
Abstract
Eleven novel clerodane-type diterpenoids, grewiifopenes A–K (1–4 and 12–18), along with nine known compounds (5–11, 19, and 20) were purified from the dichloromethane extract of the twigs and stems of Casearia grewiifolia Vent. (Salicaceae). Their spectroscopic data, including the NMR, HRESIMS, and electronic circular dichroism calculations were employed to completely characterize and elucidate the chemical structures and absolute configurations. The clerodane diterpenoids possessing a 6-OH group and no substitution at C-7 exhibited greater cytotoxic activity than others, with their IC50 values ranging from 0.3 to 2.9 μM. Isocaseamembrin E (7) exhibited antibacterial activity against Staphylococcus aureus, while isocaseamembrin E (7), corymbulosin X (8), caseargrewiin A (9), kurzipene A (10), and balanspene F (11) exhibited antibacterial activity against Bacillus cereus.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Casearia grewiifolia Vent. belongs to the Salicaceae family, previously placed in the defunct Flacourtiaceae family, and is widely distributed in the Indo-China to Queensland (Cook). In Thailand, C. grewiifolia, known as “Kruaipa”, is a perennial evergreen tree (2–20 m high), growing widely in the north and northeast of Thailand. In Thai folk medicine, the four various parts of plant, root, leaves, seeds and flowers, have been traditionally utilized to treat liver dysfunction, skin diseases, hemorrhoids, as well as antidiarrheal and antipyretic agents, a tonic and febrifuge [1, 2]. Previous chemical studies of this plant revealed that the clerodane-type diterpenes were the main components of C. grewiifolia. Most clerodane diterpenes found in the genus Casearia are bicyclic diterpenoids with a cis-fused decalin ring system and a C-11–C-16 side chain at C-9 and a five-membered ring containing oxygen atom at C-18 and C-19 [2,3,4,5,6]. These compounds have already been reported to exhibit cytotoxic, antimalarial, hypoglycemic, antiulcer, anti-inflammatory, anti-snake venom, antiparasitic, and antimycobacterial activities [3,4,5, 7,8,9]. Recently, there has been reported that the grewiifolin C, a clerodane diterpene, downregulated PCSK9 and IDOL mRNA expression [6]. In an attempt to search for structurally intriguing and bioactive clerodane diterpenes, a dichloromethane extract of C. grewiifolia has been investigated. In present work, we describe in detail of isolation and structure determination of eleven novel clerodane diterpenoids (1–4 and 12–18) along with nine known compounds (5–11, 19, and 20) and evaluation of their cytotoxicity, antibacterial activity, and aromatase inhibition.
2 Results and discussion
2.1 Structure elucidation
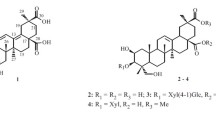
The CH2Cl2 crude extract of twigs and stems of C. grewiifolia was fractionated by chromatographic method, including silica gel and Sephadex LH20 column chromatography, and the obtained fractions were then subjected to purification by preparative TLC and HPLC, leading to the isolation and elucidation of a total of twenty compounds, which included eleven novel clerodane-type diterpenoids (grewiifopenes A–K, 1–4 and 12–18), along with nine known compounds (Fig. 1). The known compounds, corymbulosin N (5) [10], corymbulosin K (6) [10], isocaseamembrin E (7) [11], corymbulosin X (8) [12], caseargrewiin A (9) [3], kurzipene A (10) [13], balanspene F (11) [14], (−)-syringaresinol (19) [15, 16], and 4-acetonyl-3,5-dimethoxy-p-quinol (20) [17] were identified. The structure elucidation of 1–4 and 12–18 were identified by using spectroscopic data, including 1D and 2D NMR and HRESIMS data analysis. In addition, the comparisons between the calculated and the experimental electronic circular dichroism (ECD) curves were employed to determine the absolute stereochemistry of those compounds.

Chemical structures of compounds 1–20
Grewiifopene A (1) was colorless oil and gave a molecular formula of C32H46O10 by HRESIMS analysis at m/z 613.2990 [M+Na]+ (calcd for C32H46NaO10, 613.2983), corresponding to ten degrees of unsaturation. Absorption band in the IR spectrum at 1729 cm−1 suggested the present of the ester carbonyl functionality. In the 1H NMR spectrum (Table 1) of 1, the occurrence of signals for two methyl protons [δH 0.88 (d, J = 6.7 Hz, H-17) and 0.96 (s, H-20)], two acetal-acyloxy methine protons [δH 6.66 (br t, J = 1.3 Hz, H-18) and 6.47 (s, H-19)], six olefinic protons [δH 6.44 (dd, J = 17.7, 10.9 Hz, H-14), 6.00 (br dd, J = 4.1, 1.2 Hz, H-3); 5.00 (s, H-16a); 5.08 (s, H-16b); 5.15 (d, J = 17.7 Hz, H-15a); and 5.02 (d, J = 10.9 Hz, H-15b)], five aliphatic methines (including three oxygenated methines) [δH 2.37 (dd, J = 10.0, 7.3 Hz, H-10); 5.48 (br d, J = 2.4 Hz, H-2); 3.30 (d, J = 10.2 Hz, H-6); 5.19 (t-like, J = 10.9 Hz, H-7); and 1.83 (m, H-8)], three sp2 methylenes [δH 1.98 (m, H-1); 1.26 and 1.66 (m, H-11); and 2.09 (m, H-12)], three acetoxy methyls [δH 1.86 (s, at C-19); 2.06 (s, at C-18); and 2.11 (s, at C-7)], and a methoxy group [δH 3.42 (s, at C-6] were observed. Its 13C NMR spectra (Table 3) together with the help of the DEPT and HSQC data, exhibited six methyls (δC 10.9, 21.0, 21.3, 21.5, 25.9, and 62.4), five methylenes (δC 23.8, 26.9, 28.9, 112.0, and 116.1), nine methines (δC 36.0, 42.4, 65.8, 74.9, 84.5, 95.7, 98.5, 122.5, and 140.5) and seven nonprotonated carbons including three ester carbonyls (δC 169.3, 169.9, and 170.1), two olefinic carbons (δ 144.4 and 145.2), and two sp3 quaternary carbons (δC 39.0 and 53.7) (Table 3). In addition to the substituent group, a 2-methylbutanoyl side chain was detected from the resonances at δH 2.46 (sext, J = 6.9 Hz, H-2′)/δC 41.1; δH 1.57 (m, H-3′a) and 1.68 (m, H-3′b)/δC 27.0; δH 0.97 (t, J = 7.4 Hz, H-4′)/δC 11.6; δH 1.18 (d, J = 6.9 Hz, H-5′)/δC 16.6, as well as the ester carbonyl at C-1′ at δC 175.9. The data described above indicated that 1 was likely a clerodane diterpene structurally similar to the co-isolated corymbulosin N (5), which has been previously found from the bark of Laetia corymbulosa [10]. The only difference in the structure was that 1 has an additional acetoxy moiety at C-7 which could be inferred by the HMBC correlations (Fig. 2) of H-7 to the ester carbonyl (δC 169.9) and both Me-17 and H-6 to C-7 (δC 74.9). Five structural fragments were established from the 1H–1H COSY spectrum of 1 as shown in Fig. 2. The assignment of relative stereochemistry of 1 was initially attempted by using the values of 1H–1H coupling constants (Table 1) together with a NOESY experiment (Fig. 3). Consideration of the relative configurations of H-6, H-7, and H-8 in ring B were first analyzed to delineate the relationships of other protons throughout the clerodane diterpenoid 1. The strong 1,3 diaxially NOESY correlation of H-6 and H-8 and the large 3J coupling constants of H-7 (10.9 Hz) appearing as a triplet suggested the trans diaxial relationships among H-6, H-7, and H-8. In addition, the NOESY correlations of H-8 and Me-20; Me-20 and H-10; H-10 and H-12; and Me-20 and H-1 indicated cis-fused bicyclic six-membered rings, rings A and B, with an α-equatorial and an α-axial orientation for H-10 and CH2-11 of the C-11–C-16 conjugated diene moiety, respectively. The acetoxy group located at C-19 was oriented on α face, according to the NOESY cross peak between H-19/H-7. In addition, the homoallylic 5J coupling (J = 1.3 Hz) of H-18 and H-2 from the overlapping π-orbitals of the double bond with the 1s orbitals of the hydrogen atom also inferred the α-orientation for both acetoxy group at C-18 and 2-methylbutyryloxy side chain at C-2 [18,19,20]; this was also supported by the key correlations in NOESY experiment of 18-OAc/H-10, H-18/H-19, and H-18/6-OMe. Furthermore, the α-orientation of the 2-methylbutyryloxy moiety at C-2 was supported from the characteristic chemical shift at δC 65.8 (C-2); the chemical shift of C-2 with the α-oriented side chain would appear at the higher field (less than 66 ppm) than that of the β-oriented one which would appear at the lower field (greater than 70 ppm) [10, 21]. Additionally, the NOESY spectrums of H-11 and H-16; H-16 and H-14; and H-12 and H-15 suggested that the conjugated diene at C-9 was s-trans conformation. ECD comparison of its experimental with the calculated analysis (Fig. 4), thereby allowing the absolute stereochemistry of 1 as shown.

1H–1H COSY (blue) and HMBC (red) correlations of 1, 4, 13, 15, and 18

NOESY correlations of 1, 4, 15, and 18

Experimental and calculated ECD spectra of 1, 4, 13, 14, 17, and 18
Grewiifopene B (2) had the same molecular formula, C32H46O10, as that of compound 1, by HRESIMS at m/z 613.2981 [M+Na]+ (calcd for C32H46NaO10, 613.2983). Analyses of 1H and 13C NMR spectroscopic data (Tables 1 and 3) of 2 displayed close similarities to those of 1, with the only difference assignable to the presence of an isovaleryloxy moiety in 2 instead of 2-methylbutyryloxy side chain in 1, while the other moieties of 2 appear identical to those of 1. The characteristic 1H NMR resonances of an isovaleryloxy moiety at δH 2.26 (dd, J = 14.2, 6.9 Hz, H-2′a), 2.29 (dd, J = 14.2, 7.2 Hz, H-2′b), 2.13 (m, H-3′), 1.01 (d, J = 6.6 Hz, H-4′), 1.02 (d, J = 6.6 Hz, H-5′), along with the 13C NMR resonances at δC 172.4 (C-1′), 43.6 (C-2′), 26.1 (C-3′), 22.3 (C-4′), and 22.4 (C-5′) were observed in 2 and the long-range HMBC experiments (Figure S16, Supporting Information) of H-1, H-3, H-10, and H-18 to C-2 (δC 66.0); and H-2 to C-1′ (δC 172.4) suggested that this side chain is attached to C-2. Analyses of the NOESY experiment and the coupling constants in 1H NMR of 1 and 2 suggested that their relative configurations were identical (Figure S17, Supporting Information), while their closely related ECD curves of 1 established its absolute configuration (Fig. 5). Therefore, the chemical structure of 2 was purposed as shown.

Experimental ECD spectra of 1–3, 12, 15, and 16
Grewiifopene C (3) was assigned the molecular formula C33H46O11 by HRESIMS at m/z 641.2931 [M+Na]+ (calcd for C33H46NaO11, 641.2932). Analyses of 1H and 13C NMR data (Tables 1 and 3) of 3 was similar to those of 2, except for the different substituent at C-6; the appearance of an acetoxy group in 3 instead of a methoxy group in 2. This inference was implied by the downfield chemical shift of H-6 from δH 3.30 (d, J = 10.3 Hz) in 2 to 5.20 (m, overlapped) in 3, signals for 6-OAc (δH 2.07/δC 20.9) and ester carbonyl (δC 170.1) as well as the HMBC experiments of H-19, H-7, and 6-OAc to C-6 (δC 73.7) and H-6 to δC 170.1 (Figure S26, Supporting Information). Inspection of 1H–1H coupling constants and NOESY spectra of 3 and 2 and their matched ECD curves (Fig. 5) conferred the absolute stereochemistry of 3 as shown. However, the coupling constants between H-6 and H-7 of 3 in CDCl3 were difficult to measure directly due to signal overlap, but a spectrum obtained from a sample using benzene-d6 as solvent (Table 1) provided the resolution of these two positions. Therefore, the chemical structure of 3 was purposed as shown.
Grewiifopene D (4) has an identical molecular formula to that of the co-occurring corymbulosin N (5). The resonance patterns of NMR data of 4 was identical to those of corymbulosin N (5) [10], excepted for the different stereochemistry at C-2 and C-19. However, the configuration at C-2 could be deduced by comparing the 13C diagnostic shift of C-2 (δc around 66.0 for 2R; δc around 70.5 for 2S) [10, 21]. Therefore, the S-configuration at C-2 of 4 could be inferred from the 13C chemical shift at δC 70.5. The observed NOESY correlations of 19-OAc/6-OMe and H-19/H-11 (Fig. 3) implied the relative configurations of 19-OAc as β-oriented in 4. Based on these results, together with ECD comparison of its experimental with the calculated analysis (Fig. 4), its absolute stereochemistry was as shown.
Unfortunately, the stereochemistry at C-2′ of the 2-methybutyryl side chain in grewiifopenes A and D (1 and 4) could not be determined by comparing experimental ECD spectra with calculated ECD spectra method because its spectra of the 2′R and 2′S isomers of both compounds could not reveal the difference of diastereomers. These data are consistent with the previous reports for corymbulosin N (5) possessing the 2-methybutyryl side chain moiety [10]. In an effort to further study, corymbulosin N (5) was used as authentic compounds because it was available in sufficient quantity by its conversion to the corresponding 2-methylbutyric acid benzyl ester using transesterification by heating in benzyl alcohol in the presence of a catalytic amount of dimethylaminopyridine (DMAP). In addition, the two authentic samples, S- and R/S-2-methylbutyric acid benzyl ester were synthesized from S- and R/S-2-methylbutyric acid, respectively. The ester products, including the desired product from transesterification of 5 and the two benzyl-2-methylbutyrate in S- and R/S-forms, were analyzed with the chiral-phase HPLC on CHIRALPAK® AD-H column (40% CH3CN-H2O at flow rate 1 mL/min) revealing the S-configuration at C-2′ of 2-methylbutyryl side chain in corymbulosin N (5) (Fig. S118, Supporting Information). Based on the biogenesis rationale of compounds isolated from the same plant, the absolute configuration at C-2′ of 2-methylbutyryl side chain in grewiifopenes A and D (1 and 4), isocaseamembrin E (7), balanspene F (11), and grewiifopene I (16) should be established as S-configuration.
From the HRESIMS and NMR data of 12, its molecular formula was detected to be C34H50O10. The 1H and 13C NMR spectroscopic data (Tables 1 and 3) showed two sets of signals from the two isovaleryloxy groups. The comparison of the NMR data of compounds 2 and 12 indicated a structural difference by replacing the methoxy group at C-6 and an acetoxy group at C-7 in 2 with the hydroxy group and one of the isovaleryloxy groups in 12, respectively. This conclusion was in agreement with a series of the correlations of H-6/H-7/H-8/Me-17 in COSY experiment (Fig. S51, supporting information) and the characteristic correlations from H-7 to C-1′′ (δC 173.9); H-10 and H-8 to C-6 (δC 75.4); H-6 to C-19 (δC 97.9); and Me-17 to C-7 (δC 74.8), C-8 (δC 41.8), and C-9 (δC 39.1) in the HMBC experiment (Fig. S53, supporting information). The absorption in the IR spectrum at 3503 cm−1 implied the occurrence of the hydroxy moiety in 12. Based on the NOESY experiments (Fig. S54, supporting information) and 1H–1H coupling constants suggested that 12 had the same relative configurations as those of 2. Furthermore, the similarity of their ECD spectra of 12 and 2 (Fig. 5) indicated that the absolute stereochemistry of both 12 and 2 were virtually identical. Thus, compound 12 was elucidated and named as grewiifopene E.
Grewiifopene F (13) was colorless oil, and its molecular formula of C37H48O10 was determined by HRESIMS analysis, which exhibited a [M+Na]+ at m/z 675.3138 (calcd for C37H48NaO10, 675.3140). Comparison of the 1H and 13C NMR spectra (Tables 2 and 3) of 13 with 12 disclosed that compound 13 possessed a trans p-coumaroyl moiety at C-7 and a methoxy moiety at C-18 instead of the isovaleryloxy and acetoxy moieties in 12, respectively. The 1H and 13C NMR spectroscopic data (Tables 2 and 3) were consistent with the occurrence of a trans p-coumaroyl group [δH 6.27 (d, J = 15.9 Hz, H-2″), 7.62 (d, J = 15.9 Hz, H-3′′), 6.82 (d, J = 8.6 Hz, H-6′′, H-8′′), and 7.39 (d, J = 8.6 Hz, H-5′′, H-9′′); δC 167.9 (C-1″), 114.8 (C-2″), 145.39 (C-3″), 127.0 (C-4″), 130.1 (C-5″, C-9″), 115.9 (C-6″, C-8″), 158.0 (C-7″)] and a methoxy signal (δH 3.43/δC 56.3). It was evident from the HMBC experiments (Fig. 2) from H-7, H-2′′, and H-3′′ to carbonyl of ester group at δC 167.9 (C-1″) and Me-17 to C-7 (δC 75.1) that this trans p-coumaroyl group was resonated at C-7. Moreover, a methoxy group at C-18 position of 13 was evident by the correlations of H-3, 18-OMe, and H-19 to C-18 (δC 104.5). Analysis of coupling constants in 1H NMR and NOESY experiments suggested that 13 had the same relative configurations as those of 12. Furthermore, ECD comparison of its experimental with the calculated analysis (Fig. 4), and its absolute stereochemistry was assigned as shown.
From the HRESIMS and NMR spectra of 14, its molecular formula was determined to be C38H48O11. Analyses of the 1H and 13C NMR data (Tables 1 and 3) of 14 were closely similar to those of 13, excepted that a methoxy group at C-18 was substituted with an acetoxy group in 14. This evidence was verified by the downfield shift of H-18 from δH 5.49 (t, J = 1.5 Hz) in 13 to 6.74 (s) in 14. NOESY experiments (Fig. S74, supporting information) and 1H–1H coupling constants of 13 and 14 indicated the identical relative stereochemistry of 13 and 14, except for the H-18 position, which resonated as a singlet due to the disappearance of homoallylic 5J coupling between H-2 and H-18 and no cross peak between H-18 and H-19 in the NOESY spectrum. This, in turn, would suggest the α-orientation for H-18; the other configuration of 14 was assigned to be the same as 13 based on the NOESY experiments and 1H–1H coupling constants. Consequently, the absolute stereochemistry of 14 was finally deduced as shown by the ECD calculation method (Fig. 4). Therefore, the structure of 14 (grewiifopene G) was assigned.
Grewiifopene H (15) was assigned the molecular formula C33H46O11 by HRESIMS and 1H and 13C NMR and DEPT experiments. The chemical structure of 15 was nearly identical to that of 3, except that the resonances for 3-methylenepent-1-ene unit at C-9 in 3 were replaced by resonances for a 3-methylpenta-1,3-diene moiety in 15. The above assignment was further supported by the 1H NMR spectrum (Table 1), which showed some characteristic signals two double bonds [including a terminal monosubstituted olefin (δH 6.64 (dd, J = 17.2, 10.8 Hz, H-14), 5.15 (d, J = 10.8 Hz, H-15a) 5.23 (d, J = 17.2 Hz, H-15b) and one trisubstituted olefin (δH 5.25 (m, H-12)], an olefinic methyl group [δH 1.82 (s, Me-16)], and nonequivalent methylene protons [δH 1.68 (d, J = 16.1 Hz, H-11a and 2.56 (dd, J = 16.1, 9.3 Hz, H-11b)]. Further HMBC experiments (Fig. 2) from H-11 to C-9 (δC 39.1), C-10 (δC 36.9), Me-20 (δC 25.4), C-13 (δC 134.1); Me-16 to C-12 (δC 125.4), C-13, and C-14 (δC 133.2); H-14, H-15, and Me-16 to C-13 confirmed the 3-methylpenta-1,3-diene at C-9 in 15. The trisubstituted double bond at C12/C-13 for compound 15 was assigned with the Z geometry due to the NOESY cross peak between H-12 and Me-16 (Fig. 3). Moreover, the Z-configuration was confirmed by comparing the 13C NMR chemical shift at δC 20.4 for 16-Me in compound 15 with the available diagnostic data for Z-(δC 20.3) and E-(δC 12.0) double bonds [18, 19, 21]. With the similar ECD curves (Fig. 5), NOESY experiments (Fig. 3), and 1H–1H coupling constants, compound 15 was deduced to possess the same absolute configurations as those of 3.
Grewiifopene I (16) was assigned the molecular formula C29H42O8, by HRESIMS at m/z 541.2782, [M+Na]+. The IR absorptions at 3456 cm−1 for the hydroxy and 1749, and 1729 cm−1 for ester carbonyl functionalities were detected. Analyses of the 1H and 13C NMR spectra (Tables 1 and 3) of 16 showed resonances similar to those of the co-occurring isocaseamembrin E (7) [11], except for the structural differences in the C-11–C-16 side chain moiety at C-9, which was identified as (Z)-3-methylpenta-1,3-diene. The relative stereochemistry of 16 was virtually similar to that of isocaseamembrin E (7) by analyzing their coupling constants in 1H NMR and NOESY correlations (Fig. S94, supporting information). The comparison of the ECD curves of compound 16 with those of structural analogs 1, 2, 3, 12, and 15 (Fig. 5) penultimately assigned its absolute stereochemistry as shown.
Grewiifopene J (17) was isolated as a colorless oil. Its HRESIMS data gave a molecular formula of C38H48O11. Analyses of its 1H and 13C NMR spectra (Tables 2 and 3) of 17 displayed the same skeleton as 12, with the structural differences at C-9 and C-7. The NMR data exhibited resonances for (Z)-3-methylpenta-1,3-diene moiety at C-9 and trans p-coumaroyloxy group at C-7 in 17 instead of the 3-methylenepent-1-ene side chain and isovaleryloxy group, respectively, in 12. On the basis of the NOESY experiments (Fig. S104, supporting information) and 1H–1H coupling constants, compound 17 had the same relative stereochemistry as those of 12. The absolute stereochemistry of 17 was established as shown by using ECD comparison of its experimental with the calculated analysis.
Grewiifopene K (18) had the same molecular formula of C38H48O11 as 17 based on a sodium ion adduct peak in the HRESIMS spectrum, indicating that 18 is isomeric with 17. The closely matched 1H and 13C NMR spectra (Tables 2 and 3) suggested that 18 possessed the same planar structure as 17, but they have differences in the geometry of the C-12/C-13 tri-substituted alkene. The NOESY cross peaks (Fig. 3) between H-12 and H-14; H-11 and Me-16 and the characteristic 13C chemical shift of the olefinic methyl moiety (Me-16; δC 12.1) indicated its E-configuration [21]. Both the NOESY experiments (Fig. S114, supporting information) and 1H–1H coupling constants as well as ECD curves (Fig. 4) suggested that 18 had the same absolute configurations as those of 17.
2.2 Cytotoxicity, antibacterial activities, and inhibition of aromatase of some isolated compounds
The in vitro cytotoxicity assay toward a panel of human cancer cell lines of several clerodane diterpenoids has been well investigated [3,4,5, 7, 10, 21, 22]. Thus, the isolates in adequate amounts were tested against MOLT-3 (acute lymphoblastic leukemia), HuCCA-1 (cholangiocarcinoma), HepG2 (hepatocellular carcinoma), A549 (lung adenocarcinoma), HeLa (cervical carcinoma), and MDA-MB-231 (triple negative breast cancer) cancer cell lines using the MTT and XTT methods. Doxorubicin and etoposide were used as positive controls. The cytotoxicity results as shown in Table 4 revealed that the clerodane diterpenoids 7, 8, and 16, with a free hydroxy moiety at C-6 and no substituent at C-7, exhibited strong cytotoxicity with IC50 values in the range of 0.3 to 2.9 µM and exhibited a greater cytotoxic activity than those with either the methoxy or acetoxy groups at C-6. These data are consistent with the previous reports that the hydroxy substituted at C-6 was very important for enhancing cytotoxic activity [7, 23]. Cytotoxicity toward A549 cell line of compounds 7 and 16 (both with the IC50 = 0.3 μM) and toward MDA-MB-231 cell line of compounds 7, 8, and 16–18 (IC50 = 2.0–2.2 μM) was virtually comparable to that of a standard, etoposide. Furthermore, compounds 8 and 17 were two-fold more efficacious than etoposide against HuCCA-1 cell line. Interestingly, selective cytotoxicity toward MOLT-3 (IC50 = 3.7 µM) was observed for 13, while the other cell lines tested (HuCCA-1, A549, HeLa, and MDA-MB-231) were found to exhibit only weak activities. When evaluated against HepG2, the IC50 values of the tested clerodane diterpenoids, except for the compounds 2, 13, and 15, ranged from 1.9 to 14.3 μM; compound 7 (IC50 = 1.9 μM) was the most cytotoxic. The tested clerodane diterpenoids, except for 18 and 19, exhibited moderate cytotoxicity toward MOLT-3 cell line with IC50 values ranging from 1.0 to 5.0 µM. Structural differences in the six-carbon conjugated diene side chain, including (E)/(Z)-3-methylpenta-1,3-diene and 3-methylpenta-1,3-diene at C-9 in compounds 14, 17, and 18 did not significantly affect cytotoxicity. Moreover, the replacement of an acetoxy group at C-18 with a methoxy group in compounds 13 and 10 rendered them less active. (−)-Syringaresinol (19), a known lignan, was inactive against all cell lines, even at 50 µg/mL. 4-Acetonyl-3,5-dimethoxy-p-quinol (20) exhibited weak cytotoxicity against only MOLT-3 (IC50 = 10.0 μM) and MDA-MB-231 (IC50 = 24.4 μM) cell lines.
In addition, the isolates were also evaluated for their antibacterial activity towards three strains including Bacillus cereus (ATCC 11778), Staphylococcus aureus (ATCC 6538) and Staphylococcus epidermidis (ATCC 12228) using penicillin G and gentamicin as the antibiotic controls (Table 5). However, due to insufficient materials for testing, only compounds 2, 3, 7–11, 15, 19, and 20 were subjected to evaluation. Most of the tested compounds, except for compound 7, did not show significant activity towards S. aureus and S. epidermidis. Compound 7 exhibited antibacterial activity against only S. aureus with the MIC and MBC values of 25 (50) µg/mL. Compounds 7–11 exhibited antibacterial activity toward B. cereus with MIC and MBC values of 6.25 (> 200), 25 (50), 100 (100), 12.5 (12.5), and 12.5 (25.0) μg/mL, respectively. Finally, the inhibitory activity of aromatase by using a CYP19 screening kit was investigated. The aromatase (also called estrogen synthase) is responsible for converting the non-aromatic ring A of androgens to estrogens, which are related to the proliferation of breast cancer cells. However, almost all the tested compounds (3, 5, 7, 9, 11, 12, 19, and 20) did not show activity at the concentration up to 12.15 µM (Table S11, supporting information).
3 Conclusions
In summary, a total of twenty isolates, including eleven novel clerodane diterpenoids, grewiifopenes A–K, as well as nine known compounds were isolated from the dichloromethane extract of C. grewiifolia. Their structures were determined by extensive spectroscopic analysis. Meanwhile, the absolute stereochemistry was established by comparing the experimental and calculated ECD data. A biological evaluation of some isolated compounds revealed that all tested clerodane diterpenoids possessed cytotoxic activities against a panel of cancer cell lines. On the other hand, only compound 7 exhibited antibacterial activity against S. aureus, while compounds 7–11 exhibited antibacterial activity against B. cereus. None of the tested compounds inhibited the aromatase. To the best of our knowledge, this is the first report on the antibacterial activity and inhibition of aromatase of clerodane diterpenoids from C. grewiifolia.
4 Experimental
4.1 General experimental procedures
A JASCO P-1020 digital polarimeter measured at 590 nm (Na lamp D line) was used to measure the specific optical rotation values. A JASCO J-815 spectropolarimeter was utilized to provide the ECD spectra. A Perkin–Elmer Spectrum One spectrometer [attenuated total reflectance (ATR)] was used to obtain FT-IR spectra. A Bruker AVANCE 600 spectrometers was used to record 1D and 2D NMR spectra. A Bruker MicroTOFLC spectrometer was used to record the ESITOFMS (positive mode) data. The stationary phases for column chromatography (CC) are silica gel 60 (Merck, 70–200 mesh ASTM), Sephadex LH-20 gel (Ambersham Biosciences), and silica gel 60 RP-18 (Merck, 40–63 µm). Preparative TLC was performed with silica gel 60 PF254. TLC plate detection was carried out on silica gel precoated aluminum-backed plate (silica gel 60 F254, Merck). A VertiSep™ UPS RP-C18 column (21.2 × 250 mm) on Waters 600 Delta HPLC instrument connected to a Water photodiode array detector was used for purification with the eluent flow rate at 12 mL/min.
4.2 Plant materials
The twigs and stems of Casearia grewiifolia Vent. (Salicaceae) were collected from the Sai Yok National Park, Kanchanaburi Province, Thailand (14.2495, 99.0270) in December 2020. One of the authors (T.T.) authenticated the plant, and a voucher specimen (SY16) has been deposited at the Forest Herbarium, Department of National Parks, Wildlife and Plant Conservation, Thailand.
4.3 Extraction and isolation
Air dried twigs and stems powder of C. grewiifolia (8.9 kg) were macerated two times with CH2Cl2 at ambient temperature, and the resulting solvent was concentrated under vacuum to afford 50.0 g of the CH2Cl2 extract. A 48 g of CH2Cl2 extract was developed to column chromatography (CC) with silica gel with the mobile phase of hexane–acetone (a gradient from 100:0 to 0:100) and acetone-MeOH (a gradient from 100:0 to 0:100) provide sixteen fractions (A1–A16). The Sephadex LH-20 CC was then applied for fraction A6 (13.6 g) using CH2Cl2–MeOH in a ratio of 1:1 as eluent to give five fractions (B1–B5). Fraction B3 (2.7 g), was fractionated by silica gel CC with the mobile phase of hexane–EtOAc (a gradient from 100:0 to 0:100) providing seven fractions (C1–C7). A gradient elution of hexane and acetone was applied to silica gel CC for further separated fraction C5 (1.5 g) to provide eleven fractions (D1–D11). Using HPLC approach for purification of fraction D3 (69.0 mg) and eluting with a CH3CN–H2O (70:30 to 100:0 for 180 min) gave compounds 10 (2.0 mg) and 11 (2.6 mg). Compound 4 (2.0 mg) was afforded from the purification of fraction D5 (1.5 g) by HPLC, using an isocratic system of CH3CN in H2O (73:27) as eluent. Compounds 6 (1.3 mg), 9 (25.6 mg), and 5 (28.5 mg) were obtained from fraction D6 (782.5 mg) by the RP 18 HPLC column using an isocratic system of CH3CN–H2O (72:27) as mobile phase. Fraction B4 (3.4 g) was chromatographed by CC with gel filtration, Sephadex LH-20, eluting with the mobile phase of CH2Cl2–MeOH in ratio of 1:1., then followed by RP 18 HPLC column, eluting with an isocratic system of CH3CN–H2O (65:35) to give additional yields of compounds 11 (5.7 mg), 9 (21.5 mg), and 5 (42.6 mg). A 1.6 g of fraction A8 was subjected by CC with a gel filtration, Sephadex LH-20, eluting with CH2Cl2–MeOH in a ratio of 1:1 and then purified by silica gel CC with a gradient of MeOH in CH2Cl2 as eluent to afford sixteen fractions (E1–E16). After purifying fraction E10 (15.9 mg) with RP 18 HPLC column, using a mobile phase of MeOH–H2O (a gradient from 60:40 to 80:20 in 140 min), compounds 3 (19.5 mg) and 15 (8.5 mg) were obtained. The eluent of CH2Cl2–MeOH (1:1) was applied on Sephadex LH-20 CC for the fractionations of fraction E12 (231.9 mg) to give three fractions (F1–F3). Subsequently, fraction F2 (139.9 mg) was applied on preparative TLC plates, eluting with the mobile phase of hexane:acetone:EtOAc in a ratio of 8:1:1 yielding the three bands (G1–G3). Compound 12 (17.5 mg) was also obtained from band G1, while band G2 (47.4 mg) was subjected to RP 18 HPLC column using a mixture of CH3CN in H2O (a gradient from 70:30 to 80:20 for 60 min) as eluent to give compounds 2 (6.4 mg) and 1 (1.8 mg). Compounds 8 (11.0 mg), 7 (29.5 mg), and 16 (6.5 mg) was obtained from fraction E13 (181.2 mg), purifying by RP 18 HPLC column with a gradient elution of CH3CN in H2O (60:40 to 80:20 for 90 min) at flow rate of 10 mL/min. The Sephadex LH-20 CC with eluent of CH2Cl2–MeOH (1:1) and silica gel CC with a gradient elution of MeOH in CH2Cl2 were used for fractionation of fraction A12 (5.9 g), yielding twelve fractions (H1–H12). Fraction H10 (424.4 mg) was applied on preparative TLC plates, eluting with a mixture of hexane:acetone:MeOH in the ratio of 70:30:1 to provide three bands (I1–I3). Band I2 (152.4 mg) was further purified with HPLC with a gradient elution of CH3CN in H2O (65:35 to 70:30 for 140 min) to afforded compounds 18 (3.0 mg) and 14 (2.5 mg). Compound 17 (8.2 mg) was received from band I3 (198.5 mg) by HPLC, eluting with an isocratic system of CH3CN–H2O (65:35). Purification of fraction H11 (709.3 mg) with RP 18 HPLC column which was eluted with an isocratic system of CH3CN in H2O (80:20) afford compounds 14 (20.0 mg) and 13 (6.0 mg). The eluent, a mixture of CH2Cl2 and MeOH (1:1), was applied to Sephadex LH-20 CC for the purification of fraction A13 (3.06 g) provided four fractions (J1–J4). Further purification of fraction J3 (1.1 g) by silica gel CC with a gradient elution of MeOH in CH2Cl2 provided seven fractions (K1–K7) were obtained. Compounds 19 (23.5 mg) and 20 (21.0 mg) were purified from fractions K4 (52.0 mg) and K6 (35.0 mg), respectively, were applied on preparative TLC plates, eluting with MeOH:CH2Cl2 in the ratio of 1:99 and 2:98 respectively.
4.4 Compound characterization
4.4.1 Grewiifopene A (1)
Optically active colorless oil; [α] 25D + 29.6 (c 0.30, CHCl3); ECD (c 0.06 μM, acetonitrile) λmax (Δε): 204 (+ 30.94), 226 (− 0.98), 236 (+ 0.45); IR (ATR) νmax 2970, 2933, 1729, 1456, 1373, 1226, 1182, 1138, 1098, 1074, 1019, 951, 897, 733 cm−1; HRESIMS (positive mode) m/z 613.2990 (calcd for C32H46NaO10 [M+Na]+, 613.2983).
4.4.2 Grewiifopene B (2)
Optically active colorless oil; [α] 25D + 52.0 (c 0.67, CHCl3); ECD (c 0.06 μM, acetonitrile) λmax (Δε): 203 (+ 20.34), 223 (+ 0.24), 228 (− 0.29), 243 (+ 0.32); IR (ATR) νmax 2959, 2931, 1733, 1457, 1372, 1226, 1184, 1096, 1074, 1019, 998, 979, 950, 897, 734 cm−1; HRESIMS (positive mode) m/z 613.2981 (calcd for C32H46NaO10 [M+Na]+, 613.2983).
4.4.3 Grewiifopene C (3)
Optically active colorless oil; [α] 25D + 58.7 (c 2.30, CHCl3); ECD (c 0.06 μM, acetonitrile) λmax (Δε): 191 (+ 24.76), 227 (+ 0.60), 237 (+ 1.42); IR (ATR) νmax 2960, 2930, 1744, 1371, 1217, 1167, 1100, 1071, 1023, 999, 946, 917, 900, 734 cm−1; HRESIMS (positive mode) m/z 641.2931 (calcd for C33H46NaO11 [M+Na]+, 641.2932).
4.4.4 Grewiifopene D (4)
Optically active colorless oil; [α] 25D − 108.3 (c 0.07, CHCl3); ECD (c 0.28 μM, acetonitrile) λmax (Δε): 200 (− 2.33), 220 (− 0.34); IR (ATR) νmax 2965, 2930, 1755, 1731, 1454, 1374, 1225, 1179, 1151, 1100, 1076, 1024, 995, 936, 898 cm−1; HRESIMS (positive mode) m/z 555.2932 (calcd for C30H44NaO8 [M+Na]+, 555.2928).
4.4.5 Grewiifopene E (12)
Optically active colorless oil; [α] 25D + 66.2 (c 1.72, CHCl3); ECD (c 0.05 μM, acetonitrile) λmax (Δε): 200 (+ 28.49), 229 (+ 0.70), 236 (+ 1.17), 238 (+ 1.05), 250 (+ 0.62); IR (ATR) νmax 3503, 2959, 2927, 1733, 1465, 1457, 1371, 1291, 1226, 1188, 1167, 1093, 1023, 996, 956, 902, 735 cm−1; HRESIMS (positive mode) m/z 641.3303 (calcd for C34H50NaO10 [M+Na]+, 641.3296).
4.4.6 Grewiifopene F (13)
Optically active colorless oil; [α] 25D + 26.8 (c 0.41, CHCl3); ECD (c 0.06 μM, acetonitrile) λmax (Δε): 200 (+ 6.29), 229 (− 0.22), 279 (+ 0.61), 312 (+ 0.90); IR (ATR) νmax 3388, 2957, 2925, 2854, 1724, 1632, 1604, 1515, 1446, 1372, 1264, 1168, 1097, 1044, 1006, 991, 948, 900, 833, 731 cm−1; HRESIMS (positive mode) m/z 675.3138 (calcd for C37H48NaO10 [M+Na]+, 675.3140).
4.4.7 Grewiifopene G (14)
Optically active colorless oil; [α] 25D + 54.1 (c 0.39, CHCl3); ECD (c 0.07 μM, acetonitrile) λmax (Δε): 190 (+ 15.69), 201 (+ 7.82), 206 (+ 7.93), 231 (+ 0.31), 299 (+ 0.72), 340 (+ 0.02); IR (ATR) νmax 3406, 2960, 2928, 1711, 1632, 1604, 1515, 1443, 1373, 1227, 1168, 1094, 1024, 996, 903, 833, 735 cm−1; HRESIMS (positive mode) m/z 703.3098 (calcd for C38H48NaO11 [M+Na]+, 703.3089).
4.4.8 Grewiifopene H (15)
Optically active colorless oil; [α] 25D + 58.7 (c 0.98, CHCl3); ECD (c 0.06 μM, acetonitrile) λmax (Δε): 196 (+ 23.82), 239 (− 1.61), 259 (+ 0.47); IR (ATR) νmax 2963, 2930, 1748, 1434, 1370, 1216, 1105, 1071, 1060, 999, 982, 962, 931, 917 cm−1; HRESIMS (positive mode) m/z 641.2943 (calcd for C33H46NaO11 [M+Na]+, 641.2932).
4.4.9 Grewiifopene I (16)
Optically active colorless oil; [α] 25D + 40.7 (c 0.62, CHCl3); ECD (c 0.07 μM, acetonitrile) λmax (Δε): 204 (+ 19.80), 246 (− 0.30), 256 (+ 0.26); IR (ATR) νmax 3456, 2965, 2930, 2878, 1749, 1729, 1461, 1373, 1227, 1177, 1150, 1099, 1074, 1023, 1002, 959, 946, 925, 890, 735 cm−1; HRESIMS (positive mode) m/z 541.2782 (calcd for C29H42NaO8 [M+Na]+, 541.2772).
4.4.10 Grewiifopene J (17)
Optically active colorless oil; [α] 25D + 116.7 (c 0.46, CHCl3); ECD (c 0.06 μM, acetonitrile) λmax (Δε): 209 (+ 9.20), 238 (− 5.48), 290 (+ 2.36); IR (ATR) νmax 3407, 2962, 2929, 1727, 1632, 1604, 1515, 1441, 1371, 1329, 1226, 1168, 1093, 1024, 998, 981, 960, 918, 891, 833, 734 cm−1; HRESIMS (positive mode) m/z 703.3090 (calcd for C38H48NaO11 [M+Na]+, 703.3089).
4.4.11 Grewiifopene K (18)
Optically active colorless oil; [α] 25D + 74.0 (c 0.29, CHCl3); ECD (c 0.09 μM, acetonitrile) λmax (Δε): 198 (+ 8.60), 223 (+ 0.10), 236 (− 1.91); IR (ATR) νmax 3374, 2958, 2926, 1728, 1632, 1604, 1515, 1448, 1372, 1227, 1161, 1095, 1025, 998, 894, 834, 736 cm−1; HRESIMS (positive mode) m/z 703.3090 (calcd for C38H48NaO11 [M+Na]+, 703.3089).
4.5 Preparation of 2-methylbutyric acid benzyl esters
A solution of benzyl alcohol (31.8 mg, 0.3 mmol) in CH2Cl2 1 mL was added to R/S-2-methylbutanoic acid (20 mg, 0.2 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (57 mg, 0.3 mmol), and dimethylaminopyridine (DMAP) (1 mg, 0.008 mmol). The reaction mixture was stirred at room temperature for 3 h, then evaporated and purified by PTLC with 5% EtOAc-hexnae to afford R/S-2-methylbutyric acid benzyl esters (32 mg, 85%): colorless oil; 1H NMR (400 MHz, CDCl3) 7.37–7.29 (m, H-2–H-6), 5.12 (s, H2-7), 2.45 (sext, J = 7.0 Hz, H-2′), 1.70 (m, J = 7.4 Hz H-3′a), 1.49 (m, H-3′b), 1.16 (d, J = 7.0 Hz, Me-5′), 0.89 (t, J = 7.5 Hz, Me-4′); 13C NMR (100 MHz, CDCl3): 176.6 (C-1′), 136.3 (C-1), 128.5 (C-2, C-6), 128.07 (C-3, C-5), 128.1 (C-4), 66.0 (C-7), 41.1 (C-2′), 26.8 (C-3′), 16.6 (C-5′), 11.6 (C-4′) (Figs. S119 and S120, Supporting Information). The corresponding S-2-methylbutyric acid benzyl ester (30 mg, 85%) was obtained by the same procedure using (S)-2-methylbutanoic acid.
4.6 Transesterification of compound 5
Corymbulosin N (5) (28.1 mg, 0.053 mmol), benzyl alcohol (0.5 mL, 4.8 mmol), and 4-dimethylaminopyridine (0.65 mg, 0.053 mmol) was heated to 120 °C in seal tube and stirred for 48 h. The reaction was allowed to cool to room temperature and then was purified with column chromatography with 5% EtOAc in hexane as eluent to remove a large amount of benzyl alcohol. The resulting was analyzed by chiral HPLC.
4.7 ECD computational methods
Spartan’ 20 with MMFF94 molecular mechanics model (within 5 kcal/mol energy window) was used for a conformational search and Guassian 16 Rev. C.01 program was used for all DFT calculations [24, 25]. Further optimization of the obtained low-energy conformers was calculated using wB97XD/cc-PVDZ level of theory with IEFPCM of methanol solvent model. All optimized conformers were confirmed to be a true minimum of electronic potential energy by means of the vibrational frequency calculation at the same level without any detection of imaginary frequencies. Each conformer with the over 2% population was subjected to the ECD calculations, according to Boltzmann distribution law base on Gibbs free energies. Using TD-DFT calculations at the CAM-B3LYP/def2-SVP level of theory and the application of the IEFPCM of methanol solvent model [26] were computed the simulated ECD spectra of 1, 4, 8, and 15. Thirty excited states of each conformer were calculated and the resulting ECD curves were developed using SpecDis with Boltzmann average all conformers and overlapping Gaussian function with an exponential half-width (σ = 0.35) [27, 28]. For the related enantiomers of 1, 4, 8, and 15, their theoretical ECD spectra derived from the direct inversion of their simulated ECD spectra.
4.8 Cytotoxicity assays
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay [29] was employed for the cytotoxic evaluation against five human cancer cell lines, including HepG2, A549, MDA-MB-231, HeLa, and HuCCA-1, while the XTT (2,3-bis-(2-methoxy-4-nitro-5-sulphenyl)-(2H)-tetrazolium-5-carboxanilide) method [30] was used for MOLT-3 cancer cell. Doxorubicin and etoposide were used as positive controls (Table 4).
4.9 Antibacterial susceptibility testing
4.9.1 Tested microorganisms
The tested strains, Staphylococcus aureus (S. aureus) ATCC6538, Staphylococcus epidermidis (S. epidermidis) ATCC12228, and Bacillus cereus (B. cereus) ATCC11778 were obtained from the American Type Culture Collection (ATCC).
4.9.2 Inoculum preparation
All tested species were grown on Mueller Hinton Agar (MHA) and incubated at 37 °C for 18–24 h. The bacterial colonies suspension in sterile normal saline was diluted until they reached a 1 × 106 CFC/mL concentration, which was then compared to the McFarland scale. This step provided a standard bacterial suspension (1 × 106 CFU/mL), which was used for the following assays.
4.9.3 Determination of minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC)
The broth microdilution technique was used for the MIC and MBC determination, which was expressed according to Clinical and Laboratory Standards Institute (CLSI) standard M07 guideline [31]. The compounds were prepared as a stock solution by dissolving in dimethyl sulfoxide (DMSO) with the concentration ranging from 0.195 to 200 µg/mL. DMSO and penicillin G or gentamicin were used as normal and standard drug control. The minimum inhibitory concentration (MIC) refers to the lowest antibacterial compound concentration at which no visual growth was observed, while the complete death of bacteria compared to the initial bacterial inoculum refers to the MBC (minimum bactericidal concentration). Three replicates were performed for each experiment.
4.10 Aromatase (CYP19) inhibition assay
The aromatase assay used a method previously designed by Stresser and co-workers [32]. Letrozole, a positive control, exhibited an IC50 value of 1.4 ± 0.3 nM for CYP19 inhibition.
Availability of data and materials
All relevant data supporting the results are provide in the manuscript and its Additional files.
References
Chuakul W, Saralamp P, Paonil W, Temsiririrkkul R, Clayton T. Medicinal plants in Thailand, vol. II. Bangkok: Amarin Printing and Publishing Public Co., Ltd.; 1997. p. 52–3.
Xia L, Guo Q, Tu P, Chai X. The genus Casearia: a phytochemical and pharmacological overview. Phytochem Rev. 2015;14:99–135. https://doi.org/10.1007/s11101-014-9336-6.
Kanokmedhakul S, Kanokmedhakul K, Kanarsa T, Buayairaksa M. New bioactive clerodane diterpenoids from the bark of Casearia grewiifolia. J Nat Prod. 2005;68:183–8. https://doi.org/10.1021/np049757k.
Kanokmedhakul S, Kanokmedhakul K, Buayairaksa M. Cytotoxic clerodane diterpenoids fruits of Casearia grewiifolia. J Nat Prod. 2007;70:1122–6. https://doi.org/10.1021/acs.jnatprod.5b00677.
Nguyen HTT, Truong NB, Doan HTM, Litaudon M, Retailleau P, Do TT, Nguyen HV, Chau MV, Pham CV. Cytotoxic clerodane diterpenoids from the leaves of Casearia grewiifolia. J Nat Prod. 2015;78:2726–30. https://doi.org/10.1021/acs.jnatprod.5b00677.
Nhoek P, An C-Y, Son M-G, Chae H-S, Pel P, Kim Y-M, Khiev P, Choi WJ, Choi YH, Chin Y-W. Stereochemical assignment of clerodane-type diterpenes from the fruits of Casearia grewiifolia and their ability to inhibit PCSK9 expression. Phytochemistry. 2023;216: 113864. https://doi.org/10.1016/j.phytochem.2023.113864.
Shen Y-C, Wang C-H, Cheng Y-B, Wang L-T, Guh J-H, Chien C-T, Khalil AT. New cytotoxic clerodane diterpenoids from the leaves and twigs of Casearia membranacea. J Nat Prod. 2004;67:316–21. https://doi.org/10.1021/np0303658.
Bou DD, Tempone AG, Pinto ÉG, Lago JHG, Sartorelli P. Antiparasitic activity and effect of casearins isolated from Casearia sylvestris on Leishmania and Trypanosoma cruzi plasma membrane. Phytomedicine. 2014;21:676–81. https://doi.org/10.1016/j.phymed.2014.01.004.
Williams RB, Norris A, Miller JS, Birkinshaw C, Ratovoson F, Andriantsiferana R, Rasamison VE, Kingston DGI. Cytotoxic clerodane diterpenoids and their hydrolysis products from Casearia nigrescens from the rainforest of Madagascar. J Nat Prod. 2007;70(2):206–9. https://doi.org/10.1021/np0605034.
Aimaiti S, Suzuki A, Saito Y, Fukuyoshi S, Goto M, Miyake K, Newman DJ, O’Keefe BR, Lee K-H, Nakagawa-Goto K. Corymbulosins I−W, cytotoxic clerodane diterpenes from the bark of Laetia corymbulosa. J Org Chem. 2018;83:951–63. https://doi.org/10.1021/acs.joc.7b02951.
Ledoux A, Hamann C, Bonnet O, Jullien K, Quetin-Leclercq J, Tchinda A, Smadja J, Gauvin-Bialecki A, Maquoi E, Frédérich M. Bioactive clerodane diterpenoids from the leaves of Casearia coriacea Vent. Molecules. 2023;28:1197. https://doi.org/10.3390/molecules28031197.
Cai S, Risinger AL, Petersen CL, Grkovic T, O’Keefe BR, Mooberry SL, Cichewicz RH. Anacolosins A−F and corymbulosins X and Y, clerodane diterpenes from Anacolosa clarkii exhibiting cytotoxicity toward pediatric cancer cell lines. J Nat Prod. 2019;82:928–36. https://doi.org/10.1021/acs.jnatprod.8b01015.
Ma J, Yang X, Zhang Q, Zhang X, Xie C, Tuerhong M, Zhang J, Jin DQ, Lee D, Xu J, Ohizumi Y, Guo Y. Cytotoxic clerodane diterpenoids from the leaves of Casearia kurzii. Bioorg Chem. 2019;85:558–67. https://doi.org/10.1016/j.bioorg.2019.01.048.
Xu J, Zhang Q, Wang M, Ren Q, Sun Y, Jin D-Q, Xie C, Chen H, Ohizumi Y, Guo Y. Bioactive clerodane diterpenoids from the twigs of Casearia balansae. J Nat Prod. 2014;77:2182–9. https://doi.org/10.1021/np5003516.
Yamauchi S, Ichikawa H, Nishiwaki H, Shuto Y. Evaluation of plant growth regulatory activity of furofuran lignan bearing a 7,9′:7′,9-diepoxy structure using optically pure (+)- and (−)-enantiomers. J Agric Food Chem. 2015;63:5224–8. https://doi.org/10.1021/acs.jafc.5b01099.
Ito A, Kasai R, Yamasaki K, Duc NM, Nham NT. Lignan glycosides from bark of Albizzia myriophylla. Phytochemistry. 1994;37(5):1455–8. https://doi.org/10.1016/s0031-9422(00)90432-1.
Luo JR, Jiang HE, Zhao YX, Zhou J, Qian JF. Components of the heartwood of Populus euphratica from an ancient tomb. Chem Nat Compd. 2008;44:6–9. https://doi.org/10.1007/s10600-008-0003-2.
Itokawa H, Totsuka N, Morita H, Takeya K, Iitaka Y, Schenkel EP, Motidome M. New antitumor principles, casearins A–F, for Casearia sylvestris Sw. (Flacourtiaceae). Chem Pharm Bull. 1990;38(12):3384–8. https://doi.org/10.1248/cpb.38.3384.
Santos AG, Ferreira PMP, Vieira-Junior GM, Perez CC, Tininis AG, Silva GH, Bolzani VS, Costa-Lotufo LV, Pessoa C, Cavalheiro AJ. Casearin X, its degradation product and other clerodane diterpenes from leaves of Casearia sylvestris: evaluation of cytotoxicity against normal and tumor human cells. Chem Biodivers. 2010;7:205–15. https://doi.org/10.1002/cbdv.200800342.
Guittet E, Stoven V, Lallemand J-Y. Pitumbin, a novel kolavene acylal from Casearia pitumba pleumer. Tetrahedron. 1988;44:2893–901. https://doi.org/10.1016/S0040-4020(88)90026-9.
Liu F, Ma J, Shi Z, Zhang Q, Wang H, Li D, Song Z, Wang C, Jin J, Xu J, Tuerhong M, Abudukeremu M, Shuai L, Lee D, Guo Y. Clerodane diterpenoids isolated from the leaves of Casearia graveolens. J Nat Prod. 2020;83:36–44. https://doi.org/10.1021/acs.jnatprod.9b00515.
Prakash CVS, Hoch JM, Kingston DGI. Structure and stereochemistry of new cytotoxic clerodane diterpenoids from the bark of Casearia lucida from the Madagascar Rainforest. J Nat Prod. 2002;65:100–7. https://doi.org/10.1021/np010405c.
Morita H, Nakayama M, Kojima H, Takeya K, Itokawa H, Schenkel EP, Motidome M. Structures and cytotoxic activity relationship of casearins, new clerodane diterpenes from Casearia sylvestris Sw. Chem Pharm Bull. 1991;39(3):693–7. https://doi.org/10.1248/cpb.39.693.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR; Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 16, revision C.01. Wallingford: Gaussian, Inc.; 2016. https://gaussian.com/gaussian16/.
Dennington R, Keith TA, Millam JM. GaussView, version 6. Shawnee Mission: Semichem Inc.; 2016. https://gaussian.com/gaussview6/.
Yanai T, Tew D, Handy NA. New hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett. 2004;393:51–7. https://doi.org/10.1016/j.cplett.2004.06.011.
Bruhn T, Schaumlöffel A, Hemberger Y, Bringmann G. SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality. 2013;25:243–9. https://doi.org/10.1002/chir.22138.
Bruhn T, Schaumlöffel A, Hemberger Y, Pescitelli G. SpecDis, Ver. 1.71 Berlin, Germany; 2017. http://specdis-software.jimdo.com.
Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–42.
Doyle A, Griffiths JB, editors. Mammalian cell culture: essential techniques. New York: Wiley; 1997.
Clinical and Laboratory Standards Institute (CLSI). Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 11th edition. CLSI standard M07, Wayne; 2018.
Stresser DM, Turner SD, McNamara J, Stocker P, Miller VP, Crespi CL, Patten CJ. A high-throughput screen to identify inhibitors of aromatase (CYP19). Anal Biochem. 2000;284:427-f30. https://doi.org/10.1006/abio.2000.4729.
Acknowledgements
We thank Ms. Pakamas Intachote, Ms. Suchada Sengsai, and Ms. Busakorn Saimanee for evaluation the cytotoxicity and Ms. Somkid Sitthimonchai for the aromatase inhibitory assay. In addition, we are grateful to the National Park Authority’s staff for their support in collecting the samples.
Funding
The grants from Thailand Science Research and Innovation (TSRI) and Chulabhorn Research Institute (grant numbers 48296/4691995) and Center of Excellence on Environmental Health and Toxicology (EHT), OPS, Ministry of Higher Education, Science, Research and Innovation were supported the research funding.
Author information
Authors and Affiliations
Contributions
VP performed the experiment including isolation, purification and structure elucidation as well as wrote the manuscript. VP and PPa completed the identification of chemical structures. PB performed the ECD calculations. WY prepared the manuscript. TT was responsible for the identification of plant. PPl edited the manuscript. NR, PK carried out the antibacterial assays. ST, SR and CM. suggested and gave comments on this work, as well as edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest in this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
13659_2024_475_MOESM1_ESM.pdf
Additional file 1. The NMR, HRESIMS, IR, ECD spectra, chiral HPLC analysis of 2-methylbutyric acid benzyl ester derived from transesterification of 5 and synthetic samples, ECD calculation details of new compounds and the aromatase inhibition data of 3, 5, 7, 9, 11, 12, 19, and 20.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pailee, P., Batsomboon, P., Yaosanit, W. et al. Grewiifopenes A–K, bioactive clerodane diterpenoids from Casearia grewiifolia Vent.. Nat. Prod. Bioprospect. 14, 54 (2024). https://doi.org/10.1007/s13659-024-00475-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13659-024-00475-7