p inhibitor were partially reversed by si-ONECUT2. In addition, we performed a clonogenic survival assay and observed that DNAH17-AS1 promoted DSB repair to increase the radioresistance of GC cells. These effects were also achieved through the suppression of the miR-202-3p-mediated degradation of ONECUT2.
In summary, our study revealed that DNAH17-AS1 is upregulated in GC and enhances ONECUT2 expression by acting as a sponge of miR-202-3p, which promotes cell cycle progression, inhibits apoptosis to facilitate cell proliferation, and enhances GC radioresistance by facilitating DSB repair (Fig. 9). Therefore, we identified a novel DNAH17-AS1/miR-202-3p/ONECUT2 signalling axis, which may serve as a potential therapeutic target for GC.

Schematic model for the DNAH17-AS1/miR-202-3p/ONECUT2 axis in GC. It was created by Figdraw (https://www.figdraw.com, copyright code: RRSOAe3e99)
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015;385(9972):977–1010.
Diagnosis, Treatment Guidelines For Colorectal Cancer Working Group C. Chinese Society of Clinical Oncology (CSCO) diagnosis and treatment guidelines for colorectal cancer 2018 (English version). Chin J Cancer Res. 2019;31(1):117–34.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21.
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–61.
Deng P, Li K, Gu F, Zhang T, Zhao W, Sun M, Hou B. LINC00242/miR-1-3p/G6PD axis regulates Warburg effect and affects gastric cancer proliferation and apoptosis. Mol Med. 2021;27(1):9.
Zhu P, He F, Hou Y, Tu G, Li Q, Jin T, Zeng H, Qin Y, Wan X, Qiao Y, et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40(9):1609–27.
Jiang L, Wan Y, Feng Z, Liu D, Ouyang L, Li Y, Liu K. Long noncoding RNA UCA1 is related to autophagy and apoptosis in endometrial stromal cells. Front Oncol. 2020;10:618472.
Zhang C, Wang H, Liu Q, Dai S, Tian G, Wei X, Li X, Zhao L, Shan B. LncRNA CCAT1 facilitates the progression of gastric cancer via PTBP1-mediated glycolysis enhancement. J Exp Clin Cancer Res. 2023;42(1):246.
Dou R, Han L, Yang C, Fang Y, Zheng J, Liang C, Song J, Wei C, Huang G, Zhong P, et al. Upregulation of LINC00501 by H3K27 acetylation facilitates gastric cancer metastasis through activating epithelial–mesenchymal transition and angiogenesis. Clin Transl Med. 2023;13(10):e1432.
Deng H, Gao J, Cao B, Qiu Z, Li T, Zhao R, Li H, Wei B. LncRNA CCAT2 promotes malignant progression of metastatic gastric cancer through regulating CD44 alternative splicing. Cell Oncol. 2023;46(6):1675–90.
Du LJ, Mao LJ, Jing RJ. Long noncoding RNA DNAH17-AS1 promotes tumorigenesis and metastasis of non-small cell lung cancer via regulating miR-877-5p/CCNA2 pathway. Biochem Biophys Res Commun. 2020;533(3):565–72.
Xu T, Lei T, Li SQ, Mai EH, Ding FH, Niu B. DNAH17-AS1 promotes pancreatic carcinoma by increasing PPME1 expression via inhibition of miR-432-5p. World J Gastroenterol. 2020;26(15):1745–57.
Ghasemian A, Omear HA, Mansoori Y, Mansouri P, Deng X, Darbeheshti F, Zarenezhad E, Kohansal M, Pezeshki B, Wang Z, et al. Long non-coding RNAs and JAK/STAT signaling pathway regulation in colorectal cancer development. Front Genet. 2023;14:1297093.
Tang Y, Tian W, Zheng S, Zou Y, Xie J, Zhang J, Li X, Sun Y, Lan J, Li N, et al. Dissection of FOXO1-induced LYPLAL1-DT impeding triple-negative breast cancer progression via mediating hnRNPK/beta-catenin complex. Research. 2023;6:0289.
Yu Z, Tang H, Chen S, Xie Y, Shi L, Xia S, Jiang M, Li J, Chen D. Exosomal LOC85009 inhibits docetaxel resistance in lung adenocarcinoma through regulating ATG5-induced autophagy. Drug Resist Updates. 2023;67:100915.
Li Y, Xia Y, Jiang T, Chen Z, Shen Y, Lin J, Xie L, Gu C, Lv J, Lu C, et al. Long noncoding RNA DIAPH2-AS1 promotes neural invasion of gastric cancer via stabilizing NSUN2 to enhance the m5C modification of NTN1. Cell Death Dis. 2023;14(4):260.
Yang H, Hu Y, Weng M, Liu X, Wan P, Hu Y, Ma M, Zhang Y, Xia H, Lv K. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J Adv Res. 2022;37:91–106.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–8.
Wang J, He Z, Xu J, Chen P, Jiang J. Long noncoding RNA LINC00941 promotes pancreatic cancer progression by competitively binding mir-335-5p to regulate ROCK1-mediated LIMK1/Cofilin-1 signaling. Cell Death Dis. 2021;12(1):36.
Chen M, Wei X, Shi X, Lu L, Zhang G, Huang Y, Hou J. LncRNA HIF1A-AS2 accelerates malignant phenotypes of renal carcinoma by modulating miR-30a-5p/SOX4 axis as a ceRNA. Cancer Biol Med. 2021;18(2):587–603.
Yang J, Qiu Q, Qian X, Yi J, Jiao Y, Yu M, Li X, Li J, Mi C, Zhang J, et al. Long noncoding RNA LCAT1 functions as a ceRNA to regulate RAC1 function by sponging mir-4715-5p in lung cancer. Mol Cancer. 2019;18(1):171.
Zhang Y, Chen Z, Li MJ, Guo HY, Jing NC. Long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 regulates the expression of Gli2 by miR-202 to strengthen gastric cancer progression. Biomed Pharmacother. 2017;85:264–71.
Zhao Y, Li C, Wang M, Su L, Qu Y, Li J, Yu B, Yan M, Yu Y, Liu B, et al. Decrease of miR-202-3p expression, a novel tumor suppressor, in gastric cancer. PLoS ONE. 2013;8(7):e69756.
Han X, Wang Q, Wang Y, Hu B, Dong X, Zhang H, Wang W. Long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1/microRNA-202-3p/periostin axis modulates invasion and epithelial–mesenchymal transition in human cervical cancer. J Cell Physiol. 2019;234(8):14170–80.
Santos NL, Bustos SO, Reis PP, Chammas R, Andrade LNS. Extracellular vesicle-packaged mir-195-5p sensitizes melanoma to targeted therapy with kinase inhibitors. Cells. 2023;12(9):1317.
Fu Q, Tan X, Tang H, Liu J. CCL21 activation of the MALAT1/SRSF1/mTOR axis underpins the development of gastric carcinoma. J Transl Med. 2021;19(1):210.
Li K, Zhou M, Zhang Y. MicroRNA-1298-3p induces tumor-suppressive effects in human cervical cancer cells via post-transcriptional suppression of ONECUT2. Acta Biochim Pol. 2022;69(4):737–43.
Luna Velez MV, Neikes HK, Snabel RR, Quint Y, Qian C, Martens A, Veenstra GJC, Freeman MR, van Heeringen SJ, Vermeulen M. ONECUT2 regulates RANKL-dependent enterocyte and microfold cell differentiation in the small intestine; a multi-omics study. Nucleic Acids Res. 2023;51(3):1277–96.
Jiang Q, Lei Z, Wang Z, Wang Q, Zhang Z, Liu X, Xing B, Li S, Guo X, Liu Y, et al. Tumor-associated fibroblast-derived exosomal circDennd1b promotes pituitary adenoma progression by modulating the miR-145-5p/ONECUT2 axis and activating the MAPK pathway. Cancers. 2023;15(13):3375.
Wu B, Zhang L, Yu Y, Lu T, Zhang Y, Zhu W, Song Q, Lv C, Guo J, Tian Y, et al. miR-6086 inhibits ovarian cancer angiogenesis by downregulating the OC2/VEGFA/EGFL6 axis. Cell Death Dis. 2020;11(5):345.
Ma Q, Wu K, Li H, Li H, Zhu Y, Hu G, Hu L, Kong X. ONECUT2 overexpression promotes RAS-driven lung adenocarcinoma progression. Sci Rep. 2019;9(1):20021.
Funding
This work was supported by the Youth Fund of the National Natural Science Foundation of China (82102989).
Author information
Authors and Affiliations
Contributions
QS designed and supervised the research. YG and HC performed most of the experiments and drafted the manuscript. JX and HW performed some of the experiments and analyzed the data. BW drew the diagrams. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research was approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University (Permit Number: 2018-SRFA-108) and was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments. The tissue samples were obtained with written informed consent from all patients. All animal experiments were conducted according to animal protocols approved by Nanjing Medical University and were approved by the Ethics Committee of Nanjing Medical University (Permit Number: IACUC-2306037).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ge, Y., Cang, H., Xiao, J. et al. LncRNA DNAH17-AS1 promotes gastric cancer proliferation and radioresistance by sponging miR-202-3p to upregulate ONECUT2. Discov Onc 15, 432 (2024). https://doi.org/10.1007/s12672-024-01297-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01297-2