
Abstract
Nirmatrelvir is a potent and selective inhibitor of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease that is used as an oral antiviral coronavirus disease 2019 (COVID-19) treatment. To sustain unbound systemic trough concentrations above the antiviral in vitro 90% effective concentration value (EC90), nirmatrelvir is coadministered with 100 mg of ritonavir, a pharmacokinetic enhancer. Ritonavir inhibits nirmatrelvir’s cytochrome P450 (CYP) 3A4-mediated metabolism which results in renal elimination becoming the primary route of nirmatrelvir elimination when dosed concomitantly. Nirmatrelvir exhibits absorption-limited nonlinear pharmacokinetics. When coadministered with ritonavir in patients with mild-to-moderate COVID-19, nirmatrelvir reaches a maximum concentration of 3.43 µg/mL (11.7× EC90) in approximately 3 h on day 5 of dosing, with a geometric mean day 5 trough concentration of 1.57 µg/mL (5.4× EC90). Drug interactions with nirmatrelvir/ritonavir (PAXLOVIDTM) are primarily attributed to ritonavir-mediated CYP3A4 inhibition, and to a lesser extent CYP2D6 and P-glycoprotein inhibition. Population pharmacokinetics and quantitative systems pharmacology modeling support twice daily dosing of 300 mg/100 mg nirmatrelvir/ritonavir for 5 days, with a reduced 150 mg/100 mg dose for patients with moderate renal impairment. Rapid clinical development of nirmatrelvir/ritonavir in response to the emerging COVID-19 pandemic was enabled by innovations in clinical pharmacology research, including an adaptive phase 1 trial design allowing direct to pivotal phase 3 development, fluorine nuclear magnetic resonance spectroscopy to delineate absorption, distribution, metabolism, and excretion profiles, and innovative applications of model-informed drug development to accelerate development.
Similar content being viewed by others
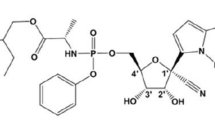

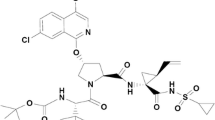
Avoid common mistakes on your manuscript.
Nirmatrelvir/ritonavir (PAXLOVIDTM) is the first approved oral antiviral drug indicated for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults who are at high risk for progression to severe COVID-19, including hospitalization or death. |
When coadministered with the pharmacokinetic enhancer ritonavir, nirmatrelvir’s cytochrome P450 3A4-mediated clearance is inhibited, allowing unbound nirmatrelvir trough concentration to exceed the antiviral in vitro 90% effective concentration throughout the dosing period. |
The clinical management of drug interactions for individual patients is a very important consideration for use of nirmatrelvir/ritonavir. |
1 Introduction
Since the start of the coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), there have been a few different approaches for the development of antiviral treatments for COVID-19. Initial efforts focused on re-purposing approved drugs with activity against some bacteria or viruses in hopes of potentially finding some overlapping antiviral activity against SARS-CoV-2 [1]. Attempted re-purposed drugs included hydroxychloroquine with or without azithromycin, favipiravir, and nelfinavir. Initial optimism of finding a repurposed antiviral drug faded as investigators tested these drugs clinically in a robust and systematic manner, as none of these potentially repurposed drugs proved to be effective against SARS-CoV-2 viral replication to treat COVID-19 [2,3,4,5].
Several pharmaceutical companies and government agencies began developing new antivirals against various potential targets within the SARS-CoV-2 viral replication cycle [6]. Remdesivir (GS-5734) was developed as an antiviral against RNA-based viruses that maintain global pandemic potential, such as the Ebola virus and the Coronaviridae family of viruses [7]. Molnupiravir was already a preclinical candidate at the onset of the COVID-19 pandemic and had been due to enter clinical trials against influenza [8]. Given its mechanism of action to terminate RNA chain elongation via viral RNA-dependent RNA polymerase (RdRp), molnupiravir showed activity against the SARS-CoV-2 virus [9]. Various intravenously administered monoclonal antibodies directed against the spike protein of SARS-CoV-2 received emergency use authorization (EUA) in the USA but have since been discontinued from clinical use due to changes in the spike protein with emerging viral variants [10].
Nirmatrelvir (PF-07321332) was developed in response to the COVID-19 pandemic as a potent and selective inhibitor of the SARS-CoV-2 main protease (Mpro), also referred to as 3-chymotrypsin-like (3CL) protease, which is a key enzyme needed for SARS-CoV-2 viral replication. Nirmatrelvir coadministered with 100 mg of ritonavir, a pharmacokinetic (PK) enhancer, is an oral antiviral COVID-19 treatment marketed as PAXLOVIDTM (Pfizer Inc, New York, NY) [11]. PAXLOVID (nirmatrelvir/ritonavir) is approved for use in the USA in adult patients with mild-to-moderate COVID-19 who are at high risk of progression to severe disease [11]. Results from a phase 2/3 clinical trial in patients with mild-to-moderate COVID-19 indicated that treatment with nirmatrelvir/ritonavir decreased the risk of progression to severe COVID-19 by 86% compared with placebo [11]. This manuscript provides a review of clinical pharmacology studies of nirmatrelvir/ritonavir conducted by the sponsor (Pfizer, Inc) as part of nirmatrelvir/ritonavir’s clinical development. It also provides a unique perspective on the rapid development of nirmatrelvir/ritonavir and innovative approaches utilized to elucidate its clinical pharmacology properties.
2 Mechanism of Action
The causative pathogen of the COVID-19 pandemic, SARS-CoV-2, is a member of the coronavirus family [12]. SARS-CoV-2 infects cells through the angiotensin-converting enzyme 2 (ACE2) receptor, with the lung and bronchial epithelial cells being the primary sites of infection [12, 13]. Like other coronaviruses, SARS-CoV-2 encodes Mpro, also referred to as 3CL protease or nonstructural protease 5 (nsp5) [14]. Mpro digests the viral pp1a and pp1ab polyproteins at multiple junctions to generate a series of proteins critical for viral replication and transcription, including RdRp, the helicase, and Mpro itself (Fig. 1) [12, 15, 16]. No close human analogs of the coronavirus Mpro are known [17]. The essential functional importance in viral replication together with the absence of closely related homologs in humans make Mpro an attractive antiviral drug target [15].

Nirmatrelvir acts on the proteolysis step of the SARS-CoV-2 coronavirus inside the host cell1. 1Reprinted from Alzyoud et al. [61]. Drug Design, Development and Therapy 2022:16 2463–2478 Originally published by and used with permission from Dove Medical Press Ltd. ACE2, angiotensin-converting enzyme 2; Mpro, main protease; PLpro, papain-like protease; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2
Nirmatrelvir is a potent and selective inhibitor of Mpro, exhibiting a broad-spectrum activity across the Coronaviridae family of 3CL proteases [18]. The mechanism of action of nirmatrelvir has been demonstrated by various biochemical, crystallographic, and cell-based methods [18, 19]. Nirmatrelvir binds to the active site of SARS-CoV-2 Mpro and forms a covalent interaction (1.90 Å C–S bond length) with the cysteine at position 145 in Mpro, as determined by the co-crystal structure [18]. Nirmatrelvir inhibited the full-length enzyme activity of SARS‑CoV-2 Mpro with a geometric mean half maximal inhibitory concentration (IC50) of 0.0192 μM and an inhibitory constant (Ki) of 0.00311 μM [18]. In cell cultures, nirmatrelvir exhibited antiviral activity against SARS-CoV-2 (USA-WA1/2020 isolate) infection of differentiated normal human bronchial epithelial (dNHBE) cells with 50% and 90% effective concentration (EC50 and EC90) values of 62 nM and 181 nM, respectively, as measured by viral replication after 3 days of nirmatrelvir exposure [19].
Nirmatrelvir retains consistent and potent in vitro antiviral activity across SARS-CoV-2 variants, including the Omicron variant [19]. Mpro is conserved in recent variants of interest, with variations occurring in the spike protein [20, 21]. Thus, nirmatrelvir/ritonavir’s efficacy is anticipated to be maintained as long as Mpro is conserved. Nirmatrelvir retained consistent and potent in vitro antiviral activity against SARS-CoV-2 Alpha, Beta, Gamma, Delta, Lambda, Mu, and Omicron BA.1 variants [11]. Additionally, in vitro antiviral activity of nirmatrelvir against the Omicron sub-variants BA.2, BA.2.12.1, BA.4, BA.4.6, BA.5, BF.7, BQ.1, BQ.1.11, and XBB.1.5 was also consistent, potent, and similar to that observed for other variants of SARS-CoV-2 [11].
No significant emergent Mpro mutations have been observed to date. In a series of in vitro experiments, mutant Mpro viruses were not viable and could not be evaluated in reverse-engineered recombinant SARS-CoV-2 assays [19, 22, 23]. The majority of Mpro amino acid substitutions that were selected by nirmatrelvir in vitro resulted in an EC50 shift of less than approximately five fold compared with wild type [19]. The greatest reduction in susceptibility occurred in virus containing a single E166V mutation [19]. This E166V mutation likely has a viral replication defect, as it either could not be generated or had low virus titer [19]. Only E166V of the identified in vitro mutations was observed in clinical development studies for nirmatrelvir/ritonavir [19]. The three patients with the E166V mutation cleared virus within 14 days of dose initiation, and none were hospitalized due to COVID-19 [19]. Additionally, as of the end of 2022, the E166V was only found in 16 out of ~13 million isolates included in the Global Initiative on Sharing Avian Influenza Data (GISAID) database, a public domain database for Mpro used for routine resistance surveillance [19].
3 Rationale for Ritonavir as a PK Enhancer
Less than dose-proportional increases in nirmatrelvir exposure were observed following single dose administration of nirmatrelvir as an oral suspension at doses of 150 mg, 500 mg, and 1500 mg without ritonavir under fasted conditions [24]. The median time of maximum observed concentration (Tmax) was 0.63–1.0 h post-dose across all doses tested without ritonavir [24]. Of the doses administered, mean half-life (t1/2) could only be calculated for the 150 mg dose, which was approximately 2 h (Table 1) [24]. Thus, to increase t1/2 to support a twice daily dosing regimen and boost systemic nirmatrelvir concentrations, a low dose of 100 mg ritonavir was chosen as a PK enhancer [11, 19]. A 100 mg ritonavir dose was chosen based on previously approved protease inhibitors that also use ritonavir as a PK enhancer, such as lopinavir, atazanavir, and darunavir [25,26,27]. Of note, ritonavir is not active against SARS-CoV-2 Mpro [11].
Coadministration of ritonavir increased nirmatrelvir concentration approximately eight fold (see Table 1 for a comparison of PK parameters of nirmatrelvir alone versus nirmatrelvir coadministered with 100 mg ritonavir) [24]. All further clinical development of nirmatrelvir was conducted with coadministration of 100 mg ritonavir [19]. The goal of coadministering ritonavir was to maintain trough concentration (Ctrough) of nirmatrelvir above the in vitro antiviral EC90 [292 ng/mL; adjusted by nirmatrelvir molecular weight (499.54 Daltons) and human plasma protein binding (fraction unbound = 0.310)] in > 90% of patients, which is believed to be important for a robust pharmacodynamic response.
4 Pharmacokinetics of Nirmatrelvir Coadministered with Ritonavir
4.1 Absorption
In a human mass balance study, approximately 55.0% of nirmatrelvir dose was recovered as unchanged drug in urine (representing drug absorbed systemically), and 27.5% was recovered in feces (potentially representing unabsorbed drug) after normalization to recovery determined by fluorine-19 nuclear magnetic resonance spectroscopy (19F-NMR )(see Section 4.3 for additional details) [28].
Following single doses ranging from 250 to 750 mg and multiple doses ranging from 75 to 500 mg of nirmatrelvir (coadministered with 100 mg ritonavir) under fasted conditions, nirmatrelvir exposure on days 1, 5, and 10 appeared to increase in a less than dose-proportional manner (Table 2) [24]. Dose-normalized area under the concentration versus time curve through the dosing interval (AUCtau) and maximum observed concentration (Cmax) values decreased as the nirmatrelvir dose increased [24]. Dose nonlinearity might be due to low permeability of nirmatrelvir [29]. As a moderately lipophilic, neutral compound, nirmatrelvir demonstrated a low passive permeability of 1.76 × 10−6 cm/s in a low-efflux Madin–Darby canine kidney (MDCK) cell assay [29]. Further, in vitro studies suggest that nirmatrelvir is a substrate for P-glycoprotein (P-gp), but not human breast cancer resistance protein (BCRP), transporters [29]. Low permeability coupled with low solubility (0.90–1.21 mg/mL throughout the physiological pH range) suggests that nirmatrelvir is classified as a Biopharmaceutics Classification System Class IV drug.
Following multiple dose administration of nirmatrelvir/ritonavir at doses of 75 mg/100 mg, 250 mg/100 mg, and 500 mg/100 mg twice daily under fasted conditions, dose-normalized geometric mean AUCtau on day 10 was 168.7, 151.1 and 79.56 ng*h/mL/mg, respectively [24]. Steady-state plasma concentrations were achieved by day 2 for all dose levels, with approximately two fold higher Ctrough on day 2 and trough values remaining similar on day 5 and day 10 [24]. Geometric mean accumulation ratios for AUCtau (Rac) ranged from 1.8 to 2.1 across all dose levels [24].
Nirmatrelvir/ritonavir PK in COVID-19 patients is generally consistent with PK observed in healthy participants. In EPIC-HR, patients with mild-to-moderate COVID-19 who received 300 mg/100 mg nirmatrelvir/ritonavir tablets twice daily reached a geometric mean Cmax of 3.43 µg/mL in approximately 3 h on day 5 of dosing, with a geometric mean day 5 Ctrough of 1.57 µg/mL [11].
4.1.1 Food effect
In an initial assessment of a food effect on nirmatrelvir/ritonavir exposure, four healthy adult volunteers were administered a single 250 mg/100 mg nirmatrelvir/ritonavir dose as an oral solution in either a fed or fasted state in different periods of a single ascending dose (SAD) study [24]. A geometric mean 1.5% and 15.3% increase in area under the concentration versus time curve from time zero extrapolated to infinity (AUCinf) and Cmax, respectively, was observed in participants in the fed state relative to the fasted state [24]. Thus, subsequent phase 2/3 studies included nirmatrelvir/ritonavir dosing without regard to food [11, 19].
The impact of a high-fat meal on the relative bioavailability of the commercial tablet formulation of nirmatrelvir was later assessed in a phase 1, open-label, randomized, single dose, two-sequence, two-period crossover study [30]. Twelve healthy adults were enrolled and randomized to receive 300 mg/100 mg single doses of nirmatrelvir/ritonavir under fed (800–1000 calories; 50% fat) and fasted conditions [11, 30]. A high-fat meal had a slight, but not clinically meaningful, impact on the systemic exposure of nirmatrelvir, with an approximately 1.2-fold increase in geometric mean AUCinf for the fed treatment compared to the fasted treatment [11]. Peak exposure was higher in the fed treatment, with a 1.6-fold increase in geometric mean Cmax compared with the fasted treatment [11]. However, since phase 2/3 studies included dosing without regard to food, there are no food restrictions for administration of nirmatrelvir/ritonavir [11, 19].
4.2 Distribution
The binding of nirmatrelvir to human plasma proteins was assessed at nirmatrelvir concentrations ranging from 0.3 to 10 µM [29]. The protein binding of nirmatrelvir in human plasma was approximately 69%, and there was no concentration dependency [29]. At a concentration of 1 µM, nirmatrelvir preferentially partitioned into human plasma relative to red blood cells, with a blood-to-plasma ratio of 0.60 [29]. The mean nirmatrelvir apparent volume of distribution was 104.7 L when coadministered with ritonavir [11].
4.3 Metabolism and Excretion
Ritonavir-mediated inhibition of cytochrome P450 (CYP) 3A4 led to a change in the primary excretion mechanism of nirmatrelvir to renal excretion [24]. Following multiple dosing of nirmatrelvir/ritonavir, nirmatrelvir mean t1/2 values on day 10 ranged from 6.8 to 8.0 h across all doses studied (Table 2) [24]. The percent of dose excreted unchanged during the nirmatrelvir dosing interval decreased with an increase in nirmatrelvir dose, with geometric mean (geometric coefficient of variability) 64% (12), 52% (4), and 23% (121) of the nirmatrelvir dose recovered in urine following 75 mg, 250 mg, and 500 mg doses (coadministered with 100 mg ritonavir), respectively, which is likely a consequence of lower bioavailability with increasing doses [24]. However, renal clearance was similar across all doses, which was 3.78, 3.43, and 2.93 L/h following 75 mg, 250 mg, and 500 mg nirmatrelvir doses (coadministered with 100 mg ritonavir), respectively [24].
A mass balance study used 19F-NMR methodology to determine the extent of nirmatrelvir-related material in urine and feces after a single 300 mg/100 mg nirmatrelvir/ritonavir dose [28]. Excretion into urine and feces was 49.6% and 35.3% of administered dose, respectively. When calculated by normalizing the recovery determined by 19F-NMR (80.7%) to a value of 95.8%, which is 100% minus 4.2%, the percentage of dose represented by hydrolytic metabolite M8 (19F-NMR silent), the percent of nirmatrelvir dose excreted unchanged in urine and feces was calculated as 55% and 27.5%, respectively [28]. In plasma, the only drug-related entity quantifiable by 19F-NMR was unchanged nirmatrelvir [28]. In excreta, unchanged nirmatrelvir was also the predominant drug-related entity, with small amounts of hydrolytic metabolites [28]. These metabolites included a carboxylic acid metabolite M5, present at 12.1% of dose and further metabolized into an acyl glucuronide secondary metabolite M7, and a carboxylic acid metabolite M9, present at < 1.0% of dose [28].
5 Drug Interactions
In vitro data indicate that nirmatrelvir is a substrate for P-gp and CYP3A4, but not for BCRP, multidrug and toxin extrusion protein (MATE) 1, MATE2K, sodium taurocholate cotransporting polypeptide (NTCP), organic anion transporter (OAT) 1, OAT2, OAT3, organic cation transporter (OCT) 1, OCT2, peptide transporter (PEPT) 1, organic anion transporter polypeptide (OATP) 1B1, OATP1B3, OATP2B1, or OATP4C1 [11, 31]. Nirmatrelvir does not reversibly inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6, nor does it induce any CYPs in vitro at clinically relevant concentrations [11, 31]. However, nirmatrelvir does have the potential to reversibly and time-dependently inhibit CYP3A4 and inhibit P-gp and OATP1B1 [11, 31]. Ritonavir has been shown in vitro to be mainly a substrate of CYP3A, although it can inhibit both CYP3A and (to a lesser extent) CYP2D6 [31, 32]. It appears to induce CYP3A, CYP1A2, CYP2C9, and CYP2B6, as well as other enzymes, such as glucuronosyltransferases (UGTs) [31, 33].
To assess victim drug interaction risk, clinical drug interaction studies were conducted with a strong CYP3A inhibitor (itraconazole) and with a strong CYP3A inducer (carbamazepine) to investigate the effects of their coadministration on the PK of nirmatrelvir/ritonavir in healthy subjects [32, 34]. To assess perpetrator drug interaction risk, two additional studies assessed the effect of nirmatrelvir/ritonavir as well as ritonavir alone on midazolam, a substrate of CYP3A4, and dabigatran, a substrate of P-gp [35]. A summary of the results from these drug interaction studies is provided in Table 3. Adding itraconazole, a second potent CYP3A4 inhibitor, on top of ritonavir, which is already used as PK enhancer of nirmatrelvir, minimally increased the exposure of nirmatrelvir beyond that caused by ritonavir [34]. Besides potently inhibiting CYP3A, itraconazole also has some P-gp inhibitory potential, and this is presumably the cause of a slight increase in nirmatrelvir AUCtau when nirmatrelvir/ritonavir is coadministered with itraconazole [34]. Carbamazepine markedly reduced nirmatrelvir and ritonavir exposure (with a 55% and 83% reduction in AUCinf for nirmatrelvir and ritonavir, respectively), and the magnitude of decrease was different such that a dose increment to counter this interaction was not clinically feasible [34]. The midazolam and dabigatran drug interaction studies suggest that interactions with concomitant medications that are CYP3A4 and P-gp substrates are mainly associated with ritonavir (Table 3) [35].
5.1 Management of Drug Interactions During Clinical Use
Clinical drug interactions with nirmatrelvir/ritonavir are primarily driven by ritonavir-mediated inhibition of CYP3A4, and to a lesser extent CYP2D6 and P-gp [11, 36]. Ritonavir is a potent, irreversible, and mechanism-based inhibitor of CYP3A4 [37]. Inhibition of CYP3A4 occurs rapidly after initiating ritonavir, with maximum inhibition occurring within 48 h [38, 39]. After ritonavir is discontinued, over 80% of CYP3A4 inhibition resolves within 3 days, which is well beyond ritonavir’s half-life of ~ 5 h, because new CYP3A4 enzyme needs to be regenerated due to irreversible inhibition [37, 39, 40]. However, the time to resolution of inhibition varies based on factors such as the patient’s age, and therefore resolution may take longer in some individuals, such as in the elderly [40]. When ritonavir is used for 5 days in combination with nirmatrelvir, its induction properties are less likely to be clinically relevant than when ritonavir is used chronically (e.g., in people who take HIV protease inhibitors), as induction is typically not observed until after 5–7 days of ritonavir dosing [39].
There are two classes of drugs that are contraindicated for use with nirmatrelvir/ritonavir: (1) drugs that are highly dependent on CYP3A for clearance and for which elevated concentrations are associated with serious and/or life-threatening reactions, and (2) drugs that are potent CYP3A inducers that reduce nirmatrelvir concentration, resulting in potential lack of efficacy [11]. It is important to note that nirmatrelvir/ritonavir cannot be started immediately after discontinuation of potent CYP3A inducers due to the delayed offset of the recently discontinued CYP3A inducer [11]. Other potentially important drug interactions can be managed by either dose reduction, increased monitoring of adverse events, monitoring concomitant drug levels when feasible, and temporary interruption of the concomitant medication [11]. These interactions must be managed during the short 5 day duration of nirmatrelvir/ritonavir dosing and for about 3–5 days after completion of nirmatrelvir/ritonavir dosing since ritonavir is an irreversible inhibitor of CYP3A4 [11]. One notable drug class that has resulted in serious drug interactions, including reported fatalities, is immunosuppressants [41]. Coadministration of nirmatrelvir/ritonavir with many immunosuppressants, such as tacrolimus, should be avoided, especially when close monitoring of immunosuppressant concentrations is not feasible [11].
The clinical management of drug interactions is a very important consideration when prescribing and dispensing nirmatrelvir/ritonavir. There are several resources available to healthcare providers to help them manage drug interactions with nirmatrelvir/ritonavir. The PAXLOVID EUA Fact Sheet and US Prescribing Information for Healthcare Providers extensively describe the risk of drug interactions, along with instructions on how to successfully manage patients on these drugs during the short duration of nirmatrelvir/ritonavir dosing [11, 42]. In addition, the US Food and Drug Administration (FDA) has provided a PAXLOVID Patient Eligibility Screening Checklist Tool for Prescribers for nirmatrelvir/ritonavir drug interactions [43]. The University of Liverpool provides a COVID-19 Drug Interactions Checker, which was revised following introduction of nirmatrelvir/ritonavir [44]. There is also an accompanying phone app, Liverpool COVID-19 iChart, available to prescribers for easy reference. The National Institution of Health has provided color-coded tables of drugs coadministered with nirmatrelvir/ritonavir based on how these interactions can be managed clinically [45]. Finally, an analysis of the top 100 drugs identified using real-world data as most likely to be prescribed to US high risk COVID-19 patients identified which of these top 100 coadministered drugs are and are not expected to interact with nirmatrelvir/ritonavir [46]. To safely prescribe nirmatrelvir/ritonavir, the potential for drug interactions needs to be considered by prescribers, both to take actions to manage these interactions as well as to determine whether nirmatrelvir/ritonavir is an appropriate treatment choice for each individual patient.
6 Bioanalytical Assays
Liquid chromatography tandem mass spectrometry (LC–MS/MS) bioanalytical methods for nirmatrelvir and ritonavir in plasma and urine were developed at Pfizer (Groton, CT) and York Bioanalytical Solutions (York, UK) as previously described [24, 47]. Briefly, the nirmatrelvir calibration curve range was 10.0–50,000 ng/mL for plasma and urine for the phase 1 study at Pfizer. Plasma and urine methods were subsequently revalidated at York with a calibration curve range of 10.0–10,000 ng/mL for nirmatrelvir and 5.0–5000 ng/mL for ritonavir in plasma and 100–200,000 ng/mL for nirmatrelvir in urine. Analytes and their internal standards (PF-07818226 and ritonavir-d6 for nirmatrelvir and ritonavir, respectively) were isolated from 100 µL of human plasma via protein precipitation with acetonitrile. Nirmatrelvir and internal standard (PF-07818226) were isolated from 50 µL of human urine sample either via protein precipitation with acetonitrile (Pfizer method) or dilution (York method) with 0.1% formic acid in acetonitrile:water 40:60 (v/v). Extracted plasma and urine samples were analyzed by LC–MS/MS in positive ionization mode. Separation was accomplished using a Waters Acquity ultra performance liquid chromatography (UPLC) BEH column (Waters Corporation, Milford, MA) (C18, 2.1 × 50 mm, 1.7 µM) and gradient elution. Both nirmatrelvir and ritonavir were quantified simultaneously in a single bioanalytical assay (i.e., in a single assay run). The overall inter-run nirmatrelvir plasma assay percent coefficient of variation and percent relative error were ≤ 8.4% and − 8.1% to − 1.0%, respectively (≤ 9.9% and − 6.4% to − 3.1%, respectively, for urine).
As previously described [24, 47], all samples were analyzed within established storage stability. Incurred sample reproducibility assessments met acceptance criteria for both plasma and urine. All analyses were conducted in concordance with industry best practices, Pfizer and contract laboratory standard operating procedures, and FDA and European Medicines Agency (EMA) requirements [48, 49].
Viral load was quantified as previously described at the University of Washington Medicine Clinical Virology Laboratory (Seattle, WA) using a validated Abbott RealTime Quantitative SARS-CoV-2 assay (Abbott, Abbott Park, IL) [50]. SARS-CoV-2 nucleic acid from nasopharyngeal or nasal swabs was quantified via detection of the RdRp and nucleocapsid genes using an Abbott m2000 System [50]. Total RNA was extracted using the Roche MagnaPure LC automated platform (Roche, Basel, Switzerland) [50].
7 Dose Selection—Model-Informed Drug Development
The therapeutic dose of 300 mg/100 mg nirmatrelvir/ritonavir twice daily for 5 days was selected based on achievement of the pharmacokinetic/pharmacodynamic (PK/PD) target. Maintaining antiviral plasma concentrations above the EC90 was considered important for effective antiviral activity [51, 52]. Therefore, in the absence of any prior exposure-response relationship for SARS-CoV-2, the PK/PD target was defined as nirmatrelvir dosing achieving protein binding-corrected Ctrough above the EC90 of 292 ng/mL (181 nM) in > 90% of patients starting after the first dose and maintaining Ctrough throughout the nirmatrelvir/ritonavir dosing duration [18, 24]. A model-informed drug development (MIDD) approach was used to support the therapeutic dose selection of nirmatrelvir/ritonavir for adults with COVID-19 and to guide dose adjustment for special patient populations [24]. Dosage and dose duration were supported by population pharmacokinetic (PopPK) modeling and quantitative systems pharmacology (QSP) modeling, respectively, which together were confirmed with physiologically based pharmacokinetic (PBPK) modeling.
7.1 Population Pharmacokinetic Modeling
Initially, a preliminary PopPK model of nirmatrelvir (coadministered with ritonavir 100 mg) was developed based on the preliminary data from the first in human (FIH) study, which included SAD and multiple ascending dose (MAD) data from 20 healthy volunteers receiving doses ranging from 75 to 750 mg [24]. The PopPK model was a two-compartment disposition model with first-order absorption and included a standard allometric model of baseline body weight (normalized to 70 kg) on apparent clearances and volumes with exponents fixed to 0.75 and 1, respectively [24]. The dose effects on the relative bioavailability (F1) and the first-order absorption rate constant (ka) were described by separate power functions [24].
Simulations were conducted utilizing the preliminary PopPK model across a dose range of 100–500 mg nirmatrelvir by 100 mg increments coadministered with 100 mg ritonavir and given twice daily for 5 days [24]. The interindividual variability on nirmatrelvir clearance was inflated to 60% to mimic variability in an outpatient population [24]. The simulation results suggested that the PK/PD target (> 90% of patients above EC90 after the first dose) was achieved with a 300 mg/100 mg dose of nirmatrelvir/ritonavir (Table 4) [24]. Nirmatrelvir doses of 100 mg and 200 mg did not reach target attainment, whereas nirmatrelvir doses of 400 mg and 500 mg offered only marginal gains in target attainment as compared to the 300 mg dose [24].
Subsequently, the preliminary PopPK model was updated with additional data in healthy adults and nonhospitalized symptomatic adults with COVID-19 who were at increased risk of progressing to severe illness [53]. The available intrinsic and extrinsic covariates were assessed with stepwise covariate modeling (forward selection and backward elimination) [53]. Other than the allometric scaling of body weight and dose-dependent absorption, nirmatrelvir clearance increased proportionally with body surface area-normalized creatinine clearance (nCLCR) up to an estimated breakpoint of 70 mL/min/1.73 m2 and was independent of nCLCR above this breakpoint [53]. Other significant covariates included carbamazepine or itraconazole coadministration as markers for drug interactions, COVID-19 on apparent clearance, formulation on F1, and age on central apparent volume of distribution [53]. Simulations were conducted again with the updated PopPK model, which confirmed that the dose recommendation remained unchanged [53].
7.2 Quantitative Systems Pharmacology Modeling
Dose duration selection of nirmatrelvir/ritonavir was informed by a published QSP model of the immune response to SARS-CoV-2 infection that quantitatively recapitulates the heterogeneity observed in clinical populations through the generation of a robust virtual population. QSP model details were previously published by Singh et al. and Rao et al. [24, 54]. Briefly, the QSP model consists of ordinary differential equations that link the within-host viral dynamics of SARS-CoV-2 to the activation of the innate and adaptive immune response and the accumulation of tissue damage as a result of proinflammatory-mediated cell death [24, 54]. Parameters of the virtual population were constrained to literature data from interventional randomized clinical trials investigating the efficacy of neutralizing antibody cocktail and antiviral therapeutics in outpatients with COVID-19, as well as published observational clinical reports in COVID-19 patients spanning viral load and immune responses in plasma and lung [55,56,57]. The resulting virtual population was used to predict the virological efficacy of 5 day and 10 day twice daily dosing of nirmatrelvir/ritonavir by incorporating the simulated nirmatrelvir/ritonavir PopPK profile and preclinical data on nirmatrelvir pharmacology in a mouse model of SARS-CoV-2 that was used to estimate the in vivo nirmatrelvir potency with the QSP model [24].
To assess the efficacy of different dose durations, QSP model simulations assumed a symptomatic outpatient COVID-19 virtual population with dosing 4 days post viral load peak/symptom onset informed by the above-mentioned published randomized clinical trials on outpatient COVID-19 therapies [24]. The QSP model simulations predicted that, on an aggregate level, the viral load decline with a 5 day and 10 day nirmatrelvir/ritonavir regimen would be similar (Fig. 2) [24]. Thus, 5 days of twice daily dosing was predicted to be sufficient for the treatment of symptomatic confirmed SARS-CoV-2 outpatients [24]. Moreover, uncertainties around the evolving disease and patient landscape with respect to the prevalent variants, seropositivity, and vaccination status, were addressed through sensitivity analysis of the model to address emerging development questions. The clinical dose and dose duration were subsequently confirmed by results of the Evaluation of Protease Inhibition for COVID-19 in High-Risk Patients (EPIC-HR) study, a pivotal phase 2/3 study, which demonstrated the safety and efficacy of 300 mg/100 mg nirmatrelvir/ritonavir administered twice daily for 5 days [50]. Therefore, the QSP model provided an expedient MIDD approach to inform emerging clinical development decisions in short time frames. Further research is warranted to determine if the 5 day dosing regimen is optimal for patients with varying degrees of immunocompromised status.

QSP-predicted viral load decline versus time with various dose durations of 300 mg/100 mg nirmatrelvir/ritonavir1. Simulation of a virtual population (n = 502) to predict viral load effect for nirmatrelvir/ritonavir 300 mg/100 mg twice daily in symptomatic patients with COVID-19. 1Reprinted from Singh et al. [24]. BID, twice daily; PI, prediction interval; QSP, quantitative systems pharmacology model
7.3 Physiologically Based Pharmacokinetic Modeling
A PBPK model of nirmatrelvir/ritonavir with first-order absorption was developed using Simcyp (Certara, Sheffield, UK) [58]. The nirmatrelvir model was developed using both preclinical and clinical data of nirmatrelvir with and without ritonavir, then verified with clinical data from nirmatrelvir/ritonavir SAD, MAD, and drug interaction studies [58]. The existing Simcyp first-order ritonavir compound file was leveraged without further modification and further verified with additional nirmatrelvir/ritonavir clinical data [58]. The combined nirmatrelvir/ritonavir PBPK model was able to predict nirmatrelvir AUC and Cmax within ± 20% of corresponding observed values and predict ritonavir PK parameters within two fold of observed values [58]. A complex absorption PBPK model was further developed for nirmatrelvir (manuscript in preparation). Both the first-order and complex absorption PBPK models were used throughout the development program to estimate perpetrator and victim drug interactions and in formulation development from FIH study to commercial supplies.
7.4 Exposure-Response Analysis
An exploratory exposure-response (ER) analysis of viral load in patients enrolled in EPIC-HR was investigated primarily through graphical examination of PK (predicted nirmatrelvir day 5 Ctrough) versus pharmacodynamic (PD) [change from baseline (CFB) in day 5 viral RNA]. Viral load over time was evaluated using viral RNA titers measured in nasopharyngeal swabs via reverse transcription–polymerase chain reaction (RT–PCR) [50]. The ER analysis included 740 and 734 patients in the placebo and nirmatrelvir/ritonavir arm, respectively, who had both PK and PD data available.
Day 5 CFB in viral load did not show any correlation with nirmatrelvir exposure across the exposure range observed (Fig. 3). Additionally, no meaningful trend in ER relationship was evident when looking at categories of clinical interest, including baseline SARS-CoV-2 serology status, baseline viral load, or treatment onset (data not shown). The lack of relationship can be attributed to several factors. First, only one dose level was studied, which did not allow a wide spread of exposure data. Secondly, a significant portion (49.9%) of available Ctrough values were > 5 × EC90 with very few values less than 3 × EC90. Lastly, there was a high degree of variability in the viral load data, both at baseline and day 5 CFB. It was observed that patients who were hospitalized had nirmatrelvir Ctrough values below the median Ctrough value. However, this observation should also be interpreted with caution due to the small sample size of six patients in the PK/PD dataset that were hospitalized while receiving nirmatrelvir/ritonavir in EPIC-HR.

Viral load CFB versus nirmatrelvir Ctrough on day 5. Blue dots represent individual data from patients enrolled in EPIC-HR, the pivotal nirmatrelvir/ritonavir phase 2/3 study, who received 300 mg/100 mg nirmatrelvir/ritonavir twice daily for 5 days [50]. Data from patients in the active treatment arm of EPIC-HR who were hospitalized are represented by red dots. Median PopPK-predicted nirmatrelvir day Ctrough is shown as a black dashed line, and nirmatrelvir concentration relative to EC90 (292 ng/mL) reference lines are shown as blue dashed lines. A linear regression is shown as a solid blue line with an associated 95% confidence interval shaded in gray. CFB, change from baseline; Cmin, minimum observed concentration; Ctrough, trough concentration; EC90, 90% effective concentration; EPIC-HR, Evaluation of Protease Inhibition for COVID-19 in High-Risk Patients; PopPK, population pharmacokinetic model
8 Special Populations
8.1 Renal Impairment
The effect of renal impairment on the PK of nirmatrelvir/ritonavir was examined in a single dose PK study [47]. This was a phase 1, nonrandomized, open-label study in participants with stable mild [estimated glomerular filtration rate (eGFR) ≥ 60 to < 90 mL/min], moderate (eGFR ≥ 30 to < 60 mL/min), or severe (eGFR < 30 mL/min and not requiring dialysis) renal impairment, and a control group of participants with normal renal function (eGFR ≥ 90 mL/min) [47]. eGFR was determined using the Chronic Kidney Disease Epidemiology Collaboration (CKD–EPI) equation [47, 59]. All study participants received a single 100 mg dose of nirmatrelvir enhanced with 100 mg ritonavir administered − 12, 0, 12, and 24 h relative to nirmatrelvir dosing to ensure maximal inhibition of CYP3A4 [47]. Eight participants each were treated in the mild, moderate, and severe renal impairment group, and ten participants were treated in the control group [47].
Nirmatrelvir exposure increased with increasing severity of renal impairment [47]. Compared with the control group, the mean AUCinf in participants with mild, moderate, and severe renal impairment was higher by 24%, 87%, and 204%, respectively [47]. The mean Cmax compared with the control group was higher by 30%, 38%, and 48% for participants with mild, moderate, and severe renal impairment, respectively [47]. Urinary recovery of unchanged nirmatrelvir was geometric mean (geometric coefficient of variability) 31% (45), 43% (23), 31% (56), and 19% (50) for the normal renal function and mild, moderate, and severe renal impairment groups, respectively [47]. Nirmatrelvir clearance increased proportionally to eGFR but appeared to plateau after ~ 70 mL/min (Fig. 4) [47].

Nirmatrelvir CL/F versus eGFR following a single 100 mg nirmatrelvir dose enhanced with 100 mg ritonavir1. The bold line is the predicted linear regression line; the shaded area represents the 90% confidence interval. The vertical lines represent the boundary criteria of the renal function groups. 1Reprinted from Toussi et al. [47]. CKD–EPI, Chronic Kidney Disease Epidemiology Collaboration equation; CL/F, apparent oral clearance; eGFR, estimated glomerular filtration rate
The preliminary PopPK model (see Section 7.1) was used to conduct simulations with the population clearance reduced by one-third and one-half to mimic mild and moderate renal impairment, respectively, included in the clinical study [47]. Mild and moderate renal impairment were simulated because these populations were enrolled in phase 3 studies with an eGFR cutoff of 45 mL/min/1.73 m2 [50]. Based on matching the predicted renal impairment nirmatrelvir exposures to those from the normal renal function group (in order to achieve the PK/PD target of > 90% of simulated patients with Ctrough > EC90), the recommended nirmatrelvir/ritonavir dose was 300 mg/100 mg for mild renal impairment (nCLCR 60 to < 90 mL/min/1.73 m2) and 150 mg/100 mg for moderate renal impairment (nCLCR 30 to < 60 mL/min/1.73 m2) (Fig. 5). Additional clinical data and simulations are needed to select a dose in patients with severe renal impairment [47].

PopPK-predicted nirmatrelvir Ctrough on day 5 of twice daily nirmatrelvir with 100 mg ritonavir dosing for simulated patients with varying degrees of renal impairment1. Open circles represent predicted Ctrough; red symbols represent group means; blue lines represent 10th and 90th percentiles; the red-dashed line is the EC90 (292 ng/mL). (A) 150 mg/100 mg nirmatrelvir/ritonavir every 12 h, with clearance reduced by one-half (i.e., moderate renal impairment); (B) 300 mg/100 mg nirmatrelvir/ritonavir every 12 h, with clearance reduced by one-third (i.e., mild renal impairment); (C) 300 mg/100 mg nirmatrelvir/ritonavir every 12 h, with no reduction in clearance (reference group). 1Reprinted from Toussi et al. [47]. Ctrough, trough concentration; EC90, 90% effective concentration; PopPK, population pharmacokinetic model
8.2 Hepatic Impairment
The effect of hepatic impairment on the plasma PK of nirmatrelvir was assessed in a phase 1, nonrandomized, open-label study [60]. A single oral dose of 100 mg nirmatrelvir enhanced with 100 mg ritonavir was administered at − 12, 0, 12 and 24 h to ensure maximal inhibition of CYP3A4 in 16 adult participants: 8 participants with stable moderate hepatic impairment (Child–Pugh class B) and 8 age- and weight-matched participants with normal hepatic function [60].
Nirmatrelvir systemic exposure was comparable between the moderate hepatic impairment group and the normal hepatic function group [60]. Geometric mean AUCinf was 15.24 µg h/mL and 15.06 µg h/mL and geometric mean Cmax was 1.886 µg/mL and 1.923 µg/mL for the normal hepatic function and moderate hepatic impairment groups, respectively [60]. Adjusted geometric mean ratio (90% confidence interval) of nirmatrelvir AUCinf and Cmax comparing moderate hepatic impairment (test) to normal hepatic function (reference) was 98.78% (70.65%, 138.12%) and 101.96% (74.20%, 140.11%), respectively [60]. Thus, no dose adjustment is needed for patients with moderate hepatic impairment (Child–Pugh class B), nor for patients with mild impairment (Child–Pugh class A). Nirmatrelvir/ritonavir has not been studied in participants with severe hepatic impairment since ritonavir is not recommended in severe hepatic impairment patients [11, 37].
8.3 Additional Subpopulation Analyses
In the PopPK covariate analysis, gender, race, and obesity status (body mass index ≥ 30 kg/m2) did not significantly impact nirmatrelvir PK parameters [53]. Apparent oral central volume of distribution decreases as age increases, but the effect was not considered to be clinically relevant [53]. Systemic exposure in a cohort of Japanese participants included in the MAD study was numerically lower, but not clinically meaningfully different, from those in other cohorts [24]. COVID-19 infection status did significantly decrease predicted apparent oral clearance in the PopPK model, but that could be a counteracting effect of the commercial formulation of nirmatrelvir 150 mg tablet on relative bioavailability [53].
Clinical studies to evaluate the use of nirmatrelvir/ritonavir in additional patient populations, including patients who are immunocompromised, pediatric, pregnant, lactating, and who have severe renal impairment, are still ongoing at the time of publication.
9 Innovation and Acceleration During the Pandemic
The rapid clinical development of nirmatrelvir/ritonavir was feasible partly due to numerous innovations in the conduct of clinical pharmacology evaluations. First, unlike traditional SAD followed by MAD studies, the FIH study employed a multipart method with flexibility built into the protocol design [24]. Coupled with rapid turnaround of PK data and automated analysis of PK and safety data, this design enabled twice weekly dose escalations. With evolving data, the protocol was modified to include additional assessments, such as a mass balance assessment by 19F-NMR and concentration-QTc evaluation to further accelerate clinical development [24]. A cohort of Japanese subjects enrolled to assess the influence of race on PK enabled Japan to participate in the nirmatrelvir/ritonavir phase 3 program and accelerated drug approval in Japan. Further, since nirmatrelvir contains a trifluoroacetyl moiety that is retained, it allowed the use of 19F-NMR to quantitate nirmatrelvir’s in vivo disposition. Accordingly, a 19F-NMR mass balance assessment was utilized to elucidate the absorption, distribution, metabolism, and excretion (ADME) profile of nirmatrelvir when coadministered with ritonavir [28]. This approach, referred to as “cold ADME” negated the need for expensive, time-consuming manufacturing of the 14C-radiolabeled drug that is typically used for such studies.
The nirmatrelvir/ritonavir development program involved innovative applications of MIDD at all stages of decision-making. PopPK modeling of data from SAD and MAD cohorts of the FIH study enabled rapid phase 3 dose selection, which ensured that systemic nirmatrelvir concentrations stayed well above the in vitro EC90 throughout nirmatrelvir/ritonavir dosing. QSP model development during the early days of the pandemic informed the 5 day dosing duration in phase 2/3 studies since QSP simulations provided confidence that viral load decline would be comparable between 5 day versus 10 day treatment durations, avoiding additional phase 2 studies to assess optimal treatment duration. PBPK modeling was used to assess drug interaction potential and to support formulation development processes. The use of a fully automated, in-house program for assessing relationship between drug concentrations and QTc interval allowed for submission-ready reports in less than 1 day, which eliminated the need for electrocardiogram monitoring in EPIC-HR after completion of a sentinel cohort. Removal of electrocardiogram monitoring from phase 2/3 studies meant patients did not have to return to the site for electrocardiograms, resulting in a significant impact on patient burden, enrollment, and trial cost. This analysis, along with additional preclinical data, served to waive a thorough QT/QTc study.
10 Conclusions
Nirmatrelvir is an oral antiviral treatment that potently and selectively inhibits SARS-CoV-2 Mpro to treat COVID-19. To prolong its half-life and maintain systemic concentrations above the target EC90, nirmatrelvir is coadministered with 100 mg of ritonavir. PopPK and QSP modeling support clinical doses of 300 mg/100 mg nirmatrelvir/ritonavir (150 mg/100 mg for patients with mild renal impairment) twice daily for 5 days to maintain Ctrough above EC90.
Nirmatrelvir demonstrates nonlinear pharmacokinetics owing to absorption-limited bioavailability. When coadministered with ritonavir at the clinical dose (300 mg/100 mg nirmatrelvir/ritonavir), ritonavir near-maximally inhibits nirmatrelvir’s CYP3A4-mediated metabolism, making renal elimination the predominant nirmatrelvir elimination pathway and increasing half-life to approximately 6 h. Following clinical dosing, nirmatrelvir reaches a Cmax of 3.43 µg/mL in approximately 3 h.
Ritonavir is the driver of nirmatrelvir/ritonavir drug interactions, which primarily occur via ritonavir-mediated CYP3A4, CYP2D6, or P-gp inhibition. Because of this, drug interaction management is an important consideration when prescribing and dispensing nirmatrelvir/ritonavir. Numerous drug interaction resources are available to healthcare providers to manage drug interactions. Most notably, the PAXLOVID EUA Fact Sheet for Healthcare Providers and US Prescribing Information for nirmatrelvir/ritonavir extensively describe the risk of interactions with instructions on how to successfully manage patients on these drugs during the short duration of nirmatrelvir/ritonavir dosing.
Rapid clinical development of nirmatrelvir/ritonavir in the face of an emerging COVID-19 pandemic was possible in part due to a number of clinical pharmacology innovations. An adaptive phase 1 trial design was used to add additional cohorts to accelerate clinical development. Using 19F-NMR mass balance “cold ADME” assessment negated the need for time-consuming manufacture of radiolabeled nirmatrelvir drug typically used for such studies. MIDD applications, including PopPK, QSP, PBPK, and concentration-QTc interval modeling, aided decision-making and hastened the nirmatrelvir/ritonavir development process.
References
Riva L, Yuan S, Yin X, Martin-Sancho L, Matsunaga N, Pache L, et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. 2020;586(7827):113–9.
Xu J, Cao B. Lessons learnt from hydroxychloroquine/azithromycin in treatment of COVID-19. Eur Respir J. 2022;59(1):1–3.
Avezum Á, Oliveira GBF, Oliveira H, Lucchetta RC, Pereira VFA, Dabarian AL, et al. Hydroxychloroquine versus placebo in the treatment of non-hospitalised patients with COVID-19 (COPE - Coalition V): a double-blind, multicentre, randomised, controlled trial. Lancet Reg Health Am. 2022;11: 100243.
Bosaeed M, Alharbi A, Mahmoud E, Alrehily S, Bahlaq M, Gaifer Z, et al. Efficacy of favipiravir in adults with mild COVID-19: a randomized, double-blind, multicentre, placebo-controlled clinical trial. Clin Microbiol Infect. 2022;28(4):602–8.
Miyazaki T, Hosogaya N, Fukushige Y, Takemori S, Morimoto S, Yamamoto H, et al. A multicenter randomized controlled trial to evaluate the efficacy and safety of nelfinavir in patients with mild COVID-19. Microbiol Spectr. 2023;e0431122.
Santos IA, Grosche VR, Bergamini FRG, Sabino-Silva R, Jardim ACG. Antivirals against coronaviruses: candidate drugs for SARS-CoV-2 treatment? Front Microbiol. 2020;11:1818.
Eastman RT, Roth JS, Brimacombe KR, Simeonov A, Shen M, Patnaik S, et al. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent Sci. 2020;6(5):672–83.
Imran M, Kumar Arora M, Asdaq SMB, Khan SA, Alaqel SI, Alshammari MK, et al. Discovery, development, and patent trends on molnupiravir: a prospective oral treatment for COVID-19. Molecules. 2021;26(19).
Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, Gonzalez A, Delos Reyes V, et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N Engl J Med. 2022;386(6):509–20.
National Institute of Health. COVID-19 treatment guidelines: anti-SARS-CoV-2 monoclonal antibodies. 2023. https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/anti-sars-cov-2-monoclonal-antibodies/. Accessed 31 May 2023.
PAXLOVID (nirmatrelvir tablets; ritonavir tablets), co-packaged for oral use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217188s000lbl.pdf. Accessed 31 May 2023.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
de Vries M, Mohamed AS, Prescott RA, Valero-Jimenez AM, Desvignes L, O’Connor R, et al. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J Virol. 2021;95(7):e01819-e1820.
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–12.
Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59(14):6595–628.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–93.
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–7.
Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586–93.
Pfizer. The Antimicrobial Drugs Advisory Meeting: PAXLOVIDTM (nirmatrelvir [PF-07321332] tablets; ritonavir tablets). Advisory Committee briefing materials. 2023. https://www.fda.gov/media/166199/download. Accessed 31 May 2023.
Chen SA, Arutyunova E, Lu J, Khan MB, Rut W, Zmudzinski M, et al. SARS-CoV-2 M(pro) protease variants of concern display altered viral substrate and cell host target galectin-8 processing but retain sensitivity toward antivirals. ACS Cent Sci. 2023;9(4):696–708.
Candido KL, Eich CR, de Fariña LO, Kadowaki MK, da Conceição Silva JL, Maller A, et al. Spike protein of SARS-CoV-2 variants: a brief review and practical implications. Braz J Microbiol. 2022;53(3):1133–57.
Iketani S, Mohri H, Culbertson B, Hong SJ, Duan Y, Luck MI, et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature. 2023;613(7944):558–64.
Zhou Y, Gammeltoft KA, Ryberg LA, Pham LV, Tjørnelund HD, Binderup A, et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in infectious cell culture system. Sci Adv. 2022;8(51).
Singh RSP, Toussi SS, Hackman F, Chan PL, Rao R, Allen R, et al. Innovative randomized phase I study and dosing regimen selection to accelerate and inform pivotal COVID-19 trial of nirmatrelvir. Clin Pharmacol Ther. 2022;112(1):101–11.
KALETRA (lopinavir and ritonavir) tablet, for oral use and KALETRA (lopinavir and ritonavir oral solution. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021251s052_021906s046lbl.pdf. Accessed 28 June 2023.
REYATAZ (atazanavir sulfate) capsules. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021567s026lbl.pdf. Accessed 28 June 2023.
PREZISTA (darunavir) oral suspension, for oral use and PREZISTA (darunavir) tablet, film coated for oral use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021976s021lbl.pdf. Accessed 28 June 2023.
Singh RSP, Walker GS, Kadar EP, Cox LM, Eng H, Sharma R, et al. Metabolism and excretion of nirmatrelvir in humans using quantitative fluorine nuclear magnetic resonance spectroscopy: a novel approach for accelerating drug development. Clin Pharmacol Ther. 2022;112(6):1201–6.
Eng H, Dantonio AL, Kadar EP, Obach RS, Di L, Lin J, et al. Disposition of nirmatrelvir, an orally bioavailable inhibitor of SARS-CoV-2 3C-like protease, across animals and humans. Drug Metab Dispos. 2022;50(5):576–90.
National Institute of Health ClinicalTrails.gov. Food effect study to evaluate the effect of high-fat meal on the relative bioavailability of PF-07321332 boosted with ritonavir in healthy adult participants. 2022. https://clinicaltrials.gov/ct2/show/NCT05129475?term=C4671019&draw=2&rank=1. Accessed 31 May 2023.
Atmar RL, Finch N. New perspectives on antimicrobial agents: molnupiravir and nirmatrelvir/ritonavir for treatment of COVID-19. Antimicrob Agents Chemother. 2022;66(8):e02404-e2421.
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–6.
Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of the HIV protease inhibitors 3: effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampin, or bupropion. Drug Metab Dispos. 2012;40(3):610–6.
Cox DS, van Eyck L, Pawlak S, Beckerman B, Linn C, Ginman K, et al. Effects of itraconazole and carbamazepine on the pharmacokinetics of nirmatrelvir/ritonavir in healthy adults. Br J Clin Pharmacol. 2023;89(9):2867–76.
Cox DS, Rehman M, Khan T, Ginman K, Salageanu J, LaBadie RR, et al. Effects of nirmatrelvir/ritonavir on midazolam and dabigatran pharmacokinetics in healthy participants. Br J Clin Pharmacol. 2023 (Online ahead of print).
Aarnoutse RE, Kleinnijenhuis J, Koopmans PP, Touw DJ, Wieling J, Hekster YA, et al. Effect of low-dose ritonavir (100 mg twice daily) on the activity of cytochrome P450 2D6 in healthy volunteers. Clin Pharmacol Ther. 2005;78(6):664–74.
NORVIR (ritonavir) tablet, for oral use and NORVIR (ritonavir) oral solution and NORVIR (ritonavir) oral powder. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209512lbl.pdf. Accessed 31 May 2023.
Katzenmaier S, Markert C, Riedel KD, Burhenne J, Haefeli WE, Mikus G. Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using a limited sampling strategy. Clin Pharmacol Ther. 2011;90(5):666–73.
Marzolini C, Kuritzkes DR, Marra F, Boyle A, Gibbons S, Flexner C, et al. Recommendations for the management of drug-drug interactions between the COVID-19 antiviral nirmatrelvir/ritonavir (Paxlovid) and comedications. Clin Pharmacol Ther. 2022;112(6):1191–200.
Stader F, Khoo S, Stoeckle M, Back D, Hirsch HH, Battegay M, et al. Stopping lopinavir/ritonavir in COVID-19 patients: duration of the drug interacting effect. J Antimicrob Chemother. 2020;75(10):3084–6.
US Food and Drug Administration. The Antimicrobial Drugs Advisory Meeting: PAXLOVIDTM (nirmatrelvir [PF-07321332] tablets; ritonavir tablets). FDA Briefing Document. 2023. https://www.fda.gov/media/166197/download. Accessed 15 Nov 2023.
Emergency use authorization for PAXLOVID (nirmatrelvir tablets; ritonavir tablets), co-packaged for oral use. https://www.fda.gov/media/155050/download. Accessed 31 May 2023.
US Food and Drug Administration. PAXLOVID patient eligibility screening checklist tool for prescribers. 2023. https://www.fda.gov/media/158165/download. Accessed 31 May 2023.
University of Liverpool. COVID-19 drug interactions checker. 2023. https://www.covid19-druginteractions.org/checker. Accessed 31 May 2023.
National Institute of Health. Drug-drug interactions between ritonavir-boosted nirmatrelvir (Paxlovid) and concomitant medications. 2023. https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/ritonavir-boosted-nirmatrelvir--paxlovid-/paxlovid-drug-drug-interactions/. Accessed 31 May 2023.
Gerhart J, Draica F, Benigno M, Atkinson J, Reimbaeva M, Francis D, et al. Real-world evidence of the top 100 prescribed drugs in the United States and their potential for drug interactions with nirmatrelvir; ritonavir. AAPS J. 2023;25(5):73.
Toussi SS, Neutel JM, Navarro J, Preston RA, Shi H, Kavetska O, et al. Pharmacokinetics of oral nirmatrelvir/ritonavir, a protease inhibitor for treatment of COVID-19, in subjects with renal impairment. Clin Pharmacol Ther. 2022;112(4):892–900.
US Food and Drug Administration. Bioanalytical method validation: guidance for industry. 2018. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf. Accessed 31 May 2023.
European Medicines Agency. Guideline on bioanalytical method validation. 2011. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed 31 May 2023.
Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N Engl J Med. 2022;386(15):1397–408.
Reddy MB, Morcos PN, Le Pogam S, Ou Y, Frank K, Lave T, et al. Pharmacokinetic/pharmacodynamic predictors of clinical potency for hepatitis C virus nonnucleoside polymerase and protease inhibitors. Antimicrob Agents Chemother. 2012;56(6):3144–56.
Bertz RJ, Persson A, Chung E, Zhu L, Zhang J, McGrath D, et al. Pharmacokinetics and pharmacodynamics of atazanavir-containing antiretroviral regimens, with or without ritonavir, in patients who are HIV-positive and treatment-naive. Pharmacotherapy. 2013;33(3):284–94.
Chan PLS, Singh RSP, Cox DS, Shi H, Damle B, Nicholas T. Dosing recommendation of nirmatrelvir/ritonavir using an integrated population pharmacokinetic analysis. CPT Pharmacometrics Syst Pharmacol. 2023 (Online ahead of print).
Rao R, Musante CJ, Allen R. A quantitative systems pharmacology model of the pathophysiology and treatment of COVID-19 predicts optimal timing of pharmacological interventions. NPJ Syst Biol Appl. 2023;9(1):13.
Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N Engl J Med. 2021;384(3):238–51.
Fischer WA 2nd, Eron JJ Jr, Holman W, Cohen MS, Fang L, Szewczyk LJ, et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci Transl Med. 2022;14(628):1–10.
Dougan M, Nirula A, Azizad M, Mocherla B, Gottlieb RL, Chen P, et al. Bamlanivimab plus etesevimab in mild or moderate Covid-19. N Engl J Med. 2021;385(15):1382–92.
Sagawa K, Lin J, Jaini R, Di L. Physiologically-based pharmacokinetic modeling of PAXLOVIDTM with first-order absorption kinetics. Pharm Res. 2023;25:1–12.
Orskov B, Borresen ML, Feldt-Rasmussen B, Østergaard O, Laursen I, Strandgaard S. Estimating glomerular filtration rate using the new CKD-EPI equation and other equations in patients with autosomal dominant polycystic kidney disease. Am J Nephrol. 2010;31(1):53–7.
Singh RSP, LaBadie RR, Toussi SS, Shi H, Berg JK, Neutel JM, et al. Effect of hepatic impairment on the pharmacokinetics of nirmatrelvir/ritonavir, the first oral protease inhibitor for the treatment of COVID-19. J Clin Pharmacol. 2023 (Online ahead of print).
Alzyoud L, Ghattas MA, Atatreh N. Allosteric binding sites of the SARS-CoV-2 main protease: potential targets for broad-spectrum anti-coronavirus agents. Drug Des Dev Ther. 2022;16:2463–78.
Acknowledgments
The authors thank all the participants and their families who volunteered for the clinical studies described in this review. The authors would also like to thank Rajendra Kadam, Narayan Cheruvu, Mohamed Shahin, Kazuko Sagawa, and Li Di for their contributions to the work presented in this review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was sponsored by Pfizer Inc.
Conflict of interest
All authors are employees of Pfizer Inc and may hold stock or stock options.
Availability of data and material
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Ethics approval
The final protocol, amendments, and informed consent documents for all clinical studies presented herein were reviewed and approved by the independent ethics committee or institutional review board for each participating investigational center. All subjects provided written, informed consent. The studies were conducted in accordance with the protocol and consensus ethical principles derived from international guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable International Council for Harmonisation Good Clinical Practice guidelines, and applicable laws and regulations, including applicable privacy laws.
Consent to participate
All subjects provided written, informed consent.
Consent for publication
Not applicable.
Code availability
Not applicable.
Author contributions
Conceptualized the manuscript: J.G. and B.D. Wrote or contributed to the writing of the manuscript: J.G., D.S.C., R.S.P.S, P.L.S.C., R.R., R.A., H.S., J.C.M., and B.D.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gerhart, J., Cox, D.S., Singh, R.S.P. et al. A Comprehensive Review of the Clinical Pharmacokinetics, Pharmacodynamics, and Drug Interactions of Nirmatrelvir/Ritonavir. Clin Pharmacokinet 63, 27–42 (2024). https://doi.org/10.1007/s40262-023-01339-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01339-y