
Abstract
The approval of targeted therapies for metastatic colorectal cancer (mCRC) has led to important improvements in patient outcomes. However, it is still necessary to increase individualisation of treatments based on tumour genetic profiles to optimise efficacy, while minimising toxicity. As such, there is currently great focus on the discovery and validation of further biomarkers in mCRC, with many new potential prognostic and predictive markers being identified alongside developments in patient molecular profiling technologies. Here, we review data for validated and emerging biomarkers impacting treatment strategies in mCRC. We completed a structured literature search of the PubMed database to identify relevant publications, limiting for English-language publications published between 1 January 2014 and 11 July 2018. In addition, we performed a manual search of the key general oncology and CRC-focused congresses to identify abstracts reporting emerging mCRC biomarker data, and of ClinicalTrials.gov to identify ongoing clinical trials investigating emerging biomarkers in mCRC and/or molecular-guided clinical trials. There is solid evidence supporting the use of BRAF status as a prognostic biomarker and DYPD, UGT1A1, RAS, and microsatellite instability as predictive biomarkers in mCRC. There are a number of emerging biomarkers that may prove to be clinically relevant in the future to have prognostic (HPP1 methylation), predictive (HER3, microRNAs, anti-angiogenic markers, and CRC intrinsic subtypes), or both prognostic and predictive values (HER2, CpG island methylator phenotype, tumour mutational load, gene fusions, and consensus molecular subtypes). As such, new biomarker-led treatment strategies in addition to anti-epidermal growth factor receptor and anti-angiogenetic treatments are being explored. Biomarkers that are not recommended to be tested in clinical practice or are unlikely to be imminently clinically relevant for mCRC include thymidylate transferase, ERCC1, PIK3CA, and PTEN. We highlight the clinical utility of existing and emerging biomarkers in mCRC and provide recommended treatment strategies according to the biomarker status. An update on ongoing molecular-guided clinical trials is also provided.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
With the approval of therapies that specifically target the molecular differences between normal cells and cancer cells, there is a strong need to ensure that the most beneficial therapeutic strategies are adopted for each patient. |
Therapies can be targeted appropriately by assessing the presence of biomarkers. |
The biomarker landscape in metastatic colorectal cancer is evolving and we provide guidance on which biomarkers currently are (DPYD, UGT1A1, RAS, microsatellite instability), and may become (BRAF, HER2, consensus molecular subtypes, CRC intrinsic subtypes, EGFR, HER3, microRNA, anti-angiogenic markers, tumour mutational load, gene fusions, CpG island methylator phenotype), most relevant for clinical practice. |
We recommend treatment strategies according to the presence or absence of biomarkers including RAS, MSI, BRAF, and HER2 and provide an update on ongoing molecular-guided clinical trials, which will further individualise therapy for patients with mCRC. |
1 Introduction
Colorectal cancer (CRC) is one of the most diagnosed cancers worldwide, with 1.84 million estimated new cases in 2018 [1]. Fluorouracil (5-FU) was the historic standard of care for patients with CRC, but the treatment landscape has evolved rapidly in the metastatic setting following the approval of several targeted therapies, leading to improvements in tumour response rates and patient survival [2]. Despite the multitude of treatments available, outcomes and toxicity with each regimen can vary markedly from patient to patient [3]. Therefore, there is a strong need to identify disease and host biomarkers that will ensure the most beneficial therapeutic strategy is adopted for each patient.
Although primary tumour location [right-sided (located in the caecum to transverse colon) or left-sided (located from the splenic flexure to rectum)] has been identified as a surrogate marker for tumour biology [4,5,6], more accurate knowledge of a patient’s tumour profile is needed to better personalise treatment. Indeed, in recent years there has been a great focus on the development of biomarkers in metastatic CRC (mCRC) [6,7,8,9], all with the aim of improving outcomes for patients, including avoiding missed treatment opportunities or unnecessary toxicity. These have included new diagnostic biomarkers (for disease detection and cancer staging or risk stratification), new predictive biomarkers (to predict patient response to therapy) and new prognostic biomarkers (to assess how the disease is likely to evolve). Based on these efforts, testing for some biomarkers is now standard practice in the mCRC setting. Newer technologies such as next-generation sequencing (NGS) and tumour panels have highlighted many more potential predictive and prognostic markers. While these techniques can provide a wealth of information, their application in clinical practice is not always straightforward [10, 11]. There is a clear need for evidence-based recommendations to guide the use of validated and emerging biomarkers in clinical practice. Here we review the clinical utility of existing and emerging biomarkers that are being used or investigated to support treatment decisions for patients with mCRC, including those who develop acquired resistance to treatment.
2 Methods
We completed a structured literature search to identify relevant publications. The PubMed database (www.ncbi.nlm.nih.gov/pubmed/) was searched using the following terms and restrictions: (“metastatic colorectal cancer”[Title/Abstract] OR “mCRC”[Title/Abstract]) AND (“biomarkers”[Title/Abstract] OR “molecular”[Title/Abstract] OR “molecular guided”[Title/Abstract] OR “tumor board”[Title/Abstract]), limiting for English-language publications (specifically of clinical trials, meta-analyses, observational studies, comparative studies, clinical studies, systematic reviews, multicentre studies, or case reports) published between 1 January 2014 and 11 July 2018. The search produced 519 hits. The titles and abstracts of these publications were reviewed and the full-text versions of manuscripts reporting emerging mCRC biomarker data were retrieved and reviewed in detail. In addition, we performed a manual search of the key general oncology and CRC-focused congresses to identify abstracts reporting emerging mCRC biomarker data (published between 1 January 2015 and 11 July 2018). We also performed a manual search of ClinicalTrials.gov to identify ongoing clinical trials investigating emerging biomarkers in mCRC and/or molecular-guided clinical trials.
3 Biomarkers and Chemotherapy in mCRC
Neoadjuvant and adjuvant chemotherapy with fluoropyrimidine-based regimens are beneficial for many patients with mCRC, and several markers of chemotherapy sensitivity or toxicity have been proposed. Dihydropyrimidine dehydrogenase (DPD) is an enzyme encoded by the DPYD gene that catalyses the inactivation of some fluoropyrimidines, and its deficiency is associated with increased chemotherapy-related toxicity [12,13,14]. DPYD allelic variants that are associated with severe toxicity include DPYD*2A and A2846T [12, 15,16,17]. Other variants have been identified but their clinical relevance remains to be confirmed [15]. The European Society for Medical Oncology (ESMO) guidelines do not recommend systematic DPD testing before 5-FU or capecitabine administration (Table 1), although testing remains a good option, with some groups calling for DPYD genotype- and/or phenotype-guided individualised dosing to be a new standard of care [3, 17]. Indeed, DPD testing is standard practice in some European countries, including France [18]. Given that fluoropyrimidine treatment can result in severe toxicity in up to 39% of patients [17], DPD testing, which is feasible in routine clinical practice, may be of value and will probably be extended to other European countries in forthcoming years. Other potential markers of toxicity or response associated with fluoropyrimidines are yet to be validated, including genetic variations in the thymidylate synthase gene and microRNA (miRNA)-143 [3, 19]. Polymorphisms in the gene encoding UDP glucuronosyltransferase 1 family, polypeptide A1 (UGT1A1) have also been linked with tolerance to chemotherapy [20]. While recent data from the PETACC-3 trial confirmed an association between UGT1A1*28 genotype and chemotherapy-dependent toxicity, other clinical parameters (including sex, age and performance status) were found to be stronger predictors of toxicity risk [21]. Further to this, a meta-analysis revealed an association between UGT1A1*6 polymorphisms and irinotecan-induced toxicity in Asian patients [22]. Patients heterozygous for UGT1A1*6 were found to be at increased risk for severe neutropenia, while patients who were homozygous for UGT1A1*6 were found to be at even higher risk for neutropenia and were also more likely to suffer from severe diarrhoea [22]. UGT1A1 genotyping/phenotyping is not recommended as a predictive biomarker in everyday practice, but remains an option and should be conducted when UGT1A1 deficiency is suspected, as indicated by low conjugated bilirubin, and when administration of > 180 mg/m2 irinotecan is planned (Table 1) [3, 23]. The frequency of UGT1A1*6 is higher, while the frequency of UGT1A1*28 genotype is lower, in Asian versus Caucasian patients [23]. Therefore, the Pan-Asian-adapted ESMO consensus guidelines for the management of patients with mCRC also recommend that a lower irinotecan threshold dose for genotyping may be used depending on the prevalence of UGT1A1 polymorphisms per country [23].
A number of studies have indicated that the excision repair cross-complementation group 1 (ERCC1) protein is a possible prognostic biomarker in platinum-based treatment of metastatic cancers [7, 24,25,26]. However, survival outcomes did not significantly differ in patients with high versus low baseline ERCC1 levels who received bevacizumab plus mFOLFOX6 (leucovorin [folinic acid], 5-FU, oxaliplatin) or FOLFIRI (leucovorin, 5-FU, irinotecan) in the MAVERICC trial, the first prospective study to investigate ERCC1 as a potential biomarker for oxaliplatin-containing regimens in patients with untreated mCRC [27, 28]. It is not recommended as a biomarker in clinical practice (Table 1) [3].
4 Biomarkers and Anti-EGFR Therapy in mCRC
The development and progression of CRC is influenced by epidermal growth factor receptor (EGFR) and its downstream signalling pathways (Fig. 1) [29]. Therefore, investigation of predictive and prognostic biomarkers historically focused on EGFR expression and subsequently on alterations in the RAS/BRAF/MEK/MAPK and PI3K/PTEN/AKT pathways. Data from clinical trials demonstrate that across all lines of therapy RAS mutations predict a lack of response to monoclonal antibodies directed against EGFR (panitumumab/cetuximab) and potentially a detrimental effect of such therapies when combined with oxaliplatin-based chemotherapy [30,31,32,33,34]. Effective first- and second-line therapies are therefore needed for patients with RAS-mutated mCRC. Chemotherapy plus bevacizumab is a standard first-line therapy for these patients (Fig. 2) [3, 35], but has limitations. Specifically, RAS mutations may be associated with lesser benefit from chemotherapy plus bevacizumab compared with RAS wild type (WT) mCRC [36,37,38], although the recent JACCRO CC-11 trial suggests that first-line mFOLFOXIRI (leucovorin, 5-FU, oxaliplatin, irinotecan) plus bevacizumab is effective for patients with RAS-mutated mCRC [39]. Treatment with aflibercept or ramucirumab (in combination with FOLFIRI) may be efficacious as second-line therapies for patients with RAS-mutated mCRC [3, 40]. Inhibitors of some mutant forms of RAS, such as KRAS G12C, are now entering clinical trials [41, 42].

Overview of the main EGFR and VEGF angiogenic signalling cascades. Upon EGFR dimerisation and autophosphorylation, the RAS/BRAF/MEK and PI3K/PTEN/AKT pathways are induced (adapted from [29] under the Creative Commons Attribution License CC BY-NC 3.0 [https://creativecommons.org/licenses/by-nc/3.0/]). Ligand binding to VEGFR-1, VEGFR-2, and VEGFR-3 activates a number of processes that drive angiogenesis. AKT AKR mouse thymoma, BRAF B-rapidly accelerated fibrosarcoma, EGFR epidermal growth factor receptor, ERK extracellular receptor kinase, HER human epidermal growth factor, MEK mitogen-activated protein kinase, mTOR mammalian target of rapamycin, P phosphorylation, PI3K phosphatidylinositol 3-kinase, PIGF phosphatidylinositol-glycan biosynthesis class F, PTEN phosphatase and tensin homolog, RAS rat sarcoma, VEGF vascular endothelial growth factor, VEGFR VEGF receptor

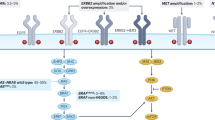
Possible treatment strategies according to biomarker status in mCRC. aWhere maximum tumour shrinkage is the goal; further confirmatory data are needed. Colours indicate possible treatment strategies for tumours with amplified HER2 (pink), mutant BRAF (green), MSI-H (light orange), WT RAS; (yellow) and mutant RAS (orange). Grey shading indicates WT/normal expression/MSS with no treatment recommendations. AMP amplified, B binimetinib, Bev bevacizumab, BRAF B-rapidly accelerated fibrosarcoma, Cmab cetuximab, CT chemotherapy, D dabrafenib, E encorafenib, EGFR epidermal growth factor receptor, FOLFIRI leucovorin, fluorouracil and irinotecan, FOLFOX leucovorin, fluorouracil and oxaliplatin, FOLFOXIRI leucovorin, fluorouracil, irinotecan and oxaliplatin, HER2 human epidermal growth factor 2, Ir irinotecan, L lapatinib, mCRC metastatic colorectal cancer, MSI-H microsatellite instability high, MSS microsatellite stable, MUT mutant, NORM normal, Pmab panitumumab, Pz pertuzumab, RAS rat sarcoma, T trametinib, Tz trastuzumab, V vemurafenib, WT wild type
Testing for RAS mutational status is recommended for all patients at the time of mCRC diagnosis (Table 1) [3, 7]. Initially only mutations in exon 2 of KRAS (which lead to constitutive activation of EGFR) were routinely tested. A prospective–retrospective biomarker analysis of the Phase III PRIME study reported that the presence of additional RAS mutations (KRAS exons 3/4 and NRAS exon 2/3/4) also predict a lack of response to panitumumab plus FOLFOX [31]. This observation was subsequently confirmed by retrospective and prospective analyses of other trials of anti-EGFR therapies [43,44,45,46,47,48]. These mutations are now tested for in extended RAS analysis [49]. As around 20% of KRAS exon 2 WT tumours harbour a different RAS mutation, extended RAS testing has significantly impacted clinical outcomes [34]. An NGS-based extended RAS panel has recently been clinically validated using formalin-fixed paraffin-embedded mCRC tumour samples [10]. Of note however, a recently reported prospective study of more than 400 patients demonstrated that testing circulating tumour DNA (ctDNA) for RAS correlated well with tissue assessment, with increased accuracy for patients with liver metastases [50]. While still not recommended by current guidelines [3, 7], ctDNA testing could therefore potentially replace tissue assessment as routine practice in these patients.
Clinical trial data suggest that the mutated BRAF V600E is a negative prognostic marker for patients with mCRC and may predict resistance to EGFR-antibody therapy, especially in heavily pre-treated patients [51,52,53]; the predictive value of BRAF V600E mutations in earlier lines of therapy is uncertain [54, 55]. A recent meta-analysis of randomised controlled trials suggested there was insufficient evidence to demonstrate that BRAF V600E mutations are a negative predictive marker of response to EGFR inhibitors [56], while a second meta-analysis demonstrated that anti-EGFR treatment did not increase progression-free survival (PFS) or overall response rate (ORR) in patients with BRAF-mutated mCRC [57]. However, the outcomes of these meta-analyses are debatable as the analyses included studies with different patient populations, lines of therapy, control arms and anti-EGFR treatment options. More recently, analysis of the VOLFI trial found an impressively increased response rate in BRAF-mutated patients receiving first-line panitumumab plus a triplet chemotherapy regimen versus triplet chemotherapy alone (86% vs 22%), although PFS was comparable in the two treatment arms [58]. Of note, the sample size was small (n = 16) and a cautious interpretation is warranted [58]. On balance, accumulating evidence suggests that anti-EGFR therapy may be of interest in patients with BRAF-mutated mCRC, if used in earlier rather than later lines of therapy, but this is not currently a first-choice therapy in this setting (Fig. 2). As demonstrated in a small subgroup analysis of the TRIBE study [36], and other patient cohorts [59, 60], FOLFOXIRI plus bevacizumab may also be a beneficial first-line treatment for these patients and is recommended by ESMO guidelines for patients with BRAF-mutated mCRC (Fig. 2) [3]. Other vascular endothelial growth factor (VEGF) targeting agents may also be efficacious in this patient population. Tumour samples were obtained for 482/1226 (39%) patients randomised in the VELOUR clinical trial, which demonstrated that aflibercept in combination with FOLFIRI is a beneficial second-line treatment for mCRC [61]. Patients with BRAF-mutated mCRC (n = 36) had a larger benefit from the addition of aflibercept versus placebo to FOLFIRI (median OS 10.3 vs 5.5 months) compared with patients with WT BRAF (13.0 vs 12.4 months) [61]. However, the difference was not significant [HR 0.49 (95% CI 0.22–1.09), p = 0.08], possibly due to the small series of patients [61]. Similar results were reported in a biomarker analysis of the RAISE trial, where the addition of ramucirumab to FOLFIRI provided a non-significant benefit in BRAF-mutated tumours [62].
Inhibition of BRAF V600E has been shown to cause rapid feedback activation of EGFR, which supports continued tumour proliferation [63]. As such, inhibition of EGFR has been shown to be strongly synergistic with BRAF V600E inhibition in CRC (Fig. 2) [63]. An open-label Phase I/II study demonstrated that the BRAF inhibitor dabrafenib plus trametinib (a MEK inhibitor) had activity in patients with BRAF V600E mutation-positive mCRC, and patients receiving triple therapy (dabrafenib, trametinib and panitumumab) had a numerically improved ORR (21%) compared with those receiving panitumumab plus either dabrafenib (10%) or trametinib (0%) and had a longer PFS (4.2 vs 3.5 or 2.6 months) [64, 65]. More recently, in a randomised trial, addition of vemurafenib to the combination of cetuximab and irinotecan resulted in prolonged PFS [4.4 vs 2.0 months, hazard ratio (HR) 0.42, 95% confidence interval (CI) 0.26–0.66; p < 0.001] and a higher disease control rate (67% vs 22%; p < 0.001) compared with cetuximab and irinotecan treatment alone in heavily pre-treated patients with BRAF-mutant mCRC [66]. The combination of encorafenib (BRAF inhibitor), binimetinib (MEK inhibitor) and cetuximab is being assessed in the BEACON trial and has previously shown encouraging clinical activity in BRAF V600E mCRC (ORR: 48%) [67]. Of note, extended (non-V600/non-V600E) BRAF mutations may have different clinical implications compared with BRAF V600E mutations [68, 69]. It is recommended that BRAF mutation status is assessed alongside that of RAS for prognostic assessment and/or selection for clinical trials (Table 1) [3, 7]. The predictive potential of BRAF mutation status is not yet confirmed.
Regarding the potential of EGFR activation as a biomarker, studies have focused on the expression of EGFR ligands and EGFR gene copy number. High expression levels of the EGFR ligands amphiregulin (AREG) and epiregulin (EREG) have positive prognostic value and are associated with a positive response to anti-EGFR therapy [70,71,72]. For example, in RAS (KRAS and NRAS) WT patients receiving anti-EGFR therapy, high AREG and EREG expression correlated with better survival outcomes [73,74,75]. High EGFR gene copy number has also been associated with improved response to anti-EGFR-targeted therapies [76,77,78], but clinical use of such a biomarker is limited by the rarity of true gene amplification and difficulties in obtaining reproducible results by fluorescence in situ hybridisation assessment [79]. Evaluation of EGFR ligands, EGFR gene copy number, or EGFR protein expression is currently not recommended for routine patient management in mCRC (Table 1) [3].
Concerning other components of EGFR downstream signalling pathways, contradictory data are reported for the prognostic and predictive role of PIK3CA and PTEN mutations in mCRC [80]. As such, according to European and US guidelines there is insufficient evidence for their use as predictive biomarkers for EGFR-antibody therapy (Table 1) [3, 7].
Other promising biomarkers include the receptors HER2 and HER3 (Table 2). Approximately 5% of mCRC tumours are driven by HER2 amplification or mutation, which can lead to resistance to EGFR-targeted treatment by activating a bypass signalling pathway [81,82,83,84]. Although the prognostic role of HER2 remains uncertain, alterations in this gene have been associated with poorer survival outcomes [81, 85]. There is also a growing interest in HER2 as a therapeutic target in mCRC (Fig. 2). Dual HER2 blockade with a monoclonal antibody (pertuzumab or trastuzumab) and a tyrosine kinase inhibitor (lapatinib) has been shown to inhibit tumour growth in patient-derived xenografts of HER2-amplified mCRC [86, 87]. Moreover, results from the HERACLES-A Phase II trial showed that dual blockade was efficacious and well tolerated in HER2-positive KRAS exon 2 WT mCRC patients refractory to current standard of care [88, 89]. HER2-targeted therapy was also effective in the Phase IIa MyPathway study that involved 57 patients with refractory HER2-amplified/overexpressing CRC treated with trastuzumab plus pertuzumab; the ORR was 32% [90]. Interestingly, this study included patients with KRAS-mutated CRC, but efficacy of HER2-directed therapy was notably higher in those with KRAS-WT tumour status [90].
With respect to HER3, a prospectively planned retrospective biomarker study of pre-treatment samples from the PICCOLO trial showed that patients with RAS WT mCRC and high HER3 mRNA expression benefited markedly from panitumumab treatment, whereas those with low HER3 mRNA expression did not [91]. There were statistically significant biomarker-treatment interactions for both PFS (p = 0.001) and OS (p = 0.004) [91].
Some miRNAs have been suggested to predict response to anti-EGFR therapy [92,93,94]. For instance, high-intensity levels of the Let-7c/miR-99a/miR-125b signature have been associated with longer PFS in KRAS WT patients receiving such therapy [95]. Low miR-181a expression has also been associated with poorer outcomes in KRAS WT patients undergoing treatment with EGFR-targeting monoclonal antibodies [93, 95], and upregulation of miR-31-5p has been shown to be predictive of shorter PFS in patients with mCRC receiving anti-EGFR treatment [92, 96]. Furthermore, a number of studies have identified miR-31-3p as a promising predictive biomarker for anti-EGFR therapy in RAS WT mCRC, with therapeutic benefit potentially restricted to patients with low miR-31-3p expression [97,98,99].
Finally, primary tumour location, as a surrogate marker for tumour molecular characteristics, is known to affect prognosis and treatment outcomes with anti-EGFR therapy. Left-sided tumours are more prevalent and associated with better prognosis than right-sided tumours [4,5,6]. Right-sided tumours are more frequently associated with mutations in BRAF, TGFβR2, and PI3KCA and are microsatellite unstable [4, 5, 100]. In contrast, amplification of EGFR and HER2, overexpression of EGFR ligands and chromosomal instability are more common in left- than right-sided tumours [4, 5]. In the first-line setting, anti-EGFR treatment combined with chemotherapy appears to be more effective than bevacizumab in left-sided RAS WT mCRC [100]. Moreover, patients with right- versus left-sided tumours benefit less from anti-EGFR therapy [100]. In patients with right-sided tumours, treatment with intensive chemotherapy plus bevacizumab may be more appropriate, although anti-EGFR therapy remains an option to achieve an objective response if cytoreduction is the treatment goal [100]. Furthermore, patients with right-sided tumours appear to benefit more from immunotherapies due to increased antigenic load, although further validation is required [101].
Critically, clinical efficacy of targeted therapy is limited by the development of acquired resistance [102]. A comprehensive analysis of mechanisms of resistance in plasma from a large cohort of patients treated with anti-EGFR therapy showed that the emergence of RAS mutations (30%) and EGFR extracellular domain (ECD) mutations (25%) were the most frequent mechanisms of resistance [103]. The dynamics of EGFR ECD mutations differ from the emergence of RAS mutations; patients who experience greater and longer responses to anti-EGFR therapy reportedly develop EGFR ECD mutations and patients with shorter PFS seem more likely to develop RAS mutations [104]. Mutations in BRAF, as well as MET and HER2, were also detected [103]. Importantly, these biomarkers of resistance appear to be heterogeneous and mostly sub-clonal, which will potentially limit the efficacy of further lines of therapy [103]. Another recent study noted that in patients receiving anti-EGFR therapy and undergoing resection, some patients (19%) gained while others (35%) lost mutations on the resection specimen as compared with previous biopsy, mainly in RAS, providing further evidence of intra-tumoural heterogeneity [105]. RAS mutations (at biopsy or resection) were associated with worse response and survival compared with tumours that were RAS WT [105]. However, in another study, RAS mutations that emerged during panitumumab-based treatment (detected by plasma analysis of ctDNA) were not associated with less favourable outcomes [106]. Of note, the emergence of acquired mutations shown to confer resistance can be detected in plasma months before morphological tumour progression [107,108,109]; the clinical utility of assessing emerging mutations requires further validation.
5 Biomarkers and Anti-angiogenic Therapy in mCRC
The role of angiogenesis in tumourigenesis is shown in Fig. 1. Despite the importance of this process in disease pathology, not all patients with mCRC derive clinical benefit from anti-angiogenic therapy, highlighting the need for biomarkers to ensure treatment is appropriately targeted. However, the discovery of universal predictive biomarkers for anti-angiogenic therapies is challenging, due to host-involvement in angiogenesis.
Many factors have been identified as being associated with better outcomes in patients treated with anti-angiogenic agents, suggesting their potential predictive value, such as the loss of chromosome 18q11.2–q12.1 [110], the transcription factor homeobox B9 [111, 112], VEGF-D [113, 114], and markers of tumour vasculature immaturity [115]. While VEGF-A was found not to be predictive of anti-angiogenic treatment efficacy in retrospective and prospective series [27, 28], it has been suggested that the VEGF-A splice isoforms 165b and 121 may predict response to bevacizumab [116, 117]. Low levels of hepatocyte growth factor have also been associated with survival benefit from bevacizumab treatment [117]. Further, several miRNAs have been identified as possible biomarkers for anti-angiogenic therapy [118]. For example, high miR-664-3p expression was significantly predictive of improved outcomes in patients with mCRC receiving bevacizumab treatment plus chemotherapy compared with those receiving chemotherapy alone [119]. However, none of these potential biomarkers have acquired sufficient evidence to recommend their use in daily practice and their clinical utility needs to be confirmed in large prospective trials.
6 Microsatellite Instability/Deficient Mismatch Repair Disease
Microsatellite instability (MSI) is a consequence of deficient mismatch repair (dMMR) and serves as a favourable prognostic marker for stage II/III CRC [120,121,122]. However, the prevalence of dMMR in mCRC is lower (5%) than in the adjuvant setting (around 15%) [123], and data on the prognostic and predictive values of MSI in the metastatic setting are scarce and conflicting [3, 124, 125]. In the metastatic setting, MSI-High (H) tumours are associated with poor prognosis, although BRAF mutations are more common in patients with MSI-H tumours versus those with proficient mismatch repair (pMMR) (p < 0.001), which may account for this prognosis [126]. While some studies indicate that MSI status does not predict the effect of chemotherapy or targeted therapy in mCRC [127, 128], a recent randomised Phase III trial has found that patients with MSI-H tumours, experienced a longer OS when treated with chemotherapy plus bevacizumab versus cetuximab (p < 0.001) [129]. Further prospective studies are warranted. However, MSI status has been shown to be predictive for the use of immunotherapy in the treatment of patients with mCRC (Table 1) [130]. In a Phase II study designed to evaluate the clinical activity of pembrolizumab (programmed cell death ligand 1 inhibitor), the immune-related objective response rate [40% (n = 4/10) vs 0% (n = 0/18)] and 20-week immune-related PFS rate [78% (n = 7/9) vs 11% (n = 2/18)] were higher for patients with dMMR versus pMMR CRCs [130]. Based on such early clinical data, nivolumab and pembrolizumab have been approved by the US Food and Drug Administration (FDA) for mCRC patients with MSI-H/dMMR disease that has progressed following treatment with a fluoropyrimidine, oxaliplatin and irinotecan (Fig. 2) [131, 132]. More recently, the FDA approved low-dose ipilimumab in combination with nivolumab for use in these patients based on the CheckMate-142 study, which demonstrated that this combination produces high response rates, encouraging survival outcomes and may provide improved efficacy relative to immuno-monotherapy (Fig. 2) [133, 134]. MSI testing for immune checkpoint inhibitors was included in the most recent ESMO guidelines, prior to FDA approval of these agents [3].
There is an ongoing need to develop new strategies to improve the efficacy of checkpoint inhibitors in microsatellite stable disease. However, the use of combination strategies, such as combining checkpoint inhibitors with MEK inhibitors to increase the number of infiltrating effector lymphocytes, or anti-angiogenic agents for their immunomodulatory properties, have not demonstrated any benefit to date [135,136,137]. Recent data suggest that inactivation of DNA repair may provide benefit by increasing the tumour neoantigen burden [138], an approach which has the potential to be therapeutically exploited by the use of alkylating agents.
7 Other Emerging Biomarkers for Predicting Therapeutic Response in mCRC
Recent studies have uncovered a number of other potentially important biomarkers for predicting therapeutic response (Table 2). Tumour mutational load, defined as the number of mutations per coding area of a tumour genome, is associated with MSI/MMR status [139]. Some studies have demonstrated that tumour mutational load may be a predictive biomarker for response to chemotherapy and immunotherapy in patients with mCRC; however, the data need to be confirmed in larger studies [129, 140,141,142]. The relationship between mutations that impair DNA polymerase epsilon (POLE) proofreading and tumour immunogenicity have been explored. In a retrospective analysis of more than 4500 patients with stage II/III CRC, the presence of POLE mutations identified a subset of CRC patients with immunogenic tumours and very good prognosis [143]. The hypermutated phenotype of these tumours suggests that they will be excellent candidates for immunotherapeutic approaches.
Tumours bearing gene fusions, including rearrangements in RET, ALK, ROS1, and NTRK1-2-3, may represent rare but clinically relevant mCRC subtypes with poor prognosis [144, 145]. Targeted strategies inhibiting RET, ALK, ROS, and TrkA-B-C have demonstrated encouraging results [144,145,146]; however, mechanisms of resistance may develop and mutations have been observed in the catalytic domain of receptors [147]. Preliminary evidence suggests these fusions may be negative predictive biomarkers for anti-EGFR therapy [144, 145, 148]. Of note, a recent post hoc analysis of the VALENTINO study evaluating the PRESSING panel, which was created to group rare genomic markers beyond RAS/BRAF, including RET, ALK, ROS1, and NTRK1-2-3, to predict anti-EGFR resistance [149], found that PRESSING-positive tumours had poorer outcomes compared with PRESSING-negative tumours in patients receiving FOLFOX plus panitumumab followed by maintenance with panitumumab ± 5-FU/leucovorin (PFS 7.7 vs 12.1 months, HR 2.07, 95% CI 1.43–2.99; p = 0.0001) [148].
Hypermethylation of CpG islands is frequently observed in CRCs, which are then classified as CpG island methylator phenotype (CIMP) positive [150, 151]. Contradictory data have been reported for the prognostic and predictive role of CIMP status in CRC [152,153,154,155,156]. In patients with stage III CRC treated with oxaliplatin-based adjuvant chemotherapy, CIMP was recently found to be associated with shorter OS (HR 1.46, 95% CI 1.02–1.94; p = 0.04) and shorter survival after recurrence (HR 1.76, 95% CI 1.20–2.56; p < 0.0004) [157]. Interestingly, there was a non-significant trend for a possible detrimental effect of cetuximab in patients with CIMP-positive tumours [157]. Promoter CpG island hypermethylation of O(6)-methylguanine-DNA-methyltransferase, a DNA repair protein, may predict clinical response to alkylating agents although further research is warranted [158, 159]. A number of other epigenetic prognostic markers have been investigated in CRC [160]. For example, a recent prospective study suggests that HPP1 methylation may be both a prognostic marker and early marker of response in mCRC [161].
Interest is growing in ctDNA as an analyte for evaluating prognosis and early treatment response [162]. For example, in addition to being of known prognostic value, ctDNA has been proposed as an early marker of response to chemotherapy in patients with mCRC [163, 164]. In a recent prospective study, patients with a high (> 10 ng/mL) versus low (≤ 0.1 ng/mL) ctDNA concentration before initiating first- or second-line chemotherapy had a shorter OS (6.8 vs 33.4 months; adjusted HR 5.64, 95% CI 2.5–12.6; p < 0.0001) [164]. Further to this, patients who did not experience ‘early normalisation’ or an ‘early decrease > 80%’ of ctDNA concentration after initiation of treatment experienced less benefit from chemotherapy [164].
Finally, classification/stratification systems for CRC, such as consensus molecular subtypes (CMS) and colorectal cancer intrinsic subtypes (CRIS), have been proposed [165, 166]. Both exploit the intrinsic gene signatures specific to CRC cells and may have predictive and prognostic value in mCRC [165,166,167,168]. Specifically, CMS has been shown to be prognostic for ORR (p = 0.023), PFS (p < 0.001), and OS (p < 0.001) [167], and a recent small retrospective study suggests that it is also predictive for the efficacy of chemotherapy in mCRC [168]. Further to this, CRIS has been shown to predict response to EGFR-targeting antibodies and to predict disease outcome independently of clinical stage and stromal infiltration [166].
8 Ongoing Molecular-Guided Clinical Trials
Several molecular-guided clinical trials in mCRC are underway (Table 3). FOCUS4, which began recruitment in 2014, is an integrated programme of parallel, molecularly stratified, randomised comparisons for patients with advanced or mCRC [169]. It is derived from a multi-arm, multi-stage design to be adjustable should new biomarker and clinical data arise during the trial, while being cost and time efficient. In this programme, novel agents are tested in patient populations defined by whether their tumours harbour mutations in BRAF, PIK3CA, RAS, and TP53 or are MSI/dMMR. Its multi-stage design provides an early efficacy signal of the new agents being assessed through a series of pre-planned interim analyses [169]. Other ongoing molecular-guided trials include a Phase III study investigating pembrolizumab versus standard-of-care chemotherapy as first-line therapy for dMMR or MSI-H mCRC (NCT02563002), and Phase II studies investigating atezolizumab and bevacizumab in patients with unresectable mCRC and MSI (NCT02982694), anti-EGFR therapy in mCRC patients with low or high ERCC1 (NCT01703390), treatment with HER2 monoclonal antibodies in HER2-amplified advanced or mCRC (NCT03365882), and Sym004 treatment in chemotherapy-refractory mCRC patients with acquired resistance to anti-EGFR therapy (NCT03549338).
Other clinical trials are exploring re-challenge with anti-EGFR therapy. Dynamic clonal competition leads to a rise in anti-EGFR-resistant mutant clones during anti-EGFR therapy and a decline upon withdrawal of anti-EGFR antibodies [170, 171]. A recent study found that, after discontinuation of anti-EGFR therapy, RAS and EGFR clones exponentially decayed with an estimated half-life of 3.4 and 6.9 months, respectively [171]. These observations provide a molecular rationale for studies that have proposed re-challenge with cetuximab [172,173,174] or panitumumab [175, 176], after a previous response to anti-EGFR therapy and may help guide timing of re-challenge therapies. Recent results from the CRICKET trial indicate that re-challenge with cetuximab following acquired resistance is more efficient in patients without RAS mutations assessed by ctDNA [177]. These first results suggest that monitoring tumour sensitivity to anti-EGFR agents by iterative ctDNA assessments may soon form part of our daily practice. Additional studies (CHRONOS, NCT03227926; RASINTRO, NCT03259009; FIRE-4, NCT02934529; A-REPEAT, NCT03311750) are investigating different challenge strategies using anti-EGFR therapy based on liquid biopsy assessment of dynamic RAS clones.
9 Concluding Remarks
Current guidelines regarding biomarkers recommend routinely making treatment decisions based on RAS, BRAF, or MSI status. In this review, we highlight several other molecular and non-molecular biomarkers that are undergoing clinical testing and we describe their possible clinical relevance (Table 4) as well as highlighting possible treatment strategies according to biomarker status (Fig. 2). For example, DPYD genotyping/phenotyping may soon become a standard of care to individualise 5-FU chemotherapy dosing. Furthermore, in addition to assessment of BRAF V600E mutations, assessment of HER2 amplification may be useful to inform physicians of their patients’ prognosis and to guide enrolment of patients into ongoing clinical trials dedicated to these rare subgroups. Knowledge of prognostic factors can be used in clinical decision making to determine the goal of treatment and to tailor treatment, for example the selection of adjuvant therapy or level of treatment intensity [3, 178]. Plasma analysis of ctDNA shows promise as a minimally invasive and sensitive method to monitor patient response, including acquired resistance to anti-EGFR agents. However, its utility has to be confirmed and crucially, simple assessment tools and positive controls are required for daily use, before ctDNA testing can become standard practice. Of note, recent studies highlight the use of serial plasma biopsies to assess tumour heterogeneity to further inform treatment decisions [179, 180].
Further clinical validation of many of the biomarkers reviewed here is still required. However, validation and translation of new biomarkers into clinical practice is a complex process involving a number of steps, each with its own set of challenges [181]. Many biomarkers are validated retrospectively, yet such studies can be affected by multiple sources of bias [181]. Thus large-scale prospective trials with well-defined protocols are needed to further identify, develop, and validate biomarkers, to standardise their use in clinical practice and inform treatment options [2, 8]. However, these are costly and time consuming [181]. Employing prospective–retrospective study designs [182] or using biobanks from randomised trials [183] provide alternative options for biomarker discovery [181], and validating molecular-guided clinical trials, including those utilising a multi-arm, multi-stage design for cost and time efficiency, are underway. Of note, the interaction between biomarkers is also likely to be clinically relevant and network biomarkers may provide further prognostic and predictive insight in the future [184].
Overall, we are optimistic that the continued prospective validation of biomarkers along with further developments in patient molecular profiling technologies will help to achieve the goal of true individualised therapy for patients with mCRC.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Patil H, Saxena SG, Barrow CJ, Kanwar JR, Kapat A, Kanwar RK. Chasing the personalized medicine dream through biomarker validation in colorectal cancer. Drug Discov Today. 2017;22:111–9.
Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422.
Boeckx N, Janssens K, Van Camp G, Rasschaert M, Papadimitriou K, Peeters M, et al. The predictive value of primary tumor location in patients with metastatic colorectal cancer: a systematic review. Crit Rev Oncol Hematol. 2018;121:1–10.
Stintzing S, Tejpar S, Gibbs P, Thiebach L, Lenz HJ. Understanding the role of primary tumour localisation in colorectal cancer treatment and outcomes. Eur J Cancer. 2017;84:69–80.
Kamran SC, Clark JW, Zheng H, Borger DR, Blaszkowsky LS, Allen JN, et al. Primary tumor sidedness is an independent prognostic marker for survival in metastatic colorectal cancer: results from a large retrospective cohort with mutational analysis. Cancer Med. 2018;7:2934–42.
Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. J Mol Diagn. 2017;19:187–225.
Peluso G, Incollingo P, Calogero A, Tammaro V, Rupealta N, Chiacchio G, et al. Current tissue molecular markers in colorectal cancer: a literature review. Biomed Res Int. 2017;2017:2605628.
Price TJ, Tang M, Gibbs P, Haller DG, Peeters M, Arnold D, et al. Targeted therapy for metastatic colorectal cancer. Expert Rev Anticancer Ther. 2018;18:991–1006.
Udar N, Lofton-Day C, Dong J, Vavrek D, Jung AS, Meier K, et al. Clinical validation of the next-generation sequencing-based Extended RAS Panel assay using metastatic colorectal cancer patient samples from the phase 3 PRIME study. J Cancer Res Clin Oncol. 2018;144:2001–10.
van der Velden DL, van Herpen CML, van Laarhoven HWM, Smit EF, Groen HJM, Willems SM, et al. Molecular Tumor Boards: current practice and future needs. Ann Oncol. 2017;28:3070–5.
Deenen MJ, Tol J, Burylo AM, Doodeman VD, de Boer A, Vincent A, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res. 2011;17:3455–68.
Dhelens C, Bonadona A, Thomas F, Chapuis C, Potton L, Marsili S, et al. Lethal 5-fluorouracil toxicity in a colorectal patient with severe dihydropyrimidine dehydrogenase (DPD) deficiency. Int J Colorectal Dis. 2016;31:699–701.
Boige V, Vincent M, Alexandre P, Tejpar S, Landolfi S, Le Malicot K, et al. DPYD genotyping to predict adverse events following treatment with flourouracil-based adjuvant chemotherapy in patients with stage III colon cancer: a secondary analysis of the PETACC-8 randomized clinical trial. JAMA Oncol. 2016;2:655–62.
Meulendijks D, Henricks LM, Sonke GS, Deenen MJ, Froehlich TK, Amstutz U, et al. Clinical relevance of DPYD variants c.1679T > G, c.1236G > A/HapB3, and c.1601G > A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16:1639–50.
Deenen MJ, Meulendijks D, Cats A, Sechterberger MK, Severens JL, Boot H, et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol. 2016;34:227–34.
Henricks LM, Lunenburg C, de Man FM, Meulendijks D, Frederix GWJ, Kienhuis E, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19:1459–67.
Loriot MA, Ciccolini J, Thomas F, Barin-Le-Guellec C, Royer B, Milano G, et al. Dihydropyrimidine dehydrogenase (DPD) deficiency screening and securing of fluoropyrimidine-based chemotherapies: update and recommendations of the French GPCO-Unicancer and RNPGx networks. Bull Cancer. 2018;105:397–407.
Simmer F, Venderbosch S, Dijkstra JR, Vink-Borger EM, Faber C, Mekenkamp LJ, et al. MicroRNA-143 is a putative predictive factor for the response to fluoropyrimidine-based chemotherapy in patients with metastatic colorectal cancer. Oncotarget. 2015;6:22996–3007.
Takano M, Sugiyama T. UGT1A1 polymorphisms in cancer: impact on irinotecan treatment. Pharmacogenom Pers Med. 2017;10:61–8.
Tejpar S, Yan P, Piessevaux H, Dietrich D, Brauchli P, Klingbiel D, et al. Clinical and pharmacogenetic determinants of 5-fluorouracyl/leucovorin/irinotecan toxicity: results of the PETACC-3 trial. Eur J Cancer. 2018;99:66–77.
Cheng L, Li M, Hu J, Ren W, Xie L, Sun ZP, et al. UGT1A1*6 polymorphisms are correlated with irinotecan-induced toxicity: a system review and meta-analysis in Asians. Cancer Chemother Pharmacol. 2014;73:551–60.
Yoshino T, Arnold D, Taniguchi H, Pentheroudakis G, Yamazaki K, Xu R-H, et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: a JSMO–ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncol. 2017;29:44–70.
El-Baiomy MA, El-Kashef WF. ERCC1 expression in metastatic triple negative breast cancer patients treated with platinum-based chemotherapy. Asian Pac J Cancer Prev. 2017;18:507–13.
Ryu H, Song IC, Choi YS, Yun HJ, Jo DY, Kim JM, et al. ERCC1 expression status predicts the response and survival of patients with metastatic or recurrent cervical cancer treated via platinum-based chemotherapy. Medicine (Baltimore). 2017;96:e9402.
Choueiri MB, Shen JP, Gross AM, Huang JK, Ideker T, Fanta P. ERCC1 and TS expression as prognostic and predictive biomarkers in metastatic colon cancer. PLoS One. 2015;10:e0126898.
ClinicalTrials.gov. Study of bevacizumab + mFOLFOX6 versus bevacizumab + FOLFIRI with biomarker stratification in participants with previously untreated metastatic colorectal cancer (mCRC) (MAVERICC). NCT01374425. 2011. https://clinicaltrials.gov/ct2/show/NCT01374425. Accessed 11 July 2019.
Parikh AR, Lee FC, Yau L, Koh H, Knost J, Mitchell EP, et al. MAVERICC, a randomized, biomarker-stratified, phase 2 study of mFOLFOX6-bevacizumab vs FOLFIRI-bevacizumab as first-line chemotherapy in metastatic colorectal cancer. Clin Cancer Res. 2019;25:2988–95.
Saletti P, Molinari F, De Dosso S, Frattini M. EGFR signaling in colorectal cancer: a clinical perspective. Gastrointest Cancer. 2015;5:21–38.
Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:4706–13.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-RAS mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65.
Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol. 2015;26:13–21.
Masuda T, Hayashi N, Kuroda Y, Ito S, Eguchi H, Mimori K. MicroRNAs as biomarkers in colorectal cancer. Cancers. 2017;9:124.
Cremolini C, Loupakis F, Antoniotti C, Lupi C, Sensi E, Lonardi S, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16:1306–15.
Modest D, von Fischer WL, Decker T, Vehling-Kaiser U, Uhlig J, Schenk M, et al. Randomized phase III study of fluoropyrimidine (FP) plus bevacizumab (BEV) vs. FP plus irinotecan (IRI) and BEV as first-line therapy for metastatic colorectal cancer (mCRC): German AIO KRK0110 (ML22011) study [abstract O-026]. Ann Oncol. 2017;28:mdx262.025.
Kubicka S, Greil R, Andre T, Bennouna J, Sastre J, Van Cutsem E, et al. Bevacizumab plus chemotherapy continued beyond first progression in patients with metastatic colorectal cancer previously treated with bevacizumab plus chemotherapy: ML18147 study KRAS subgroup findings. Ann Oncol. 2013;24:2342–9.
Satake H, Sunakawa Y, Miyamoto Y, Nakamura M, Nakayama H, Shiozawa M, et al. A phase II trial of 1st-line modified-FOLFOXIRI plus bevacizumab treatment for metastatic colorectal cancer harboring RAS mutation: JACCRO CC-11. Oncotarget. 2018;9:18811–20.
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology. Colon cancer. Version 2.2019. 2019. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed 11 July 2019.
ClinicalTrials.gov. A phase 1, study evaluating the safety, tolerability, PK, and efficacy of AMG 510 in subjects with solid tumors with a specific KRAS mutation. NCT03600883. 2018. https://clinicaltrials.gov/ct2/show/NCT03600883. Accessed 11 July 2019.
ClinicalTrials.gov. MRTX849 in patients with cancer having a KRAS G12C mutation. NCT03785249. 2018. https://clinicaltrials.gov/ct2/show/NCT03785249. Accessed 11 July 2019.
Brodowicz T, Vrbanec D, Kaczirek K, Ciuleanu T-E, Knittelfelder R, Lindner E, et al. FOLFOX4 plus cetuximab administered weekly or every two weeks in first-line treatment of patients with KRAS and NRAS wild-type (wt) metastatic colorectal cancer (mCRC). J Clin Oncol. 2014;32:LBA391.
Bokemeyer C, Kohne C-H, Ciardiello F, Lenz H-J, Heinemann V, Klinkhardt U, et al. Treatment outcome according to tumor RAS mutation status in OPUS study patients with metastatic colorectal cancer (mCRC) randomized to FOLFOX4 with/without cetuximab. J Clin Oncol. 2014;32:3505.
Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32:2240–7.
Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–75.
Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, et al. Analysis of KRAS/NRAS mutations in phase 3 study 20050181 of panitumumab (pmab) plus FOLFIRI versus FOLFIRI for second-line treatment (tx) of metastatic colorectal cancer (mCRC). J Clin Oncol. 2014;32:LBA387.
Kim TW, Elme A, Park JO, Udrea AA, Kim SY, Ahn JB, et al. Final analysis of outcomes and RAS/BRAF status in a randomized phase 3 study of panitumumab and best supportive care in chemorefractory wild type KRAS metastatic colorectal cancer. Clin Colorectal Cancer. 2018;17:206–14.
Al-Shamsi HO, Alhazzani W, Wolff RA. Extended RAS testing in metastatic colorectal cancer-refining the predictive molecular biomarkers. J Gastrointest Oncol. 2015;6:314–21.
Bachet JB, Bouche O, Taieb J, Dubreuil O, Garcia ML, Meurisse A, et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: the AGEO RASANC prospective multicenter study. Ann Oncol. 2018;29:1211–9.
Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19:1902–12.
Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14:749–59.
Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O’Callaghan CJ, Marginean C, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer–results from NCIC CTG/AGITG CO.17. Clin Cancer Res. 2014;20:744–53.
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–75.
Oliner KS, Douillard J-Y, Siena S, Tabernero J, Burkes RL, Barugel ME, et al. Analysis of KRAS/NRAS and BRAF mutations in the phase III PRIME study of panitumumab (pmab) plus FOLFOX versus FOLFOX as first-line treatment (tx) for metastatic colorectal cancer (mCRC). J Clin Oncol. 2013;31:3511.
Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer. 2015;112:1888–94.
Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51:587–94.
Geissler M, Klingler T, Riera Knorrenschild J, Tannapfel A, Greeve J, Seufferlein T, et al. 1st-line mFOLFOXIRI + panitumumab vs FOLFOXIRI treatment of RAS WT mCRC: a randomized phase II VOLFI trial of the AIO (KRK-0109) [453PD]. Ann Oncol. 2018;29:mdy281.001.
Loupakis F, Cremolini C, Salvatore L, Masi G, Sensi E, Schirripa M, et al. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur J Cancer. 2014;50:57–63.
Masi G, Loupakis F, Salvatore L, Fornaro L, Cremolini C, Cupini S, et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: a phase 2 trial. Lancet Oncol. 2010;11:845–52.
Wirapati P, Pomella V, Vandenbosch B, Kerr P, Maiello E, Jeffery GM, et al. Velour trial biomarkers update: impact of RAS, BRAF, and sidedness on aflibercept activity. J Clin Oncol. 2017;35:3538.
Yoshino T, Portnoy DC, Obermannová R, Bodoky G, Prausová J, Garcia-Carbonero R, et al. Biomarker analysis beyond angiogenesis: RAS/RAF mutation status, tumour sidedness, and second-line ramucirumab efficacy in patients with metastatic colorectal carcinoma from RAISE—a global phase III study. Ann Oncol. 2018;30:124–31.
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3.
Corcoran RB, Andre T, Atreya CE, Schellens JHM, Yoshino T, Bendell JC, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov. 2018;8:428–43.
Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol. 2015;33:4023–31.
Kopetz S, McDonough SL, Morris VK, Lenz H-J, Magliocco AM, Atreya CE, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG 1406). J Clin Oncol. 2017;35:520.
Van Cutsem E, Cuyle P, Huijberts S, Schellens J, Elez E, Yaeger R, et al. BEACON CRC study safety lead-in: assessment of the BRAF inhibitor encorafenib + MEK inhibitor binimetinib + anti-epidermal growth factor receptor antibody cetuximab for BRAF V600E metastatic colorectal cancer [O-027]. Ann Oncol. 2018;29:mdy149.026.
Jones JC, Renfro LA, Al-Shamsi HO, Schrock AB, Rankin A, Zhang BY, et al. (Non-V600) BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J Clin Oncol. 2017;35:2624–30.
Taieb J, Balogoun R, Le Malicot K, Tabernero J, Mini E, Folprecht G, et al. Adjuvant FOLFOX ± cetuximab in full RAS and BRAF wildtype stage III colon cancer patients. Ann Oncol. 2017;28:824–30.
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–7.
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27:5068–74.
Stintzing S, Ivanova B, Ricard I, Jung A, Kirchner T, Tannapfel A, et al. Amphiregulin (AREG) and epiregulin (EREG) gene expression as predictor for overall survival (OS) in oxaliplatin/fluoropyrimidine plus bevacizumab treated mCRC patients-analysis of the phase III AIO KRK-0207 trial. Front Oncol. 2018;8:474.
Jing C, Jin YH, You Z, Qiong Q, Jun Z. Prognostic value of amphiregulin and epiregulin mRNA expression in metastatic colorectal cancer patients. Oncotarget. 2016;7:55890–9.
Stahler A, Heinemann V, Giessen-Jung C, Crispin A, Schalhorn A, Stintzing S, et al. Influence of mRNA expression of epiregulin and amphiregulin on outcome of patients with metastatic colorectal cancer treated with 5-FU/LV plus irinotecan or irinotecan plus oxaliplatin as first-line treatment (FIRE 1-trial). Int J Cancer. 2016;138:739–46.
Seligmann JF, Elliott F, Richman SD, Jacobs B, Hemmings G, Brown S, et al. Combined epiregulin and amphiregulin expression levels as a predictive biomarker for panitumumab therapy benefit or lack of benefit in patients with RAS wild-type advanced colorectal cancer. JAMA Oncol. 2016;2:633–42.
Sartore-Bianchi A, Moroni M, Veronese S, Carnaghi C, Bajetta E, Luppi G, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25:3238–45.
Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6:279–86.
Kato S, Okamura R, Mareboina M, Lee S, Goodman A, Patel SP, et al. Revisiting epidermal growth factor receptor (EGFR) amplification as a target for anti-EGFR therapy: analysis of cell-free circulating tumor DNA in patients with advanced malignancies. JCO Precis Oncol. 2019. https://doi.org/10.1200/PO.18.00180.
Sartore-Bianchi A, Fieuws S, Veronese S, Moroni M, Personeni N, Frattini M, et al. Standardisation of EGFR FISH in colorectal cancer: results of an international interlaboratory reproducibility ring study. J Clin Pathol. 2012;65:218–23.
Martini G, Troiani T, Cardone C, Vitiello P, Sforza V, Ciardiello D, et al. Present and future of metastatic colorectal cancer treatment: a review of new candidate targets. World J Gastroenterol. 2017;23:4675–88.
Siena S, Sartore-Bianchi A, Marsoni S, Hurwitz HI, McCall SJ, Penault-Llorca F, et al. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann Oncol. 2018;29:1108–19.
Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832–41.
Raghav KPS, Overman MJ, Yu R, Meric-Bernstam F, Menter D, Kee BK, et al. HER2 amplification as a negative predictive biomarker for anti-epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. J Clin Oncol. 2016;34:3517.
Raghav K, Loree JM, Morris JS, Overman MJ, Yu R, Meric-Bernstam F, et al. Validation of HER2 amplification as a predictive biomarker for anti-epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. JCO Precis Oncol. 2019. https://doi.org/10.1200/PO.18.00226.
Laurent-Puig P, Balogoun R, Cayre A, Le Malicot K, Tabernero J, Mini E, et al. ERBB2 alterations a new prognostic biomarker in stage III colon cancer from a FOLFOX based adjuvant trial (PETACC8). Ann Oncol. 2016;27:459O.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–23.
Leto SM, Sassi F, Catalano I, Torri V, Migliardi G, Zanella ER, et al. Sustained inhibition of HER3 and EGFR is necessary to induce regression of HER2-amplified gastrointestinal carcinomas. Clin Cancer Res. 2015;21:5519–31.
Siena S, Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, et al. Final results of the HERACLES trial in HER2 amplified colorectal cancer. Cancer Res. 2017;17(13 Suppl):abstract CT005.
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:738–46.
Meric-Bernstam F, Hurwitz H, Raghav KPS, McWilliams RR, Fakih M, VanderWalde A, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019;20:518–30.
Seligmann JF, Hatch AJ, Richman SD, et al. Association of tumor HER3 messenger rna expression with panitumumab efficacy in advanced colorectal cancer. JAMA Oncol. 2017;4:564–8.
Mosakhani N, Lahti L, Borze I, Karjalainen-Lindsberg ML, Sundstrom J, Ristamaki R, et al. MicroRNA profiling predicts survival in anti-EGFR treated chemorefractory metastatic colorectal cancer patients with wild-type KRAS and BRAF. Cancer Genet. 2012;205:545–51.
Pichler M, Winter E, Ress AL, Bauernhofer T, Gerger A, Kiesslich T, et al. miR-181a is associated with poor clinical outcome in patients with colorectal cancer treated with EGFR inhibitor. J Clin Pathol. 2014;67:198–203.
Xie T, Huang M, Wang Y, Wang L, Chen C, Chu X. MicroRNAs as regulators, biomarkers and therapeutic targets in the drug resistance of colorectal cancer. Cell Physiol Biochem. 2016;40:62–76.
Cappuzzo F, Sacconi A, Landi L, Ludovini V, Biagioni F, D’Incecco A, et al. MicroRNA signature in metastatic colorectal cancer patients treated with anti-EGFR monoclonal antibodies. Clin Colorectal Cancer. 2014;13(37–45):e4.
Igarashi H, Kurihara H, Mitsuhashi K, Ito M, Okuda H, Kanno S, et al. Association of microRNA-31-5p with clinical efficacy of anti-EGFR therapy in patients with metastatic colorectal cancer. Ann Surg Oncol. 2015;22:2640–8.
Manceau G, Imbeaud S, Thiebaut R, Liebaert F, Fontaine K, Rousseau F, et al. Hsa-miR-31-3p expression is linked to progression-free survival in patients with KRAS wild-type metastatic colorectal cancer treated with anti-EGFR therapy. Clin Cancer Res. 2014;20:3338–47.
Pugh S, Thiebaut R, Bridgewater J, Grisoni ML, Moutasim K, Rousseau F, et al. Association between miR-31-3p expression and cetuximab efficacy in patients with KRAS wild-type metastatic colorectal cancer: a post hoc analysis of the New EPOC trial. Oncotarget. 2017;8:93856–66.
Laurent-Puig P, Grisoni ML, Heinemann V, Liebaert F, Neureiter D, Jung A, et al. Validation of miR-31-3p expression to predict cetuximab efficacy when used as first-line treatment in RAS wild-type metastatic colorectal cancer. Clin Cancer Res. 2019;25:134–41.
Gallois C, Pernot S, Zaanan A, Taieb J. Colorectal cancer: why does side matter? Drugs. 2018;78:789–98.
Baran B, Mert Ozupek N, Yerli Tetik N, Acar E, Bekcioglu O, Baskin Y. Difference between left-sided and right-sided colorectal cancer: a focused review of literature. Gastroenterol Res. 2018;11:264–73.
Van Emburgh BO, Sartore-Bianchi A, Di Nicolantonio F, Siena S, Bardelli A. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol Oncol. 2014;8:1084–94.
Montagut C, Argiles G, Ciardiello F, Poulsen TT, Dienstmann R, Kragh M, et al. Efficacy of Sym004 in patients with metastatic colorectal cancer with acquired resistance to anti-EGFR therapy and molecularly selected by circulating tumor DNA analyses: a phase 2 randomized clinical trial. JAMA Oncol. 2018;4:e175245.
Van Emburgh BO, Arena S, Siravegna G, Lazzari L, Crisafulli G, Corti G, et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun. 2016;7:13665.
Gollins S, West N, Sebag-Montefiore D, Myint AS, Saunders M, Susnerwala S, et al. Preoperative chemoradiation with capecitabine, irinotecan and cetuximab in rectal cancer: significance of pre-treatment and post-resection RAS mutations. Br J Cancer. 2017;117:1286–94.
Kim TW, Peeters M, Thomas AL, Gibbs P, Hool K, Zhang J, et al. Impact of emergent circulating tumor DNA RAS mutation in panitumumab-treated chemoresistant metastatic colorectal cancer. Clin Cancer Res. 2018;24:5602–9.
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–73.
Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6.
van Dijk E, Biesma HD, Cordes M, Smeets D, Neerincx M, Das S, et al. Loss of chromosome 18q11.2-q12.1 is predictive for survival in patients with metastatic colorectal cancer treated with bevacizumab. J Clin Oncol. 2018;36:2052–60.
Carbone C, Piro G, Simionato F, Ligorio F, Cremolini C, Loupakis F, et al. Homeobox B9 mediates resistance to anti-VEGF therapy in colorectal cancer patients. Clin Cancer Res. 2017;23:4312–22.
Zhan J, Niu M, Wang P, Zhu X, Li S, Song J, et al. Elevated HOXB9 expression promotes differentiation and predicts a favourable outcome in colon adenocarcinoma patients. Br J Cancer. 2014;111:883–93.
Weickhardt AJ, Williams DS, Lee CK, Chionh F, Simes J, Murone C, et al. Vascular endothelial growth factor D expression is a potential biomarker of bevacizumab benefit in colorectal cancer. Br J Cancer. 2015;113:37–45.
Tabernero J, Hozak RR, Yoshino T, Cohn AL, Obermannova R, Bodoky G, et al. Analysis of angiogenesis biomarkers for ramucirumab efficacy in patients with metastatic colorectal cancer from RAISE, a global, randomized, double-blind, phase III study. Ann Oncol. 2018;29:602–9.
Noonan SA, Morrissey ME, Martin P, Biniecka M, Ó’Meachair S, Maguire A, et al. Tumour vasculature immaturity, oxidative damage and systemic inflammation stratify survival of colorectal cancer patients on bevacizumab treatment. Oncotarget. 2018;9:10536–48.
Bates DO, Catalano PJ, Symonds KE, Varey AH, Ramani P, O’Dwyer PJ, et al. Association between VEGF splice isoforms and progression-free survival in metastatic colorectal cancer patients treated with bevacizumab. Clin Cancer Res. 2012;18:6384–91.
Bai L, Wang F, Zhang DS, Li C, Jin Y, Wang DS, et al. A plasma cytokine and angiogenic factor (CAF) analysis for selection of bevacizumab therapy in patients with metastatic colorectal cancer. Sci Rep. 2015;5:17717.
Wang Y, Wang L, Chen C, Chu X. New insights into the regulatory role of microRNA in tumor angiogenesis and clinical implications. Mol Cancer. 2018;17:22.
Boisen MK, Dehlendorff C, Linnemann D, Nielsen BS, Larsen JS, Osterlind K, et al. Tissue microRNAs as predictors of outcome in patients with metastatic colorectal cancer treated with first line capecitabine and oxaliplatin with or without bevacizumab. PLoS One. 2014;9:e109430.
Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, et al. ESMO consensus guidelines for management of patients with colon and rectal cancer. A personalized approach to clinical decision making. Ann Oncol. 2012;23:2479–516.
Zaanan A, Shi Q, Taieb J, Alberts SR, Meyers JP, Smyrk TC, et al. Role of deficient DNA mismatch repair status in patients with stage III colon cancer treated with FOLFOX adjuvant chemotherapy: a pooled analysis from 2 randomized clinical trials. JAMA Oncol. 2018;4:379–83.
Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–18.
Oncology Pro. Microsatellite instability—defective DNA mismatch repair: ESMO biomarker factsheet. 2016. https://oncologypro.esmo.org/Education-Library/Factsheets-on-Biomarkers/Microsatellite-Instability-Defective-DNA-Mismatch-Repair. Accessed 11 July 2019.
Sinicrope FA, Shi Q, Allegra CJ, et al. Association of DNA mismatch repair and mutations in BRAF and KRAS with survival after recurrence in Stage III colon cancers: a secondary analysis of 2 randomized clinical trials. JAMA Oncol. 2017;3:472–80.
Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20:5322–30.
Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–32.
Kim ST, Lee SJ, Lee J, Park SH, Park JO, Lim HY, et al. The impact of microsatellite instability status and sidedness of the primary tumor on the effect of cetuximab-containing chemotherapy in patients with metastatic colorectal cancer. J Cancer. 2017;8:2809–15.
Des Guetz G, Uzzan B, Nicolas P, Schischmanoff O, Morere JF. Microsatellite instability: a predictive marker in metastatic colorectal cancer? Target Oncol. 2009;4:57–62.
Innocenti F, Ou F-S, Qu X, Zemla TJ, Niedzwiecki D, Tam R, et al. Mutational analysis of patients with colorectal cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol. 2019;37:1217–27.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
FDA press release. FDA grants nivolumab accelerated approval for MSI-H or dMMR colorectal cancer. 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm569366.htm. Accessed 11 July 2019.
FDA press release. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560040.htm. Accessed 11 July 2019.
FDA press release. FDA grants accelerated approval to ipilimumab for MSI-H or dMMR metastatic colorectal cancer. 2018. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm613227.htm. Accessed 11 July 2019.
Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9.
Roche press release. Roche provides update on Phase III study of Tecentriq (atezolizumab) and Cotellic (cobimetinib) in people with heavily pre-treated locally advanced or metastatic colorectal cancer. 2018. https://www.roche.com/media/releases/med-cor-2018-05-10.htm/. Accessed 11 July 2019.
Grothey A, Tabernero J, Arnold D, De Gramont A, Ducreux MP, O’Dwyer PJ, et al. Fluoropyrimidine (FP) + bevacizumab (BEV) + atezolizumab vs FP/BEV in BRAF WT metastatic colorectal cancer (mCRC): findings from Cohort 2 of MODUL—a multicentre, randomized trial of biomarker-driven maintenance treatment following first-line induction therapy [LBA19]. Ann Oncol. 2018;29:mdy424.020.
Bendell J, Ciardiello F, Tabernero J, Tebbutt N, Eng C, Di Bartolomeo M, et al. Efficacy and safety results from IMblaze370, a randomised phase III study comparing atezolizumab + cobimetinib and atezolizumab monotherapy vs regorafenib in chemotherapy-refractory metastatic colorectal cancer [LBA-004]. Ann Oncol. 2018;29:mdy208.003.
Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–20.
Cesano A, Warren S. Bringing the next generation of immuno-oncology biomarkers to the clinic. Biomedicines. 2018;6:E14.
Pai SG, Carneiro BA, Chae YK, Costa RL, Kalyan A, Shah HA, et al. Correlation of tumor mutational burden and treatment outcomes in patients with colorectal cancer. J Gastrointest Oncol. 2017;8:858–66.
George TJ, Frampton GM, Sun J, Gowen K, Kennedy M, Greenbowe JR, et al. Tumor mutational burden as a potential biomarker for PD1/PD-L1 therapy in colorectal cancer. J Clin Oncol. 2016;34:3587.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
Domingo E, Freeman-Mills L, Rayner E, Glaire M, Briggs S, Vermeulen L, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016;1:207–16.
Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Tejpar S, Sartore-Bianchi, et al. AALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J Natl Cancer Inst. 2017;109:djx089.
Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Morano F, Fucà G, et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol. 2018;29:1394–401.
Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A, et al. Sensitivity to entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal cancer. J Natl Cancer Inst. 2016;108:djv306.
Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016;6:36–44.
Morano F, Corallo S, Di Bartolomeo M, Lonardi S, Cremolini C, Rimassa L, et al. Negative hyper-selection of RAS wild-type (wt) metastatic colorectal cancer (mCRC) patients randomized to first-line FOLFOX plus panitumumab (Pan) followed by maintenance therapy with either 5FU/LV plus pan or single-agent pan: translational analyses of the VALENTINO study [abstract LBA22]. Ann Oncol. 2018;29:mdy424.023.
Cremolini C, Morano F, Moretto R, Berenato R, Tamborini E, Perrone F, et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: the PRESSING case–control study. Ann Oncol. 2017;28:3009–14.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–6.
Rashid A, Shen L, Morris JS, Issa JP, Hamilton SR. CpG island methylation in colorectal adenomas. Am J Pathol. 2001;159:1129–35.
Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014;25:2314–27.
Jia M, Jansen L, Walter V, Tagscherer K, Roth W, Herpel E, et al. No association of CpG island methylator phenotype and colorectal cancer survival: population-based study. Br J Cancer. 2016;115:1359–66.
Jia M, Gao X, Zhang Y, Hoffmeister M, Brenner H. Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: a systematic review. Clin Epigenet. 2016;8:25.
Gallois C, Laurent-Puig P, Taieb J. Methylator phenotype in colorectal cancer: a prognostic factor or not? Crit Rev Oncol Hematol. 2016;99:74–80.
Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9:2898–903.
Gallois C, Taieb J, Le Corre D, Le Malicot K, Tabernero J, Mulot C, et al. Prognostic value of methylator phenotype in stage III colon cancer treated with oxaliplatin-based adjuvant chemotherapy. Clin Cancer Res. 2018;24:4745–53.
Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, et al. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin Cancer Res. 2013;19:2265–72.
Calegari MA, Inno A, Monterisi S, Orlandi A, Santini D, Basso M, et al. A phase 2 study of temozolomide in pretreated metastatic colorectal cancer with MGMT promoter methylation. Br J Cancer. 2017;116:1279–86.
Draht MXG, Goudkade D, Koch A, Grabsch HI, Weijenberg MP, van Engeland M, et al. Prognostic DNA methylation markers for sporadic colorectal cancer: a systematic review. Clin Epigenet. 2018;10:35.
Herbst A, Vdovin N, Gacesa S, Ofner A, Philipp A, Nagel D, et al. Methylated free-circulating HPP1 DNA is an early response marker in patients with metastatic colorectal cancer. Int J Cancer. 2017;140:2134–44.
Kidess-Sigal E, Liu HE, Triboulet MM, Che J, Ramani VC, Visser BC, et al. Enumeration and targeted analysis of KRAS, BRAF and PIK3CA mutations in CTCs captured by a label-free platform: comparison to ctDNA and tissue in metastatic colorectal cancer. Oncotarget. 2016;7:85349–64.
Khakoo S, Georgiou A, Gerlinger M, Cunningham D, Starling N. Circulating tumour DNA, a promising biomarker for the management of colorectal cancer. Crit Rev Oncol Hematol. 2018;122:72–82.
Garlan F, Laurent-Puig P, Sefrioui D, Siauve N, Didelot A, Sarafan-Vasseur N, et al. Early evaluation of circulating tumor DNA as marker of therapeutic efficacy in metastatic colorectal cancer patients (PLACOL study). Clin Cancer Res. 2017;23:5416–25.
Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6.
Isella C, Brundu F, Bellomo SE, Galimi F, Zanella E, Porporato R, et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat Commun. 2017;8:15107.
Stintzing S, Wirapati P, Lenz H-J, Neureiter D, von Weikersthal LF, Decker T, et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. J Clin Oncol. 2017;35:3510.
Okita A, Takahashi S, Ouchi K, Inoue M, Watanabe M, Endo M, et al. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget. 2018;9:18698–711.
FOCUS4. A molecularly stratified, multi-site randomised trial programme for patients with colorectal cancer. 2019. http://www.focus4trial.org/. Accessed 11 July 2019.
Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801.
Parseghian CM, Loree JM, Morris VK, Pereira AAL, Sanchez EV, Kee BK, et al. Anti-EGFR resistant clones decay exponentially after progression: implications for anti-EGFR rechallenge. J Clin Oncol. 2018;36:3511.
Santini D, Vincenzi B, Addeo R, Garufi C, Masi G, Scartozzi M, et al. Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann Oncol. 2012;23:2313–8.
Osawa H, Satake H, Kobayashi T, Suto T, Kitano S, Ohashi Y, et al. Phase II study of cetuximab rechallenge in patients with RAS wild-type metastatic colorectal cancer: e-rechallenge trial [481P]. Ann Oncol. 2018;29:mdy281.029.
Cremolini C, Rossini D, Dell’Aquila E, Lonardi S, Conca E, Del Re M, et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: a phase 2 single-arm clinical trial. JAMA Oncol. 2018;5:343–50.
Wadlow RC, Hezel AF, Abrams TA, Blaszkowsky LS, Fuchs CS, Kulke MH, et al. Panitumumab in patients with KRAS wild-type colorectal cancer after progression on cetuximab. Oncologist. 2012;17:14.
Siena S, George P, Gerald P, Andrea S-B, Aziz Z, Jean-Yves D, et al. Rechallenge with EGFR inhibitors in patients with metastatic colorectal cancer: effect on outcomes [P-320]. Ann Oncol. 2017;28:mdx261.317.
Rossini D, Cremolini C, Conca E, Del Re M, Busico A, Pietrantonio F, et al. Liquid biopsy allows predicting benefit from rechallenge with cetuximab(cet) + irinotecan(iri) in RAS/BRAF wild-type mCRC patients(pts) with resistance to 1st-line cet + iri: final results and translational analyses of the CRICKET study by GONO [O-007]. Ann Oncol. 2018;29:mdy149.006.
Gospodarowicz M, Mackillop W, O’Sullivan B, Sobin L, Henson D, Hutter RV, et al. Prognostic factors in clinical decision making. Cancer. 2001;91:1688–95.
Siena S, Sartore-Bianchi A, Garcia-Carbonero R, Karthaus M, Smith D, Tabernero J, et al. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann Oncol. 2018;29:119–26.
Siravegna G, Lazzari L, Crisafulli G, Sartore-Bianchi A, Mussolin B, Cassingena A, et al. Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell. 2018;34(148–62):e7.
Goossens N, Nakagawa S, Sun X, Hoshida Y. Cancer biomarker discovery and validation. Transl Cancer Res. 2015;4:256–69.
Patterson SD, Cohen N, Karnoub M, Truter SL, Emison E, Khambata-Ford S, et al. Prospective–retrospective biomarker analysis for regulatory consideration: white paper from the industry pharmacogenomics working group. Pharmacogenomics. 2011;12:939–51.
Kinkorová J. Biobanks in the era of personalized medicine: objectives, challenges, and innovation: overview. EPMA J. 2016;7:4.
Liu R, Wang X, Aihara K, Chen L. Early diagnosis of complex diseases by molecular biomarkers, network biomarkers, and dynamical network biomarkers. Med Res Rev. 2014;34:455–78.
Barry GS, Cheang MC, Chang HL, Kennecke HF. Genomic markers of panitumumab resistance including ERBB2/HER2 in a phase II study of KRAS wild-type (wt) metastatic colorectal cancer (mCRC). Oncotarget. 2016;7:18953–64.
Acknowledgements
Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact checking and referencing) was provided by Emma Evans PhD, CMPP and Louise Niven DPhil, CMPP at Aspire Scientific (Bollington, UK), and funded by Amgen (Europe) GmbH (Rotkreuz, Switzerland).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Medical writing support was funded by Amgen (Europe) GmbH.
Conflict of interest
Julien Taieb has acted in consultancy and/or advisory roles for, and received honoraria from, Amgen, Celgene, Lilly, Merck, MSD, Roche, Sanofi, Servier and Sirtex. Andreas Jung has acted in advisory roles for, and received honoraria and travel support from, Amgen, AstraZeneca, Biocartis, Boehringer Ingelheim, Bristol-Myers Squibb, Merck, Novartis and Roche. Andrea Sartore-Bianchi has acted in consultancy and/or advisory roles for, and received honoraria from, Amgen, Bayer, Lilly and Sanofi. Marc Peeters has received research funding from Amgen and Roche, and honoraria from Amgen, Lilly, Merck Serono, Remedus, Roche, Sanofi-Aventis, Servier, Sirtex and Terumo. Jenny Seligmann has acted in an advisory role for Roche and received honoraria from Merck Serono. Aziz Zaanan has acted in consultancy and/or advisory roles for Amgen, Baxter, Lilly, Merck Serono, MSD, Roche, Sanofi and Servier. Peter Burdon is an employee of Amgen (Europe) GmbH and owns shares in Amgen. Clara Montagut has acted in consultancy and/or advisory roles for Amgen, Merck Serono, Roche, Sanofi-Aventis, Servier and Symphogen, and received research funding from Amgen, Merck Serono and Symphogen. Pierre Laurent-Puig has acted in consultancy and/or advisory roles for Amgen, Biocartis, Boehringer Ingelheim, Bristol-Myers Squibb, Lilly, MDS, Merck Serono, Roche, Sanofi, and holds a patent for miR-31-3p.
Data sharing
All information generated as part of the literature review for this paper has been included in the publication.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Taieb, J., Jung, A., Sartore-Bianchi, A. et al. The Evolving Biomarker Landscape for Treatment Selection in Metastatic Colorectal Cancer. Drugs 79, 1375–1394 (2019). https://doi.org/10.1007/s40265-019-01165-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01165-2