
Abstract
Epidermolysis bullosa (EB) comprises rare genetic disorders characterized by skin and mucosal membrane blistering induced by mechanical trauma. Molecularly, pathogenic variants affect genes encoding proteins crucial for epidermal–dermal adhesion and stability. Management of severe EB is multidisciplinary, focusing on wound healing support, ensuring that patients thrive, and complication treatment. Despite extensive research over 30 years, novel therapeutic approaches face challenges. Gene therapy and protein therapy struggle with efficacy, while regenerative cell-based therapies show limited effects. Drug repurposing to target various pathogenic mechanisms has gained attention, as has in vivo gene therapy with drugs for dystrophic and junctional EB that were recently approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA). However, their high cost limits global accessibility. This review examines therapeutic advancements made over the past 5 years, exploiting a systematic literature review and clinical trial data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Targeted corrective therapies are the ultimate way of treating epidermolysis bullosa (EB). |
In vivo gene therapy is entering the stage of clinical application. Beremagene geperpavec (B-VEC) has been approved by the FDA and has become the first gene therapy available to patients with dystrophic EB. This can be considered a significant advancement. |
Treatment through drug repurposing (repositioning) is gaining attention. The application of existing drugs to target pathogenic mechanisms such as wound healing, inflammation, pruritus, and fibrosis is being considered. This approach is regarded as relatively inexpensive and highly feasible. |
It is pointed out that IL-4/13 inhibitors and JAK inhibitors, which are used to treat atopic dermatitis, show promise for treating pruritus in EB. They may provide new options for managing pruritus and skin lesions in EB. |
Regarding cell therapy, it has been shown that long-term effects are limited, and clinical application has not been achieved. Challenges remain in regenerative medicine approaches. |
New treatment concepts, such as gene editing and RNA therapy, are emerging but are still in the preclinical stage. They are mentioned as future possibilities. |
Introduction
Epidermolysis bullosa (EB) encompasses rare genetic disorders that manifest with mechanically induced blistering of the skin and mucosal membranes. Molecular pathology involves pathogenic variants in genes that code for proteins contributing to epidermal–dermal adhesion and stability [1]. The most common type, EB simplex (EBS), involving skin cleavage within the basal epidermal layer, is also the most heterogeneous genetically and clinically [2]. The second most common is dystrophic EB (DEB), characterized by a split in the upper dermis due to genetic defects of type VII collagen (C7). Junctional EB (JEB) is very rare; it is caused by mutations in genes encoding transmembrane proteins of the hemidesmosomes, focal adhesions, or the basement membrane laminin 332 [2]. Kindler EB is ultra-rare, with a few hundred individuals affected worldwide.
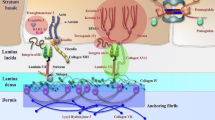
Mucocutaneous fragility causes blisters and wounds, tissue damage, breakage of the cutaneous barrier, chronic bacterial colonization of the skin, inflammation, and, depending on the level of cleavage, scarring. Thus, wounds are the central element in the pathogenesis of long-term disease manifestations and complications including failure to thrive, anemia, scarring, strictures, and skin cancer in some of the EB types (such as dystrophic, junctional, and Kindler EB) (Fig. 1). The management of severe EB subtypes is symptomatic and multidisciplinary and aims at supporting wound healing, ensuring that patients thrive, and treating complications [3, 4].

Schematic representation of the timescale of therapy development for epidermolysis bullosa (EB)
Extensive effort has been put into research into novel therapeutic approaches for EB during the past 30 years (Fig. 2). The repair or replacement of EB-associated genes or proteins by gene or protein therapy has proven to be more challenging than expected. Regenerative cell-based therapies have been comprehensively studied [5]. They showed only a limited effect, mainly due to the short survival of the cells and protein turnover. Repurposing drugs for EB treatment came into the focus of researchers around 15 years ago. There are various mechanisms by which such drugs interfere with EB pathogenesis: read-through of nonsense mutations, suppression of inflammation, suppression of fibrosis, etc. [6]. The first drugs for the treatment of EB wounds in dystrophic and junctional EB have recently achieved US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval, marking an important milestone in EB research. However, they are still extremely expensive and are not yet available for all patients worldwide [7].

The pathogenesis chain in severe EB, in particular DEB. Skin fragility and wounds are the initial manifestations of EB and lead to cutaneous and systemic complications. Thus, corrective therapies aiming at the addition of type VII collagen into the skin are a meaningful approach
Here we review the development of these therapeutic approaches, based on systematic research of the literature covering the past 5 years.
Method
A comprehensive search was carried out on studies published on PubMed and ClinicalTrials.gov by the end of March 2024. The following MeSH (Medical Subject Headings) terms were used: “epidermolysis bullosa and therapy,” “epidermolysis bullosa and gene therapy,” “epidermolysis bullosa and cell-based therapy,” “epidermolysis bullosa and protein therapy,” “epidermolysis bullosa and repurposed drugs,” “epidermolysis bullosa and read-through therapies,” “epidermolysis bullosa and anti-inflammatory therapy,” “epidermolysis bullosa and anti-fibrotic therapy,” and “epidermolysis bullosa and anti-pruritic therapy.” The initial search yielded 123 studies. Eighty-nine studies were found to be relevant to the research topic. The eligibility criteria included articles published in English. Eligible studies were observational, retrospective, prospective, or cohort studies or single reports. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Gene Replacement Therapy
Numerous studies have focused on gene replacement strategies aiming at the correction of the genetic defect (reviewed in [8]).
In Vivo Gene Replacement
Krystal Biotech have developed a topical gene therapy known as beremagene geperpavec (B-VEC) that uses a non-integrative replication-defective herpes simplex virus-1 (HSV-1) containing two copies of the COL7A1 gene [9]. This approach facilitates the expression of proα1 (VII) polypeptides in the skin, enabling their integration into trimeric C7 molecules and subsequent assembly into functional anchoring fibrils. In 2022, findings from a randomized, placebo-controlled phase I/II clinical trial (NCT03536143) were disclosed [10]. Additionally, results from a phase III investigation of B-VEC in Recessive Dystrophic Epidermolysis Bullosa (RDEB) (NCT04917874) were released, demonstrating improvements in wound healing. B-VEC received full licensing approval for clinical application in the United States on May 19, 2023 [11, 12]. Consequently, B-VEC became the first gene therapy treatment accessible to individuals with DEB. A continuing observational study with long-term follow-up of all patients treated with an HSV-1 vector is currently underway and has an anticipated completion date in 2028 (NCT04917887).
Ex Vivo Gene Therapy
The most exciting and technically challenging gene therapy combined with stem cell therapy has been developed by the group of Michele de Luca for intermediate junctional EB with LAMB3 mutations. Life-saving transplantation of transgenic keratinocytes on 80% of the body surface was groundbreaking in 2017 [13]. Five years of follow-up demonstrated that genetically repaired stem cells survived and continued to regenerate an almost normal epidermis [14].
Abeona Therapeutics, Inc., cultures autologous keratinocytes, genetically corrects them ex vivo, and grafts epidermal sheaths onto denuded skin areas. They use a lentiviral vector to integrate COL7A1 cDNA into recipient cells' genomes. Initial findings suggest that corrected keratinocytes express C7 and show evidence of anchoring fibril assembly. Concerns have arisen regarding the durability of this approach, with early data indicating that collagen gene expression may diminish over time due to factors like viral promoter use or graft integration issues [15].
Castle Creek Biosciences, Inc., developed D-Fi (dabocemagene autoficel) by correcting autologous fibroblasts with a vector integrating the transgene into the genome. Two trials, phase I (NCT02493816—Lenticol-F) and phase I/II (NCT02810951—FCX-007, later named dabocemagene autoficel, D-Fi), used a lentiviral vector to deliver COL7A1 cDNA into autologous fibroblasts [16, 17]. Post-treatment, the correct localization of C7 at the dermal–epidermal junction (DEJ) and enhanced wound healing were reported. However, one trial (NCT02493816) showed low C7 expression levels and no anchoring fibrils initially, contrasting with the other trial (NCT02810951), which showed fibril formation. A phase 3 trial (NCT04213261) assessing this approach is ongoing. D-Fi has FDA Orphan Drug, Rare Pediatric Disease, Fast Track, and Regenerative Medicine Advanced Therapy designations.
Recent research has emphasized the correction of both keratinocytes and fibroblasts for optimal fibril assembly. Grafts with both corrected cell types showed functional assembly, contrasting with mouse model findings [18]. Ongoing efforts are aimed at the development of GENEGRAFTs (gene therapy for RDEB through the transplantation of autologous skin equivalents that have been genetically corrected using a self-inactivating retroviral vector encoding COL7A1) that correct both cell types using a viral vector expressing C7 [19].
Natural Gene Therapy or Revertant Mosaicism
Revertant mosaicism, or "natural gene therapy," shows promise for cell-based therapy in EB, where certain skin cells spontaneously reverse mutations, resulting in patches of normal skin. Mechanisms include mitotic recombinations, back mutations, and second-site mutations. While primarily observed in RDEB keratinocytes, it has also been noted in fibroblasts [20]. Challenges in long-term culture stem from stem cell depletion, but punch grafting revertant skin onto lesions has led to limited re-epithelialization. Expansion can be achieved by generating induced pluripotent stem cells (iPSCs) from revertant keratinocytes and differentiating them into corrected keratinocytes. These revertant iPSCs offer the potential for cell-based therapy in EB, although their personalized nature limits wide clinical application [21, 22].
Gene Editing
Gene editing techniques like clustered regularly interspaced short palindromic repeats (CRISPR/Cas9) offer the potential for treating EB by correcting mutations and improving gene correction efficacy in patient-derived cells, enabling the development of functional collagen-synthesizing skin grafts [23]. Challenges include the need for a large quantity of cultured cells, which is addressed through solutions like iPSCs [24, 25]. Transcription activator-like effector nucleases (TALENs) and CRISPR/Cas9 have been used successfully in gene editing for EB to generate improved stability in gene-edited skin equivalents (SEs) [26, 27]. Base editing techniques and prime editing show promise in correcting point mutations. However, challenges in clinical translation remain, including off-target effects and effectively targeting unwounded skin. While significant progress has been made in genome editing technology, no gene editing therapies for human skin diseases have advanced to clinical trials [28, 29].
Exon Skipping and Other RNA-Based Therapies
Exon skipping utilizes antisense oligonucleotides (ASOs) to alter the splicing of pre-messenger RNA (mRNA), thereby eliminating the mutation responsible for a disease. Initially developed for Duchenne muscular dystrophy (DMD), antisense-mediated exon skipping has progressed to clinical trials targeting recurrently mutated exons in the dystrophin gene [30]. While direct delivery of ASOs to wound cells is feasible, efforts are also directed towards enhancing delivery into intact skin using cationic liposomes.
The rationale for applying exon skipping in EB stems from genotype–phenotype correlation studies indicating its potential to alleviate the severity of JEB and DEB. Furthermore, given that most exons of COL17A1 and COL7A1 are in frame, several preclinical investigations have illustrated that skipping certain exons, such as 13, 70, 73, 80, or 105 in COL7A1, yields slightly truncated yet partially functional proteins deposited at the DEJ [31, 32]. A clinical trial assessing the topical administration of QR-313, a water-based gel (hydrogel) containing an ASO targeting exon 73 that is directly applied onto DEB wounds for COL7A1 correction, concluded in 2021 (Wings Therapeutics; NCT03605069), but results have not been published yet [33].
Protein Therapy
Treatment with recombinant proteins shows promise for recessive forms of EB characterized by loss-of-function mutations, such as C7 in RDEB and laminin-332 in JEB. Mice studies demonstrated that injecting recombinant C7 into the skin induced anchoring fibril formation without significant antibody binding. Intravenously administered recombinant C7 targeted the skin and promoted anchoring fibril formation, with minimal antibody binding to the skin basement membrane [7].
A phase I/II clinical trial conducted by Phoenix Tissue Repair, Inc., assessed recombinant C7's safety, tolerability, and clinical efficacy in adults with RDEB (NCT03752905—completed). Systemic administration offers advantages in distribution to extracutaneous tissues affected by RDEB, facilitating repair in these areas.
Building on this, a phase II clinical trial (NCT04599881) evaluating the efficacy of intravenous recombinant C7 injections in patients with RDEB began in 2019. While some improvements were noted, outcomes were modest. Nonetheless, recombinant C7 therapy shows the potential to complement local therapies, improving innate immune function and reducing infection in RDEB wounds, which enhances the effectiveness of other localized skin treatments [34].
Cell-Based Therapies
Cell-based therapy encompasses a range of treatments involving primary keratinocytes, fibroblasts, hematopoietic cells, and mesenchymal stem/stromal cells. Important considerations include whether these cells are autologous or allogeneic, whether they possess true stem cell properties or are primarily stromal cells, and the method of administration to patients with RDEB, such as skin grafts, local injections, or intravenous infusions [35,36,37,38]. Although the first studies were performed more than 10 years ago, cell-based therapies have not yet entered clinical application.
Fibroblasts
Multiple human clinical trials have investigated the efficacy of intradermal injections of allogeneic fibroblasts into chronic RDEB wounds. Results have shown variable outcomes, with wound healing improvement ranging from 30 to 80%. However, the efficacy has not consistently surpassed that of placebo injections. The potential benefits of allogeneic fibroblasts stem from the enhancement of the recipient's own mutant COL7A1 mRNA expression, leading to transient increased production of partially functional C7. Additionally, there might be a minor direct secretion of C7 by the injected allogeneic fibroblasts [17, 39].
Bone Marrow Transplantation
In murine RDEB models, hematopoietic stem cells differentiated into epidermal cells, generating C7 and extending the lifespan. Epidermal grafting using a minimally invasive automated device (CELLUTOME) to harvest epidermal micrografts from the same donor post-allogeneic hematopoietic cell transplantation showed prolonged engraftment, lasting up to 3 years without rejection [40].
Bone marrow cells aid skin repair and address C7 deficiency in RDEB mouse models. Clinical trials of bone marrow transplantation (BMT) started in 2010. Initial trials involved seven RDEB children undergoing myeloablative chemotherapy and allogeneic stem cell transplantation, resulting in long-term donor cell expression and new C7 at the DEJ in five patients. Subsequently, reduced-intensity conditioning regimens were introduced to mitigate risks. The precise mechanisms of BMT's effectiveness in RDEB remain unclear, as some patients showed clinical improvement despite lacking new C7 expression. In contrast, BMT in JEB has not shown significant clinical benefits and carries substantial mortality risks [41].
Mesenchymal Stem Cells
Mesenchymal stem cells (MSCs), which possess self-renewal and differentiation capabilities, offer potential benefits in DEB cell therapy due to their immunomodulatory and anti-inflammatory properties. Allogeneic MSCs sourced from unrelated donors show low immunogenicity, ensuring safety in human trials without the need for aggressive pre-conditioning regimens [42].
MSCs have been administered intradermally or intravenously to patients with RDEB. Intradermal injection offers localized delivery, potentially enhancing the interaction with host cells, while intravenous infusion allows for a systemic impact. However, the fate of MSCs in both skin and blood remains uncertain [43]. MSCs exhibit heterogeneity and include multilineage-differentiating stress-enduring (MUSE) cells and ABCB5+ MSCs, offering therapeutic options for RDEB. MUSE cells (SSEA-3(+) bone marrow cells) can differentiate into keratinocytes and fibroblasts in vitro. An ongoing phase III trial aims to validate their efficacy in RDEB [44,45,46,47,48].
Bone-marrow-derived MSCs release extracellular vesicles (EVs) containing COL7A1 mRNAs and C7 protein, which can induce diverse phenotypic responses. While the influence of the systemic administration of EVs on C7 expression in RDEB skin remains uncertain, the direct application of EVs to chronic wounds is being assessed [49].
Advancements in our understanding of EVs may lead to the development of cell-free therapy for RDEB, which would sustain therapeutic efficacy without incurring the complexities of living cell product manufacturing and transportation [50].
A non-randomized phase I/IIa trial (NCT04173650) assessing the topical application of EVs from normal donors to chronic EB wounds may provide further clinical insights. Additionally, components derived from MSCs, like mitochondria and apoptotic bodies, hold the potential for stimulating recipient tissue stem cells.
Tissue‑Engineered Skin Graft Therapy
A pilot study to evaluate a temporary skin substitute (Spincare® Matrix) for wound healing in patients with RDEB (NCT05944250) is ongoing. It uses the Spincare device created by Nanomedic. This portable device—the first of its kind—is designed to administer a non-invasive, non-therapeutic electrospun nanofibrous matrix dressing to wounds, facilitating the healing process.
Pathogenesis-Based Therapies and Repurposed Drugs
Such approaches may include repurposing existing drugs or creating novel therapies, aiming to alleviate disease severity and enhance patient well-being. It is crucial to note that these treatments are not designed to cure EB but rather to enhance the overall quality of life for affected individuals.
Drugs that Enhance Wound Healing
High Mobility Group Box-1 (HMGB1)
The bone marrow comprises diverse hematopoietic and non-hematopoietic stem cells, including epithelial progenitors implicated in tissue repair. High mobility group box 1 (HMGB1) mobilizes these progenitors, playing a crucial role in wound healing. The A box domain of HMGB1 stimulates the release of Lin−/PDGFRα+ bone marrow cells capable of differentiating into keratinocytes and fibroblasts and producing C7 [51].
Animal models have demonstrated the systemic administration of the HMGB1 fragment to be beneficial, preserving cardiac function in delta-sarcoglycan-deficient hamsters and improving manifestations in DEB model mice.
StemRIM developed a recombinant peptide of the A box domain (redasemtide) for potential human use in tissue regeneration. Safety assessments in a phase I trial involving intravenous infusion in 48 healthy volunteers were conducted. In RDEB, a phase II clinical trial evaluated the effects of intravenous HMGB1 fragment administration in nine patients, with reduced blistering and enhanced wound healing experienced by the majority.
While the intravenous HMGB1 peptide awaits clinical approval in Japan, current data suggest its potential utility, particularly in intermediate forms of RDEB and possibly dominant DEB (DDEB). For severe RDEB cases, an alternative approach could involve developing HMGB1-peptide-soaked nets for subcutaneous implantation, which would capture crucial progenitor cells for genetic modification before their re-administration as autologous cell therapy [4].
Oleogel-S10
Amyrt Pharma developed Oleogel-S10, containing 10% birch triterpene extracts, which has been shown to accelerate wound healing by modulating inflammation, promoting keratinocyte migration, and exerting antimicrobial effects. Previously used in burn and split-thickness skin graft wounds, these extracts expedite wound closure [52, 53]. A recent phase III trial, EASE (Efficacy And Safety of Oleogel-S10 in patients with EB), revealed that topical Oleogel-S10 application every 4 days resulted in complete wound healing in 41% of patients within 45 days, compared to 29% in the control group, with minimal adverse effects [54, 55]. Despite promising results, the FDA initially considered the wound healing data insufficient for product approval. However, in June 2022, the EMA approved Oleogel-S10 for wound healing in JEB and DEB, and the FDA approved it in December 2023.
Calcipotriol
Guttman-Gruber et al. explored the potential of calcipotriol in RDEB wound healing. Their preclinical findings demonstrated that at low doses (100 nM), calcipotriol boosted the antimicrobial peptide cathelicidin (hCAP18), fostering wound healing and enhancing antimicrobial defense. Subsequently, in a randomized, placebo-controlled trial involving six patients with RDEB, calcipotriol-treated lesions exhibited a significant reduction in wounded area (88%) compared to placebo (66%). Additionally, itch scores decreased significantly within 2–4 weeks of treatment initiation. While promising, there are currently no specific recommendations or formulations of calcipotriol for RDEB use. Repurposing existing calcipotriol creams for RDEB is not advised due to concentration differences between psoriasis and RDEB applications [56, 57].
Read‑Through Therapies
The read-through approach utilizes small-molecular-weight compounds to enable the translational machinery to suppress nonsense mutations by inserting an amino acid in place of a premature stop codon, thereby facilitating the synthesis of full-length proteins [58].
The original prototype of such read-through molecules, PTC124 (ataluren), has shown the ability to bypass pathogenic premature termination codons (PTCs), but it fails to efficiently override naturally occurring endogenous stop codons of translation. PTC124 has been extensively tested on various genes, particularly the dystrophin gene in DMD, and proved to be effective in alleviating the severity of junctional EB in a case report [59].
Studies have shown that gentamicin effectively bypasses COL7A1 and LAMB3 PTCs, with promising results observed in clinical trials for RDEB [60]. However, long-term systemic administration of gentamicin poses potential issues, including the risk of renal toxicity, prompting the consideration of topical application as a more favorable delivery route [61]. Despite these considerations, clinical investigations assessing the impact of intravenous injections of gentamicin were performed for RDEB (NCT03392909) and JEB (NCT03526159) [62].
The optimal dosing, treatment intervals, and long-term toxicity of gentamicin therapy remain to be precisely determined. An ongoing clinical trial (NCT04140786) assessing higher gentamicin concentrations (10 mg/kg/day) and longer treatment durations (biweekly intravenous injections for 3 months) may provide valuable insights. The therapy with gentamicin has been associated with side effects like ototoxicity, nephrotoxicity, and neurotoxicity. Safe use involves intravenous or intramuscular administration at doses of 7.5 mg/kg/day for 3 weeks; topically, as 0.1% ointment applied 3 times daily for 2 weeks; or intradermal injections (8 mg for 2 days) [60,61,62].
A strategy for the optimization of read-through employed a combination of low-dose gentamicin and paromomycin together with enhancers (CC90009 and NMDI14) and antioxidants (melatonin and apocynin) and achieved a significant increase in read through of type XVII collagen in JEB keratinocytes [63].
Anti-inflammatory Drugs
Methotrexate, Small Molecules, and Biologics
Transcriptome profiling of RDEB wound skin revealed the upregulation of cytokine and chemokine signaling pathways like Janus kinases/signal transducer and activator of transcription protein (JAK/STAT), IL-6, and IL-20. Computational analysis identified methotrexate as a promising candidate for reversing these gene expression patterns, suggesting its potential to alleviate systemic inflammation and reduce itch in RDEB. Clinical trials are needed to validate these findings. Additionally, the increased IL-17A expression observed in DEB patients suggests that small molecules and biologics targeting IL-17A, which are commonly used in psoriasis treatment, may also benefit individuals with DEB [64, 65].
Anti-fibrotic Therapy
Losartan
The repetitive tissue damage and progressive scarring in RDEB are mainly driven by Transforming growth factor-β (TGF-β), suggesting that therapies that reduce TGF-β signaling could slow disease progression and alleviate morbidity. Losartan, an anti-hypertensive medication, can attenuate TGF-β signaling. Its efficacy was assessed in a col7a1 hypomorphic mouse model, where it delayed paw scarring and digit fusion, indicating a potential for human RDEB therapy [66]. A case series of six patients with RDEB treated with oral losartan (0.7 mg/kg) for 6 weeks showed clinical benefits and improved skin histology [67]. In the phase I/II REFLECT trial, 29 pediatric patients with RDEB receiving losartan for 10 months demonstrated positive safety outcomes in interim analysis, with final results awaited. Although some small case reports support oral losartan use in RDEB, definitive clinical recommendations should await the results of larger trials [68,69,70].
Decorin
Another approach to reduce TGF-β activity in the skin involves increasing the expression of decorin, a stromal proteoglycan that inhibits TGF-β by binding to its core protein, preventing its interaction with receptors. Injection of a lentiviral vector carrying human decorin core protein cDNA into col7a1 hypomorphic mice increased decorin expression, prolonging survival, delaying paw deformities and digit fusion, and reducing skin fibrosis. Intraperitoneal administration of decorin fused with the skin-homing peptide Cysteine-rich receptor-like kinases (CRK) containing the CendR domain notably decreased fibrotic gene expression and mitigated fibrosis in RDEB mouse skin. FIBRX Derm Inc. is developing a topical gel with recombinant decorin for evaluation in human RDEB [71].
Anti-pruritic Therapies
Biologics and Janus Kinase Inhibitors
Itch is a distressing symptom that significantly impacts the quality of life for many individuals with EB, especially those with a particularly itchy form known as EB pruriginosa, which is challenging to manage using conventional anti-inflammatory treatments. Dupilumab, a monoclonal antibody targeting the IL-4 receptor to reduce the activity of cytokines IL-4 and IL-13, has been investigated for EB pruritus [72, 73]. Several anecdotal reports suggest promising results with dupilumab, with rapid reductions in itch severity and skin inflammation seen in patients with EB pruriginosa but also in those with other types of EB [74,75,76,77]. Additionally, elevated IgE levels have been observed, and anti-IgE therapy with omalizumab has shown some efficacy in reducing skin inflammation and itch in certain individuals [78]. Other options include oral baricitinib and tofacitinib, which are JAK inhibitors that have demonstrated the ability to alleviate itch and skin inflammation in DEB [79]. However, structured recommendations for drug selection and dosing in individual patients are currently lacking due to the absence of placebo-controlled clinical trials for biologics or JAK inhibitors in DEB.
Serlopitant, a neurokinin-1 receptor antagonist that disrupts substance P signaling, holds promise as a potential treatment for itch. A phase II trial involving 14 EB patients showed no adverse effects during an 8-week treatment period followed by a 4-week washout period. However, the serlopitant group reported only minor itch relief compared to placebo, with a marginal reduction that was not significantly different from placebo on a numeric rating scale. A randomized phase II trial (NCT03836001) was performed to further evaluate serlopitant's efficacy [80].
Repurposed Drugs for EBS
Diacerein, derived from rhubarb root, acts as a prodrug of IL-1 converting enzyme inhibitor, which helps to reduce IL-1β signaling. Topical application of 1% diacerein was found to be beneficial in stabilizing keratin filaments and decreasing skin blisters. Subsequently, a phase II/III placebo-controlled crossover clinical trial was conducted, involving 15 EBS participants treated for 4 weeks. Results showed that more individuals in the diacerein group experienced a reduction in blisters of over 40%, with the efficacy lasting for more than 3 months [81]. A larger multicenter trial failed to show a difference between 1% diacerein and placebo [82].
Thymosin β4, a naturally occurring polypeptide with various wound-healing properties, such as anti-inflammatory, anti-fibrotic, pro-angiogenic, and stem cell recruitment abilities, has been studied for its potential for treating EB. In a study involving 30 patients with JEB/RDEB, topical application of thymosin β4 gel at different concentrations (0.01%, 0.03%, and 0.1%) for 56 consecutive days showed promising results, with a tendency towards accelerated healing. Notably, the mid-range dosage (0.02–0.03%) demonstrated the most significant efficacy, while higher and lower concentrations showed less pronounced benefits. A phase II clinical trial (NCT03578029) was performed to further evaluate the effectiveness of topical thymosin β4 dermal gel in JEB/DEB patients, but no publications about the results are available [83].
Apremilast, an anti-Th17 molecule, resulted in a significant improvement in the skin condition of Epidermolysis bullosa simplex generalized severe (EBS-gen sev) patients. The significance of inflammation in patients with severe EBS and the crucial role played by Th17 cells in its pathogenesis have been demonstrated [84].
mTOR inhibitors and phosphatidylinositol 3-kinase inhibitors (rapamycin, sirolimus) showed promising results in treating painful plantar keratoderma resulting from chronic blistering in patients with EBS [85] .
A summary of all therapies described in this review is given in Table 1.
Conclusion
Genetic defects in severe EB have high penetrance and expressivity and a strong impact on cutaneous homeostasis. Targeted corrective therapies and regenerative medicine based on the replacement of deficient genes, proteins, or cells offer the only effective treatments, but they remain challenging and expensive. Thus, efforts should be focused on implementing current developments such as in vivo and ex vivo gene therapy into the clinical care of patients. Adjunctive therapies utilizing less expensive repurposed drugs may help alleviate disease severity, in particular in less severe EB subtypes, and enhance the quality of life for affected individuals. These therapies could complement strategies targeting the underlying pathologic processes of skin and mucous membrane fragility.
Proving the efficacy of such measures and designing clinical trials with appropriate endpoints and participant numbers poses numerous challenges. Nevertheless, patients with EB and their families eagerly anticipate effective treatments in the near future.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primer. 2020;6:78.
Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614–27.
Bruckner-Tuderman L. Skin fragility: perspectives on evidence-based therapies. Acta Derm Venereol. 2020;100(5):adv0053.
Hou PC, Wang HT, Abhee S, Tu WT, McGrath JA, Hsu CK. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22(6):801–17.
Mellerio JE, Uitto J. Meeting report: the first global congress on epidermolysis bullosa, EB2020 London: toward treatment and cure. J Invest Dermatol. 2020;140(9):1681–7.
Has C, South A, Uitto J. Molecular therapeutics in development for epidermolysis bullosa: update 2020. Mol Diagn Ther. 2020;24(3):299–309.
Hou PC, Del Agua N, Lwin SM, Hsu CK, McGrath JA. Innovations in the treatment of dystrophic epidermolysis bullosa (DEB): current landscape and prospects. Ther Clin Risk Manag. 2023;14(19):455–73.
Bischof J, Hierl M, Koller U. Emerging gene therapeutics for epidermolysis bullosa under development. Int J Mol Sci. 2024;25(4):2243.
Guide SV, Gonzalez ME, Bağcı IS, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387(24):2211–9.
Gurevich I, Agarwal P, Zhang P, et al. In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: a phase 1 and 2 trial. Nat Med. 2022;28(4):780–8.
Khan A, Riaz R, Ashraf S, Akilimali A. Revolutionary breakthrough: FDA approves Vyjuvek, the first topical gene therapy for dystrophic epidermolysis bullosa. Ann Med Surg (Lond). 2023;85(12):6298–301.
Koller U, Bauer JW. Gene replacement therapies for genodermatoses: a status quo. Front Genet. 2021;30(12): 658295.
Hirsch T, Rothoeft T, Teig N, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680):327–32.
Kueckelhaus M, Rothoeft T, De Rosa L, et al. Transgenic epidermal cultures for junctional epidermolysis bullosa—5-year outcomes. N Engl J Med. 2021;385(24):2264–70.
Subramaniam KS, Antoniou MN, McGrath JA, Lwin SM. The potential of gene therapy for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2022;186(4):609–19.
Marinkovich M, Lane A, Sridhar K, Keene DR, Malyala A, Maslowski J. A phase 1/2 study of genetically-corrected, collagen VII expressing autologous human dermal fibroblasts injected into the skin of patients with recessive dystrophic epidermolysis bullosa (RDEB). J Invest Dermatol. 2018;138(5):S100.
Lwin SM, Syed F, Di WL, et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(11): e126243.
Conradt G, Hausser I, Nyström A. Epidermal or dermal collagen VII is sufficient for skin integrity: insights to anchoring fibril homeostasis. J Invest Dermatol. 2024;144(6):1301–1310.e7.
Gaucher S, Lwin SM, Titeux M, et al. EBGene trial: patient preselection outcomes for the European GENEGRAFT ex vivo phase I/II gene therapy trial for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2020;182(3):794–7.
Twaroski K, Eide C, Riddle MJ, et al. Revertant mosaic fibroblasts in recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2019;181(6):1247–53.
Matsumura W, Fujita Y, Shinkuma S, et al. Cultured epidermal autografts from clinically revertant skin as a potential wound treatment for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2019;139(10):2115–2124.e11.
van den Akker PC, Bolling MC, Pasmooij AMG. Revertant mosaicism in genodermatoses: natural gene therapy right before your eyes. Biomedicines. 2022;10(9):2118.
Thompson EL, Pickett-Leonard M, Riddle MJ, Chen W, Albert FW, Tolar J. Genes and compounds that increase type VII collagen expression as potential treatments for dystrophic epidermolysis bullosa. Exp Dermatol. 2022;31(7):1065–75.
Bonafont J, Mencía Á, García M, et al. Clinically relevant correction of recessive dystrophic epidermolysis bullosa by dual sgRNA CRISPR/Cas9-mediated gene editing. Mol Ther. 2019;27(5):986–98.
Bonafont J, Mencía A, Chacón-Solano E, et al. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol Ther. 2021;29(6):2008–18.
Bischof J, March OP, Liemberger B, et al. Paired nicking-mediated COL17A1 reframing for junctional epidermolysis bullosa. Mol Ther. 2022;30(8):2680–92.
Sproule TJ, Wilpan RY, Low BE, et al. Functional analysis of Collagen 17a1: a genetic modifier of junctional epidermolysis bullosa in mice. PLoS ONE. 2023;18(10): e0292456.
Sheriff A, Guri I, Zebrowska P, et al. ABE8e adenine base editor precisely and efficiently corrects a recurrent COL7A1 nonsense mutation. Sci Rep. 2022;12(1):19643.
Osborn MJ, Newby GA, McElroy AN, et al. Base editor correction of COL7A1 in recessive dystrophic epidermolysis bullosa patient-derived fibroblasts and iPSCs. J Invest Dermatol. 2020;140(2):338–47.
Shimizu-Motohashi Y, Murakami T, Kimura E, Komaki H, Watanabe N. Exon skipping for Duchenne muscular dystrophy: a systematic review and meta-analysis. Orphanet J Rare Dis. 2018;13(1):93.
Vermeer FC, Bremer J, Sietsma RJ, et al. Therapeutic prospects of exon skipping for epidermolysis bullosa. Int J Mol Sci. 2021;22(22):12222.
Bremer J, van der Heijden EH, Eichhorn DS, et al. Natural exon skipping sets the stage for exon skipping as therapy for dystrophic epidermolysis bullosa. Mol Ther Nucleic Acids. 2019;18:465–75.
Bornert O, Hogervorst M, Nauroy P, et al. QR-313, an antisense oligonucleotide, shows therapeutic efficacy for treatment of dominant and recessive dystrophic epidermolysis bullosa: a preclinical study. J Invest Dermatol. 2021;141(4):883-893. e6.
Bruckner A, Tang J, Chung W, et al. Collagen 7 (C7) protein replacement therapy (PTR-01) durably reduces wound size and symptoms in patients with recessive dystrophic epidermolysis bullosa (RDEB). J Invest Dermatol. 2022;142(8):S50.
Blau HM, Daley GQ. Stem cells in the treatment of disease. N Engl J Med. 2019;380(18):1748–60.
Naso G, Petrova A. Cellular therapy options for genetic skin disorders with a focus on recessive dystrophic epidermolysis bullosa. Br Med Bull. 2020;136(1):30–45.
Rashidghamat E, Kadiyirire T, Ayis S, et al. Phase I/II open-label trial of intravenous allogeneic mesenchymal stromal cell therapy in adults with recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol. 2020;83(2):447–54.
Niti A, Koliakos G, Michopoulou A. Stem cell therapies for epidermolysis bullosa treatment. Bioengineering (Basel). 2023;10(4):422.
Shams F, Rahimpour A, Vahidnezhad H, et al. The utility of dermal fibroblasts in treatment of skin disorders: a paradigm of recessive dystrophic epidermolysis bullosa. Dermatol Ther. 2021;34(4): e15028.
Ebens CL, McGrath JA, Riedl JA, et al. Immune tolerance of allogeneic haematopoietic cell transplantation supports donor epidermal grafting of recessive dystrophic epidermolysis bullosa chronic wounds. Br J Dermatol. 2021;184(6):1161–9.
Ebens CL, McGrath JA, Tamai K, et al. Bone marrow transplant with post-transplant cyclophosphamide for recessive dystrophic epidermolysis bullosa expands the related donor pool and permits tolerance of nonhaematopoietic cellular grafts. Br J Dermatol. 2019;181(6):1238–46.
Riedl J, Popp C, Eide C, Ebens C, Tolar J. Mesenchymal stromal cells in wound healing applications: role of the secretome, targeted delivery and impact on recessive dystrophic epidermolysis bullosa treatment. Cytotherapy. 2021;23(11):961–73.
Gostyńska KB, Yenamandra VK, Lindemans C, et al. Allogeneic haematopoietic cell transplantation for epidermolysis bullosa: the Dutch experience. Acta Derm Venereol. 2019;99(3):347–8.
Niebergall-Roth E, Dieter K, Daniele C, et al. Kinetics of wound development and healing suggests a skin-stabilizing effect of allogeneic ABCB5+ mesenchymal stromal cell treatment in recessive dystrophic epidermolysis bullosa. Cells. 2023;12(11):1468.
Riedl J, Pickett-Leonard M, Eide C, et al. ABCB5+ dermal mesenchymal stromal cells with favorable skin homing and local immunomodulation for recessive dystrophic epidermolysis bullosa treatment. Stem Cells. 2021;39(7):897–903.
Fujita Y, Komatsu M, Lee SE, et al. Intravenous injection of muse cells as a potential therapeutic approach for epidermolysis bullosa. J Invest Dermatol. 2021;141(1):198-202.e6.
Kiritsi D, Dieter K, Niebergall-Roth E, et al. Clinical trial of ABCB5+ mesenchymal stem cells for recessive dystrophic epidermolysis bullosa. JCI Insight. 2021;6(22): e151922.
Dieter K, Niebergall-Roth E, Daniele C, et al. ABCB5+ mesenchymal stromal cells facilitate complete and durable wound closure in recessive dystrophic epidermolysis bullosa. Cytotherapy. 2023;25(7):782–8.
O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21(10):585–606.
Bray ER, Kirsner RS, Badiavas EV. Mesenchymal stem cell-derived extracellular vesicles as an advanced therapy for chronic wounds. Cold Spring Harb Perspect Biol. 2022;14(10): a041227.
Goto T, Miyagawa S, Tamai K, et al. High-mobility group box 1 fragment suppresses adverse post-infarction remodeling by recruiting PDGFRα-positive bone marrow cells. PLoS ONE. 2020;15(4): e0230392.
Schwieger-Briel A, Ott H, Kiritsi D, Laszczyk-Lauer M, Bodemer C. Mechanism of Oleogel-S10: a triterpene preparation for the treatment of epidermolysis bullosa. Dermatol Ther. 2019;32(4): e12983.
Torres Pradilla M, Álvarez E, Novoa M, Lozano I, Trujillo M. Oleogel-S10 in dystrophic epidermolysis bullosa: a case series evaluating the impact on wound burden over two years. Adv Ther. 2024;41(2):867–77.
Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes) for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188(1):12–21.
Kern JS, Schwieger-Briel A, Löwe S, Sumeray M, Davis C, Martinez AE. Oleogel-S10 phase 3 study “EASE” for epidermolysis bullosa: study design and rationale. Trials. 2019;20(1):350.
Bolton L. New options to manage epidermolysis bullosa. Wounds. 2022;34(12):297–9.
Guttmann-Gruber C, Piñón Hofbauer J, Tockner B, et al. Impact of low-dose calcipotriol ointment on wound healing, pruritus and pain in patients with dystrophic epidermolysis bullosa: a randomized, double-blind, placebo-controlled trial. Orphanet J Rare Dis. 2021;16(1):473.
Wally V, Reisenberger M, Kitzmüller S, Laimer M. Small molecule drug development for rare genodermatoses—evaluation of the current status in epidermolysis bullosa. Orphanet J Rare Dis. 2020;15(1):292.
Orlowski GM, Amano SU, Flanagan KE, Rieger KE, Marinkovich MP, Wiss K. Treatment with ataluren for wound healing and health complications in a patient with junctional epidermolysis bullosa. JAMA Dermatol. 2023;159(10):1145–7.
Hammersen J, Neuner A, Wild F, Schneider H. Attenuation of severe generalized junctional epidermolysis bullosa by systemic treatment with gentamicin. Dermatology. 2019;235(4):315–22.
Osipowicz K, Wychowanski P, Nieckula P, et al. Efficacy of gentamicin 0.3% solution of oral erosions healing in patients with severe generalized recessive dystrophic epidermolysis bullosa and its impact on the expression of type VII collagen. Postepy Dermatol Alergol. 2021;38(6):979–84.
Mosallaei D, Hao M, Antaya RJ, et al. Molecular and clinical outcomes after intravenous gentamicin treatment for patients with junctional epidermolysis bullosa caused by nonsense variants. JAMA Dermatol. 2022;158(4):366–74.
Saya SB, Has C. Strategy for the optimization of read-through therapy for junctional epidermolysis bullosa with COL17A1 nonsense mutation. J Invest Dermatol. 2024. https://doi.org/10.1016/j.jid.2024.02.027.
Onoufriadis A, Proudfoot LE, Ainali C, et al. Transcriptomic profiling of recessive dystrophic epidermolysis bullosa wounded skin highlights drug repurposing opportunities to improve wound healing. Exp Dermatol. 2022;31(3):420–6.
Haghighi Javid A, Li D, Technau-Hafsi K, Has C. IL-17A immune pattern across genetic acantholytic and blistering disorders. Clin Exp Dermatol. 2023;48(5):llad012.
Uitto J, Vahidnezhad H. Losartan for treatment of epidermolysis bullosa: a new perspective. Dermatol Ther. 2021;34(1): e14638.
Pourani MR, Vahidnezhad H, Mansouri P, Youssefian L, Rakhshan A, Hajimoradi B, Abdollahimajd F, Uitto J. Losartan treatment improves recessive dystrophic epidermolysis bullosa: a case series. Dermatol Ther. 2022;35(7): e15515.
Relvas M, Figueiredo AC, Calado R, Calvão J, Ramos L, et al. Losartan as therapy for recessive dystrophic epidermolysis bullosa: report of three cases. Dermatol Ther. 2022;35(9): e15678.
Inamadar AC. Losartan as disease modulating therapy for recessive dystrophic epidermolysis bullosa. Dermatol Ther. 2020;33(6): e14279.
Elezaj V, Lura A, Canha L, Breitkreutz J. Pharmaceutical development of film-coated mini-tablets with losartan potassium for epidermolysis bullosa. Pharmaceutics. 2022;14(3):570.
Cianfarani F, De Domenico E, Nyström A, et al. Decorin counteracts disease progression in mice with recessive dystrophic epidermolysis bullosa. Matrix Biol. 2019;81:3–16.
Wu XG, Yan S, Jiang JQ, et al. Successful treatment of epidermolysis bullosa pruriginosa by dupilumab. J Dermatol. 2023;50(6):837–42.
Xará J, Relvas M, Figueiredo C, Calvão J, Batista M. Ramos L Dupilumab in the treatment of dystrophic epidermolysis bullosa: off-label use in a pediatric patient. Int J Dermatol. 2023;62(12):e617–8.
Roque Quintana B, Piqué Durán E, Pérez Cejudo JA. Successful control of recalcitrant pruritus in epidermolysis bullosa pruriginosa with dupilumab. Actas Dermosifiliogr. 2024;115(2):184–6.
Shehadeh W, Sarig O, Bar J, Sprecher E, Samuelov L. Treatment of epidermolysis bullosa pruriginosa-associated pruritus with dupilumab. Br J Dermatol. 2020;182(6):1495–7.
Zhou AG, Little AJ, Antaya RJ. Epidermolysis bullosa pruriginosa treated with dupilumab. Pediatr Dermatol. 2021;38(2):526–7.
Gewert S, Davidovic M, Has C, Kiritsi D. Dupilumab improves itch and blistering in different subtypes of epidermolysis bullosa. J Dtsch Dermatol Ges. 2024. https://doi.org/10.1111/ddg.15416.
Chen F, Guo Y, Zhou K, et al. The clinical efficacy and safety of anti-IgE therapy in recessive dystrophic epidermolysis bullosa. Clin Genet. 2022;101(1):110–5.
Kwon IJ, Kim SE, Kim SC, Lee SE. Efficacy of oral JAK1 or JAK1/2 inhibitor for treating refractory pruritus in dystrophic epidermolysis bullosa: a retrospective case series. J Dermatol. 2024;51(3):441–7.
Chiou AS, Choi S, Barriga M, et al. Phase 2 trial of a neurokinin-1 receptor antagonist for the treatment of chronic itch in patients with epidermolysis bullosa: a randomized clinical trial. J Am Acad Dermatol. 2020;82(6):1415–21.
Limmer AL, Nwannunu CE, Shah R, Coleman K, Patel RR, Mui UN, Tyring SK. Topical diacerein ointment for epidermolysis bullosa simplex: a review. Skin Therapy Lett. 2019;24(3):7–9.
Teng J, Paller AS. Bruckner AL. Diacerein 1% ointment for the treatment of epidermolysis bullosa simplex: a randomized controlled trial. J Drugs Dermatol. 2023;22(6):599–604.
Yang WS, Kang S, Sung J, Kleinman HK. Thymosin β4: potential to treat epidermolysis bullosa and other severe dermal injuries. Eur J Dermatol. 2019;29(5):459–67.
Castela E, Tulic MK, Rozières A, et al. Epidermolysis bullosa simplex generalized severe induces a T helper 17 response and is improved by apremilast treatment. Br J Dermatol. 2019;180(2):357–64.
Lee GH, Lekwuttikarn R, Tafoya E, Martin M, Sarin KY, Teng JM. Transcriptomic repositioning analysis identifies mTOR inhibitor as potential therapy for epidermolysis bullosa simplex. J Invest Dermatol. 2022;142(2):382–9.
Mayr E, Ablinger M, Lettner T, et al. 5’RNA trans-splicing repair of COL7A1 mutant transcripts in epidermolysis bullosa. Int J Mol Sci. 2022;23(3):1732.
Liemberger B, Bischof J, Ablinger M, et al. COL7A1 editing via RNA trans-splicing in RDEB-derived skin equivalents. Int J Mol Sci. 2023;24(5):4341.
So JY, Nazaroff J, Iwummadu CV, et al. Long-term safety and efficacy of gene-corrected autologous keratinocyte grafts for recessive dystrophic epidermolysis bullosa. Orphanet J Rare Dis. 2022;17(1):377.
Funding
No funding or sponsorship was received for this study or the publication of this article.
Author information
Authors and Affiliations
Contributions
Conceptualization: Cristina Has and Sorina Danescu; methodology: Cristina Has, Sorina Danescu, Mircea Negrutiu; software: Cristina Has, Sorina Danescu, Mircea Negrutiu; validation: Cristina Has; formal analysis: Cristina Has; investigation: Mircea Negrutiu; resources: Sorina Danescu; data curation: Cristina Has, Sorina Danescu; writing—original draft preparation: Sorina Danescu; writing: Sorina Danescu, Mircea Negrutiu, Cristina Has; supervision: Cristina Has.
Corresponding author
Ethics declarations
Conflict of Interest
Cristina Has has served as an advisor for Amryt Pharma and Krystal Biotech. Sorina Danescu and Mircea Negrutiu have nothing to disclose.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Danescu, S., Negrutiu, M. & Has, C. Treatment of Epidermolysis Bullosa and Future Directions: A Review. Dermatol Ther (Heidelb) 14, 2059–2075 (2024). https://doi.org/10.1007/s13555-024-01227-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-024-01227-8