
Abstract
Background and Objective
The immunosuppressant everolimus is increasingly applied in renal transplantation. Its extensive pharmacokinetic variability necessitates therapeutic drug monitoring, typically based on whole-blood trough concentrations (C0). Unfortunately, therapeutic drug monitoring target attainment rates are often unsatisfactory and patients with on-target exposure may still develop organ rejection. As everolimus displays erythrocyte partitioning, haematocrit-normalised whole-blood exposure has been suggested as a more informative therapeutic drug monitoring marker. Furthermore, model-informed precision dosing has introduced options for more sophisticated dose adaptation. We have previously developed a mechanistic population pharmacokinetic model, which described everolimus plasma pharmacokinetics and enabled estimation of haematocrit-normalised whole-blood exposure. Here, we externally evaluated this model for its utility for model-informed precision dosing.
Methods
The retrospective dataset included 4123 pharmacokinetic observations from routine clinical therapeutic drug monitoring in 173 renal transplant recipients. Model appropriateness was confirmed with a visual predictive check. A fit-for-purpose analysis was conducted to evaluate whether the model accurately and precisely predicted a future C0 or area under the concentration–time curve (AUC) from prior pharmacokinetic observations. Bias and imprecision were expressed as the mean percentage prediction error (MPPE) and mean absolute percentage prediction error (MAPE), stratified on 6 months post-transplant. Additionally, we compared dose adaptation recommendations of conventional C0-based therapeutic drug monitoring and C0- or AUC-based model-informed precision dosing, and assessed the percentage of differences between observed and haematocrit-normalised C0 (∆C0) and AUC (∆AUC) exceeding ± 20%.
Results
The model showed adequate accuracy and precision for C0 and AUC prediction at ≤ 6 months (MPPEC0: 8.1 ± 2.5%, MAPEC0: 26.8 ± 2.1%; MPPEAUC: − 9.7 ± 5.1%, MAPEAUC: 13.3 ± 3.9%) and > 6 months post-transplant (MPPEC0: 4.7 ± 2.0%, MAPEC0: 25.4 ± 1.4%; MPPEAUC: − 0.13 ± 4.8%, MAPEAUC: 13.3 ± 2.8%). On average, dose adaptation recommendations derived from C0-based and AUC-based model-informed precision dosing were 2.91 ± 0.01% and 13.7 ± 0.18% lower than for conventional C0-based therapeutic drug monitoring at ≤ 6 months, and 0.93 ± 0.01% and 3.14 ± 0.04% lower at > 6 months post-transplant. The ∆C0 and ∆AUC exceeded ± 20% on 13.6% and 14.3% of occasions, respectively.
Conclusions
We demonstrated that our population pharmacokinetic model was able to accurately and precisely predict future everolimus exposure from prior pharmacokinetic measurements. In addition, we illustrated the potential added value of performing everolimus therapeutic drug monitoring with haematocrit-normalised whole-blood concentrations. Our results provide reassurance to implement this methodology in clinical practice for further evaluation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Everolimus therapy is challenged by extensive between- and within-patient pharmacokinetic variability, which necessitates a personalised treatment approach. While most clinics rely on conventional therapeutic drug monitoring based on everolimus whole-blood trough concentrations, model-informed precision dosing and dose adaptation based on haematocrit-normalised everolimus exposure have been suggested as options to further optimise everolimus therapy. |
We performed an extensive external validation for a previously published mechanistic population pharmacokinetic model, intended for model-informed precision dosing of everolimus, which provides a practical methodology for sophisticated dose adaptation based on haematocrit-normalised everolimus exposure. |
The findings of this work demonstrate the utility of the model for model-informed precision dosing and illustrate its potential added value over conventional therapeutic drug monitoring. Furthermore, our results provide reassurance that this methodology can be implemented in clinical practice for further evaluation. |
1 Introduction
Everolimus is an antiproliferative and immunosuppressive drug from the class of mammalian target of rapamycin (mTOR) inhibitors. The inhibition of mTOR exerts various immunosuppressive effects, including the inhibition of dendritic cell maturation and function and T cell proliferation [1]. Furthermore, various cancers rely on mTOR activation to drive tumour growth [2, 3]. Therefore, everolimus is used for the prophylaxis of allograft rejection after transplantation and for the treatment of a variety of malignant diseases, including breast cancer, renal cell carcinoma, neuroendocrine tumours and sub-ependymal giant cell astrocytoma with tuberous sclerosis complex.
Although everolimus therapy is generally effective, it is not often without treatment-limiting toxicity, including interstitial pneumonitis, dyspnoea, fever, anaemia, hypertension and hyperlipidaemia. In sub-ependymal giant cell astrocytoma, assessment of whole-blood trough concentrations to individualise everolimus treatment by means of therapeutic drug monitoring (TDM) is included in the European and US drug labels [4, 5]. Additionally, an international consensus exists to perform TDM-guided everolimus dose individualisation for prophylaxis of allograft rejection [6]. Although everolimus therapy in cancer is typically toxicity driven only [5], TDM is increasingly advocated to optimise treatment in this setting as well [7, 8], arguing from the clear exposure-efficacy and exposure-toxicity relationship for this indication [9, 10].
The term model-informed precision dosing (MIPD) has recently been coined as the natural evolution of the classical TDM methodology [11]. Whereas classical TDM typically comprises an assessment of pharmacokinetics and concurrent dose adaptation, MIPD brings pharmacometrics to the bedside of the patient. It captures drug, disease, and patient characteristics in a mathematical model and can be used to perform Bayesian forecasting and dose optimisation. Amongst others, important advantages of MIPD are that it allows for the extrapolation of existing dosing regimens to other populations and that it facilitates patient-friendly blood sampling schemes.
In previous work, we have developed a mechanistic population pharmacometric model for everolimus in cancer and solid organ transplantation [12]. Here, we performed an extensive external validation of this model in a retrospective cohort of renal transplant recipients, as an important next step towards implementation of this model in a MIPD tool for routine clinical care.
2 Methods
2.1 General Approach
Below, we first describe the external validation cohort and the previously published mechanistic population pharmacokinetic model. Subsequently, the three-step methodology for the external validation analysis is described. First, we evaluated whether the model adequately described the pharmacokinetics of everolimus in the external cohort and thus is appropriate for application in this population. Second, we assessed the ability of the full model and two reduced model versions to predict future everolimus exposure based on prior pharmacokinetic information. This was performed to evaluate whether the model is fit for its intended clinical purpose and to explore options to optimise its clinical feasibility. Third, we applied the model to illustrate the potential added value of our MIPD approach, by comparing MIPD-based dose adaptation recommendations and MIPD-based haematocrit-normalised everolimus TDM with conventional TDM.
2.2 External Validation Cohort
The retrospective external dataset comprised pooled everolimus whole-blood trough concentrations (C0) and short pharmacokinetic curves sampled at C0 and at approximate times of 1, 2, 3, 4, 5, and 6 h after drug administration. These originated from routine everolimus whole-blood TDM performed by a standard phlebotomy protocol in adult renal transplant recipients treated at Leiden University Medical Center between July 2014 and June 2019. This retrospective cohort thus provided a cross-sectional representation of a real-world clinical setting. Everolimus was quantified using a validated multiplex liquid chromatography-tandem mass spectrometry assay, capable of determining everolimus, tacrolimus, sirolimus and cyclosporine A in whole-blood simultaneously [13]. The lower and upper limits of quantification for everolimus were 0.5 µg/L and 50 µg/L, respectively. Patient characteristics were extracted from clinical records, including sex, age, height, weight, haematocrit, date of the transplant procedure and immunosuppressant regime.
2.3 Pharmacometric Modelling
2.3.1 Description of the Pharmacokinetic Model
Details on the previously developed mechanistic population pharmacometric model, including the population, development and evaluation are provided elsewhere [12]. In short, the study was based on rich clinical pharmacokinetic data of 126 patients treated with everolimus for prophylaxis of renal allograft rejection, metastatic thyroid cancer or breast cancer. The model was developed with NONMEM® version 7.3, using the stochastic approximation expectation–maximisation algorithm followed by Monte Carlo Importance Sampling for objective function assessment (SAEM-IMP). The model accounted for saturable erythrocyte binding of everolimus in whole-blood and estimated the pharmacokinetic parameters in plasma from paired observations of whole-blood concentrations and haematocrit.
This model is referred to as Model 1 in this article. In Model 1, the pharmacokinetics were scaled allometrically to fat-free body mass to account for body composition [14, 15]. Measures of body composition are, however, not always collected routinely in the outpatient setting. Individualised dose forecasting without incorporation of body size characteristics may, therefore, be more pragmatic. For this purpose, a population pharmacokinetic model without allometric scaling is required. Hence, the allometric scaling was removed from Model 1 and fitted to the same dataset to yield a reduced version of Model 1, which is hereinafter referred to as Model 2. Last, the utility of model-based dose individualisation is challenged by within-subject variability [16]. As the mean absorption time of everolimus shows extensive within-subject variability, Model 2 was further reduced through removal of the within-subject variability on mean absorption time and re-estimation of all other parameters. This model was used to investigate the importance of within-subject variability in dose forecasting of everolimus and is further referred to as Model 3. A schematic of the base model, parameter estimates, prediction-corrected visual predictive checks (pcVPCs) and the NONMEM code for these models are provided in the Electronic Supplementary Material (ESM).
2.3.2 Haematocrit-Normalised Therapeutic Drug Monitoring (TDM)
The mechanistic pharmacokinetic model allowed for haematocrit correction of everolimus whole-blood concentrations. In routine clinical care, everolimus pharmacokinetics are typically assessed in whole-blood. Everolimus, however, displays extensive accumulation in erythrocytes. In therapeutically relevant concentrations, approximately 75% of everolimus partitions in erythrocytes and displays saturable binding [17, 18], whereas protein binding is concentration independent with very limited variability [19]. Hence, changes in haematocrit affect the whole-blood concentrations without impacting the actual pharmacologically active (unbound) fraction in the plasma [19]. As everolimus therapy is associated with anaemia, haematocrit often undergoes profound changes after solid organ transplantation [20, 21]. Similarly, highly variable haematocrit is often observed in patients with cancer [22]. Hence, the haematocrit fraction is highly variable in the population receiving everolimus. Consequently, when whole-blood pharmacokinetics are assessed in samples in which the haematocrit differs substantially from the average haematocrit in the population in which the therapeutic ranges were established, this may lead to incorrect clinical interpretation and decisions. In patients with cancer and renal transplant recipients, the mean haematocrit fraction is approximately 0.38 [19, 23, 24]. Haematocrit-corrected whole-blood concentrations normalised to a fraction of 0.38 could, therefore, be more informative for TDM-guided clinical decision making in these settings. In the pharmacokinetic model, everolimus plasma and erythrocyte concentrations are calculated from paired observations of haematocrit and whole-blood concentrations, assuming that everolimus binds to erythrocytes in a specific and a non-specific manner, as described with Eq. 1 [12].
In Eq. 1, Crb represents the everolimus erythrocyte concentration, Bmax the maximal everolimus concentration bound specifically to erythrocytes, Kd the predicted dissociation constant, Kns the constant expressing non-specific binding of everolimus to erythrocytes and Cp the everolimus plasma concentration. Details on the derivation of these binding constants are provided elsewhere [12]. Furthermore, it is assumed that the whole-blood concentration can be described with Eq. 2, in which Cwb represents the whole-blood concentration and Ht the haematocrit fraction.
In this article, both the everolimus whole-blood concentration corresponding with the observed haematocrit fraction and the whole-blood concentration normalised to a haematocrit fraction of 0.38 are presented, with the latter calculated using Eq. 3.
2.4 External Validation
2.4.1 Visual Predictive Check
A pcVPC based on a 1000 simulated datasets was constructed for each model [25]. A pcVPC allows for visual inspection of the concordance between the 5th percentile, median, and 95th percentile of the observed and predicted everolimus concentrations over time. The extent of overlay between the observations and the model simulations provided information on the appropriateness of the model to describe the population pharmacokinetics of everolimus in the external cohort.
2.4.2 Fit-for-Purpose Validation
A fit-for-purpose validation was performed for each model. This procedure was aimed to capture, as best as possible, the fitness of each model for its intended purpose [26]. Namely, to accurately predict (a posteriori) a future everolimus C0 or area under the concentration–time curve from time zero to 12 h post-dose (AUC0–12) based on the patient covariates at that timepoint and the C0 or AUC0–12 from the prior TDM instance.
The fit-for-purpose validation was performed for C0 and AUC0–12, separately, to allow for assessment of model performance for both TDM approaches. The paired C0 and AUC0–12 were stratified according to the time after transplantation to distinguish between model performance in the pharmacokinetically unstable (≤ 6 months) and stable (> 6 months) post-transplant periods. The extent of within-subject pharmacokinetic variability of everolimus typically decreases from approximately 50% in the first 6 months after transplantation [27], to approximately 20% thereafter [28]. Hence, model performance was expected to vary between these periods. Last, model performance for the a priori prediction of the initial everolimus C0 and AUC0–12 was evaluated to provide insight on the added value of performing TDM early after transplantation.
All predicted everolimus concentrations were individual predictions derived from maximum a posteriori Bayesian forecasting using Models 1–3. The predictive performance was expressed with the mean percentage prediction error (MPPE) for bias, and the mean absolute percentage prediction error (MAPE) and normalised root mean squared error for imprecision [29]. In addition, the percentages of predictions within ± 10%, ± 20% and ± 30% of the observations (P10, P20 and P30) were assessed to aid interpretation. The maximal tolerable discordance was set at ± 25%, as variability below this threshold is expected to arise from normal within-subject pharmacokinetic variability [30].
2.4.3 Evaluation of the Potential Added Value of Model-Informed Precision Dosing (MIPD) over Conventional TDM
In addition, we substantiated the potential added value of our MIPD approach over conventional TDM. In conventional TDM, everolimus dose adaptation is typically performed based solely on C0, relying on an assumption of linear pharmacokinetics in the therapeutic concentration range. For example, this means that if a C0 at a given TDM instance is half of the desired C0, the everolimus dose is doubled in an effort to achieve an on-target subsequent C0.
The dose adaptation recommendations aimed at achieving an on-target C0 and AUC0–12 as derived from our MIPD approach were compared to those derived using conventional C0-based TDM. This analysis was performed solely with Model 1, as any differences between Models 1–3 are captured adequately in the fit-for-purpose analysis. The following target ranges for everolimus TDM in renal transplantation were applied: a C0 of 6 µg/L or 7 µg/L; and AUC0–12 of 100 µg/L × h or 120 µg/L × h, for patients with or without concomitant tacrolimus therapy, respectively.
2.4.4 Evaluation of the Potential Added Value of Haematocrit-Normalised TDM
Last, we illustrated the potential added value of TDM based on haematocrit-corrected everolimus whole-blood concentrations, by evaluating the differences between TDM based on the observed and haematocrit-corrected C0 (ΔC0) and AUC0–12 (ΔAUC0–12). For this purpose, all observed everolimus whole-blood concentrations were normalised to the aforementioned mean haematocrit fraction of 0.38, using Eq. 3 with Model 1. This allowed for assessment of ΔC0. For the AUC0–12, this allowed for estimation of the haematocrit-corrected AUC0–12 via addition of a virtual compartment in the model code and subsequent assessment of ΔAUC0–12. For ΔC0 and ΔAUC0–12, a change exceeding ± 20% was considered clinically relevant, as changes in C0 or AUC0–12 exceeding ± 20% typically result in different dose adaptation recommendations in clinical TDM practice. This analysis was performed solely with Model 1, as any differences between Models 1–3 are captured adequately in the fit-for-purpose analysis. For additional insight into the impact of our approach for extreme cases, we also evaluated the ΔC0 and ΔAUC0–12 separately for those occasions in which patients displayed very low (5th percentile) or very high (95th percentile) haematocrit.
2.5 Software
The pharmacometric modelling and external validation were performed with the non-linear mixed-effects modelling software package NONMEM® version 7.3 (Icon Development Solutions, Ellicott City, MD, USA), using Pirana 2.9.7 and Perl-speaks-NONMEM (PsN) Toolkit 4.8.1 as the modelling interface. Data handling, visualisation and statistics were performed in R 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria) and RStudio 1.2.5001 (RStudio Inc., Boston, MA, USA).
3 Results
3.1 External Validation Cohort
Pharmacokinetic data from 173 renal transplant recipients were available, yielding 4123 everolimus whole-blood concentrations including 2933 C0 and 322 AUC0–12. All calculated AUC0–12 included the C0 and at least three additional concentrations up to 6 h after everolimus intake. The characteristics of the external cohort and the pharmacokinetic data are summarised in Table 1.
3.2 External Validation
3.2.1 Prediction-Corrected Visual Predictive Check
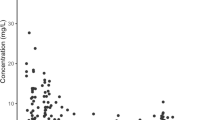
The pcVPCs of Models 1–3 on the external dataset are depicted in Fig. 1a–c. The pcVPCs showed a good overlay between the median, 5th, and 95th percentiles of the simulations of all three models and the observations, indicative of adequate description of the population pharmacokinetics of everolimus in the external cohort. Furthermore, the pcVPCs of Models 1–3 showed an overall identical trend, which indicated that exclusion of allometric scaling or inter-occasion variability did not significantly affect model appropriateness.

Prediction-corrected visual predictive checks of a Model 1, b Model 2 and c Model 3 on the external dataset. The solid black lines depict the 5th, median and 95th percentiles of the observed everolimus concentrations. The dashed black lines and blue-shaded areas depict the 5th, median, and 95th percentiles of the model-simulated everolimus concentrations and their respective 95% confidence intervals
3.2.2 Fit-for-Use Validation
The predictive performances of Models 1–3 for estimation of the initial (a priori) and future (a posteriori) C0 or AUC0–12 in the unstable and stable post-transplant periods are graphically depicted in Fig. 2a–d and summarised in Table 2. Scatter plots of the individual predicted and observed C0 and AUC0–12 are provided in the ESM.

Predictive performance of Models 1, 2 and 3 in terms of bias (prediction error) and imprecision (absolute prediction error) for (a–b) everolimus trough concentrations (C0) and (c–d) area under the everolimus concentration–time curves from time zero to 12 h after administration (AUC0–12) for the fit-for-purpose analysis. The solid black line represents the line of equality, whereas the grey-shaded area depicts the predefined tolerable extent of bias or imprecision
The a priori C0 prediction showed a relatively low bias (MPPE: 5.4–7.4%, 95% confidence interval − 4.7, 17.6) but unacceptable extent of imprecision (MAPE: > 30%, normalised root mean squared error: 78.1–82.7%). Furthermore, the a priori AUC0–12 prediction was profoundly biased and imprecise as indicated by the MPPE (42.7–49.3%), MAPE (52.7–58.0%) and the normalised root mean squared error (66.8–75.2%) exceeding 40%.
The a posteriori C0 prediction with Model 1 showed a low mean positive bias in the unstable (MPPE: 8.12% ± 2.54%) and stable (MPPE: 4.74% ± 1.97%) post-transplant periods, indicative of accurate model predictions for the average renal transplant recipient. The MAPE for C0 prediction in the unstable (26.8% ± 2.09%) and stable (25.4% ± 1.41%) post-transplant periods slightly exceeded the imprecision limit of 25%. Overall, 71.1% and 70.7% of C0 predictions fell within 30% of the observations. The a posteriori AUC0–12 prediction with Model 1 showed a relatively low mean negative bias in the unstable (MPPE: − 9.70% ± 5.10%) and the stable (MPPE: − 0.13% ± 4.79%) post-transplant periods, indicative of accurate model predictions for the average renal transplant recipient. Furthermore, AUC0–12 were predicted precisely, with a MAPE of 13.3% ± 3.87% and 13.3% ± 2.80% for the unstable and stable post-transplant periods, respectively. Overall, 90.6% and 95.8% of predicted AUC0–12 fell within 30% of the observations.
For C0, extreme prediction errors (absolute prediction error > 100%) were observed on 3.6%, 2.7% and 2.1% of occasions for the initial C0 and the unstable and stable periods, respectively. For the AUC0–12, extreme prediction errors were observed on 3.6% of occasions for the initial AUC0–12, whereas none occurred for the unstable and stable periods. Inconsistent small deviations between the predictive performances of Models 1–3 within the various fit-for-purpose scenarios were observed, with differences in MPPE, MAPE and normalised root mean squared error between the models with ranges of 0.0–5.4%, 0.0–3.3% and 0.1–2.8%, respectively.
3.2.3 Evaluation of the Potential Added Value of MIPD over Conventional TDM
The relations between the MIPD-based and conventional C0-based dosing recommendations are graphically depicted in Fig. 3a, b. For C0-based MIPD, the dosing recommendations were on average 0.25 ± 0.03 mg (2.91% ± 0.01%) lower than for conventional C0-based dosing in the unstable period, and 0.07 ± 0.02 mg (0.93% ± 0.01%) lower for the stable period. For AUC0–12-based MIPD, the dosing recommendations were on average 1.25 ± 0.62 mg (13.7% ± 0.18%) lower than for conventional C0-based dosing in the unstable period, and 0.14 ± 0.09 mg (3.14% ± 0.04%) lower in the stable period. Overall, a trend towards lower dosing recommendations for MIPD as compared with conventional TDM was apparent for the unstable period, whereas differences were limited for the stable period.

Potential added value of (a) everolimus trough concentration (C0)-based model-informed precision dosing (MIPD) and (b) area under the everolimus concentration–time curve from time zero to 12 h after administration (AUC0–12)-based MIPD over conventional C0-based therapeutic drug monitoring. Each hexagram depicts the conventional dosing advice and concurrent MIPD dosing advice, shaded according to the number of observations for that particular combination. The solid black line represents the loess regression line, whereas the solid grey line depicts the line of equality
3.2.4 Evaluation of the Potential Added Value of Haematocrit-Normalised TDM
The relationships between the observed and the haematocrit-normalised C0 and AUC0–12 are depicted in Fig. 4a, b. The average ΔC0 was 4.01% ± 0.47% (range − 34.3% to 52.6%) and the ΔAUC0–12 was 2.92% ± 1.43% (range − 30.8 to 51.2%). For patients with very low (5th percentile: 0.280; n = 147) and very high (95th percentile: 0.472; n = 148) haematocrit, the average ΔC0 was 31.0% ± 0.8% and − 20.9% ± 0.7%, respectively. Similarly, an average ΔAUC0–12 of 32.1% ± 3.2% and − 21.5% ± 2.6% was observed for patients with very low (5th percentile: 0.279; n = 17) and very high (95th percentile: 0.461; n = 17) haematocrit.

Potential added value of haematocrit normalisation of the whole-blood (a) everolimus trough concentration (C0) and (b) area under the everolimus concentration–time curve from time zero to 12 h after administration (AUC0–12) to a haematocrit of 0.38 over the haematocrit range. The data points depict the absolute haematocrit-normalised C0 (∆C0) or ∆AUC0–12 over the haematocrit range, shaded according to the relative ∆C0 or ∆AUC0–12. The dots and triangles represent a ΔC0 or ΔAUC0–12 of ≤ ±20% and > ±20%, respectively. The solid grey lines represent the lines of equality, whereas the dashed black lines indicate the haematocrit fraction of 0.38
The ΔC0 and ΔAUC0–12 exceeded the predefined ± 20% threshold on 13.6% and 14.3% of occasions, respectively. This roughly translates to the situation in which in one in every seven to eight TDM instances the patient displayed such a divergent haematocrit that the pharmacologically active everolimus plasma fraction is affected by over ± 20%, but not accounted for in the clinical decision.
4 Discussion
In this study, a previously published population pharmacokinetic model for everolimus was externally validated in a retrospective cohort of renal transplant recipients and evaluated for its utility for MIPD. Whereas other population pharmacokinetic models for everolimus in renal transplantation and various malignant diseases are available [10, 18, 23, 31,32,33,34,35,36], our model is, to the best of our knowledge, the first to be thoroughly validated externally.
First, we showed that our model adequately described the population pharmacokinetics of everolimus in the external cohort and thus is appropriate for this population. Second, the model was unable to accurately and precisely predict a priori the optimal initial everolimus dosage based solely on the baseline patient characteristics. This was expected, as the model was intended specifically for a posteriori prediction purposes and relies heavily on the input of prior pharmacokinetic information to guide its predictions. These findings underline the need to perform TDM after renal transplantation to provide pharmacokinetic input for the model. Third, we demonstrated that our model was able to predict a posteriori a future everolimus C0 or AUC0–12 with a bias of < 15% and < 10% and imprecision of ≤ 30% and < 15%, respectively. Model performance for a posteriori C0 prediction was lower than for AUC0–12, as most clearly reflected in their respective P20 of approximately 50% and 80%. However, as approximately 70% of the a posteriori C0 predictions were within 30% of the observations, the data demonstrated that the model is fit for a posteriori estimation of both AUC0–12 and C0. Whereas extreme prediction errors occurred on approximately 3% of occasions, we suspected that these outliers mainly arose from premature everolimus intakes prior to the C0 blood draw or missed everolimus intakes during the dosing interval prior to C0 assessment. This could, however, not be confirmed owing to the retrospective nature of the study. Fourth, model performance varied only very slightly between Models 1, 2 and 3, as indicated by comparable visual diagnostics, prediction bias and prediction imprecision across the three models. Hence, removal of allometric scaling from Model 1 could be considered to improve clinical feasibility and exclusion of within-subject variability on the mean absorption time seems possible without giving in on model performance. In addition, we performed an explorative comparison of our MIPD-derived and conventional TDM-derived dosing recommendations, in an effort to substantiate the potential added value of the MIPD approach. Here, we observed that the MIPD approach on average yielded slightly lower dosing recommendations. This finding was most pronounced for the unstable post-transplant period, which could be indicative of slight overdosing of everolimus early after transplantation when relying on conventional TDM. This also indicated that, for the stable post-transplant period, performing AUC0–12-based MIPD may be of limited added value as compared with conventional C0-based TDM. It is important to note, however, that the discrepancy in the monitoring frequencies for AUC0–12 (annually) and C0 (every 3 months) in the stable post-transplant period may have influenced this comparison. In addition, AUC0–12 monitoring in the stable period may still be warranted for patients whom display a variable C0–AUC0–12 correlation, for which the C0 is of limited informative value for everolimus exposure assessment [37]. In any case, these results should be interpreted with caution as the retrospective nature of our study complicated translation to the real-world clinical setting. Last, we explored the potential added value of performing everolimus TDM using haematocrit-normalised whole-blood concentrations. Here, we found that in roughly one in every seven to eight TDM instances, the patient displayed such a divergent haematocrit that the everolimus plasma fraction was affected by over ± 20%, which is not accounted for in current clinical decision making.
When interpreting the predictive performance of our model, one should take into account that maximum a posteriori Bayesian dose adaptation is challenged by within-subject variability. Namely, the extent of within-subject variability of a given drug ultimately determines the maximal degree of target attainment to be expected. Holford and Buclin have illustrated that if 90% of the population is to achieve exposure within 80–125% of the target for a given drug, this can only be achieved if the within-subject variability is below 13.6% [38]. The within-subject variability of everolimus C0 and AUC0–12 in stable renal transplant recipients has been reported to range from 10 to 19% and from 14 to 16%, respectively [28]. For unstable patients, however, variability of C0 and AUC0–12 is reported to be in the range of 45% and 27% [27]. A significant portion of the discrepancy between the variability in the unstable and stable post-transplant periods can, likely, be explained by haematocrit-dependent erythrocyte partitioning. Whereas the haematocrit typically changes profoundly over the unstable period, this is not corrected for in current TDM practice. It might, therefore, be more informative to focus on the within-subject variability across the entire post-transplant period without stratification, which has been reported to be approximately 25% [30]. A significant portion of this variability is expected to arise from within-subject variability in bioavailability, which has been reported to be approximately 25% [23]. When taking into account these phenomena and the residual unexplained variability of 17.9% of our model, the observed prediction bias of < 15% and imprecision of ≤ 30% are reassuring that the model in general is performing well.
For tacrolimus, which displays similar erythrocyte partitioning as everolimus, various authors have advocated for the implementation of TDM based on haematocrit-normalised whole-blood concentrations across different fields of transplantation [39,40,41,42,43]. Additionally, a recent consensus paper on tacrolimus TDM included recommendations for further investigation of alternative TDM strategies, including haematocrit normalisation, to explain and resolve the highly variable tacrolimus efficacy in patient subpopulations [37]. As TDM based on intracellular everolimus concentrations or pharmacodynamic monitoring remain distant prospects, haematocrit-normalised whole-blood concentrations might pose the most informative marker for patient and graft outcomes, which is currently available. Our findings add to the support and provide a pragmatic methodology for clinical implementation of this approach to further optimise everolimus therapy.
Our study has some limitations. We included renal transplant recipients exclusively, whereas the evaluated model was also intended for application in patients with cancer. We postulate, however, that our results can be extrapolated to the oncology setting, as we demonstrated that the pharmacokinetics of everolimus is similar for this patient population [12]. Furthermore, no full pharmacokinetic curves were available to unequivocally quantify the observed AUC0–12. Alternatively, these were estimated from the observed short pharmacokinetic curves using model-based extrapolation. This may have rendered the a posteriori AUC0–12 estimation slightly biased towards more favourable model performance. This method was, however, the most pragmatic approach to capture this aspect in this retrospective cohort. To enable clinically feasible AUC0–12-based everolimus MIPD for routine patient care, an important next step would be to develop a pragmatic limited sampling schedule. In addition, we included the pharmacokinetic information from only the prior TDM instance in the fit-for-purpose validation. Although this was the most stringent evaluation given the limited data provided to the model, which thus ensured that our model is fit for purpose, we did not investigate whether inclusion of additional historic TDM data yielded improved model performance. However, incorporation of, for instance, an autoregressive model component for weighted inclusion of additional historic TDM data would have notably complexified the model and possibly limited its clinical feasibility owing to excessive run times. Furthermore, inclusion of additional historic TDM data may introduce additional bias from within-subject pharmacokinetic variability, which is minimised when taking into account only the most recent observation. Last, our model was developed and validated in patients without concomitant cyclosporine therapy or strong cytochrome P450 enzyme 3A-inducing or inhibiting agents. While the combination of everolimus with these agents is often avoided in clinical practice and maximum a posteriori Bayesian estimation is able to capture divergent pharmacokinetic behaviour if provided sufficient a posteriori pharmacokinetic data [44], there currently is no solid evidence that, if provided with only a limited number of samples, our pharmacokinetic model would be able to adequately capture the pharmacokinetic profile for patients who do concomitantly use these types of drugs. Hence, until it is demonstrated that this phenomenon does not compromise its predictive performance, individual predictions of our model for these patients should be interpreted with caution.
We demonstrated that our MIPD approach poses a valid tool to predict future individual everolimus exposure and can be implemented in clinical practice for further evaluation. Whereas it is expected that MIPD-guided everolimus therapy yields improved C0 or AUC0–12 target attainment as compared with conventional TDM, it is important to evaluate its clinical feasibility and cost effectiveness.
5 Conclusions
We have demonstrated that our previously published mechanistic population pharmacokinetic model adequately described the population pharmacokinetics of everolimus and was able to accurately and precisely predict future everolimus exposure from prior TDM measurements, in this retrospective cohort of adult renal transplant recipients. In addition, we have illustrated the potential added value of performing everolimus TDM based on haematocrit-normalised whole-blood concentrations, which poses a promising strategy to further optimise everolimus therapy for patients with extreme haematocrit values. The results of the current study provide adequate reassurance to implement this methodology in clinical practice for further evaluation.
Availability of Data and Material
The datasets generated during and/or analysed during this study are available from the corresponding author upon reasonable request.
References
Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9(5):324–37.
Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22.
de Fijter JW. Cancer and mTOR inhibitors in transplant recipients. Transplantation. 2017;101(1):45–55.
Novartis. Afinitor: prescribing information. 2012. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/022334s036lbl.pdf. Accessed 17 June 2019.
European Medicines Agency. Afinitor: EPAR: product information. 2019. https://www.ema.europa.eu/en/documents/product-information/afinitor-epar-product-information_en.pdf. Accessed 17 June 2019.
Shipkova M, Hesselink DA, Holt DW, Billaud EM, van Gelder T, Kunicki PK, et al. Therapeutic drug monitoring of everolimus: a consensus report. Ther Drug Monit. 2016;38(2):143–69.
Verheijen RB, Yu H, Schellens JHM, Beijnen JH, Steeghs N, Huitema ADR. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin Pharmacol Ther. 2017;102(5):765–76.
Deppenweiler M, Falkowski S, Saint-Marcoux F, Monchaud C, Picard N, Laroche ML, et al. Towards therapeutic drug monitoring of everolimus in cancer? Results of an exploratory study of exposure-effect relationship. Pharmacol Res. 2017;121:138–44.
Ravaud A, Urva SR, Grosch K, Cheung WK, Anak O, Sellami DB. Relationship between everolimus exposure and safety and efficacy: meta-analysis of clinical trials in oncology. Eur J Cancer. 2014;50(3):486–95.
de Wit D, Schneider TC, Moes DJ, Roozen CF, den Hartigh J, Gelderblom H, et al. Everolimus pharmacokinetics and its exposure-toxicity relationship in patients with thyroid cancer. Cancer Chemother Pharmacol. 2016;78(1):63–71.
Darwich AS, Ogungbenro K, Vinks AA, Powell JR, Reny JL, Marsousi N, et al. Why has model-informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin Pharmacol Ther. 2017;101(5):646–56.
Ter Heine R, van Erp NP, Guchelaar HJ, de Fijter JW, Reinders MEJ, van Herpen CM, et al. A pharmacological rationale for improved everolimus dosing in oncology and transplant patients. Br J Clin Pharmacol. 2018;84(7):1575–86.
Moes DJ, van der Bent SA, Swen JJ, van der Straaten T, Inderson A, Olofsen E, et al. Population pharmacokinetics and pharmacogenetics of once daily tacrolimus formulation in stable liver transplant recipients. Eur J Clin Pharmacol. 2016;72(2):163–74.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44(10):1051–65.
Holford NHG, Anderson BJ. Allometric size: the scientific theory and extension to normal fat mass. Eur J Pharm Sci. 2017;109S:S59–64.
Abrantes JA, Jonsson S, Karlsson MO, Nielsen EI. intercessionvariability in model-based dose individualization using therapeutic drug monitoring data. Br J Clin Pharmacol. 2019;85(6):1326–36.
Kovarik JM, Kahan BD, Kaplan B, Lorber M, Winkler M, Rouilly M, et al. Longitudinal assessment of everolimus in de novo renal transplant recipients over the first post-transplant year: pharmacokinetics, exposure-response relationships, and influence on cyclosporine. Clin Pharmacol Ther. 2001;69(1):48–56.
US Food and Drug Administration. Center for Drug Evaluation and Research. Afinitor: clinical pharmacology and biopharmaceutics review. 2008. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022334s000_ClinPharmR.pdf. Accessed 17 June 2019.
van Erp NP, van Herpen CM, de Wit D, Willemsen A, Burger DM, Huitema AD, et al. A semi-physiological population model to quantify the effect of hematocrit on everolimus pharmacokinetics and pharmacodynamics in cancer patients. Clin Pharmacokinet. 2016;55(11):1447–56.
Sanchez Fructuoso A, Calvo N, Moreno MA, Giorgi M, Barrientos A. Study of anemia after late introduction of everolimus in the immunosuppressive treatment of renal transplant patients. Transpl Proc. 2007;39(7):2242–4.
Vlahakos DV, Marathias KP, Agroyannis B, Madias NE. Posttransplant erythrocytosis. Kidney Int. 2003;63(4):1187–94.
Knight K, Wade S, Balducci L. Prevalence and outcomes of anemia in cancer: a systematic review of the literature. Am J Med. 2004;116(Suppl. 7A):11s–26s.
Moes DJ, Press RR, den Hartigh J, van der Straaten T, de Fijter JW, Guchelaar HJ. Population pharmacokinetics and pharmacogenetics of everolimus in renal transplant patients. Clin Pharmacokinet. 2012;51(7):467–80.
Verheijen RB, Atrafi F, Schellens JHM, Beijnen JH, Huitema ADR, Mathijssen RHJ, et al. Pharmacokinetic optimization of everolimus dosing in oncology: a randomized crossover trial. Clin Pharmacokinet. 2018;57(5):637–44.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51.
Keizer RJ, Ter Heine R, Frymoyer A, Lesko LJ, Mangat R, Goswami S. Model-informed precision dosing at the bedside: scientific challenges and opportunities. CPT Pharmacometr Syst Pharmacol. 2018;7(12):785–7.
Kovarik JM, Kaplan B, Silva HT, Kahan BD, Dantal J, McMahon L, et al. Pharmacokinetics of an everolimus-cyclosporine immunosuppressive regimen over the first 6 months after kidney transplantation. Am J Transpl. 2003;3(5):606–13.
Budde K, Fritsche L, Waiser J, Glander P, Slowinski T, Neumayer HH. Pharmacokinetics of the immunosuppressant everolimus in maintenance renal transplant patients. Eur J Med Res. 2005;10(4):169–74.
Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9(4):503–12.
Valero R, Sanchez-Fructuoso A, Rodrigo E, Belmar L, Cos MA, Perez-Flores I, et al. Variability of the mammalian target of rapamycin inhibitors is correlated with long-term renal graft survival. Transpl Proc. 2019;51(2):341–3.
Robertsen I, Debord J, Asberg A, Marquet P, Woillard JB. A limited sampling strategy to estimate exposure of everolimus in whole blood and peripheral blood mononuclear cells in renal transplant recipients using population pharmacokinetic modeling and Bayesian estimators. Clin Pharmacokinet. 2018;57(11):1459–69.
Combes FP, Baneyx G, Coello N, Zhu P, Sallas W, Yin H, et al. Population pharmacokinetics-pharmacodynamics of oral everolimus in patients with seizures associated with tuberous sclerosis complex. J Pharmacokinet Pharmacodyn. 2018;45(5):707–19.
Tanaka A, Yano I, Shinsako K, Sato E, Fukudo M, Masuda S, et al. Population pharmacokinetics of everolimus in relation to clinical outcomes in patients with advanced renal cell carcinoma. Ther Drug Monit. 2016;38(6):663–9.
Saba NF, Hurwitz SJ, Magliocca K, Kim S, Owonikoko TK, Harvey D, et al. Phase 1 and pharmacokinetic study of everolimus in combination with cetuximab and carboplatin for recurrent/metastatic squamous cell carcinoma of the head and neck. Cancer. 2014;120(24):3940–51.
Lemaitre F, Bezian E, Goldwirt L, Fernandez C, Farinotti R, Varnous S, et al. Population pharmacokinetics of everolimus in cardiac recipients: comedications, ABCB1, and CYP3A5 polymorphisms. Ther Drug Monit. 2012;34(6):686–94.
Kovarik JM, Hsu CH, McMahon L, Berthier S, Rordorf C. Population pharmacokinetics of everolimus in de novo renal transplant patients: impact of ethnicity and comedications. Clin Pharmacol Ther. 2001;70(3):247–54.
Brunet M, van Gelder T, Asberg A, Haufroid V, Hesselink DA, Langman L, et al. Therapeutic drug monitoring of tacrolimus-personalized therapy: second consensus report. Ther Drug Monit. 2019;41(3):261–307.
Holford NH, Buclin T. Safe and effective variability: a criterion for dose individualization. Ther Drug Monit. 2012;34(5):565–8.
Storset E, Holford N, Hennig S, Bergmann TK, Bergan S, Bremer S, et al. Improved prediction of tacrolimus concentrations early after kidney transplantation using theory-based pharmacokinetic modelling. Br J Clin Pharmacol. 2014;78(3):509–23.
de Jonge H, Vanhove T, de Loor H, Verbeke K, Kuypers DR. Progressive decline in tacrolimus clearance after renal transplantation is partially explained by decreasing CYP3A4 activity and increasing haematocrit. Br J Clin Pharmacol. 2015;80(3):548–59.
Limsrichamrern S, Chanapul C, Mahawithitwong P, Sirivatanauksorn Y, Kositamongkol P, Asavakarn S, et al. Correlation of hematocrit and tacrolimus level in liver transplant recipients. Transpl Proc. 2016;48(4):1176–8.
Sikma MA, Hunault CC, Huitema ADR, De Lange DW, Van Maarseveen EM. Clinical pharmacokinetics and impact of hematocrit on monitoring and dosing of tacrolimus early after heart and lung transplantation. Clin Pharmacokinet. 2020;59(4):403–8.
Sikma MA, Van Maarseveen EM, Hunault CC, Moreno JM, Van de Graaf EA, Kirkels JH, et al. Unbound plasma, total plasma, and whole-blood tacrolimus pharmacokinetics early after thoracic organ transplantation. Clin Pharmacokinet. 2020;59(6):771–80.
Sheiner LB, Beal S, Rosenberg B, Marathe VV. Forecasting individual pharmacokinetics. Clin Pharmacol Ther. 1979;26(3):294–305.
Author information
Authors and Affiliations
Contributions
TCZ, DJARM, NPE, JWF, H-JG, RJK and RH contributed to the study conception and design. PJMB assisted in the collection of patient data. TCZ, DJARM, RJK and RH analyzed the data. The initial draft of the manuscript was written by TCZ, DJARM, RJK and RH, and all authors commented on previous versions of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflicts of Interest
Nielka P. van Erp’s institution has received research grants from Astellas, Novartis, Ipsen, and Janssen-Cilag and consulting fees from Bayer and Sanofi, none of which were related to the content of this article. Ron J. Keizer is a founder, employee and stockholder for InsightRX. Tom C. Zwart, Dirk Jan A.R. Moes, Paul J.M. van der Boog, Johan W. de Fijter, Henk-Jan Guchelaar and Rob ter Heine have no conflicts of interest that are directly relevant to the content of this article.
Code Availability
The NONMEM code used to generate and/or analyse the data described in this study is provided in the ESM.
Funding
No funding was received for this study.
Ethics Approval
This research study was conducted retrospectively with data obtained for clinical purposes. We consulted with the scientific committee of the Department of Internal Medicine of Leiden University Medical Center, who determined that our study did not need ethical approval or informed consent.
Electronic supplementary material
Below is the link to the electronic supplementary material.
40262_2020_925_MOESM1_ESM.eps
Fig. S1 Schematic depiction of the population pharmacokinetic model. All parameters are expressed as plasma pharmacokinetic parameters, with the plasma concentrations calculated from the paired observations of whole-blood concentrations and haematocrit
40262_2020_925_MOESM2_ESM.eps
Fig. S2 Prediction-corrected visual predictive checks of a Model 1, b Model 2 and c Model 3 on the original model development dataset. The solid black lines depict the 5th, median and 95th percentiles of the observed everolimus concentrations. The dashed black lines and blue-shaded areas depict the 5th, median and 95th percentiles of the model-simulated everolimus concentrations and their respective 95% confidence intervals
40262_2020_925_MOESM3_ESM.eps
Fig. S3 Scatter plots of the observed versus the individual predicted a everolimus trough concentrations (C0) and b area under the everolimus concentration-time curves from time zero to 12 h after administration (AUC0-12), for Model 1-3 for the initial therapeutic drug monitoring (TDM) instance (a priori) and the unstable and stable posttransplant periods (a posteriori). The solid grey line represents the line of equality, whereas the solid blue and orange lines and blue- and orange-shaded areas represent the loess regression lines and their 95% confidence intervals, respectively
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zwart, T.C., Moes, D.J.A.R., van der Boog, P.J.M. et al. Model-Informed Precision Dosing of Everolimus: External Validation in Adult Renal Transplant Recipients. Clin Pharmacokinet 60, 191–203 (2021). https://doi.org/10.1007/s40262-020-00925-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00925-8