
Abstract
Justification
Gaucher disease (GD) is amongst the most frequently occurring lysosomal storage disorder in all ethnicities. The clinical manifestations and natural history of GD is highly heterogeneous with extreme geographic and ethnic variations. The literature on GD has paucity of information and optimal management guidelines for Indian patients.
Process
Gaucher Disease Task Force was formed under the auspices of the Society for Indian Academy of Medical Genetics. Invited experts from various specialties formulated guidelines for the management of patients with GD. A writing committee was formed and the draft guidelines were circulated by email to all members for comments and inputs. The guidelines were finalized in December 2016 at the annual meeting of the Indian Academy of Medical Genetics.
Objectives
These guidelines are intended to serve as a standard framework for treating physicians and the health care systems for optimal management of Gaucher disease in India and to define unique needs of this patient population.
Recommendations
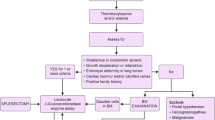
Manifestations of GD are protean and a high index of suspicion is essential for timely diagnosis. Patients frequently experience diagnostic delays during which severe irreversible complications occur. Leucocyte acid β-glucosidase activity is mandatory for establishing the diagnosis of Gaucher disease; molecular testing can help identify patients at risk of neuronopathic disease. Enzyme replacement therapy for type 1 and type 3 Gaucher disease is the standard of care. Best outcomes are achieved by early initiation of therapy before onset of irreversible complications. However, in setting of progressive neurological symptoms such as seizures and or/neuroregression, ERT is not recommended, as it cannot cross the blood brain barrier. The recommendations herein are for diagnosis, for initiation of therapy, therapeutic goals, monitoring and follow up of patients. We highlight that prevention of recurrence of the disease through genetic counseling and prenatal diagnosis is essential in India, due to uniformly severe phenotypes encountered in our population.
Article PDF
Similar content being viewed by others


Avoid common mistakes on your manuscript.
References
Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type1 Gaucher disease: The need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82:697–701.
Cox TM, Schofield JP. Gaucher’s disease: clinical features and natural history. Baillieres Clin Haematol. 1997;10:657–89.
Nair S, Boddupalli CS, Verma R, Liu J, Yang R, Pastores GM, et al. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood. 2015;125:1256–71.
Mistry PK, Belmatoug N, Vom Dahl S, Giugliani R. Understanding the natural history of Gaucher disease. Am J Hematol. 2015;90:S6–S11.
Ankleshwaria C, Mistri M, Bavdekar A, Muranjan M, Dave U, Tamhankar P. Novel mutations in the glucocerebrosidase gene of Indian patients with Gaucher disease. J Hum Genet. 2014;59:223–8.
Agarwal S, Lahiri, K, Muranjan M, Solanki N. The face of lysosomal storage disorders in India: a need for early diagnosis. Indian J Pediatr. 2015;82:525–9.
Sheth JJ, Ankleshwaria CM, Mistri MA, Nanavaty N, Mehta SJ. Splenomegaly, cardiomegaly, and osteoporosis in a child with Gaucher disease. Case Rep Pediatr. 2011;2011:564868.
Weiss K, Gonzalez A, Lopez G, Pedoeim L, Groden C, Sidransky E. The clinical management of type 2 Gaucher disease. Mol Genet Metab. 2015; 114:110–22.
Nagral A, Mewawalla P, Jagadeesh S, Phadke SR, Verma IC, Puri RD, et al. Recombinant macrophage targeted enzyme replacement therapy for Gaucher disease in India. Indian Pediatr. 2011;48:779–84.
Nagral A. Gaucher disease. J Clin Exp Hepatol. 2014;4: 37–50.
Burrow TA, Barnes S, Grabowski GA. Prevalence and management of Gaucher disease. Pediatric Health Med Ther. 2011;2:59–73.
Aggarwal S, Jain SJMN, Das BA, Tandon A, Dalal A. Molecular studies on parents after autopsy identify recombinant GBA gene in a case of Gaucher disease with ichthyosis phenotype. Am J Med Genet A. 2015;11:2858–60.
Wenstrup RJ, Roca-Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol. 2002;75:A2–12.
Lachmann RH, Wight DG, Lomas DJ, Fisher NC, Schofield JP, Elias E, et al. Massive hepatic fibrosis in Gaucher’s disease: Clinico-pathological and radiological features. QJM. 2000;93:237–44.
Mistry PK, Taddei T, vom Dahl S, Rosenbloom BE. Gaucher Disease and malignancy: A model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncog. 2013;18:235–46.
Arends M, van Dussen L, Biegstraaten M, Hollak CE. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol. 2013;161:832–42.
Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11:986–98.
Abrahamov A, Elstein D, Gross-Tsur V, Farber B, Glaser Y, Hadas-Halpern I, et al. Gaucher’s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet. 1995;346:1000–03.
Verma J, Thomas DC, Sharma S, Jhingan G, Singh A, Hsiao KJ, et al. Inherited metabolic disorders: Quality management for laboratory diagnosis. Clinica Chimica Acta. 2015;447:1–7.
Schmitz M, Alfalah M, Aerts JM, Naim HY, Zimmer KP. Impaired trafficking of mutants of lysosomal glucocerebrosidase in Gaucher’s disease. Int J Biochem Cell Biol. 2005;37:2310–20.
Lo SM, McNamara J, Seashore MR, Mistry PK. Misdiagnosis of Niemann-Pick disease type C as Gaucher disease. J Inherit Metab Dis. 2010;33:S429–33.
Gort L, Coll MJ. Diagnosis, biomarkers and biochemical alterations in Gaucher’s disease. Med Clin (Barc). 2011;137:12–6.
Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29:567–83.
Cozzolino I, Picardi M, Pagliuca S, Ciancia G, Luigia L, Pettinato G, et al. B-cell non-Hodgkin lymphoma and pseudo-Gaucher cells in a lymph node fine needle aspiration. Cytopathology. 2016;27:134–6.
Sharma P, Das R, Bansal D, Trehan A. Congenital dyserythropoietic anemia, type II with SEC23B exon 12 c.1385 A’ ! G mutation, and pseudo-Gaucher cells in two siblings. Hematology. 2015;20:104–7.
Chatterjee T, Dewan K, Ganguli P, Das S, Sharma A, Sahni AK, et al. A Rare case of hemoglobin E eemoglobinopathy with Gaucher ‘s disease. Indian J Hematol Blood Transfus. 2013;29:110–2.
Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: The need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82: 697–701.
Cox TM, Aerts JM, Belmatoug N, Cappellini MD, vom Dahl S, Goldblatt J, et al. Management of nonneuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. Inherit Metab Dis. 2008; 31:319–36.
Aerts JMFG, Hollak CEM, van Breemen M, Maas M, Groener JEM, Boot R. Identification and use of biomarkers in Gaucher disease and other lysosomal storage diseases. Acta Pædiatrica. 2005;94:43–6.
Stein P, Yang R, Liu J, Pastores GM, Mistry PK. Evaluation of high density lipoprotein as a circulating biomarker of Gaucher disease activity. J Inherit Metab Dis. 2011;34:429–37.
Murugesan V, Chuang WL, Liu J, Lischuk A, Kacena K, Lin H, et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am J Hematol. 2016;91:1082–9.
Elstein D, Hadas-Halpern I, Azuri Y, Abrahamov A, Bar-Ziv Y, Zimran A. Accuracy of ultrasonography in assessing spleen and liver size in patients with Gaucher disease: comparison to computed tomographic measurements. J Ultrasound Med. 1997;16:209–11.
Lo SM, Liu J, Chen F, Pastores GM, Knowles J, Boxer M, et al. Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and longterm outcomes of therapy. J Inherit Metab Dis. 2011;34:643–50.
Hollak CE, Maas M, Aerts JM. Clinically relevant therapeutic endpoints in type I Gaucher disease. Inherit Metab Dis. 2001;2:97–105.
Hughes DA, Gonzalez DE, Lukina EA. Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: Long-term data from phase III clinical trials. Am J Hematol. 2015:90:584–91.
Zimran A, Gonzalez-Rodriguez DE, Abrahamov A, Elstein D, Paz A, Brill-Almon E, et al. Safety and efficacy of two dose levels of taliglucerase alfa in pediatric patients with Gaucher disease. Blood Cells Mol Dis. 2015;54:9–16.
Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR, Hangartner T. Osteopenia in Gaucher disease develops early in life: Response to Imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis. 2011;46:66–72.
Weinreb N, Barranger J, Packman S, Prakash-Cheng A, Rosenbloom B, Sims K. Imiglucerase (cerezyme) improves quality of life in patients with skeletal manifestations of Gaucher disease. Clin Genet. 2007;71:576–88.
Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002;113:112–9.
Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. 2008;372:1263–71.
Mistry PK, Deegan P, Vellodi A, Cole JA, Yeh M, Weinreb NJ. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: Effect on incidence of avascular necrosis. Br J Haematol. 2009;147:561–70.
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr. 2004;144:112–20.
Mistry PK, Cappellini MD, Lukina E, Ozsan H, Mach Pascual S, Rosenbaum H. et al. A reappraisal of Gaucher disease-Diagnosis and disease management algorithms. Am J Hematol. 2011;86:110–5.
Kaplan P, Baris H, De Meirleir L, Di Rocco M, El-Beshlawy A, Huemer M, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013;172:447–58.
Pastores GM. Recombinant Glucocerebrosidase (Imiglucerase) as a therapy for Gaucher disease. Bio Drugs. 2010;24:41–7.
Zimran A, Pastores GM, Tylki-Szymanska A, Hughes DA, Elstein D, Mardach R, et al. Safety and efficacy of velaglucerase alfa in Gaucher disease type 1 patients previously treated with imiglucerase. Am J Hematol. 2013;88: 172–8.
Muranjan M, Patil S. Outcome for Gaucher disease in India: Lessons from prevalent diagnostic and therapeutic practices. Indian Pediatr. 2016;53:685–8.
Lukina E, Watman N, Arreguin EA, Dragosky M, Iastrebner M, Rosenbaum H, et al. Improvement in haematological, visceral and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study. Blood. 2010;116:4095–8.
Vashishtha VM, Choudhury P, Kalra A, Bose A, Thacker N, Yewale VN, et al. Indian Academy of Pediatrics (IAP) recommended immunization schedule for children aged 0 through 18 years—India, 2014 and updates on immunization. Indian Pediatr. 2014;51:785–800.
Weinreb N, Taylor J, Cox T, Yee J, vom Dahl S. A benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imiglucerase. Am J Hematol. 2008;83:890–5.
Somaraju UR, Tadepalli K. Hematopoietic stem cell transplantation for Gaucher disease. Cochrane Database Syst Rev. 2012;7:CD006974.
Ito S, Barrett AJ. Gauchers disease–a reappraisal of hematopoietic stem cell transplantation. Pediatr Hematol Oncol. 2013;30:61–70.
Prajnya R, Rehder C, Phadke SR, Bali D. Prenatal diagnosis of Pompe disease: enzyme assay or molecular testing? Indian Pediatr. 2011;48:901–2.
Verma J, Thomas DC, Sharma S, Jhingan G, Saxena R, Kohli S, et al. Inherited metabolic disorders: prenatal diagnosis of lysosomal storage disorders. Prenat Diagn. 2015;35:1137–47.
Author information
Authors and Affiliations
Consortia
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Puri, R.D., Kapoor, S., Kishnani, P.S. et al. Diagnosis and Management of Gaucher Disease in India – Consensus Guidelines of the Gaucher Disease Task Force of the Society for Indian Academy of Medical Genetics and the Indian Academy of Pediatrics. Indian Pediatr 55, 143–153 (2018). https://doi.org/10.1007/s13312-018-1249-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13312-018-1249-9