
Abstract
Background
Two main types of galenic formulation, immediate release and prolonged release, have been developed to optimize melatonin bioavailability. We recently described the kinetic profile of a prolonged-release form generating a peak of plasma melatonin 1 h (Tmax) after intake, followed by a prolonged decay over time. We have developed a new oral form of melatonin with the aim of producing a melatonin peak several hours after intake.
Objective
The objective is to investigate melatonin bioavailability after administration of this new delayed-release form (DR form).
Methods
In this single-centre open-label study, 12 healthy male volunteers received one tablet of the DR form containing 1.9 mg melatonin, 10 mg zinc and 200 mg lemon balm extract (Melissa officinalis L aerial parts). Blood samples were collected for 12 h, beginning at 8:00 am. Plasma concentrations of melatonin and 6-sulfatoxymelatonin (6-SMT), the main hepatic melatonin metabolite, were determined by radioimmunoassay.
Results
A progressive increase in plasma melatonin concentrations was observed from 20 min and a peak about 3 h after intake (Cmax 740 ± 824 pg/mL; Tmax 179 ± 60 min). Concentrations remained high between 140 and 220 min, the concentration remaining physiologically significant (over 100 pg/mL) up to 7 h after intake. The DR form was well tolerated.
Conclusions
The melatonin release profile was consistent with what was anticipated for the DR form. The DR form generated a 2 h delayed Tmax compared with a prolonged-release form previously evaluated. This suggests that the DR form is suitable for the treatment of certain sleep disorders such as short sleep duration or early awakening.
Trial Registry
Registration number: NCT05419466.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Kinetics consistent with those expected for the delayed-release melatonin form evaluated. |
Plasma peak observed 3 h after ingestion of one tablet. |
Concentrations of physiological meaning up to 7 h. |
1 Introduction
Melatonin, a hormone secreted at night by the pineal gland, displays a very marked nychthemeral rhythm, the result of exposure to the natural light dark cycle [1]. The secretion occurs over 8–10 h, with a peak around 2:00–4:00 am, varying according to the subjects’ chronotype. In physiological conditions, the blood melatonin profile results from hormone infusion by the pineal gland reinforced by episodes of secretion. Experimental data in humans showed that the durations of melatonin secretion and sleep are directly influenced by the length of the artificial night [2]. In that sense, melatonin should be considered the ‘night hormone’ rather than the ‘sleep hormone’.
For people who have difficulty falling asleep or remaining asleep, oral melatonin supplementation has been considered as one approach to treatment. The benefits of exogenous melatonin are now recognized in the treatment of sleep disorders. Melatonin supplementation has been shown to reduce sleep onset latency and to improve sleep duration and quality at all stages of life [3,4,5,6,7,8]. The European Health Authorities have granted a favourable opinion for the claim ‘melatonin contributes to the reduction of time taken to fall asleep’ that can be used only for food supplements containing 1 mg of melatonin per quantified portion [9]. In addition, melatonin is well tolerated [10]. Melatonin is not associated with impaired psychomotor function, memory or driving ability and it is also devoid of side-effects such as hangover, nocturnal confusion, adverse effects on cognitive performance and dependence. For these reasons, melatonin may be considered a better-tolerated alternative, better adapted to some target populations than certain highly-prescribed drugs such as benzodiazepines, chronic use of which is a major public health concern [11].
Numerous galenic formulations on the market have been developed in attempt to optimize or adapt melatonin bioavailability. There are two main types of formulations available: immediate-release and prolonged (or extended)-release. Immediate-release formulations are rapidly absorbed and eliminated by the body, making it difficult to cover an entire sleep period, whereas prolonged-release formulations are developed to mimic the endogenous melatonin secretion and display increased efficacy [12]. Depending on the timing and duration of melatonin release, the formulations are recommended for the treatment of different sleep disorders. Immediate-release preparations are mainly useful for the treatment or prevention of sleep–wake cycle disorders (jet-lag syndrome, delayed sleep phase syndrome or free running in the blind) or delayed sleep onset [13]. Clinical trials in patients aged more 55 years with primary insomnia have demonstrated the efficacy of a prolonged-release form at improving sleep and next-day morning alertness and quality of life [3, 14]. These data were validated by recent recommendations of the European Sleep Research Society (ESRS) and European Insomnia Network (EIN) and of the French Medical and Research Sleep Society (SFRMS) stating that the prolonged-release melatonin form can be used for up to 3 months in patients ≥ 55 years [15, 16].
We recently described the bioavailability of a prolonged-release form of melatonin (tablet) [17]. Its melatonin release profile was in line with that expected for this type of formulation: the melatonin peak (Cmax 1151 ± 565 pg/mL, Tmax of 64 ± 44 min) was followed by a prolonged decay over time, the concentration remaining physiologically significant (i.e. over 100 pg/mL [18]) up to 6 h after intake. A galenic preparation generating a melatonin peak several hours after intake, i.e. with a Tmax beyond that of 1 h observed with the prolonged-release form, could be particularly beneficial for reinforcing the endogenous melatonin concentrations especially at the end of the night. Elderly insomniac patients with short sleep duration or early awakening, as well as patients with abnormal behaviour during late sleep are the main candidates for this supplementation. Therefore, we have developed a new form of melatonin generating such a delayed-release of melatonin, the formulation of which has been patented [19]. In vitro dissolution tests conducted in accordance with the European Pharmacopeia showed a peak release of melatonin between 3 and 5 h after administration [19, 20]. As melatonin bioavailability depends on the galenic form, route of administration, dosage and on individual absorption and rate of hepatic metabolism, bioavailability should be investigated for each new galenic formulation brought to the market [10, 21, 22]. However, only a few studies have reported on the pharmacokinetics of melatonin forms. Most of these were performed in young male subjects during daytime. Here, we conducted a first study to document melatonin bioavailability after administration of the new delayed-release form.
In addition to melatonin determination, we measured the metabolite 6-sulphatoxymelatonin, which is not routinely measured in melatonin pharmacokinetic studies; although, it is an important indicator of hepatic transformation [1, 23]. Melatonin is a lipophilic compound (octanol/water coefficient of partition of approximately 13, close to that of oestradiol) [21, 24]. Similar to all lipophilic hormones, it is highly inactivated in the liver, leading to 6-hydroxymelatonin, which contributes to low bioavailability after oral administration [1, 25]. This product then undergoes further conjugation with either sulphate catalysed by sulfotransferase to form 6-sulphatoxymelatonin or glucuronic acid catalysed by UDP-glucuronosyltransferase to form 6-hydroxymelatonin glucuronide. Over 80% of endogenous melatonin is eliminated as 6-SMT in the urine. Also, due to an entero-hepatic cycle, 6-SMT is also detected and can be measured in the blood [26].
2 Material and Methods
2.1 Study Product
The delayed-release form of melatonin tested (hereafter named DR form, D for delayed, R for release) is a food supplement containing 1.9 mg of melatonin, 10 mg of zinc (in the form of Hypro-ri® Zinc composed of zinc sulphate and a rice protein hydrolysate) and 200 mg of a lemon balm extract (Melissa officinalis L., aerial parts) [27] per tablet. It also contains baobab pulp (Adonsonia digitata L.) and sodium alginate for delayed release [19]. The formulation as well as the ingredients Hypro-ri® Zinc and the lemon balm extract were conceived and developed by PiLeJe. In vitro tests have been carried out in accordance with the European Pharmacopoeia (under different pH conditions) to determine the composition and proportion of ingredients allowing delayed release of melatonin [19, 20]. These tests showed that in addition to baobab pulp and sodium alginate, lemon balm contributes to the delayed release (preparation process and granulometry of the extract have an impact on release). It is the combination of the three ingredients that creates a matrix effect and enables the modified release. The food supplement is marketed under the names Chronobiane Protect LD 1.9 mg and Chronobiane LD Protect 1.9 mg by PiLeJe Laboratoire. The tablet contains 1.9 mg of melatonin in accordance with the French regulation which allows the marketing of food supplements providing less than 2 mg of melatonin per day.
2.2 Study Design and Ethics Statement
The study was a single-centre, open-label, bioavailability trial and was conducted in accordance with Good Clinical Practices. The protocol was approved by Personal Protection Committee (CPP Ile de France VIII, Hôpital Ambroise Paré, Boulogne-Billancourt, France; Approval number: A02632-41) on January 10, 2023. Registration number on the ClinicalTrials.gov site is: NCT05419466.
2.3 Study Methodology
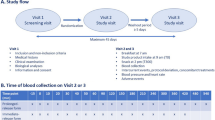
The study consisted of a screening visit and a study visit for pharmacokinetic assessment separated by a maximum of 45 days (Fig. 1).

Study flow (A) and time of blood collection on visit 2 (B)
Screening visit Volunteers taking part in the study had to meet the following inclusion criteria: men aged between 18 and 45 years, weighing more than 70 kg, with a body mass index (BMI) between 18.5 and 24.9 kg/m2, in good general health (i.e. without any chronic pathology and not taking any medication at the time of inclusion and/or on a long-term basis), able and willing to participate in the research by complying with the protocol, affiliated to a social security system and having freely signed the informed consent.
This study was conducted in men, since a study in women could add heterogeneity to the results. Indeed, endogenous or exogenous sexual steroids may influence melatonin secretion in women (related to follicular or luteal phase, contraceptive pill, menopausal status, etc.). Also, evening-type male subjects (4-point Likert scale) were excluded from the study since they usually display a residual morning melatonin secretion.
The volunteers had to present none of the following exclusion criteria:
-
Smoking, drug consumption, drinking more than two glasses of alcohol per day
-
Taking any drug, melatonin, zinc or a product containing them was forbidden from the 48 h preceding the study visit for pharmacokinetic assessment
-
Known organic or functional abnormality of the urinary tree
-
Treatments or medical conditions that would imply a modification of the melatonin metabolism including drug intake (fluvoxamine, 5- or 8-metoxypsoralen, cimetidine, carbamazepine and rifampin, analgesics), known liver abnormality or detected during the screening visit, or known autoimmune disease
-
Any condition that could lead to a zinc deficiency or excess
-
Insomnia, working on atypical schedules (night or shift work)
-
Epilepsia, asthma, migraine according to the criteria of the International Headache Society [28]
-
Hypertension (> 140/90), thyroid dysfunction, hyperglycaemia or anaemia
-
Organic or psychological abnormality (including history of severe depression) that may bias the study results in the judgment of the investigator
During this screening visit, socio-demographic data, general medical history and cardiovascular parameters (blood pressure and heart rate) were recorded by a physician (Fig. 1). Blood samples (3 × 5 mL) were collected for standard assays (blood count and platelets, glucose, creatinine, urea, liver tests, TSH and C-reactive protein). If the results were within the normal range, the volunteers could be included in the study.
Study visit Food rich in melatonin or tryptophan (a melatonin precursor) should be restricted the day before and during the study visit for pharmacokinetic assessment. In addition, volunteers were instructed not to engage in prolonged, high-intensity physical exercise during the 24 h preceding visit. On the day of the visit, breakfast was to be eaten at home 2 h before the intake of the DR tablet (i.e. at 6:00 am). To standardize food intake, volunteers were asked to eat two pastries or three slices of bread with butter and jam and to exclude tea, coffee, mate or any energy drink.
At the study centre, compliance with the protocol was verified. Two initial blood samples were collected at T − 15 min (minus 15 min) and T0 to determine baseline (Fig. 1). The DR form was taken at 8:00 am (T0). Blood samples were collected every 20 min from T0 to T240, every 30 min from T240 to T480 and every 60 min from T480 to T720. Blood pressure and heart rate were monitored every hour. Any intercurrent event, deviation from the protocol, concomitant treatment, or adverse event was recorded in the case report form. A standardized snack (approximately 500 calories, identical for all volunteers) was served at 1:00 pm (T300). Tolerance of the DR form was evaluated. Possible adverse events were collected during the study.
2.4 Biological Sample Preparation and Analyses
A total of 130 mL of blood (26 × 5 mL) were used for melatonin and 6-SMT measurement. Each blood sample (5 mL) was centrifuged at 4 °C, then the plasma was decanted and separated into two aliquots which were stored at −20 °C before treatment. Each aliquot was used for melatonin and 6-SMT assay respectively. Melatonin and 6-SMT concentrations were determined by radioimmunoassay (RIA) using an anti-melatonin or anti-6-SMT antibody and the corresponding radioactive tracers (labelled with iodine-125) [29]. Plasma melatonin and 6-SMT assays had been previously published, in particular antibody specificity and physiological validation [30, 31]. These assays are submitted to a permanent internal quality control. Briefly, for the melatonin assay, the detection limit is routinely 4 pg/mL. The intra- and inter-assay coefficients of variation are 9.5% and 12.5%, respectively, at a concentration of 50 pg/mL. For the 6-SMT assay, the detection limit is 4 pg/mL and the intra- and inter-assay coefficients of variation are 8% and 12% respectively at a concentration of 100 pg/mL. Samples from a same subject were processed in a single assay run to overcome the inter-assay variability of the technique. Quality control samples were added in each series. Raw data processing (radioactivity counting) was performed using the Rialisme® software.
2.5 Study Endpoints
The primary endpoint was the evolution of plasma melatonin concentration over 720 min (12 h) after taking the DR form. The area under the curve (AUC), Cmax, Tmax and T1/2 values were calculated. Secondary endpoints were the evolution of plasma 6-SMT concentration (AUC, Cmax, Tmax and T1/2) and tolerance.
2.6 Statistical Analysis
The inclusion of 12 subjects was sufficient for statistical validation, in accordance with previous studies [32,33,34,35,36]. Two additional subjects, leading to total of 14 subjects, were included to balance the possible dropouts during the course of the study, an incomplete blood sampling or unexploitable data.
Data were recorded using the eNNOV® Clinical system and analysed with SAS® software version 9.4. Cmax, Tmax, T1/2 and AUC values were obtained. Since melatonin and its metabolite can be present in the blood before supplementation, the AUC attributable to supplementation is obtained by subtraction of the baseline (T − 15 min) value. Results are expressed in terms of mean ± standard deviation (SD) except in Fig. 2 (and in Supplementary Fig. 1 in Supplementary Information), where standard error of the means (SEM) are shown. The Wilcoxon signed-rank test for paired samples was used to compare mean melatonin and 6-SMT concentrations at each time point of the kinetics to basal concentrations. The threshold for statistical significance is defined as alpha risk of 0.05.
3 Results
3.1 Characteristics of Volunteers and Safety
Fourteen men were included in the study. Mean age was 30.9 ± 9.4 years, ranging from 18 to 45 years. The mean BMI was 23.6 ± 1.4 kg/m2 (median [range]: 24 [20.9–24.9]), and blood parameters were within normal ranges. Volunteers had a satisfactory quality of sleep (mean VAS: 7.3 ± 0.5).
Two volunteers did not finish the study due to difficulties in obtaining blood samples at T 270 or T 300 min. Therefore, they were excluded from the study and the pharmacokinetic parameters were obtained from 12 volunteers with no blood samples missing for assays and calculations. No significant adverse events occurred during the study.
3.2 Melatonin and 6-SMT Pharmacokinetics
The observed profile was consistent with that expected for this delayed-release form (Fig. 2). Plasma melatonin concentrations showed a quick and progressive increase, which was significant from 20 min compared with baseline (58 ± 62 versus 9.2 ± 8.3 pg/mL, p < 0.01). Cmax (740 ± 824 pg/mL) was reached about 3 h after the tablet intake (Tmax: 179 ± 60 min) (Fig. 2). The Cmax was approximately seven times higher than the value of the physiological nocturnal peak (100 pg/mL) up to 7 h after intake, which corresponds to a peak value in young subjects. Melatonin concentrations remained high between 140 and 220 min (between 494 and 602 pg/mL). Melatonin levels remained above 100 pg/mL up to 7 h after the DR form intake. Elimination half-life (T1/2: 110 ± 51 min) was of the same order of magnitude as the values obtained with low-dose (0.4 mg) and high-dose (4 mg) oral melatonin preparations including 25% immediate release + 75% controlled release (T1/2: 108 min and 126 min, respectively) [37]. Melatonin relative AUC0/720 was 2328 ± 2721 pg × h/mL.

Evolution of plasma melatonin (A) and 6-sulfatoxymelatonin (B) concentrations (mean ± SEM) over time after administration of the delayed-melatonin release form of melatonin (1.9 mg) in healthy volunteers (n = 12); § p < 0.01, # p < 0.001, when comparing concentrations at each time with the basal concentration (T0 min) (Wilcoxon signed-rank test).
For plasma 6-SMT, the Cmax was 7635 ± 1161 pg/mL, ten times the Cmax of melatonin. It was reached on average at Tmax 185 ± 32 min, very close to that of melatonin. Relative AUC0/720 was 27657 ± 4178 pg × h/mL. The ratio of relative AUC0/720 of plasma melatonin to relative AUC0/720 of plasma 6-SMT was 0.08 ± 0.65. Taken together, these results are in accordance with high first-pass hepatic metabolism.
4 Discussion
In this study, plasma melatonin and 6-SMT pharmacokinetic parameters were evaluated in healthy men aged 18–45 years who received a dose of 1.9 mg melatonin by taking a tablet of a newly developed delayed-release form of melatonin. The results observed for melatonin and 6-SMT were consistent with those anticipated for this galenic form. The administration of the tablet initially produced a slow increase in plasma melatonin levels which was significant as soon as 20 min. The peak was obtained 3 h after intake, followed by a prolonged decay over time. The DR form released melatonin at a concentration of physiological meaning (over 100 pg/mL or 0.4 nmol/L) up to 7 h after intake. This value is close to the upper value of potency (EC50) of the melatonin receptor [18].
We compared the pharmacokinetic parameters of melatonin obtained in the present study with the DR form and those previously published for a prolonged-release form [17]. Although the results should be confirmed by comparison of the two forms in the same trial, they are statistically sound. The two studies were performed in the same investigation centre and the experimental conditions were very similar between the two studies. Sample size was the same, 12 healthy subjects in each study with very close characteristics. Mean ages ± SD were 33.8 ± 8.3 years (20–45) and 30.9 ± 9.4 years (18–45), mean BMI was 24.0 ± 0.8 kg/m2 (median [range]: 24.3 [22.3–24.9]) and 23.6 ± 1.4 kg/m2, (median [range]: 24 [20.9–24.9]), and sleep quality indexes (mean VAS ± SD) were 7.8 ± 0.7 and 7.3 ± 0.5 for the prolonged-release and DR forms, respectively. The products were administered early in the morning at the same dose of melatonin (1.9 mg). Finally, determination of melatonin and 6-SMT concentrations was performed in the same laboratory using identical RIAs. Consequently, data of the two groups of volunteers were considered as dependent and mean values were compared by the Wilcoxon signed-rank test (Supplementary Table 1 and Supplementary Fig. 1 in Supplementary Information). When comparing plasma melatonin profiles of the two melatonin forms, the peak of the DR form was significantly lower (Cmax 740 ± 824 versus 1151 ± 565 pg/mL, p < 0.05) and delayed (Tmax 179 ± 60 versus 64 ± 44 min, p < 0.001) compared with the prolonged-release form. One can hypothesize that the administration of the DR form in the evening, around 10:00–11:00 pm, would lead to a blood peak closer to the endogenous nocturnal peak, which is located around 2:00–4:00 am. In comparison, after administration of the prolonged-release form the peak would occur early in the night, around 11:00 pm to 12:00 am. Consequently, compared with the prolonged-release form, the DR form may be particularly suitable for patients suffering from sleep maintenance problems and early awakening. With ageing, such sleep disorders may appear [38]. In addition, the early increase in plasma melatonin obtained with the DR form, although lower than that observed with the prolonged-release form, is sufficient to promote sleep onset.
Melatonin AUC for the DR form was significantly decreased, compared with the prolonged-release form (2295 ± 2649 versus 3070 ± 1452 pg × h/mL, p < 0.05, [AUC0–540 as prolonged-release form was evaluated over 540 min]) (Supplementary Table 1 in Supplementary Information). On the contrary, the melatonin/6-SMT AUC ratios and elimination periods T1/2 were not significantly different between the two forms, in agreement with a similar hepatic metabolism. Together, these results suggest that the intestinal melatonin absorption is decreased for the DR form, although melatonin concentrations remained of therapeutic interest (over 100 pg/mL). An explanation for such a result could be the influence on melatonin bioavailability of the other ingredients in the formulation, zinc in the form of Hypro-ri® zinc and lemon balm extract, in addition to baobab pulp and sodium alginate included to achieve delayed release. The influence of the lemon balm extract was observed in the in vitro tests we carried out during product development [19]. In addition, it was shown that the formation of a complex between melatonin and zinc hampers the melatonin passage across the intestinal barrier [39]. Such a complex between these compounds has been described in vitro [40]. This interaction could also occur in vivo, especially zinc is present in very high concentration in the pineal gland [41]. On the other hand, melatonin efficacy on sleep could be reinforced by combining it with such active ingredients. Beneficial effects of zinc on sleep in adults (reduced latency to sleep, improved sleep efficiency) have been reported [42, 43]. Many experimental data also suggest that zinc plays a modulatory activity on sleep possibly via interaction with glutamatergic transmission [44]. Further, a controlled trial with a fast-release melatonin preparation including zinc and magnesium showed efficacy on primary insomnia in long-term care facility residents [45]. As regards lemon balm, clinical studies have shown an improvement in sleep (including quality, efficiency and duration of sleep) in young adults and the elderly [46,47,48]. In addition, neuroprotective effects have been reported for zinc and lemon balm, notably through antioxidant effects. Antioxidant effects were shown for the lemon balm extract included in the DR form [27]. This type of neuroprotective action is also of interest in the target population with sleep maintenance alteration or early awakening due to advancing age. The effects of the combination on sleep will have to be explored in future studies.
A limitation of our study is that the results were obtained only in male volunteers. As with any initial proof-of-concept study, we included young male subjects receiving melatonin in the morning to limit the heterogeneity of the results (male versus female). Future studies should include volunteers or patients from the general population, women and also older people. Changes in melatonin pharmacokinetic parameters may occur with ageing due to age-related alterations in hepatic and renal clearances as well as changes in body composition (e.g. fat and water) and melatonin-based treatments mainly involve the elderly.
5 Conclusions
The melatonin release profile of the newly developed delayed-release form is in line with what was anticipated for this formulation, with a peak observed 3 h after intake and significant release up to 7 h after intake. This result suggests that the DR form would be suitable for the treatment of certain sleep disorders such as short sleep duration or early awakening. The addition of zinc and lemon balm, known for their neuroprotective effects, reinforces its potential interest for treatment of age-related sleep disorders.
References
Quera-Salva M-A, Claustrat B. Mélatonine: Aspects physiologiques et pharmacologiques en relation avec le sommeil, intérêt d’une forme galénique à libération prolongée (Circadin®) dans l’insomnie. Encéphale. 2018;44:548–57. https://doi.org/10.1016/j.encep.2018.06.005.
Wehr TA. The durations of human melatonin secretion and sleep respond to changes in daylength (photoperiod). J Clin Endocrinol Metab. 1991;73:1276–80. https://doi.org/10.1210/jcem-73-6-1276.
Wade AG, Crawford G, Ford I, McConnachie A, Nir T, Laudon M, Zisapel N. Prolonged release melatonin in the treatment of primary insomnia: evaluation of the age cut-off for short- and long-term response. Curr Med Res Opin. 2011;27:87–98. https://doi.org/10.1185/03007995.2010.537317.
Bartlett DJ, Biggs SN, Armstrong SM. Circadian rhythm disorders among adolescents: assessment and treatment options. Med J Aust. 2013;199:S16-20. https://doi.org/10.5694/mja13.10912.
Lähteenmäki R, Puustinen J, Vahlberg T, Lyles A, Neuvonen PJ, Partinen M, et al. Melatonin for sedative withdrawal in older patients with primary insomnia: a randomized double-blind placebo-controlled trial. Br J Clin Pharmacol. 2014;77:975–85. https://doi.org/10.1111/bcp.12294.
Wade AG, Farmer M, Harari G, Fund N, Laudon M, Nir T, et al. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: a 6-month, randomized, placebo-controlled, multicenter trial. Clin Interv Aging. 2014;9:947–61. https://doi.org/10.2147/CIA.S65625.
Amstrup AK, Sikjaer T, Mosekilde L, Rejnmark L. The effect of melatonin treatment on postural stability, muscle strength, and quality of life and sleep in postmenopausal women: a randomized controlled trial. Nutr J. 2015;14:102. https://doi.org/10.1186/s12937-015-0093-1.
Chang Y-S, Lin M-H, Lee J-H, Lee P-L, Dai Y-S, Chu K-H, et al. Melatonin supplementation for children with atopic dermatitis and sleep disturbance: a randomized clinical trial. JAMA Pediatr. 2016;170:35–42. https://doi.org/10.1001/jamapediatrics.2015.3092.
EFSA. Scientific Opinion on the substantiation of a health claim related to melatonin and reduction of sleep onset latency (ID 1698, 1780, 4080) pursuant to Article 13(1) of Regulation (EC) No 1924/2006. EFS2. 2011;9:2241. https://doi.org/10.2903/j.efsa.2011.2241.
Biggio G, Biggio F, Talani G, Mostallino MC, Aguglia A, Aguglia E, Palagini L. Melatonin: from neurobiology to treatment. Brain Sci. 2021. https://doi.org/10.3390/brainsci11091121.
Dumur J, Csajka C, Pavec O, Messaoudi S, Cretignier T, Gaspar F, Lang PO. Quelle alternative aux benzodiazépines, Z-pills et autres hypnotiques pour les personnes âgées ?: Mélatonine, valériane ou clométhiazole. Rev Med Suisse. 2018;14:2018–23. https://doi.org/10.53738/REVMED.2018.14.626.2018.
Mun JG, Wang D, Doerflein Fulk DL, Fakhary M, Gualco SJ, Grant RW, Mitmesser SH. A randomized, double-blind, crossover study to investigate the pharmacokinetics of extended-release melatonin compared to immediate-release melatonin in healthy adults. J Diet Suppl. 2023. https://doi.org/10.1080/19390211.2023.2206475.
Skene DJ, Deacon S, Arendt J. Use of melatonin in circadian rhythm disorders and following phase shifts. Acta Neurobiol Exp (Wars). 1996;56:359–62. https://doi.org/10.55782/ane-1996-1139.
Lemoine P, Zisapel N. Prolonged-release formulation of melatonin (Circadin) for the treatment of insomnia. Expert Opin Pharmacother. 2012;13:895–905. https://doi.org/10.1517/14656566.2012.667076.
Vecchierini MF, Kilic-Huck U, Quera-Salva MA. Melatonin (MEL) and its use in neurological diseases and insomnia: Recommendations of the French Medical and Research Sleep Society (SFRMS). Rev Neurol (Paris). 2021;177:245–59. https://doi.org/10.1016/j.neurol.2020.06.009.
Riemann D, Espie CA, Altena E, Arnardottir ES, Baglioni C, Bassetti CLA, et al. The European Insomnia Guideline: an update on the diagnosis and treatment of insomnia 2023. J Sleep Res. 2023;32: e14035. https://doi.org/10.1111/jsr.14035.
Ait Abdellah S, Raverot V, Gal C, Guinobert I, Bardot V, Blondeau C, Claustrat B. Bioavailability of melatonin after administration of an oral prolonged-release tablet and an immediate-release sublingual spray in healthy male volunteers. Drugs R D. 2023;23:257–65. https://doi.org/10.1007/s40268-023-00431-9.
Dubocovich ML, Delagrange P, Krause DN, Sugden D, Cardinali DP, Olcese J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, Classification, and Pharmacology of G Protein-Coupled Melatonin Receptors. Pharmacol Rev. 2010;62:343–80. https://doi.org/10.1124/pr.110.002832.
Dubourdeaux M, Bardot V. Composition for delivery to a human or animal subject, advantageously an oral; 24 May 2023.
European Pharmacopeia 11.0. 2.9.3 Dissolution tests for solid dosage forms.
Claustrat B, Brun J, Chazot G. The basic physiology and pathophysiology of melatonin. Sleep Med Rev. 2005;9:11–24. https://doi.org/10.1016/j.smrv.2004.08.001.
Harpsøe NG, Andersen LPH, Gögenur I, Rosenberg J. Clinical pharmacokinetics of melatonin: a systematic review. Eur J Clin Pharmacol. 2015;71:901–9. https://doi.org/10.1007/s00228-015-1873-4.
Román Martinez M, García Aguilar E, Martin Vílchez S, González García J, Luquero-Bueno S, Camargo-Mamani P, et al. Bioavailability of Oniria®, a melatonin prolonged-release formulation, versus immediate-release melatonin in healthy volunteers. Drugs R D. 2022;22:235–43. https://doi.org/10.1007/s40268-022-00394-3.
Pardridge WM, Mietus LJ. Transport of albumin-bound melatonin through the blood-brain barrier. J Neurochem. 1980;34:1761–3. https://doi.org/10.1111/j.1471-4159.1980.tb11272.x.
Leone AM, Francis PL, Silman RE. The isolation, purification, and characterisation of the principal urinary metabolites of melatonin. J Pineal Res. 1987;4:253–66. https://doi.org/10.1111/j.1600-079x.1987.tb00863.x.
Arendt J, Bojkowski C, Franey C, Wright J, Marks V. Immunoassay of 6-hydroxymelatonin sulfate in human plasma and urine: abolition of the urinary 24-hour rhythm with atenolol*. J Clin Endocrinol Metab. 1985;60:1166–73. https://doi.org/10.1210/jcem-60-6-1166.
Bardot V, Escalon A, Ripoche I, Denis S, Alric M, Chalancon S, et al. Benefits of the ipowder® extraction process applied to Melissa officinalis L: Improvement of antioxidant activity and in vitro gastro-intestinal release profile of rosmarinic acid. Food Funct. 2020;11:722–9. https://doi.org/10.1039/C9FO01144G.
Olesen J. The international classification of headache disorders. 2nd edition. Rev Neurol (Paris). 2005;161:689–91. https://doi.org/10.1016/s0035-3787(05)85119-7.
Brun J, Chamba G, Khalfallah Y, Girard P, Boissy I, Bastuji H, et al. Effect of modafinil on plasma melatonin, cortisol and growth hormone rhythms, rectal temperature and performance in healthy subjects during a 36 h sleep deprivation. J Sleep Res. 1998;7:105–14. https://doi.org/10.1046/j.1365-2869.1998.00100.x.
Brun J, Claustrat B, Harthé C, Vitte P, Cohen R, Chazot G. Melatonin RIA: Analytical and physiological criteria of validity. In: Brown G, Wainwright S, editors. The pineal gland, endocrine aspects. Advances in the biosciences. Pergamon Press; 1985. p. 41–5.
Harthé C, Claustrat B, Brun J, Chazot G. Direct radioimmunoassay of 6-sulfatoxymelatonin in plasma with use of an iodinated tracer. Clin Chem. 1991;37:536–9.
Mistraletti G, Sabbatini G, Taverna M, Figini MA, Umbrello M, Magni P, et al. Pharmacokinetics of orally administered melatonin in critically ill patients. J Pineal Res. 2010;48:142–7. https://doi.org/10.1111/j.1600-079X.2009.00737.x.
Härtter S, Nordmark A, Rose D-M, Bertilsson L, Tybring G, Laine K. Effects of caffeine intake on the pharmacokinetics of melatonin, a probe drug for CYP1A2 activity. Br J Clin Pharmacol. 2003;56:679–82. https://doi.org/10.1046/j.1365-2125.2003.01933.x.
Aldhous M, Franey C, Wright J, Arendt J. Plasma concentrations of melatonin in man following oral absorption of different preparations. Br J Clin Pharmacol. 1985;19:517–21. https://doi.org/10.1111/j.1365-2125.1985.tb02679.x.
Lopez-Gamboa M, Canales-Gomez JS, Sandoval TJC, Tovar EN, Meja MA, Palma-Aguirre J. Bioavailability of long acting capsules of melatonin in mexican healthy volunteers. J Bioequiv Biovail. 2010;2:116–9. https://doi.org/10.4172/jbb.
DeMuro RL, Nafziger AN, Blask DE, Menhinick AM, Bertino JS. The absolute bioavailability of oral melatonin. J Clin Pharmacol. 2000;40:781–4. https://doi.org/10.1177/00912700022009422.
Gooneratne NS, Edwards AYZ, Zhou C, Cuellar N, Grandner MA, Barrett JS. Melatonin pharmacokinetics following two different oral surge-sustained release doses in older adults. J Pineal Res. 2012;52:437–45. https://doi.org/10.1111/j.1600-079X.2011.00958.x.
Ohayon MM, Reynolds CF. Epidemiological and clinical relevance of insomnia diagnosis algorithms according to the DSM-IV and the International Classification of Sleep Disorders (ICSD). Sleep Med. 2009;10:952–60. https://doi.org/10.1016/j.sleep.2009.07.008.
Tran HTT, Tran PHL, Lee B-J. New findings on melatonin absorption and alterations by pharmaceutical excipients using the Ussing chamber technique with mounted rat gastrointestinal segments. Int J Pharm. 2009;378:9–16. https://doi.org/10.1016/j.ijpharm.2009.05.024.
Limson J, Nyokong T, Daya S. The interaction of melatonin and its precursors with aluminium, cadmium, copper, iron, lead, and zinc: an adsorptive voltammetric study. J Pineal Res. 1998;24:15–21. https://doi.org/10.1111/j.1600-079x.1998.tb00361.x.
Ebadi M, Iversen PL, Hao R, Cerutis DR, Rojas P, Happe HK, et al. Expression and regulation of brain metallothionein. Neurochem Int. 1995;27:1–22. https://doi.org/10.1016/0197-0186(94)00164-p.
Saito H, Cherasse Y, Suzuki R, Mitarai M, Ueda F, Urade Y. Zinc-rich oysters as well as zinc-yeast- and astaxanthin-enriched food improved sleep efficiency and sleep onset in a randomized controlled trial of healthy individuals. Mol Nutr Food Res. 2017. https://doi.org/10.1002/mnfr.201600882.
Gholipour Baradari A, Alipour A, Mahdavi A, Sharifi H, Nouraei SM, Emami ZA. The effect of zinc supplementation on sleep quality of ICU nurses: a double blinded randomized controlled trial. Workplace Health Saf. 2018;66:191–200. https://doi.org/10.1177/2165079917734880.
Cherasse Y, Urade Y. Dietary zinc acts as a sleep modulator. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18112334.
Rondanelli M, Opizzi A, Monteferrario F, Antoniello N, Manni R, Klersy C. The effect of melatonin, magnesium, and zinc on primary insomnia in long-term care facility residents in Italy: a double-blind, placebo-controlled clinical trial. J Am Geriatr Soc. 2011;59:82–90. https://doi.org/10.1111/j.1532-5415.2010.03232.x.
Cases J, Ibarra A, Feuillere N, Roller M, Sukkar SG. Pilot trial of Melissa officinalis L. leaf extract in the treatment of volunteers suffering from mild-to-moderate anxiety disorders and sleep disturbances. Med J Nutrition Metab. 2011;4:211–8.
Haybar H, Javid AZ, Haghighizadeh MH, Valizadeh E, Mohaghegh SM, Mohammadzadeh A. The effects of Melissa officinalis supplementation on depression, anxiety, stress, and sleep disorder in patients with chronic stable angina. Clin Nutr ESPEN. 2018;26:47–52. https://doi.org/10.1016/j.clnesp.2018.04.015.
Soltanpour A, Alijaniha F, Naseri M, Kazemnejad A, Heidari MR. Effects of Melissa officinalis on anxiety and sleep quality in patients undergoing coronary artery bypass surgery: a double-blind randomized placebo controlled trial. Eur J Integr Med. 2019;28:27–32. https://doi.org/10.1016/j.eujim.2019.01.010.
Acknowledgements
The authors thank CEN Nutriment for carrying out the statistical analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was funded by PiLeJe Laboratoire.
Conflict of Interest
S.A.A., C.G., I.G., V.B., A.V. and C.B. are employees of PiLeJe Laboratoire. B.C. is a consultant for PiLeJe. V.R. declared that she has no conflicts of interest.
Ethical Approval
The study received a favourable opinion from a Personal Protection Committee The protocol was approved by Personal Protection Committee (CPP Ile de France VIII, Hôpital Ambroise Paré, Boulogne-Billancourt, France) on 10 January 2023.
Availability of Data and Material
Data that support the findings of the study may be requested from the corresponding author.
Code Availability
Not applicable.
Consent to Participate
Informed consent was obtained from all individual participants included in the study.
Consent for Publication
Not applicable.
Authors’ Contributions
Conceptualization: S.A.A., I.G., A.V., B.C.; methodology: S.A.A., I.G., V.B., C.G., V.R., B.C.; formal analysis and investigation: S.A.A., I.G., V.B., C.G., V.R., C.B., B.C.; writing—original draft preparation: B.C., C.B.; writing—review and editing: C.B., S.A.A., I.G., V.B., C.G., A.V., V.R., B.C.; resources: C.G., S.A.A.; supervision: C.G., S.A.A. All authors read and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
40268_2024_482_MOESM1_ESM.tif
Supplementary Fig. 1 Comparison of plasma melatonin concentrations over time in healthy subjects taking a tablet of a delayed-release form (orange line) or prolonged-release form (blue line). The concentrations are expressed as mean ± SEM
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ait Abdellah, S., Gal, C., Guinobert, I. et al. Melatonin Bioavailability After Oral Administration of a New Delayed-Release Form in Healthy Male Volunteers. Drugs R D (2024). https://doi.org/10.1007/s40268-024-00482-6
Accepted:
Published:
DOI: https://doi.org/10.1007/s40268-024-00482-6