
Abstract
Introduction
Three studies compared the bioequivalence (BE) of new generic tablet formulations of sitagliptin (100 mg; fasting) and the fixed-dose combination (FDC) of sitagliptin/metformin (50/850 mg, 50/1000 mg; both fed) in healthy volunteers with the same tablet strengths of the reference products Januvia and Janumet.
Methods
The study design was open-label, single-dose, randomized with two-way crossover periods. Blood sampling was performed for 72/48 h in the sitagliptin/FDC studies, respectively. Primary pharmacokinetic (PK) parameters for sitagliptin and metformin were area under the plasma concentration–time curve from time 0 to last timepoint of measurable concentration (AUC0–t) and maximum plasma concentration (Cmax). Test (T) and reference (R) formulations proved bioequivalent if 90% confidence interval (CI) of geometric least-squares mean ratio for AUC0–t and Cmax were within BE acceptance range of 80.00–125.00%. Safety evaluations included vital signs, clinical laboratory tests, and adverse events (AEs).
Results
Treated/evaluable volunteers for BE per study were: 30/28 (sitagliptin 100 mg), 26/25 (FDC 50/850 mg), and 26/24 (FDC 50/1000 mg). The 90% CI of the geometric means of T/R ratios for primary PK parameters were within predefined BE limits: CI for AUC0–t and Cmax were 95.83–100.37% and 91.85–109.56% (sitagliptin 100 mg); 100.84–103.69% and 93.44–105.10% (FDC 50/850 mg), and 101.26–105.20% and 98.71–112.89% (FDC 50/1000 mg); respective values for metformin were 94.23–101.89% and 91.66–99.38% (FDC 50/850 mg) and 98.45–104.89% and 96.79–105.62% (FDC 50/1000 mg). All AEs were nonserious, transient, and mostly mild. Safety evaluations did not reveal any relevant difference between T and R formulations.
Conclusions
The new generic tablet formulations of sitagliptin 100 mg and the FDCs sitagliptin/metformin 50/850 mg and 50/1000 mg demonstrated bioequivalence to originator reference products. Therefore, the new products are expected to provide efficacy and tolerability similar to those of the reference products in the treatment of patients with type 2 diabetes (T2D).
Trial registration
EudraCT EU Clinical Trials Registry (2014-005437-31); ClinicalTrials.gov Registry (NCT05549570 and NCT05549583, both retrospectively registered on 20 September 2022).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out these studies? |
Metformin and the DPP-4 inhibitor sitagliptin are well established for the management of type 2 diabetes, and their combination is widely used to improve glycemic control. The generic sitagliptin 100 mg tablet and sitagliptin/metformin fixed-dose combination tablets can be effective alternative treatment options for glycemic control in patients with type 2 diabetes. |
Three studies in healthy volunteers assessed the bioequivalence of new generic formulations of sitagliptin and sitagliptin/metformin fixed-dose combination to the approved reference products Januvia and Janumet. |
What was learned from the studies? |
The studies demonstrated bioequivalence of the new generic formulations, sitagliptin 100 mg and the fixed-dose combination of sitagliptin and metformin at 50/850 mg and 50/1000 mg strengths, to the reference products Januvia and Janumet in healthy volunteers. |
Therefore, the new generic products are expected to provide similar clinical benefits for patients with type 2 diabetes to the approved reference products. |
Introduction
The oral antidiabetic agent metformin is a member of the biguanide drug class that was originally described in 1922 and introduced into the pharmacologic management of type 2 diabetes (T2D) in 1958 [1, 2]. Since then, initial pharmacologic therapy of T2D has generally included metformin as a cornerstone, either alone or in combination with one or more antihyperglycemic agents [3, 4]. The aim of such treatments is to provide patients with T2D with effective therapy, tailored to their individual needs. Sitagliptin is a dipeptidyl peptidase-4 (DPP-4) inhibitor and was the first in its class approved in October 2006 by the Food and Drug Administration (FDA) [5, 6]. Dipeptidyl peptidase-4 (DPP-4) inhibitors, like sitagliptin, provide glycemic control with a low inherent risk of hypoglycemia and are classified as cardiovascular and body-weight neutral. Sodium–glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide 1 receptor agonists (GLP-1 RA) have expanded the treatment options for T2D, offering cardiovascular and/or renal risk reduction along with glycemic control. The emerging clinical evidence in the past two decades led to continuous updates of the major guidelines for the management of T2D; these included recommendations to refocus the use of DPP-4 inhibitors towards patients without atherosclerotic cardiovascular disease, high cardiovascular risk, heart failure, or chronic kidney disease but in need for glycemic control with minimal hypoglycemic risk [3, 7].
Sitagliptin is a highly selective inhibitor of the DPP-4 enzyme [8]. DPP-4 limits the activity of the incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), that are involved in glucose homeostasis and are released from enteroendocrine cells upon meal intake. DPP-4 inhibition with sitagliptin slows the inactivation of GLP-1 and GIP, thereby elevating their physiological levels and prolonging their action, i.e., glucose-dependent increase of insulin secretion from pancreatic β-cells and lowered glucagon release from pancreatic β-cells leading to reduced hepatic glucose production. Both effects contribute to the reduction of HbA1c plasma levels, and fasting and postprandial glucose, in patients with T2D who have hyperglycemia [5, 8,9,10].
Metformin’s primary mechanism of action as an insulin sensitizer is to improve glycemic control by increasing glucose utilization within the body and decreasing glucose uptake from food, predominantly via adenosine monophosphate-activated protein kinase (AMPK) activation [11, 12].
Combination therapy with drugs that have complementary mechanism of action is key to the management of T2D, to ensure appropriate glycemic control and prevent or delay T2D-related comorbidities.
The different mechanisms of action of metformin and sitagliptin are complementary and lead to improved glycemic control, with a low risk of hypoglycemia, and acceptable tolerability without overlapping side effects [13, 14]. Pharmacokinetics (PK) of sitagliptin and metformin are not altered by coadministration [15]. The combination of sitagliptin and metformin has proven to be efficacious and tolerable either as a free combination of the products or as a fixed-dose combination (FDC) [13, 16, 17].
We conducted PK phase I studies in healthy volunteers with two new generic formulations, i.e., of sitagliptin and the FDC of sitagliptin with metformin. The primary objective was to evaluate bioavailability and to demonstrate the bioequivalence of the generic formulations after a single oral dose compared with the same dose of the approved originator reference products. Secondary objectives were the evaluation of safety and tolerability.
Methods
Study Drugs and Study Design
The generic test products sitagliptin 100 mg film-coated tablet and the FDC of sitagliptin/metformin 50/850 mg and 50/1000 mg film-coated tablets were developed by Galenicum Health, Spain. The generic test products comprised sitagliptin hydrochloride monohydrate either alone or in combination with metformin hydrochloride. Three bioequivalence studies were conducted in healthy male and female volunteers respectively for three dosage forms. All three studies were performed under the responsibility of the sponsor (Galenicum Health) and conducted by a qualified clinical research unit/organization (CRO): one located in Madrid, Spain (sitagliptin study) and another CRO in Quebec, Canada (FDC studies).
All study procedures were performed in accordance with the 1964 Helsinki Declaration and its later amendments and National Guidelines for Biomedical Research on Human Subjects, Good Clinical Practices for Clinical Research in India, International Council for Harmonisation (ICH, step 5) Guidance on Good Clinical Practice, and related EU guidelines. Informed consent was obtained from all individual participants included in the studies. Informed consent included the intention to present or publish the study results, without disclosing the participants’ identity. Ethics approval was obtained from the corresponding committees of the CROs for all the sitagliptin and FDC studies. In addition, applicable local regulations were followed, e.g., the Spanish regulation, the FDA GCP Code of Federal Regulations Title 21 (part 56), European regulation EU 536/2014, and the Tri-Council Policy Statement (Canada).
The sitagliptin study evaluated the bioequivalence of the generic test product “Sitagliptin 100 mg film coated tablet” (containing sitagliptin hydrochloride) with the commercially available originator reference product Januvia 100 mg tablet, distributed by Merck Sharp & Dohme Ltd., Spain (containing sitagliptin phosphate monohydrate). The two FDC studies evaluated the bioequivalence of the active ingredients sitagliptin hydrochloride and metformin hydrochloride from the sitagliptin/metformin FDC at two strengths, 50/850 mg and 50/1000 mg, with the respective tablet strength of the commercially available originator reference product Janumet (containing sitagliptin phosphate monohydrate and metformin hydrochloride), distributed by Merck Sharp & Dohme Ltd., UK.
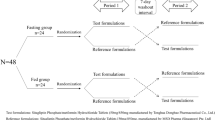
The study design was the same for all three studies, i.e., randomized, single-center, open-label, single-dose, two-treatment and two-period crossover design with a washout period of 7 days between drug administrations to ensure sufficient time for drug elimination, to minimize carry-over effects. However, the drug intake conditions were fasting conditions for the sitagliptin study and fed conditions for the FDC studies, accounting for the labeled administration of metformin.
Participants were randomized to one of the two treatment sequences, reference–test product or test–reference product in the two study periods, by using randomization code lists. The treatments were allocated in a balanced manner on each inclusion day in five blocks of six participants for the sitagliptin study and in a single block in the FDC studies. In line with ICH guidance [18], the BE studies followed the standard design, which does not require double blinding. The open-label design is appropriate considering that the primary endpoints are pharmacokinetic parameters and that the study drugs are tested in a crossover design. However, in our BE studies, the personnel of the bioanalytical facilities was kept blinded until analysis completion.
Study Drug Administration
Sitagliptin study: Participants were required to take a single oral dose of either the test or reference product with 240 mL of water after overnight fasting for at least 10 h before dosing and until at least 4 h after dosing. No water was allowed within 1 h before and until 5 h after dosing. The following meals were provided after drug intake: a standard meal after 5 h, a snack after 9 h, and an evening meal 12 h after drug intake.
FDC sitagliptin/metformin studies: After a supervised overnight fast (at least 10 h) participants received a standardized high-fat, high-calorie breakfast 30 min before a single oral dose of the assigned formulation was administered with about 240 mL of water at room temperature. Water was provided ad libitum until 1 h pre-dose and was allowed ad libitum starting 1 h after drug intake. A standardized lunch was served at least 4 h after dosing; a supper and a light snack were served at appropriate times thereafter, but not before 9 h after dosing.
Participants were to remain seated following drug administration (5 h in the sitagliptin study and 4 h in the FDC sitagliptin/metformin studies). Participants in each of the three studies were allowed to leave the clinical site after the 24-h blood draw and were asked to return before each of the two remaining blood samples were taken (48 and 72 h after sitagliptin and 36 and 48 h after sitagliptin/metformin drug intake).
Participant Selection
Healthy male and female volunteers were enrolled on the basis of inclusion and exclusion criteria of the approved protocol for each study. The FDC studies shared the same eligibility criteria, which were similar to those in the sitagliptin study. All three studies included healthy female and male volunteers who provided written informed consent for participation. Participants were: 18–55 years and ≤ 65 years of age in the sitagliptin study and the FDC studies, respectively; and non- or ex-smokers with a body mass index (BMI) between 18.5 and 30 kg/m2. Participants had: no history of clinically significant disease, nor evidence of clinically significant findings on physical examination and/or clinical laboratory evaluations [hematology, general biochemistry, electrocardiogram (ECG) and urinalysis in the normal range]; negative serology results (hepatitis B surface antigen, hepatitis C antibodies, human immunodeficiency virus antibodies), negative pregnancy results; (women); and no evidence of drug and/or alcohol abuse.
Blood Sampling and Handling
Blood sampling: Blood samples were drawn before dosing and up to 72 h and 48 h after each dosing period in the sitagliptin study and FDC studies, respectively. The total volume of blood collected per participant was considered to have a negligible or no impact on the PK profiles of the drugs, on the assessment of bioequivalence (BE) and negligible effect on participants’ safety. In the sitagliptin study blood sampling was performed in each study period within 1 h before first dosing and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 7, 8, 10, 12, 16, 20, 24, 48, and 72 h after drug dosing. In the FDC studies, blood samples were drawn in each study period prior to first drug administration and at 0.5, 1, 1.5, 2, 2.33*, 2.5** 2.67*, 3, 3.5, 4, 5, 5.5**, 6, 8, 10, 12, 16, 24, 36, and 48 h after drug administration (*collected for sitagliptin measurement only; ** collected for metformin measurement only).
Sample handling: Blood samples were collected in di- and tri-potassium ethylenediaminetetraacetic acid (K2EDTA and K3EDTA) tubes in the sitagliptin and metformin samples, respectively. Samples were centrifuged at 4 °C and at ~ 1900 g for 10 min. The plasma obtained was separated into duplicate polypropylene culture tubes. The labeled tubes (not revealing formulation identity) were retained in the site’s freezers at –20 °C until transit on dry ice to the bioanalytical facility for assay.
Plasma concentrations of sitagliptin and metformin were estimated individually by high-performance liquid chromatography with detection by sequential mass spectrometry (HPLC–MS/MS). The analytes were extracted from human EDTA plasma by protein precipitation (sitagliptin) and solid phase extraction (metformin) techniques. These estimations were performed by respective validated bioanalytical methods with a linear range of 1–500 ng/mL for sitagliptin and 2–2000 ng/mL for metformin and lower limit of quantification of 1 ng/mL for sitagliptin and 2 ng/mL for metformin.
PK and Statistical Analyses, and Sample Size Considerations
The primary PK parameters for assessment of bioequivalence in each study were area under the concentration curve from time 0 to the last timepoint of measurable concentration (AUC0–t) and maximal plasma concentration (Cmax).
Sample sizes were determined on the basis of literature estimates, to provide at least 80% power for the conclusion of bioequivalence. The test-to-reference ratios were assumed to be ≥ 90% and ≤ 110%. For the sitagliptin study, an intra-subject variation of 17.5% for Cmax and AUC0–t was used for sample size calculation, whereas for the FDC studies an intra-subject variation of 19% was applied for Cmax and AUC0–t of metformin and sitagliptin. As a result, 30 and 26 participants were required in the sitagliptin and in the two FDC studies, respectively, to conclude bioequivalence with approximately 80% power. The test and reference formulations were considered bioequivalent if the geometric least-squares mean ratio for Cmax and AUC0–t and its 90% confidence interval were within the bioequivalence acceptance range of 80.00–125.00%, in line with European guidelines [18].
Secondary PK parameters for each study were AUC extrapolated to infinite time (AUC0–inf), to reach maximal plasma concentration (Tmax), terminal elimination half-life (t1/2), and extrapolated/residual AUC. Additional secondary parameters were mean residence time (MRT), total plasma clearance (Cl), and distribution volume (Vd) in the sitagliptin study and apparent elimination rate constant (λZ) in both FDC studies. All PK parameters were calculated on the basis of the respective drug concentration in plasma. PK analyses and statistical analyses were generated using validated PK software, i.e., Phoenix WinNonlin version 6.3 (sitagliptin study) and Phoenix WinNonlin version 6.3, Phoenix ConnectTM version 1.3.1 and SAS version 9.4 (general linear models procedure) (FDC sitagliptin/metformin studies).
In all studies, logarithmically transformed PK parameters were statistically analyzed using an analysis of variance model. The fixed factors included in this model were the subject effect (nested within sequence), the treatment received, the period at which it was given, and the sequence in which each treatment was received.
The PK population included samples from all participants who received at least one of the study products. Participants who experienced emesis within 5 h (FDC study 50/850 mg) or 5.5 h (FDC study 50/1000 mg) after drug administration were withdrawn from the study, while for participants with emesis within 5–6 h or 5.5–7 h, respectively, only blood sampling for sitagliptin was collected and analyzed for this study period. No participants were removed for this reason.
Safety Evaluation
Safety assessments comprised physical examinations, vital signs, ECG, glycemia, clinical laboratory tests (hematology, clinical biochemistry, serology, urine analysis, pregnancy test for women), and adverse event (AE) monitoring by means of open questions. AE monitoring was performed at each visit; vital signs were reported following 5 min of rest in a supine position; ECG assessments were performed before dosing and 2 h post-dosing. Laboratory determinations were performed at screening and at the end of the follow-up period. Follow-up visits were conducted 5–10 days after the second dose in the sitagliptin study and 10 days after the second dose in both FDC sitagliptin/metformin studies.
Descriptive statistics were used to summarize treatment-emergent AEs (TEAEs), safety results, and demographic variables (age, height, weight, and body mass index).
Results
Demographic Characteristics and Discontinuations
Sitagliptin Study
Thirty healthy volunteers, 15 males and 15 females, were randomized as planned with a mean age of 28 years and a mean BMI of 23.5 kg/m2 (Table 1). Twenty-four participants were Caucasian and six were Latin-American. Twenty-eight participants completed both study periods, while two participants quit the study for personal reasons after period 1. Therefore, 28 participants were evaluable for PK statistical assessment.
FDC Sitagliptin/Metformin Studies
FDC sitagliptin/metformin 50/850 mg tablet study: Twenty-six healthy participants (12 males, 14 females) with a mean age of 42 years and a mean BMI of 25.6 kg/m2 (Table 1) were randomized, of whom 22 participants were Caucasian, 9 were Hispanic or Latin-American, and 3 were African-American. Twenty-five participants completed both study periods and were included in the PK statistical assessment. One female participant discontinued before study end in period 1 due to an adverse event (increased body temperature).
FDC sitagliptin/metformin 50/1000 mg tablet study: Twenty-six healthy participants (7 males and 19 females) were enrolled as planned with a mean age of 44 years and a mean BMI of 24.8 kg/m2 (Table 2). Twenty-four participants were Caucasian, six were Hispanic or Latin-American, and two were Asian. Twenty-four participants completed both study periods and were included in the PK statistical assessment. One participant withdrew consent (because she was not feeling well), and one was withdrawn due to the investigator decision (due to pain in the left knee); none of these events was considered to be relevant to the treatment.
Bioequivalence Assessment and PK
Sitagliptin Study
Bioequivalence between the test product versus the approved originator reference product was demonstrated for AUC0–t and Cmax because the 90% confidence intervals for the corresponding mean ratios (test over reference) were within the predefined bioequivalence acceptance range of 80.00–125.00% (Table 2). The mean values (SD) for AUC0–t and Cmax and other secondary PK parameters of the test product, sitagliptin 100 mg film-coated tablets, and the reference product were similar; the extrapolated portion of the AUC was on average 1.19% and ranged from 0.44% to 2.17% (Table 3). Mean sitagliptin plasma concentrations over time were similar for the test and reference product (Fig. 1).

Sitagliptin mean plasma concentration over 72 h following a single dose of a 100 mg tablet of the generic test product (black triangle) and the originator reference product (blue circle) under fasted conditions (N = 29). Test product: Sitagliptin 100 mg film-coated tablet; Reference product: Januvia 100 mg
FDC Sitagliptin/Metformin Studies
Statistical evaluation: the geometric mean ratios of the test and reference product for the primary PK parameters AUC0–t and Cmax for sitagliptin were close to 100% and for metformin slightly below (98% and 95%, respectively), and 90% confidence intervals were within the required range of 80–125% for bioequivalence (Table 4). The geometric mean ratios of the primary PK parameters of the 50/1000 mg tablets were around or slightly above 100% (105% for Cmax of sitagliptin), and the 90% confidence intervals were within the required acceptance range of 80–125% (Table 5).
The mean plasma concentration–time curves for the sitagliptin and metformin component from the generic test products at 50/850 mg and 50/1000 mg tablet strength were similar to the respective strengths of the originator reference FDC products (Figs. 2 and 3).

Mean plasma concentration over 48 h for sitagliptin (a) and metformin (b) following a single dose of a 50/850 mg tablet of the generic test product (black triangle) and the originator reference product (blue circle) under fed conditions (n = 25 for each drug). Test product: sitagliptin/metformin 50/850 mg film-coated tablet; reference product: Janumet 50/850 mg

Mean plasma concentration over 48 h for sitagliptin (a) and metformin (b) following a single dose of a 50/1000 mg tablet of the generic test product (black triangle) and the originator reference product (blue circle) under fed conditions (n = 24 for each drug). Test product: sitagliptin/metformin 50/1000 mg film-coated tablet; reference product: Janumet 50/1000 mg
The average values of secondary PK parameters were comparable between the formulations for sitagliptin and metformin in the respective FDC studies; the mean residual area values for the test and reference formulations were 3.50% and 0.99% (50/850 mg; Table 6) and 3.44% and 1.73% (50/1000 mg; Table 7), respectively.
Safety and Tolerability
Sitagliptin Study
Thirty participants who received at least one study drug were included in the safety analyses. Two participants withdrew consent and discontinued after a single 100 mg dose in the first study period. No serious AEs were reported. Three participants experienced a nonserious TEAE that resolved during the study: headache in two participants (mild n = 1; moderate n = 1) with the reference product and dysmenorrhea (mild) in another participant treated with the test product. The two AEs, mild headache and mild dysmenorrhea, were considered possibly related to the study drug.
Clinically significant alterations in laboratory parameters did not occur. In female participants, a small hemoglobin decrease of 0.30 g/dL was observed, which is to be expected due to the blood sampling, although it did not occur in male participants. No clinically relevant alterations in the physical examination, vital signs (blood pressure, heart rate), or ECG occurred.
FDC Sitagliptin/Metformin Studies
The safety population in the FDC sitagliptin/metformin 50/850 mg study comprised 26 participants, who received at least one study drug. No serious AEs were reported. Eight participants (31%) reported at least one TEAE: four participants (15%) reported ten TEAEs with the test product, and five participants (20%) reported eight TEAEs with the reference product. All events were mild except one of moderate intensity with the test product. The most common TEAE experienced in more than one participant (N = 2) was nausea, reported with the test product only. Drug-related TEAEs were nausea and headache (reported for the test product) and abdominal discomfort, abdominal pain, and somnolence (reported for the reference product).
The safety population in the FDC sitagliptin/metformin 50/1000 mg study consisted of 26 participants, who received at least one study drug. No serious AE was reported. Eight participants (31%) reported at least one TEAE: three participants (13%) reported 11 TEAEs with the test product, and six participants (23%) reported 16 TEAEs with the reference product. All events were mild, except one of moderate intensity with the test product and four moderate events with the reference product. The most common TEAEs reported in more than one participant (N = 2) were dyspepsia with the test product, and diarrhea and somnolence with the reference product. Dyspepsia, nausea, and dizziness were reported, related to the test and reference product. Vomiting, fatigue, and skin reactions were reported related to the test product only, while diarrhea and somnolence were reported related to the reference product only.
Discussion
New generic formulations of sitagliptin tablet (100 mg) and the fixed-dose combination tablet of sitagliptin and metformin (50/850 mg and 50/1000 mg) were evaluated in three phase I studies and demonstrated to be bioequivalent to the same tablet strengths of the respective reference products Januvia (Merck Sharp & Dohme Ltd., Spain) and Janumet (Merck Sharp & Dohme Ltd., UK). This proven bioequivalence allows the transfer (extrapolation) of clinical efficacy and safety data from the originator products to the new generic formulations. The bioequivalence assessment was based on the 90% confidence intervals of the ratios for sitagliptin and metformin, which met the acceptance range of 80–125% for the primary PK parameter AUC0–t and Cmax, in line with the EU guidance [18].
The phase I BE studies were designed in accordance with the ICH guideline, i.e., two-period crossover design, sufficient blood sampling timepoints, and an adequate sample size powered for bioequivalence assessment of the primary PK parameters AUC0–t and Cmax. In each study, the sample size for the BE assessment was robust as only one or two participants had to be excluded due to noncompletion of a study period. Considering the terminal half-life of sitagliptin (around 12 h) and of metformin (about 6.5 h), the washout period of 7 days applied in each study was considered appropriate to avoid carry-over effects across periods [19, 20]. Sitagliptin’s bioavailability increases in a dose-proportional manner, and PK is not influenced by food [5, 20, 21]. Therefore, and in line with the European Medicines Agency guidance on evaluation of BE, the highest recommended strength of 100 mg sitagliptin was tested under the most sensitive (i.e., fasting) condition. Due to the metformin component, the FDC tablets of sitagliptin/metformin had to be administered under standardized fed conditions.
The selection criteria applied in the presented studies were reasonable as sitagliptin’s and metformin’s PKs are not relevantly influenced by age, gender, or BMI [5, 20]. The bioequivalence of the sitagliptin test formulation with the reference product Januvia, for the key PK parameter AUC0–t and Cmax, is further supported by similarity of both formulations regarding Tmax and terminal half-life: median Tmax was 2.50 h, and mean terminal half-life was close to 12 h for the test and reference product; both results were consistent with published data for the reference drug [5]. Furthermore, the mean extrapolated AUC in the sitagliptin study and the mean residual area in the FDC studies for the test and reference products were clearly below the 20% limit defined by regulatory authorities with maximum values of 7.49% for sitagliptin and 8.36% for metformin analyte, which underlines the appropriateness of the plasma sampling schedule for a reliable estimate of the extent of exposure [18].
The new generic test products, sitagliptin and the FDC sitagliptin/metformin, were well tolerated, and serious AEs did not occur. The few AEs reported for the generic test and originator reference products were of mild intensity and included nausea, dyspepsia, abdominal pain/discomfort, headache, and somnolence. The frequency of AEs was similar for both sitagliptin formulations and slightly lower for the test product compared with the reference product in the combination studies. The AE profile, as well as laboratory parameters, vital signs, and ECG, did not reveal any relevant difference between the test and reference formulation in any of the studies. This is in line with the known safety profile of both substances.
Bioequivalence demonstration between medicinal products is key for extrapolation of the established clinical efficacy and safety from the originator reference product to the new generic product. Sitagliptin is approved for the improvement of glycemic control in patients with T2D as monotherapy in patients inadequately controlled by diet and exercise alone, and in whom metformin is inappropriate due to contraindications or intolerance and as combination therapy with other oral antidiabetics and/or insulin on top of diet and exercise, i.e., as dual therapy add-on to metformin, a sulfonylurea, or a peroxisome-proliferator-activated receptor gamma (PPARγ) agonist or as triple therapy add-on to metformin + sulfonylurea or PPARγ or insulin [5, 22]. Patients with T2D who are already treated with the individual products can be switched to the FDC of sitagliptin/metformin [22]. In the past decade the use of combination tablets for the treatment of T2D has increased [23]. Using FDC products can reduce the complexity of drug intake by decreasing the number of tablets to be taken, which can improve treatment adherence [24, 25] and ultimately treatment outcomes; as such, reducing the number of tablets to be taken may be beneficial for patients on polypharmacy. FDCs for the management of T2D have also been reported to increase patients’ treatment satisfaction [26].
A general limitation of BE studies is their conduct in healthy volunteers under strict control, and therefore the findings cannot always be extrapolated one-to-one to patients with concomitant diseases and comedications. In addition, a comparison of the safety profile is limited due to the small sample size and single dose administration.
Conclusions
The new generic formulation sitagliptin 100 mg film-coated tablet and the FDC tablets of sitagliptin with metformin with 50/850 mg and 50/1000 mg strengths proved to be bioequivalent to the respective originator reference products at the same tablet strengths. The safety and tolerability results were overall similar for the test and reference products and revealed no new findings. On the basis of the comparative bioavailability and PK, the new generic products are expected to provide therapeutic effects and tolerability similar to those of the reference products and are therefore interchangeable in the approved indications.
References
Bailey CJ. Metformin: historical overview. Diabetologia. 2017;60:1566–76. https://doi.org/10.1007/s00125-017-4318-z.
Pasik C. Diabetes and the biguanides: the mystery of each. In: Pasik C, editors. Glucophage: serving diabetology for 40 years. Lyon: Groupe Lipha; 1997. p. 79
ADA–American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes - 2022 Diabetes Care. 2022;45(Suppl 1):S125–3. https://doi.org/10.2337/dc06-0703.
Davies MJ, Aroda VR, Collins BS, et al. Management of hyperglycemia in type 2 diabetes, 2022. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2022. https://doi.org/10.1007/s00125-022-05787-2.
Januvia® European Public Assessment Report (EPAR) – Product Information, Annex I Summary of Product Characteristics updated 05Oct2021. https://www.ema.europa.eu/en/documents/product-information/januvia-epar-product-information_en.pdf.
FDA. Januvia Drug Approval Package. www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021995s000TOC.cfm. Accessed 15 Aug 2022.
NICE – National Institute for Health and Care Excellence. Summary of advice in the NICE guideline on type 2 diabetes in adults: management. https://www.nice.org.uk/guidance/ng28/resources/visual-summary-full-version-choosing-medicines-for-firstline-and-further-treatment-pdf-10956472093 based on NICE guideline [NG298] last updated 29 June 2022. https://www.nice.org.uk/guidance/ng28.
Lyseng-Williamson KA. Sitagliptin. Drugs. 2007;67(4):587–97. https://doi.org/10.2165/00019053-200826080-00006.
Aschner P, Kipnes MS, Lunceford JK, et al. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006;29:2632–7. https://doi.org/10.2337/dc06-0703.
Vardarli I, Arndt E, Deacon CF, Host JJ, Nauck MA. Effects of sitagliptin and metformin treatment on incretin hormone and insulin secretory responses to oral and “isoglycemic” intravenous glucose. Diabetes. 2014;63:663–74. https://doi.org/10.2337/db13-0805.
Bailey CJ, Campbell IW, Chan JCN, Davidson JA, Howlett HCS, Ritz, P, editors. Metformin: the gold standard. A scientific handbook. Wiley: Chichester (UK); 2007.
Campbell IW, Howlett HCS, Holman RR, Bailey CJ, editors. Metformin: 60 years of clinical experience: addendum to the scientific handbook. Weinheim: WILEY-VCH Verlag GmbH & Co. KGaA; 2017.
Charbonnel B, Karasik A, Liu J, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care. 2006;29:2638–43. https://doi.org/10.2337/dc06-0706.
Onge ELS, Miller S, Clements E. Sitagliptin/metformin (Janumet) as combination therapy in the treatment of type-2 diabetes mellitus. Pharmacy Therapeutics. 2012;37(12):699–708.
Herman GA, Bergman A, Yi B, Kipnes for the Sitagliptin Study 012 Group. Tolerability and pharmacokinetics of metformin and the dipeptidyl peptidase-4 inhibitor sitagliptin when co-administered in patients with type 2 diabetes. Curr Med Res Opin. 2006;22(10):1939–47. https://doi.org/10.1185/030079906X132587.
Ballav C, Gough SCL. Safety and efficacy of sitagliptin–metformin in fixed combination for the treatment of type 2 diabetes mellitus. Clin Med Insights: Endocrinol Diabetes. 2013;6:25–37. https://doi.org/10.4137/CMED.S7314.
Chwieduk CM. Sitagliptin/metformin fixed-dose combination. In patients with type 2 diabetes mellitus. Drugs. 2011;71(3):349–61.
EMA - European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on the investigation of bioequivalence. London, 20Jan2010; Doc. Ref.: CPMP/EWP/QWP/1401/98 Rev. 1/ Corr https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
Bergman A, Mistry GC, Lupo W-L, et al. Dose-proportionality of a final market image sitagliptin formulation, an oral dipeptidyl peptidase-4 inhibitor, in healthy volunteers. Biopharm Drug Dispos. 2007;28:307–13. https://doi.org/10.1002/bdd.559.
Glucophage 850 mg film coated tablets. SmPC. Merck Serono Ltd. UK, March 2022. https://www.medicines.org.uk/emc/product/7759/smpc. March 2015. Accessed 22 Aug 2022.
Bergman AJ, Stevens C, Zhou YY, et al. Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin Therap. 2006;28(1):55–72. https://doi.org/10.1016/j.clinthera.2006.01.015.
Janumet® European Public Assessment Report (EPAR) – Product Information, Annex I Summary of Product Characteristics updated 12Oct2021. https://www.ema.europa.eu/en/documents/product-information/janumet-epar-product-information_en.pdf
Cieslik LK, Cresswell NR, Fineberg D, Mariani JA, Patel HC. Prescription trends and costs of diabetes medications in Australia between 2003 and 2019: an analysis and review of the literature. Int Med J. 2022;52:841–7. https://doi.org/10.1111/imj.15137.
Han S, Iglay K, Davies MJ, Zhang Q, Radican L. Glycemic effectiveness and medication adherence with fixed-dose combination or coadministered dual therapy of antihyperglycemic regimens: a meta-analysis. Curr Med Res Opin. 2012;28(6):969–77. https://doi.org/10.1185/03007995.2012.684045.
Hutchins V, Zhang B, Fleurence RL, Krishnarajah G, Graham J. A systematic review of adherence, treatment satisfaction and costs, in fixed-dose combination regimens in type 2 diabetes. Curr Med Res Opin. 2011;27(6):1157–68. https://doi.org/10.1185/03007995.2011.570745.
Benford M, Milligan G, Pike J, Anderson P, Piercy J, Fermer S. Fixed-dose combination antidiabetic therapy: real-world factors associated with prescribing choices and relationship with patient satisfaction and compliance. Adv Ther. 2012;29(1):26–40. https://doi.org/10.1007/s12325-011-0096-z.
Acknowledgements
We thank the participants of the study.
Author Contributions
Olga Ribot contributed to the conception and design of the study and to the data analysis. Luis Gómez and Javier Torrejón were the responsible colleagues at Galenicum Health S.L.U, for pharmaceutical development of the product. Laura Rodriguez, Kanthikiran VS Varanasi and Olga Ribot were responsible for data interpretation. Yvonne Schnaars, Wolfgang Uhl were responsible for drafting the manuscript. All authors contributed to writing the final manuscript.
Funding
The studies were funded by Galenicum Health S.L.U., Barcelona, Spain. The journal’s rapid service fee was funded by Merck (CrossRef Funder ID: https://doi.org/10.13039/100009945) and Galenicum Health S.L.U., Barcelona, Spain.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Yvonne Schnaars, Sumedh Gaikwad, and Ulrike Gottwald-Hostalek, are employees of Merck Healthcare KGaA, Darmstadt, Germany which holds marketing authorizations of the generic products evaluated in the bioequivalence studies. Wolfgang Uhl is a former employee of Merck Healthcare KGaA, Darmstadt, Germany. Luis Gómez, Javier Torrejón, Laura Rodriguez and Olga Ribot are employees of Galenicum Health S.L.U., Spain which developed the generic products and sponsored the clinical studies presented here. VS Kanthikiran Varanasi is an employee of Galenicum Health India Pvt. Ltd. Hyderabad, India.
Compliance with Ethics Guidelines
The study protocols were approved by the local ethics committee “Ethics Committee for Clinical Research Hospital Universitario de La Princesa" for the sitagliptin study and the Institutional Review Board (IRB) Services “FDA/OHRP Registration IRB00000776 Institutional Review Board Services” and “FDA/OHRP Registration IRB00005290 Institutional Review Board Services” for both FDC sitagliptin/metformin studies. All study procedures were performed in accordance with the 1964 Helsinki declaration and its later amendments and National Guidelines for Biomedical Research on Human Subjects, Good Clinical Practices for Clinical Research in India, ICH (step 5) Guidance on Good Clinical Practice, and related EU guidelines. Informed consent was obtained from all individual participants included in the studies. Informed consents included the intention to present or publish the study results, without disclosing the participants’ identity.
Data Availability
Any requests for data by qualified scientific and medical researchers for legitimate research purposes should be submitted in writing to Galenicum Health S.L.U., Sant Gabriel 50, 08,950, Esplugues de Llobregat, Barcelona, Spain.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schnaars, Y., Gaikwad, S., Gottwald-Hostalek, U. et al. Bioequivalence Evaluation in Healthy Volunteers: New Generic Formulations of Sitagliptin and Sitagliptin–Metformin Fixed-Dose Combination Compared with the Originator Products. Diabetes Ther 14, 347–362 (2023). https://doi.org/10.1007/s13300-022-01349-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-022-01349-2