
Abstract
Background and Objectives
Elagolix is an orally active, gonadotropin-releasing hormone receptor antagonist approved for the management of endometriosis-associated pain and heavy menstrual bleeding associated with uterine fibroids. Elagolix population pharmacokinetics and factors affecting elagolix exposure in healthy women and women with endometriosis have been reported previously. The purpose of this study was to extend the population pharmacokinetics model with additional modifications to incorporate data from phase III studies of elagolix with hormonal add-back therapy in women with uterine fibroids.
Methods
Data from 13 clinical studies (a total of 2168 women) consisting of six phase I studies in healthy premenopausal women, four phase III studies in premenopausal women with endometriosis, and three phase III studies in premenopausal women with uterine fibroids were analyzed using a non-linear mixed-effects modeling approach.
Results
Elagolix population pharmacokinetics was best described by a two-compartment model with first-order absorption, lag time in absorption, and first-order elimination. Out of the covariates tested on elagolix apparent clearance, apparent volume of distribution, and/or relative bioavailability, only organic anion transporting polypeptide 1B1 genotype status and body weight had a statistically significant but no clinically meaningful effect on elagolix relative bioavailability and apparent volume of distribution, respectively. There were no clinically meaningful differences in elagolix population pharmacokinetics in healthy women or women with endometriosis or uterine fibroids.
Conclusions
Elagolix population pharmacokinetics modeling did not reveal any patient-related factors or clinical parameters that would require dose adjustments for the approved dosage of 300 mg twice daily with estradiol 1 mg /norethindrone acetate 0.5 mg daily, in women with heavy menstrual bleeding associated with uterine fibroids.
Clinical Trial Registration
ClinicalTrials.gov identifiers: NCT01620528 (EM-1), NCT01760954 (EM-1-Extend), NCT01931670 (EM-2), NCT02143713 (EM-2-Extend), NCT02654054 (UF-1), NCT02691494 (UF-2), NCT0295494 (UF-Extend).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Elagolix population pharmacokinetics in 175 healthy women, 1310 women with endometriosis, and 683 women with uterine fibroids was best described by a two-compartment model with a lag time in absorption. |
Statistically significant covariates (organic anion transporting polypeptide 1B1 genotype status on relative bioavailability, and body weight on apparent volume of distribution) that affected elagolix pharmacokinetics did not result in clinically meaningful changes. |
The elagolix population pharmacokinetics analysis supported the approved dosage for the management of heavy menstrual bleeding in women with uterine fibroids. |
1 Introduction
Elagolix is an orally active, nonpeptide, gonadotropin-releasing hormone (GnRH) antagonist that competitively binds to GnRH receptors resulting in a dose-dependent suppression of ovarian sex hormones and gonadotropins without stimulating the GnRH receptor [1]. Elagolix is approved for the management of moderate-to-severe pain associated with endometriosis (EM, Orilissa™) [2] and heavy menstrual bleeding (HMB) associated with uterine fibroids (UF, Oriahnn™) [3]. Fast-acting elagolix reaches maximum plasma concentrations in 1.0–1.5 h with a half-life of 4–6 h [1]. Pharmacodynamic effects are also rapid, suppressing luteinizing hormone and follicle-stimulating hormone within 4–6 h followed by suppression of estradiol levels completely [> 200 mg twice daily (BID)] or partially [150 mg once daily (QD)] within 24 h [1]. Discontinuation enables a return of baseline estrogen levels within 24–48 h [1], thus the pharmacodynamic effects, and hence efficacy and safety, are highly dependent on elagolix pharmacokinetics.
The ability to titrate estrogen levels into a ‘safe’ yet effective concentration window reduces hypoestrogenic effects (i.e., hot flushes and bone mineral density loss) associated with estrogen suppression [4, 5]. Titration of suppression can be achieved by dose and in combination with low-dose hormonal add-back therapy. Because of the mechanism of action and the relationship of the pharmacodynamics and pharmacokinetics of elagolix, a stable and robust population pharmacokinetics (PK) model is essential to support the approval of phase III dosage. Population PK models enable the prediction of drug exposure and the assessment of patient-related or clinical factors that may require dose adjustments to maintain adequate exposure for efficacy and/or safety. During the drug development process, the predictive accuracy of a population PK model improves as more data are acquired (increase in sample size) and new populations (expansion of covariates) participate in the drug candidate clinical programs. With the new approval of elagolix following phase III clinical studies in women with UF, drug exposures from three different populations of premenopausal women (healthy women and women with EM or UF) have been acquired consisting of dosages from 150 mg QD up to 400 mg BID. With these new data, we present the expansion of our previous population PK model [6] developed to support the US Food and Drug Administration approval of elagolix for the treatment of EM-associated pain, to support approval in women with HMB associated with UF.
2 Methods
2.1 Populations and Clinical Study Designs
Adult premenopausal female individuals (N = 2168) enrolled in six phase I studies (Studies 1–6) and seven Elaris phase III UF [7, 8] and EM [9] studies (EM-1, EM-2, EM-1-Ext, EM-2-Ext, UF-1, UF-2, and UF-Ext) were included in this population PK analysis. Study designs and treatment regimens for all 13 clinical studies are described in Table 1. All studies were conducted in accordance with their respective protocols, International Council for Harmonization Good Clinical Practice guidelines, applicable regulations and guidelines governing clinical study conduct, and ethical principles that have their origin in the Declaration of Helsinki. The study protocols were approved by the institutional review boards/ethics committees of the study sites (see Table 1), and all the participants gave written informed consent prior to participation in the studies.
Women in phase I studies were of age 18–49 years, inclusive, in general good health, and met the following criteria: a history of regular menstrual cycles (24–32 days with at least 3 and no more than 7 days of bleeding per month for 3 months), utilized two forms of non-hormonal contraception, negative urine pregnancy test results on the day of study drug administration and were > 6 months post-partum, post-abortion, or post-lactation, had not received GnRH agonists or antagonists in the previous 6 months, follicle-stimulating hormone level of < 35 mIU/mL, body mass index of 18–35 kg/m2, passed all clinical laboratory testing, and had normal liver function. Inclusion and exclusion criteria for phase III studies have been reported previously [7, 9].
Elagolix treatment regimens included in this analysis consisted of single doses of 150 mg, 200 mg, and 300 mg, and a multiple-dose range from 150 mg QD to 400 mg BID with a dosing duration range from 9 days to 12 months (see Table 1). In the UF phase III studies, elagolix 300 mg BID was administered alone and in combination with estradiol/norethindrone acetate (E2/NETA) 1/0.5 mg QD.
2.2 Study Drug Dosing and PK Sampling
During phase I studies, on-site study drug administration enabled acute blood draws typically at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 h post-dose with some extended blood draws occurring at 30, 36, and 48 h post-dose. Blood sample collection times are summarized in Table 1 for all studies. In phase III studies, blood samples were obtained during monthly visits. Dosing times in phase I studies were noted at the time of study drug administration, whereas a detailed profile of patients’ dosing behavior and exact dosing times for the majority of doses in the phase III population were obtained from electronic compliance packaging kits that dispensed the study drug to study patients. Kits were received monthly by patients and contained weekly blister packs. Compliance kits were returned during monthly visits; the kits were sent to Information Mediary Corporation (Ottawa, ON, Canada) and scanned to obtain study-specific and patient-specific compliance reports.
2.3 Bioanalysis
Pharmacokinetic samples were processed for plasma as described previously [6]. Plasma was analyzed for elagolix plasma concentrations by AbbVie Bioanalysis Lab (North Chicago, IL, USA) using a validated salt-assisted protein precipitation extraction, liquid chromatography method with tandem mass spectrometric detection [10]. Precision (coefficient of variation), accuracy (expressed as percent bias), and the lower limit of quantitation (LLOQ) for each study are provided in Table 1. Across studies, the LLOQ for elagolix established in each of the studies ranged between 0.0995 and 1.57 ng/mL with analytical precision ≤ 19.7% and a bias between − 7.3 and 8.5%.
2.4 Pharmacogenetic Testing
Pharmacogenetic testing for nucleotide polymorphisms in organic anion transporting polypeptide (OATP) 1B1 (rs4149056) was performed as described previously [6]. Consistent with the previous model, participants were classified into three different OATP1B1 transporter status categories consisting of poor transporter (PT), intermediate transporter (IT), and extensive transporter (ET) based on homozygous variant 521T>C(s5), heterozygous for 521T>C(s5), and homozygous wild-type 521T>C(s5), respectively [6].
2.5 Population PK Modeling
The elagolix population PK model was built using non-linear mixed-effects modeling based on NONMEM 7.4.2 (Icon Development Solutions, Hanover, MD, USA) compiled with the GNU Fortran compiler (Version 4.8.3). The infrastructure for model development and evaluation of the final model was a cluster featuring 47 Hewlett-Packard ProLiant servers under the OpenSUSE operating system with MOSIX Cluster and Grid Management (Version 4.4.0).
The PK model was constructed to describe the observed population pharmacokinetics of elagolix and relationships between elagolix dose, dosing compliance, elagolix plasma concentration–time profiles, and relevant covariates. Based on former knowledge of the elagolix population PK model developed for approval of elagolix for EM, a two-compartment model with lag time, combined residual error and inter-individual variability (IIV) on apparent clearance (CL/F), and apparent volume of distribution in the central compartment (Vc/F) with block matrix to estimate correlation between random effects was used as a starting model [6].
The PK model parameter estimation was conducted using the first-order conditional estimation method with interaction between IIV and residual variability (first-order conditional estimation with η-ε INTERACTION) employed within NONMEM. Relevant covariate-parameter relationships were investigated using forward inclusion/backward elimination procedures. The covariates investigated for influence on the elagolix PK parameters, CL/F and Vc/F, included age, body weight, body mass index, race, ethnicity, tobacco use, alcohol use, albumin, bilirubin, aspartate aminotransferase, alanine aminotransferase, and OATP1B1 genotype status. Creatinine, creatinine clearance, and the addition of E2/NETA were tested on CL/F, and OATP1B1 genotype status was tested on relative bioavailability (F1).
The model that best described the observed concentration–time data was selected based on a significant improvement in the objective function value, physiologically reasonable, precise, and statistically significant parameter estimates (95% confidence interval does not include reference values), adequate goodness-of-fit plots as well as minimal or no systematic bias in conditional weighted residuals (CWRES). In addition, the likelihood ratio test was used for hypothesis testing to discriminate among alternative nested models using the difference in objective function value between the different models. All statistical tests were performed at the 0.01 level of significance except for the backward elimination step of the covariate selection, which was performed at the 0.001 level of significance. Additional details can be found in the Electronic Supplementary Material (ESM).
2.6 Model Evaluation
Models were evaluated both during and after their development. Models were evaluated using goodness-of-fit plots, visual predictive checks, and bootstrap evaluation. Details of model evaluations are described in the ESM.
2.7 Prediction of Average Plasma Exposures and Evaluation of Covariates
The final PK model was used to estimate the empirical Bayesian individual participant PK parameters and average elagolix plasma concentration (Cavg). Individual average taken dose ratio (number of doses taken by the participant divided by the number of doses that the participant was prescribed) compliance scores for the pivotal and extension studies were used to calculate elagolix Cavg for individuals in the NONMEM dataset according to the following equation:
where TDOR represents the average taken dose ratio score for the pivotal and extension studies, F1 represents the relative bioavailability term, D represents the per protocol daily dose, and CL/F represents the apparent clearance. The relative bioavailability term F1 was introduced into the model to evaluate the OATP1B1 genotype status and fixed to 1 otherwise.
After identifying statistically significant covariates [i.e., body weight and OATP1B1 genotype status (Table 3)] for elagolix PK parameters, simulations were carried out to evaluate their impact on elagolix Cavg at the clinical regimen of elagolix 300 mg BID. With an average taken dose ratio compliance of 87.9% in the phase III studies, simulations were performed to compare elagolix exposures in a clinically relevant subset of participants based on the covariate of interest to the reference group. For an elagolix dose of 300 mg BID, a total of three body weight scenarios were simulated (median body weight of 76 kg ± 25 kg) to evaluate the effect of body weight on elagolix exposures. For each scenario of body weight, the individual transporter status was sampled from a multinominal distribution with a probability of having an IT or PT status of 15.5 or 1.5% (Table 2), respectively. These were compared to ET status. The final dataset included 1000 virtual participants for each scenario, and 100 replicates were simulated (total N = 100,000 for each scenario). Average plasma concentration ratio compared to the median Cavg of the reference group was calculated for each replicate and median and 5th and 95th percentiles of the ratios were compared across the different scenarios.
3 Results
3.1 Data Disposition and Exclusions
The population PK analysis included a total of 4511 data points from 175 healthy women in phase I studies, 8685 data points from 1310 women in phase III EM studies, and 4719 data points from 683 women in phase III UF studies totaling 17,915 plasma concentration data points from a total of 2168 participants across all studies (Table 1). A summary of the baseline demographics of all participants included in this analysis is provided in Table 2. For studies 1–6 (phase I), all participants receiving elagolix were included in the analysis. For the EM and UF phase III studies, data from 68 and 17 participants were excluded, respectively, because they did not have any measurable elagolix plasma concentrations above the LLOQ. As described previously [6], a data exclusion rule was used to remove implausible measurements from further analysis. No participant was completely excluded from the PK analysis because of the data exclusion rule. Handling of data below the LLOQ and additional information on data exclusion are provided in the ESM.
3.2 Population PK Model Results
Progression from the starting model [6] to a base model was achieved by accounting for differences in sampling frequency between phase I and phase III studies (e.g., intensive PK sampling for phase I vs sparse sampling in phase III) by testing different estimates for the error model for phase I and phase III studies. This resulted in a base model consisting of a two-compartment model with lag time, combined residual error model with different estimates for phase I and III, and IIV on CL/F and Vc/F (incorporating correlation using a block matrix).
The OATP1B1 genotype status on F1 was the most significant covariate found in the univariate inclusion. In addition, body weight on Vc/F was identified as significant covariate in the subsequent forward inclusion procedure. Addition of body weight to the model resulted in a minor decrease in IIV on Vc/F from 50.5 to 48.1%. Key intrinsic factors such as age (18–53 years) and race (White, Black, and other), and extrinsic factor of coadministration with E2/NETA (yes or no), were not statistically significant covariates for elagolix PK parameters.
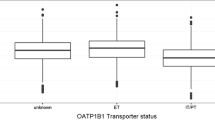
Evaluation of post-hoc CL/F and Vc/F estimates for differences in the population status showed no clear trend for differences in CL/F between healthy women or women with EM or UF (see Fig. 1). However, Vc/F estimates for healthy women were estimated slightly lower compared with women with EM or UF, which is mainly due to differences in sampling frequency between phase I and phase III studies. The estimated PK parameters from the final model including covariates and their associated variability for the selected final PK model are listed in Table 3.

Covariate relationships in healthy women and patients. a Elagolix apparent clearance and b elagolix apparent central volume of distribution. In the figure, the box shows the interquartile range with a median line. Lower/upper whiskers extend to the lowest/highest value within the 1.5× interquartile range. Data beyond the end of the whiskers are shown as filled circles
3.3 Evaluation of the Final Population PK Model
The goodness of fit for the final model was evaluated graphically and is displayed in Fig. 2. The plots of predicted and observed concentrations indicated that the model adequately described the observations over the entire range of elagolix plasma concentrations with a slight underprediction of high values. Several approaches (e.g., additive residual error model, IIV on absorption rate and lag time) were tested to improve the maximum concentration predictions, but none of them was able to better describe the high elagolix concentration values without worsening the area under the plasma concentration–time curve predictions, which is the relevant exposure needed in future applications such as exposure-efficacy and exposure-safety analyses.

Goodness-of-fit plots for the final population-pharmacokinetic model. a Population-predicted elagolix concentrations vs observed concentrations; b individual-predicted elagolix concentrations vs observed concentrations; c conditional weighted residuals vs time; and d conditional weighted residuals vs population predictions. Note: goodness-of-fit plots showing conditional weighted residuals are cut off at − 6 to 6, resulting in two data points not shown in the plot. EM endometriosis, UF uterine fibroids
The CWRES did not show any major trends when plotted against sampling times or population predictions, indicating that the model was appropriately unbiased. Outlying elagolix concentrations measured at day 1 for two participants in the UF-1 study that were not captured by the data exclusion rule did result in high CWRES (not shown in the plot) [see the ESM].
Based on 500 simulations, the visual predictive checks for the elagolix plasma concentration–time profiles showed that the model accurately describes the central tendency and variability of the data for all studies as well as for each population (i.e., healthy women and patients with UF or EM) separately. Some deviations occurred for the absorption phase because of individual variability that was not captured in the model or because of outlying measurements not captured by the implemented data exclusion rules (see ESM). The visual predictive checks for the final model are shown in Fig. 3.

Visual predictive checks for the final population-pharmacokinetic model. a phase I; b phase III, patients with endometriosis; and c phase III, patients with uterine fibroids. Note: visual predictive checks are cut off at 30 h after the last dose, as data are too sparse beyond. The gray dots represent observed data, the lines represent observed median (solid orange) and observed 5th and 95th percentiles (dashed orange), and the shaded blue regions represent the 90% prediction intervals for the simulated median and 5th and 95th percentiles (solid blue)
The final population PK model was used to estimate confidence intervals of the model parameters. A total of 880 out of 1000 bootstrap replicates plus the original dataset converged successfully. The estimated PK parameter values based on the original dataset were in good agreement with the medians of the parameter values estimated from the bootstrap replicates and none of the confidence intervals included the reference value (Table 3).
3.4 Elagolix Plasma Exposures and the Impact of Covariates in Patients with UF
Predicted elagolix exposures using the final PK model in women with UF were determined for the clinical regimen of elagolix 300 mg BID. Median (5th, 95th percentile) Cavg concentrations were 189 ng/mL (97.2, 391 ng/mL). The impact of the statistically significant covariates on elagolix exposures with 300-mg BID treatment were evaluated using simulations. A comparison of elagolix Cavg exposures based on the covariates of interest (i.e., OATP1B1 transporter status and body weight) is presented in Fig. 4. For the effect of OATP1B1 transporter status, participants with the IT genotype status had elagolix Cavg 1.45-fold higher compared with patients with the ET genotype status; while patients with the PT genotype status had elagolix exposures 2.09-fold higher compared with the reference ET genotype status. Missing OATP1B1 transporter status because of non-consent to pharmacogenetic testing was not considered as separate status during simulations. Despite these differences in elagolix Cavg across the OATP1B1 genotype status, the exposures greatly overlapped. In addition, body weight was found to not have a clinically relevant impact on elagolix exposures.

Effects of organic anion transporting polypeptide (OATP) 1B1 genotype status and body weight on elagolix average plasma concentration. Effects were determined following an elagolix dose of 300 mg twice daily. Dots and error bars represent median and 5th and 95th percentiles for simulated ratios of elagolix average concentrations stratified by covariate subgroup
4 Discussion
A population PK model for elagolix has been developed using data from six phase I studies in healthy women, four phase III studies in women with EM-associated pain, and three phase III studies in women with HMB associated with UF. With the addition of the elagolix PK data from the UF phase III studies, a broader range of PK, patient-related, and clinical data from 2168 participants across dose ranges of 150 mg QD to 400 mg BID in healthy women, 150 mg QD and 200 mg BID in women with EM, and 300 mg BID in women with UF was utilized to advance and re-evaluate the previous population PK model used to support US Food and Drug Administration approval of elagolix for EM [2, 6].
Consistent with the previously developed model, inclusion of data from several phase I and III studies enabled the development of a robust population PK model, with all structural parameters estimated with high precision (relative standard error: 0.5–4%). The overall PK parameter estimates of the final model developed using data from women with UF were similar to the parameter estimates of the EM analysis [6], suggesting that elagolix PK behavior is similar across healthy women and patients with EM or UF. This was also supported by the comparison of post-hoc CL/F and Vc/F estimates between healthy women and patients with EM or UF that demonstrated similarity between these two populations. Although the comparison was not formally tested during the covariate analysis because of the differences in sampling frequency between phase I and III studies (e.g., intensive PK sampling for phase I vs sparse sampling in phase III), and the lack of physiological basis for PK differences, the observed similarity in PK parameter estimates indicates that elagolix pharmacokinetics and exposures are comparable in healthy women and patients with EM or UF.
Of the covariates investigated for influence on CL/F, Vc/F or F1, OATP1B1 genotype status on F1 and body weight on Vc/F were the only significant covariates on elagolix PK parameters. Women with a PT and IT genotype status had an approximately 2.1-fold and 1.5-fold higher exposures (i.e., Cavg), respectively, compared with women with an ET genotype status. Overall, the Cavg for the IT and PT genotype status overlapped significantly with the range of values for ET genotypes and is not considered to have a clinically meaningful impact. This agrees with the previous model developed for EM that showed no clinical relevance of the OATP1B1 genotype status; however, in this model transporter status was added on F1 instead of CL/F as was done previously, as this is more physiologically relevant [6]. Body weight was identified as a significant covariate on Vc/F; however, the overall effect on elagolix Cavg was < 1%.
Limitations of this analysis included the constraints associated with utilizing sparse sampling data for a drug that has a short half-life and outpatient dosing to develop population PK models. These challenges were addressed by using a substantial intensive PK dataset along with the data exclusion rule in order to remove concentration data that are not physiologically plausible.
5 Conclusions
Consistent with the previous model [6], elagolix population pharmacokinetics are adequately described by a two-compartment model with a lag time in absorption and showed similar PK parameters across all populations analyzed. While OATP1B1 genotype status on F1 and body weight on Vc/F were the only statistically significant covariates on elagolix PK parameters, neither of these resulted in clinically relevant changes in exposure. None of the other tested covariates or key intrinsic factors such as age (18–53 years), race (66.7% White vs 30.4% Black), and coadministration with E2/NETA (yes or no) was found to be statistically correlated with elagolix PK parameters. These analyses suggest that patient demographics including intrinsic factors do not necessitate dose adjustment for elagolix in combination with low-dose E2/NETA as prescribed for patients with UF.
References
Ng J, Chwalisz K, Carter DC, Klein CE. Dose-dependent suppression of gonadotropins and ovarian hormones by elagolix in healthy premenopausal women. J Clin Endocrinol Metab. 2017;102(5):1683–91.
Orilissa™ (elagolix) [United States package insert]. North Chicago: AbbVie Inc.; 2018.
Oriahnn™ (elagolix, estradiol, and norethindrone acetate capsules; elagolix capsules) [United States package insert]. North Chicago: AbbVie Inc.; 2020.
Barbieri RL. Hormone treatment of endometriosis: the estrogen threshold hypothesis. Am J Obstet Gynecol. 1992;166(2):740–5.
Surrey ES. Gonadotropin-releasing hormone agonist and add-back therapy: what do the data show? Curr Opin Obstet Gynecol. 2010;22(4):283–8.
Winzenborg I, Nader A, Polepally AR, Liu M, Degner J, Klein CE, et al. Population pharmacokinetics of elagolix in healthy women and women with endometriosis. Clin Pharmacokinet. 2018;57(10):1295–306.
Schlaff WD, Ackerman RT, Al-Hendy A, Archer DF, Barnhart KT, Bradley LD, et al. Elagolix for heavy menstrual bleeding in women with uterine fibroids. N Engl J Med. 2020;382(4):328–40.
Simon JA, Al-Hendy A, Archer DF, Barnhart KT, Bradley LD, Carr BR, et al. Elagolix treatment for up to 12 months in women with heavy menstrual bleeding and uterine leiomyomas. Obstet Gynecol. 2020;135(6):1313–26.
Taylor HS, Giudice LC, Lessey BA, Abrao MS, Kotarski J, Archer DF, et al. Treatment of endometriosis-associated pain with elagolix, an oral GnRH antagonist. N Engl J Med. 2017;377(1):28–40.
Ng J, Duan WR, Marbury T, Schmidt JM, Klein CE. Elagolix pharmacokinetic profiles in women with renal or hepatic impairment. Clin Pharmacol Drug Dev. 2019;8(8):1053–61.
Shebley M, Polepally AR, Nader A, Ng JW, Winzenborg I, Klein CE, et al. Clinical pharmacology of elagolix: an oral gonadotropin-releasing hormone receptor antagonist for endometriosis. Clin Pharmacokinet. 2020;59(3):297–309.
Polepally AR, Ng JW, Salem AH, Dufek MB, Parikh A, Carter DC, et al. Assessment of clinical drug–drug interactions of elagolix, a gonadotropin-releasing hormone receptor antagonist. J Clin Pharmacol. 2020;60(12):1606–16.
Taylor HS, Dun EC, Chwalisz K. Clinical evaluation of the oral gonadotropin-releasing hormone-antagonist elagolix for the management of endometriosis-associated pain. Pain Manag. 2019;9(5):497–515.
Acknowledgements
We thank AbbVie employees Esteban Hernandez Maldonado for programming support and Stormy Koeniger, PhD for medical writing support of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by AbbVie. AbbVie contributed to the study design, research, and interpretation of the data and the writing, review, and approval of the manuscript.
Conflict of interest
DB, IW, ML, JD, NMM, PN, and MS are employees of AbbVie Inc. and may hold AbbVie stock and/or stock options.
Ethics approval
The studies reported herein were conducted in accordance with the International Council for Harmonisation guidelines, applicable regulations, guidelines governing clinical study conduct, and the ethical principles that have their origin in the Declaration of Helsinki. Approval was granted by institutional review boards and independent ethics committees at participating institutions.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
All individual participants signed an informed consent regarding the publishing of their data.
Availability of data and material
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols and clinical study reports), provided the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan and execution of a data sharing agreement. Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Code availability
Not applicable.
Author contributions
All authors contributed to the study design, analysis, and interpretation of the data. All authors participated in the drafting and revising of the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Beck, D., Winzenborg, I., Liu, M. et al. Population Pharmacokinetics of Elagolix in Combination with Low-Dose Estradiol/Norethindrone Acetate in Women with Uterine Fibroids. Clin Pharmacokinet 61, 577–587 (2022). https://doi.org/10.1007/s40262-021-01096-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01096-w