
Abstract
The application of Genotyping-by-Sequencing (GBS) approaches is often restricted in wildlife monitoring and conservation genetics, as those fields often rely on noninvasively collected samples with low DNA content. Here we selected a subset of informative single-nucleotide polymorphisms (SNPs) from genome-wide data for lineage discrimination of a locally endangered Eurasian rodent, the hazel dormouse (Muscardinus avellanarius), and designed a microfluidic 96 SNP genotyping assay suitable for noninvasively collected samples. Analyses of 43 samples from different European countries confirmed successful discrimination of the Eastern and Western lineage and local substructure within those lineages, proving the suitability of the developed panel for identifying evolutionary significant units and conservation units. Application with 94 hair and scat samples collected in a recent monitoring study on the hazel dormouse in Southern Germany resulted in > 99.5% amplification success showing the applicability of the new tool in genetic wildlife monitoring and conservation studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
While the genomic revolution has enabled in-depth assessments of genome-wide population structure in endangered non-model species (Hohenlohe et al. 2021), applied genetic wildlife monitoring still largely relies on microsatellites optimised for the applicability in noninvasive samples with low DNA content (von Thaden et al. 2017). Genotyping platforms based on microfluidic SNP detection have recently been optimised for noninvasively collected samples (Kraus et al. 2015; von Thaden et al. 2017). These panels typically allow for reliable parallel genotyping of up to 96 SNPs, preselected from genome-wide data to serve specific purposes such as individual identification, lineage discrimination, or hybrid detection (von Thaden et al. 2020).
Here we describe the development of a reduced microfluidic SNP assay for the locally endangered hazel dormouse (Muscardinus avellanarius, 1758), which suffers from reduction and fragmentation of suitable habitats across its European distribution (Mortelliti et al. 2010). Due to declining numbers, the species is listed in Annex IV of the European Union’s Habitats Directive (92/43/EEC, Council of Europe 1992). We aimed to design a rapid and cost-effective assay for hazel dormouse lineage discrimination applicable for noninvasively collected samples.
192 SNPs were initially chosen from a set of 24,903 SNPs obtained from a normalised Genotyping-by-Sequencing (nGBS) approach (Leyhausen et al. 2022). Selected SNPs were filtered for bi-allelic state, single SNP per RAD locus, 40pb flanking region on both ends, GC content ≤ 0.65, linkage r2 < 0.3, MAF > 0.1, missing data < 0.2, and Ho within sampled populations > 0.8. Respective SNPtype assays were designed using the web-based D3 assay design tool (Standard Biotools Inc., formerly Fluidigm Corp.) Each assay consists of one specific target amplification primer (STA, forward primer for preamplification), two allele-specific primers (ASPs, for distinguishing the alleles in SNP genotyping PCR) and one locus-specific primer (LSP, reverse primer for both, preamplification and SNP genotyping PCR).
Wet lab assay testing followed the recommendations provided in von Thaden et al. (2020), including initial testing of PCR performance, followed by a test for multiplex compatibility in the preamplification step (STA), and assessment of assay performance with low DNA concentrations (up to 0.2 ng/µl template DNA) for the STA step. SNP genotyping was performed on 96.96 Dynamic Arrays (Standard Biotools Inc.) with integrated fluidic circuits (Wang et al. 2009) that allow for distinct amplification reactions for each combination of SNPtype assay and sample in nano-PCR wells. The genotyping procedures followed the manufacturer’s protocols with modifications according to von Thaden et al. (2020). Allele-specific fluorescence data was analysed with the SNP Genotyping Analysis Software (version 4.1.2, Standard Biotools Inc.). All experiments were run along with four NTCs (no template controls) to monitor for potential contamination.
Testing PCR performance and preamplification compatibility resulted in 139 SNPs suitable for samples with low DNA concentrations (samples see Online Resource. Tab. S1). We selected the 96 most diverse SNPs among both lineages (Tab. S2). Genotyping this combination resulted in accurate genotyping across all markers. The PCoA (Fig. 1) resulted in two main clusters matching the expected split between the Eastern and Western lineage (Mouton et al. 2017), similar to the results in Leyhausen et al. (2022) . While the western samples clustered closely, the eastern samples formed three small clusters: one cluster with all Lithuanian samples, one with samples from Germany (west of the Elbe river) and an intermediate cluster containing few samples from Germany east of the Elbe river.

PCoA results from 51 hazel dormouse individuals. Orange coloured symbols represent populations from the Eastern lineage (LT = Lithuania, SL = Slovenia, SW = Sweden, German regions: BW = Baden-Wuerttemberg, HE = Hesse, MV = Mecklenburg-Western Pomerania, SH = Schleswig-Holstein, SN = Saxony, ST = Saxony-Anhalt, FR = Franconia) and blue coloured symbols represent populations from the Western lineage (BE = Belgium, NL = the Netherlands, IT = Italy, German States: NW = North Rhine-Westphalia)
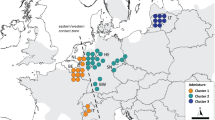
To test the applicability of the assay, we genotyped hair and scat samples of 94 individuals (pre-analysed with microsatellites) collected from nest tubes in an ongoing monitoring project in Central Franconia (Bavaria, Germany) to assess the genetic population structure in fragmented hazel dormouse populations. SNP genotyping resulted in high amplification success across hair and scat samples (99.5%, not shown), with one sample failing amplification. For K = 2, all samples clustered to the Eastern lineage (Mouton et al. 2017) using Bayesian population assignment implemented in STRUCTURE 2.3.4. (Pritchard et al., 2000) with a burn-in of 100,000 and 200,000 MCMC replicates with default settings (Fig. 2a). Higher K’s resulted in assignment to reference samples from Central Germany (Fig. 2b).

Structure results for 33 hazel dormouse individuals. (a) The most likely K (K = 2) shows a split between the Western and Eastern lineage. (b) K = 5 shows a split within both lineages. For abbreviations see Fig. 1. (DE = Germany)
The division of the hazel dormouse distribution in Europe into a highly divergent Western and Eastern cluster, potentially forming cryptic species, has been proposed on the basis of mitochondrial DNA analysis (Mouton et al. 2017) and genome-wide SNP data (Leyhausen et al. 2022). Regardless of the taxonomic status, those lineages represent evolutionary significant units and should thus be treated as separate conservation units following Moritz (1994). The new SNP panel will provide useful information for future genetic wildlife monitoring and conservation studies for lineage determination and population assignment in the hazel dormouse.
References
Council of Europe (1992) Council Directive 92/43/EEC of 21 May 1992 on the conservation of natural habitats and of wild fauna and flora. European Union
Hohenlohe PA, Funk WC, Rajora OP (2021) Population genomics for wildlife conservation and management. Mol Ecol 30:62–82. https://doi.org/10.1111/mec.15720
Kraus RHS, von Holdt B, Cocchiararo B, Harms V, Bayerl H, Kühn R, Förster DW, Fickel J, Roos C, Nowak C (2015) A single-nucleotide polymorphism-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol Ecol Resour 15:295–305. https://doi.org/10.1111/1755-0998.12307
Leyhausen J, Cocchiararo B, Nowak C, Ansorge H, Bertolino S, Büchner S, Fietz J, Foppen R, Juškaitis R, La Haye M, Lang J, Michaux J, Verbeylen G, von Thaden A, Mueller SA (2022) Genotyping-by-sequencing based SNP discovery in a non-model rodent, the endangered hazel dormouse. Conserv Genet Resour 14:195–201. https://doi.org/10.1007/s12686-022-01253-8
Moritz C (1994) Defining ‘Evolutionarily significant units’ for conservation. Trends Ecol Evol 9:373–375. https://doi.org/10.1016/0169-5347(94)90057-4
Mortelliti A, Amori G, Capizzi D, Rodiini C, Boitani L (2010) Experimental design and taxonomic scope of fragmentation studies on European mammals: current status and future priorities. Mam Rev 40:125–154. https://doi.org/10.1111/j.1365-2907.2009.00157.x
Mouton A, Mortelliti A, Grill A, Sara M, Kryštufek B, Juškaitis R, Latinne A, Amori G, Randi E, Büchner S, Schulz B, Ehlers S, Lang J, Adamik P, Verbeylen G, Dorenbosch M, Trout R, Elmeros M, Aloise G, Mazzoti S, Matur F, Poitevin F, Michaux JR (2017) Evolutionary history and species delimitations: a case study of the hazel dormouse, Muscardinus avellanarius. Conserv Genet 18:181–196. https://doi.org/10.1007/s10592-016-0892-8
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
von Thaden A, Cocchiararo B, Jarausch A, Jüngling H, Karamanlidis AA, Tiesmeyer A, Nowak C, Muñoz-Fuentes V (2017) Assessing SNP genotyping of noninvasively collected wildlife samples using microfluidic arrays. Sci Rep 7:10768. https://doi.org/10.1038/s41598-017-10647-w
von Thaden A, Nowak C, Tiesmeyer A, Reiners TE, Alves PC, Lyons LA, Mattucci F, Randi E, Cragnolini M, Galián J, Hegyeli Z, Kitchener AC, Lambinet C, Lucas JM, Mölich T, Ramos L, Schockert V, Cocchiararo B (2020) Applying genomic data in wildlife monitoring: Development guidelines for genotyping degraded samples with reduced single nucleotide polymorphism panels. Mol Ecol Resour 20:662–680. https://doi.org/10.1111/1755-0998.13136
Wang J, Lin M, Crenshaw A, Hutchinson A, Hicks B, Yeager M, Berndt S, Huang W-Y, Hayes RB, Chanock SJ, Jones RC, Ramakrishnan R (2009) High-throughput single nucleotide polymorphism genotyping using nanofluidic dynamic arrays. BMC Genomics 10:561. https://doi.org/10.1186/1471-2164-10-561
Acknowledgements
We thank Senckenberg Museum Görlitz, Ruud Foppen, Rimvydas Juškaitis, Boris Krystufek, Maurice La Haye, Jeroen van der Kooij, for providing samples from different European countries and Doris Jensch, Karsten Horn for providing samples from Franconia (Germany).
Funding
This study was partially funded by the Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz of the German Federal State of Hesse (LOEWE).
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Tobias Beez and Berardino Cocchiararo. The first draft of the manuscript was written by Carsten Nowak and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beez, T., Leyhausen, J., Mueller, S. et al. Development of a microfluidic SNP assay for lineage discrimination in the endangered hazel dormouse. Conservation Genet Resour (2024). https://doi.org/10.1007/s12686-024-01367-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12686-024-01367-1