
Abstract
Background and objective
The history of clinical trials is fraught with unethical practices. Since 1945, robust frameworks have evolved to standardise the collection and reporting of safety data, most notably, the Common Terminology Criteria for Adverse Events (CTCAE) from the National Cancer Institute; used by investigators to report side effects experienced by participants. As medicine moves into the patient-centred model, interest has been growing to collect data on adverse events directly from participants (patient-reported adverse events). The aim of this systematic scoping review was to investigate the inclusion of patient-reported adverse event data within safety/tolerability analyses and explore the collection and reporting of patient-reported adverse event data.
Methods and results
A database search was undertaken and the Covidence platform was used to manage the review; results were analysed descriptively. Sixty-eight studies were included in the analysis. An increase in the number of studies that incorporate patient-reported adverse event data was seen by year. Seventy instruments were used for the collection of patient-reported adverse event data with recall period, mode, frequency and site of administration varying across studies; the duration of data collection ranged from 28 days to 6 years. Frequently, information on these details was omitted from publications. The number of instruments used by studies to collect patient-reported adverse event data ranged from one to seven instruments.
Conclusions
Despite growing calls for the inclusion of patient-reported adverse events, this has not yet translated into published reports. The collection and reporting of these data were variable and conducted using instruments that were not designed for purpose. To address these inconsistencies, standardisation of data collection and reporting using a purpose-built validated instrument is required.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The number of clinical trials collecting symptomatic data from the patient as a patient-reported outcome is increasing over time. |
The instruments and methods used to collect these data are varied and often utilise instruments that were not designed or validated for this purpose. |
Standardisation in the collection and reporting of these data would enable a better comparison of these data across clinical trials. |
1 Introduction
The history of clinical trials has been fraught with unethical practices. The atrocities of World War II are well documented [1] but from these tragedies came the first concrete steps towards patient consent and safety in clinical trials. The writing of the Nuremberg Code in 1947 was the first step to establishing an international code of ethics around human trials; the Code focussed primarily on the importance of voluntary consent of participants [2]. Another tragedy, the birth defects caused by thalidomide [3], resulted in the 1962 Kafauver–Harris amendments to the US Food and Drug Administration Act requiring evidence, obtained via clinical trials, of the efficacy and safety of a drug prior to approval [4]. The Nuremberg Code was expanded by the Helsinki Declaration in 1964, which further solidified the rights of participants and the responsibilities of researchers to conduct research ethically [5]. Although the Declaration of Helsinki is not in itself legally binding, it has been regularly revised and is used as the basis for laws governing medical research in countries around the world [6]. The requirement for institutional review boards (human research and ethics committees in the Australian context) followed closely in 1966 [7] and in 1996 the International Council for Harmonisation published the E6 Good Clinical Practice guidelines (ICH-GCP) [8]. It is these guidelines that govern the conduct of clinical trials to this day.
With the requirement to provide adequate safety data prior to drug approval, frameworks have developed to report adverse events in cancer clinical trials. In 1982, the Common Toxicity Criteria was published by the National Cancer Institute (NCI) to enable the comparison of adverse events across cancer clinical trials. The Common Toxicity Criteria have since undergone several revisions with the current version, now named Common Terminology Criteria for Adverse Events (CTCAE), standing at Version 5 containing 26 categories and 837 adverse event terms [9]. The adverse event terminology is harmonised with the Medical Dictionary for Medical Affairs and it contains three general categories of adverse events: laboratory-based events, observable or measurable events, and symptomatic events. Each adverse event is graded on a 5-point scale based on clinical criteria [10]. The term adverse event as it applies to clinical trials is defined within ICH-GCP as “any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have a causal relationship with this treatment. An adverse event (AE) can therefore be any unfavourable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product” [8] (p2).
Since the first version of the CTCAE was published, the importance of the patient’s voice has been gaining traction and patient-reported outcomes (PROs) have been increasingly included in clinical trials. In any discussion on PROs, a definition of terms is required owing to the often interchangeable usage of terms in PRO discussions. The Federal Drug Administration (FDA) defines PRO as “a measurement based on a report that comes directly from the patient … about the status of a patient’s health condition without interpretation of the patient’s response by a clinician or anyone else” [11] whereas a clinician-reported outcome is “a measurement based on a report that comes from a trained health-care professional after observation of a patient’s health condition.” [11] The traditional adverse event reporting process in clinical trials is one in which the patient reports symptoms to the clinician who then interprets and grades the symptom according to the CTCAE. The causality of the adverse event, whether disease related, treatment related or because of other causes, is assessed by the treating clinician. These clinician-reported adverse events (CRAEs) are subsequently published as part of the safety and toxicity analysis of the intervention under investigation [10, 12].
In contrast, PROs have traditionally taken the form of quality-of-life (QOL) data, defined by the FDA as “a general concept that implies an evaluation of the effect of all aspects of life on general well-being.” [13] Over time, QOL instruments have included more items on symptoms and the impact of symptoms on a patient’s QOL (health-related QOL [HRQOL]). Health-related QOL is then defined as “a multidomain concept that represents the patient’s general perception of the effect of illness and treatment on physical, psychological, and social aspects of life.” [13] The FDA has published statements that “clinician-reported outcomes cannot directly assess symptoms that are known only to the patient” [11] and these symptoms “can only be measured by patient-reported outcome measures” [11].
As a relatively new concept, patient-reported adverse events (PRAEs) are a form of PRO that are distinct from QOL and HRQOL; Banerjee et al. [14] define PRAEs as “any untoward medical occurrence, whether or not considered treatment/intervention related, that is reported or transmitted directly by the patient without interpretation by a clinician or anyone else.” Subsequently, a plethora of articles have been published examining the correlation of PRAEs and CRAEs [15,16,17,18,19,20,21]. These have shown that there is low correlation between both the number and types of symptoms reported as well as severity. Basch et al. [22] report that agreement is higher in more observable symptoms and lower for more subjective symptoms. It has, however, since been acknowledged that concordance between patient-reported and clinician-reported severity is not expected as they are differing measures [16, 23, 24]. Importantly, it should also be noted that the FDA does not consider PRAEs to be safety data, rather, they inform on the tolerability of a treatment and are complementary to safety data [25]. With increasing discussion around PRAE data and how such data should be collected, published and included in labelling [14, 23, 26,27,28,29,30], the question arises as to the extent to which these data are currently included and reported in clinical trials.
Scoping reviews are generally conducted to examine the extent, range and nature of evidence on a topic or question; to summarise findings from knowledge bases with heterogenous methods; or to identify gaps in knowledge [31]. As clinical trials can be conducted in a variety of formats, with adverse event data collection occurring in a multitude of ways, we felt a systematic scoping review would be best placed to explore this topic. Therefore, the aim of this systematic scoping review was to investigate the number of trials publishing PRAE data within safety and/or tolerability analyses of interventions during cancer clinical trials. We also explored the methods by which PRAE data were collected and how these data were reported.
2 Methods
We followed the Preferred Reporting Items for Systematic reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) for this study [31]. The PRISMA-ScR checklist is available from the Electronic Supplementary Material (ESM).
2.1 Study Protocol
A protocol for the review was developed prior to the selection of articles for inclusion and published on Prospero (CRD42022330736) on 25 July, 2022.
2.2 Search Strategy
A search of the PubMed, EMBASE, Web of Science and Cochrane Central Register of Controlled Trials (CENTRAL) databases was undertaken without a date limit through to 31 December, 2023. Language was limited to English, and the search string was: (“clinical trial”) AND ((“adverse event*”) OR (“adverse effect*”)) AND ((“patient-report*”) OR (“patient report*”)) AND (cancer).
2.3 Selection Strategy
Criteria for the inclusion and exclusion of studies are outlined in Table 1.
2.4 Screening Process
References were imported to the Covidence platform [32] for the screening and review process; duplicate articles were automatically removed by the Covidence platform upon import. Title and abstract screening were undertaken by a single investigator; a full-text review was completed by two investigators. Conflicting votes were discussed and agreed upon by both investigators. No reference search of included articles was undertaken because of the nature of the review.
During title and abstract screening, articles that clearly did not meet the above criteria were excluded. In addition, any abstracts that did not reference the collection of PRO data in any form or that specified that PRO data were collected specifically for a QOL analysis or for single-symptom reporting were also excluded. In the full-text review, the text was reviewed to confirm the collection of PRO data, if symptomatic PRO data were collected, and if those symptomatic data were collected for specific symptoms or a range of symptoms. The results section was then reviewed to determine how that symptomatic PRO data were analysed. If the data were analysed as a QOL outcome measure, the article was excluded. The distinction between tolerability and safety data as it relates to symptomatic PRO data as defined by the FDA was not always clear from published reports, as a result, articles that published PRO data as either a safety or tolerability outcome measure were included.
2.5 Data Extraction
A single reviewer extracted data from included articles using the Covidence extraction 2.0 template.
Extracted data included:
-
1.
Study characteristics (date of publication, location the study was conducted, phase of study, study design, intervention type, cancer type, patient population);
-
2.
Details of PRAE data collection (instrument name/description used to collect data, mode of administration (electronic/paper), site of administration (on-site or in patient’s own home), frequency of administration, recall period and duration of data collection;
-
3.
Details of PRAE reporting (format in which data were reported, terminology used in data reporting, analysis of PRAE data)
2.6 Analysis
The studies identified in this review were analysed descriptively. Data on the number of studies including PRAE data over time, the instruments used in the collection of PRAE data and a narrative overview of the findings are presented.
3 Results
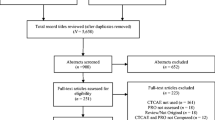
The search strategy identified 4190 articles and 2821 articles remained for screening after import to Covidence and removal of duplicates (n = 1369). During the title and abstract screening, 2128 articles were removed, and 619 articles were excluded during the full-text review leaving 74 articles for data extraction. During data extraction, multiple reports from the same clinical trial were identified. To prevent the same data being reported in this review multiple times, nine articles were merged into three studies using Covidence’s merge as a study function, leaving a total of 68 studies for analysis (Fig. 1). Details of included studies including cancer type, patient population, intervention, phase, study design and location where the study was conducted can be found in the ESM.

Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) flow diagram for the selection and inclusion of articles
An overall increase in the number of studies that incorporate PRAE data was seen by year (Fig. 2) with radiation oncology and medical oncology (systemic therapies) leading the field, with each discipline publishing 29 studies. Haematology published seven studies that incorporated PRAE data and medical oncology trials investigating localised therapies published three studies.

Number of studies published each year that incorporate patient-reported adverse events
Seventy different instruments were used for the collection of PRAE data in the studies included in this review (Table 2), with recall periods varying according to the instrument (today to 6 months) or not specified. The most commonly used instrument was the EORTC QLQ-C30 (n = 26), while 12 studies used a custom instrument or customised version of an existing instrument. Two studies did not specify the instrument used in the collection of data nor the recall period (ESM).
Twenty-seven of the 68 studies included in this review utilised a single instrument to collect PRAE data; 36 studies used between two and four instruments, while five studies used between five and seven instruments (ESM). Mode of administration was, for the most part, omitted from publications (n = 48), electronic instruments were utilised in ten studies and paper instruments were administered to patients in ten studies (ESM). For the majority of included publications, the site of administration was not reported (n = 46), while 13 studies administered instruments on-site and nine studies allowed patients to complete instruments at home (ESM).
Frequency of administration varied widely from daily data collection to weekly/monthly/3-monthly data collection (ESM). One study had only a single data collection point, while eight studies did not specify how frequently instruments were administered to patients (ESM). Duration of data collection varied from 28 days to 6 years; 14 studies did not specify the duration of data collection (ESM).
Twenty studies reported PRAE data in the results section in a separate section from CRAE and QOL/HRQOL data with a specific sub-heading. Twenty-two studies reported PRAE data alongside CRAE or QOL/HRQOL data, while 26 studies reported PRAE data without any CRAE data. The majority of these (n = 14) were radiation oncology studies. Patient-reported adverse event data were reported using terminology including “safety”, “toxicity” or “side effect/symptom/adverse event”. Three studies reported under the term “safety”, 20 studies reported using the term “toxicity” and 45 studies reported using one of the terms “side effect”, “symptom” or “adverse event”. The term “symptom” was used in reference to treatment-related symptoms. Results were primarily presented in text/descriptive format addressing changes in PRAEs over time; a comparison of PRAEs between the control and intervention; and the frequency of reported symptoms. Six studies presented PRAEs in a tabular format.
4 Discussion
This systematic scoping review aimed to investigate the use of PROs in cancer clinical trials adverse event reporting. As the value of PROs in research and clinical practice has increased, the current review has found the use of PROs in cancer clinical trials to report adverse events has also increased. A variety of instruments were used to collect PRAE data, with reporting of methodology inconsistent and in many cases lacking detail or absent altogether. Multiple instruments were frequently used in studies in order to capture the required data. The distinction between tolerability and safety data as it relates to PROs as defined by the FDA [25] was not always clear from the published reports identified in this review. Likewise, the causality of PRAEs, whether disease related, treatment related or other cause related was not clearly reported.
This review found that a variety of instruments were used to collect PRAE data; the majority being instruments designed to collect QOL and HRQOL data. The above definitions of QOL and HRQOL highlight that these instruments are not designed or validated to collect PRAE data. Basch et al. [33] in their White Paper on tolerability in cancer clinical trials states that “patient-reported data must be obtained from PRO instruments that are well-defined and reliable and that are fit-for-purpose”. Another paper by Basch et al. [30] details approaches that should be considered when selecting or developing an instrument for PRAE data collection. Namely, that a set of symptoms common to the disease or intervention should be selected during protocol development, and a free-text field considered for patients to provide unsolicited symptom reporting. Patient-reported adverse event data should be collected systematically at baseline, regularly during active treatment, with consideration given to collection during the post-treatment follow-up. Baseline assessment of symptoms is critical to accurate adverse event reporting. Symptoms present at baseline that worsen after starting treatment must be reported as adverse events. Likewise, a symptom that is present but not recorded at baseline may erroneously be reported as an adverse event [30]. The recall period should reflect the frequency at which the instrument is administered, to ensure continuous data collection without gaps in time during which symptoms may appear and resolve. The 7-day recall with an instrument administered weekly has been found to be a time frame that allows for robust data collection, without loss of information associated with longer recall periods [34]. Basch et al. [30] also detail a preference for electronic data collection, with automatic reminders to patients to self-report to enhance data completeness. In particular, web-based or phone-based methods are recommended to provide flexibility to the participants and allow for systematically timed reporting.
In contrast to the recommendations above, the instruments designed for QOL and HRQOL data collection utilised by studies identified in this review include items on only limited or specific symptoms. They also generally did not provide free-text fields for patients to provide information on additional symptoms outside of those captured within the instrument. To combat this limitation, many of the studies identified in this review included multiple surveys in order to collect a wider variety of symptom data; as many as seven instruments were used by one study [35]. Survey fatigue is a real and acknowledged issue in the collection of PRO data in clinical trials and can contribute to missed items and exacerbate incomplete data collection [36, 37]. To enable the most effective collection of QOL, HRQOL and PRAE data while limiting the burden on participants and clinical trial staff, the most appropriate instruments should be utilised. Quality-of-life, HRQOL and PRAE data each provide distinct and valuable insights into the patient’s treatment experience and the collection of each type of data is worthwhile.
This review also highlights that the included studies did not meet the above recommendations for regular data collection time frames with no gaps in recall periods. While 45 of the 68 studies collected symptomatic data at baseline, eight studies [38,39,40,41,42,43,44,45] neglected to report on the frequency of data collection entirely. Twenty-nine studies reported on the frequency of data collection in sufficient detail to correlate data collection in terms of treatment duration. In 17 of the studies, reporting on the frequency of data collection was ambiguous so we could not ascertain if reporting occurred during treatment or after treatment ceased. Recall periods matched the frequency of administration per recommendations in only seven of the studies [35, 46,47,48,49,50,51] included in this review; while seven studies [39, 52,53,54,55,56,57] did not include information on recall at all. In terms of mode of administration, it is difficult for this review to determine adherence to the recommendation for electronic methods. The majority of studies included neglected to report on the mode of administration of instruments; only ten studies [35, 46, 58,59,60,61,62,63,64,65] reported using electronic administration. In fact, only two studies [35, 46] included in this review met all the recommendations outlined by Basch et al. [30] for the collection and reporting of PRAE data.
It has been 14 years since the FDA released their guidance document recommending the inclusion of PRO measures in medical product development to support labelling claims [13]. Since then, there has been much discussion as to how PRO data should be collected, reported and subsequently utilised in the labelling of medicines [23, 26]. This review identified several articles that included PRAE data in a safety or tolerability analysis that predates the release of the guidance document [57, 66,67,68]. The earliest occurrence of an article reporting PRAE data included in the safety analysis of an intervention was in 1998 when Bailey et al. [66] investigated continuous hyperfractionated accelerated radiation therapy in non-small cell lung cancer. In fact, radiation oncology has led the way in incorporating the patient’s voice in safety and tolerability analyses, with the earliest articles identified in the review coming from that discipline. Since the release of the above guidance document, the FDA has clarified that they do not consider PRAE data to be safety data, rather they are tolerability data that are complementary to safety data [25]. The reporting of PRAE data in the articles identified in this review were not always clear in this distinction. Articles often reported these data as safety or toxicity outcome data. Clarification and standardisation of reporting of PRAE data will assist in cementing this distinction to prevent any misconceptions or misreporting of these data.
The increasing discussion and importance being placed on the inclusion of PRAEs is reflected in the steady increase in the number of articles incorporating PRAE data over time. Specific catalysts for this increase are not clear from this review but may include an increased uptake of PRAEs as an endpoint, an increase of trials incorporating PROs or improved reporting standards; this remains an area for future investigation. Despite the increase of publications incorporating PRAE data, this increase is notably slow. In 2020, the FDA launched Project Patient Voice [69], a platform for patients to access PRAE data collected during clinical trials directly. The platform remains in the pilot stage with data from a single clinical trial available for review; however, it provides an alternative to making PRAE data available outside of peer-reviewed journal articles. Publications in peer-reviewed journals face challenges in terms of limited publication space, and as a result, authors are restricted as to the information they are able to include in reports. Discussion in this arena, however, is now moving on to address standardisation and criteria for the publication of PRO and PRAE data [14, 29, 30, 70]. The variation in reporting methodology used to collect PRAE data, not to mention in some cases the absence of methodological information in the studies identified in this review, supports the efforts to standardise reporting. The Declaration of Helsinki [5] states that researchers “are accountable for the completeness and accuracy of their reports.” At a minimum, it can therefore be expected that researchers publish information on the mode, site, recall, frequency and duration of administration of any PRO instrument.
In the papers identified in this review, results of PRAE data were reported in either their own section under the results separate to CRAE and QOL/HRQOL data or in the same section as, and alongside, CRAE and/or QOL/HRQOL data. A small subset of papers, primarily from radiation oncology, reported PRAE data alone without inclusion of any CRAE data. Whether this was because of CRAE data not being collected, or reported in a publication separate to PRAE data was outside the scope of this review. Parameters included in the analysis of PRAE data were similar to those used in reporting and analysing CRAE data including changes in symptoms over time, the development of new symptoms post-treatment start, the frequency of reported symptoms and the dosing effect on PRAEs. Three papers [41, 71, 72] compared the prevalence of symptoms between CRAE and PRAE data while two papers [41, 73] included an analysis of a QOL association with PRAEs and CRAEs. Hall et al. [74] also compared PRAEs with QOL; however, they did not include CRAE data in their report. Reporting was primarily in a text/descriptive format, similar to the reporting of CRAE data, with only six studies reporting PRAE data in a tabular format [48, 49, 63, 75,76,77]. Terminology used to report PRAE data was variable across studies including “safety”, “toxicity”, “side effects”, “symptoms” and “adverse events”. Terms such as “safety” and “toxicity” are not advised, per the FDA advice that these data are not safety data but rather inform on the tolerability of interventions and can be misleading. The use of instruments designed for the collection of QOL/HRQOL data to report PRAE data certainly blurred the lines between these distinct outcome measures. The distinction in reports on the data collected and the intent in the reporting of that data, whether QOL/HRQOL data or PRAE data, was frequently unclear. In the same way that the formation of the CTCAE in 1982 allowed for equal comparison of CRAE data across clinical trial reports, standardising the reporting of PRAE data can allow the same. This would also provide clarity between reporting of QOL/HRQOL and PRAE data. Basch et al. [30] has made a start in this regard, outlining considerations for the reporting of PRAEs including how to display data, whether to report alongside CRAE data or separately, and methods for statistical analysis.
While 70 unique instruments were used by studies included in the current review, only one PRO instrument identified in this study was designed and validated specifically for the collection of PRAE data. The patient-reported outcomes version of the CTCAE (PRO-CTCAE) was developed by the NCI in 2014 [10]. This instrument was designed for direct patient reporting of the approximately 10% of items in the CTCAE that relate to symptomatic adverse events and was validated in the cancer patient population [10, 78]. The PRO-CTCAE is composed of plain language terms and symptoms are rated by patients in relation to attributes of amount, frequency, severity, interference and presence/absence [10]. This instrument incorporates the recommendations outlined above, allowing for customisable building of selected terms, a free-text field for unsolicited symptom reporting and a 7-day recall period. The item library currently consists of 78 symptom terms and careful consideration should be given by researchers using this instrument to item selection. The NCI provides advice on this on the Frequently Asked Questions page on their PRO-CTCAE website [79]. Namely, that existing data on the intervention should be reviewed to identify any known, likely or anticipated adverse events. Authors publishing PRAE data in the future could also consider publishing their rationale for item selection. From this current review, this information was not well reported. Although an electronic version of the PRO-CTCAE is not available, the NCI includes guidance on building electronic platforms to deliver PRO-CTCAE to patients [79]. The PRO-CTCAE is freely available from the NCI website and has been translated to 53 languages [80]. In 1982, the NCI developed the Common Toxicity Criteria to standardise adverse event reporting across cancer clinical trials, in the same vein, the same approach is required for PRAEs and the PRO-CTCAE provides that capability. A second instrument identified in this study is the PROMIS (Patient-Reported Outcomes Measurement Information System) platform available from HealthMeasures [81]; while not specifically developed for the collection of PRAE data, it provides an alternative to the PRO-CTCAE. PROMIS contains items for 13 symptom terms, is also freely available and is available for digital administration via various platforms. It does not, however, contain a free-text field for patients to provide additional symptoms they may have experienced during the reporting period.
A major limitation of the current review is that the lack of detail in reporting adverse event data collection methodology has likely resulted in this review missing articles that may otherwise have been included. In addition, the limited space provided by journals for abstracts may have resulted in articles being excluded at the title and abstract screening phase despite including PRAE data in the full text. Another limitation is that this review did not cross-check studies with registry information, to identify studies that collected PRAE data but did not report it. This fell outside the scope of this review; however, it is an area for future investigation that can be considered. It is certainly clear from this review that the use of instruments designed for collecting QOL/HRQOL data blurs the lines in the reporting of symptomatic data as PRAEs, which presents another limitation in this review. With a lack of clarity between the reporting of QOL/HRQOL and PRAE data, it is likely that some studies were excluded from this review. It is clear from this review, and the limitations identified, that current methods for the inclusion of PRAE data in tolerability analyses are not meeting current recommendations. Significant work is still required to ensure that PRAE data included in publications are complete and reliable. Feasibility studies [82,83,84,85,86] have shown that the collection of PRAE data during clinical trials is possible. Next steps require buy-in from sponsors, collaborative groups and investigators to include PRAEs in future protocol development.
5 Conclusions
Despite the growing calls for the inclusion of PRAEs in clinical trials, and the beginnings of efforts to standardise reporting of the same, PRAEs have not yet translated into published reports on clinical trials. In the same way clinician reporting of adverse events was standardised by the creation of CTCAE in 1982, standardisation of patient reporting of adverse events is comparatively needed. Utilising instruments designed to collect QOL and HRQOL information to collect PRAE data leaves doubt as to the appropriateness and accuracy in the use of these data in tolerability analyses of an intervention. The practice of using multiple surveys by researchers to overcome the limitations of these surveys to collect PRAE data is likely contributing to survey fatigue experienced by patients. Likewise, the variation in the mode, method and frequency of data collection results in unreliable and incomplete data. To combat the variation in the collection of PRAE data using sub-optimal instruments and to lower the burden of data collection to participants, a purpose-built validated instrument is required. Adoption of standardised data collection methods and reporting of the same offer the next iteration in patient safety in clinical trials.
References
Annas GJ, Grodin MA, editors. The Nazi doctors and the Nuremberg Code. Oxford: Oxford University Press; 1992.
Allied Control Council, No. 10. Permissable medical experiments. In: Trials of war criminals before the Nurenberg Military Tribunals under Control Council Law No. 10 “Green Series”; Volume II. Periodical. Nurenberg: U.S. Government Printing Office; 1949: p. 181–2. https://www.loc.gov/item/2011525364_NT_war-criminals_Vol-II/. Accessed 5 Feb 2022.
Knightley P, Evans H, editors. Suffer the children: the story of thalidomide. New York: The Viking Press; 1979.
Tripathi N, Davis K, Bodine C, Benjamin Jr DK. A brief history of clinical trials, drug regulations, and the Food and Drug Administration. In: Lopes R, Harrington R, editors. Understanding clinical research. New York: McGraw Hill; 2013. https://accessmedicine.mhmedical.com/content.aspx?bookid=674§ionid=45407239. Accessed 5 Feb 2022.
World Medical Association. Declaration of Helsinki: ethical principles for medical research involving human subjects. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/. Accessed 5 Feb 2022.
Nellhaus EM, Davies TH. Evolution of clinical trials throughout history. Marshall J Med. 2019;3(1):41–8. https://doaj.org/article/64eb01aafeb24846bf00f970cd84fc9c. Accessed 5 Feb 2022.
Gallin JI. A historical perspective on clinical research. In: Gallin JI, Ognibene FP, editors. Principles and practice of clinical research. 3rd ed. San Diego: Elsevier; 2012: p. 1–15. https://www.sciencedirect.com/science/article/pii/B9780123821676000011. Accessed 26 Jan 2022.
International Council for Harmonisation. Guideline for Good Clinical Practice E6(R2). 2016. https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf. Accessed 5 Feb 2022.
National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. 2017. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_50. Accessed 30 Jun 2023.
Basch E, Reeve BB, Mitchell SA, Clauser SB, Minasian LM, Dueck AC, et al. Development of the National Cancer Institute’s patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). J Natl Cancer Inst. 2014;106(9):dju244.
US Food and Drug Administration. Patient-focused drug development glossary. 2018. https://www.fda.gov/drugs/development-approval-process-drugs/patient-focused-drug-development-glossary. Accessed 5 Feb 2022.
Basch E, Artz D, Dulko D, Scher K, Sabbatini P, Hensley M, et al. Patient online self-reporting of toxicity symptoms during chemotherapy. J Clin Oncol. 2005;23(15):3552–61.
US Food and Drug Administration. Guidance for industry patient-reported outcome measures: use in medical product development to support labeling claims. 2009. https://www.web.archive.org/web/20160324162258, http://www.fda.gov/downloads/Drugs/Guidances/UCM193282pdf. Accessed 24 Mar 2023.
Banerjee AK, Okun S, Edwards IR, Wicks P, Smith MY, Mayall SJ, et al. Patient-reported outcome measures in safety event reporting: PROSPER consortium guidance. Drug Saf. 2013;36(12):1129–49.
Wilkie JR, Hochstedler KA, Schipper MJ, Matuszak MM, Paximadis P, Dominello MM, et al. Association between physician- and patient-reported symptoms in patients treated with definitive radiation therapy for locally advanced lung cancer in a statewide consortium. Int J Radiat Oncol Biol Phys. 2022;112(4):942–50.
Veitch ZW, Shepshelovich D, Gallagher C, Wang L, Abdul Razak AR, Spreafico A, et al. Underreporting of symptomatic adverse events in phase I clinical trials. J Natl Cancer Inst. 2021;113(8):980–8.
Tom A, Bennett AV, Rothenstein D, Law E, Goodman KA. Prevalence of patient-reported gastrointestinal symptoms and agreement with clinician toxicity assessments in radiation therapy for anal cancer. Qual Life Res. 2018;27(1):97–103.
Sparano F, Aaronson NK, Cottone F, Piciocchi A, La Sala E, Anota A, et al. Clinician-reported symptomatic adverse events in cancer trials: are they concordant with patient-reported outcomes? J Comp Eff Res. 2019;8(5):279–88.
Nyrop KA, Deal AM, Reeve BB, Basch E, Chen YT, Park JH, et al. Congruence of patient- and clinician-reported toxicity in women receiving chemotherapy for early breast cancer. Cancer. 2020;126(13):3084–93.
Liu L, Suo T, Shen Y, Geng C, Song Z, Liu F, et al. Clinicians versus patients subjective adverse events assessment: based on patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). Qual Life Res. 2020;29(11):3009–15.
Atkinson TM, Dueck AC, Satele DV, Thanarajasingam G, Lafky JM, Sloan JA, et al. Clinician vs patient reporting of baseline and postbaseline symptoms for adverse event assessment in cancer clinical trials. JAMA Oncol. 2020;6(3):437–9.
Basch E, Iasonos A, McDonough T, Barz A, Culkin A, Kris MG, et al. Patient versus clinician symptom reporting using the National Cancer Institute Common Terminology Criteria for Adverse Events: results of a questionnaire-based study. Lancet Oncol. 2006;7(11):903–9.
Kim J, Singh H, Ayalew K, Borror K, Campbell M, Johnson LL, et al. Use of PRO measures to inform tolerability in oncology trials: implications for clinical review, IND safety reporting, and clinical site inspections. Clin Cancer Res. 2018;24(8):1780–4.
Atkinson TM, Ryan SJ, Bennett AV, Stover AM, Saracino RM, Rogak LJ, et al. The association between clinician-based common terminology criteria for adverse events (CTCAE) and patient-reported outcomes (PRO): a systematic review. Support Care Cancer. 2016;24(8):3669–76.
US Food and Drug Administration. FDA perspective: patient self-reporting in the evaluation of cancer drug tolerability. 2021. https://www.fda.gov/media/148468/download. Accessed 25 Nov 2023.
Basch E. Beyond the FDA PRO guidance: steps toward integrating meaningful patient-reported outcomes into regulatory trials and US drug labels. Value Health. 2012;15(3):401–3.
Kluetz PG, O’Connor DJ, Soltys K. Incorporating the patient experience into regulatory decision making in the USA, Europe, and Canada. Lancet Oncol. 2018;19(5):e267–74.
Basch E. Patient-reported outcomes: an essential component of oncology drug development and regulatory review. Lancet Oncol. 2018;19(5):595–7.
Coens C, Pe M, Dueck AC, Sloan J, Basch E, Calvert M, et al. International standards for the analysis of quality-of-life and patient-reported outcome endpoints in cancer randomised controlled trials: recommendations of the SISAQOL Consortium. Lancet Oncol. 2020;21(2):e83-96.
Basch E, Rogak LJ, Dueck AC. Methods for implementing and reporting patient-reported outcome (PRO) measures of symptomatic adverse events in cancer clinical trials. Clin Ther. 2016;38(4):821–30.
Tricco AC, Lillie E, Zarin W, O’Brien KK, Colquhoun H, Levac D, et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169(7):467–73.
Veritas Health Innovation. Covidence systematic review software. Melbourne, Australia. https://www.covidence.org. Accessed 27 Mar 2024.
Basch EM, Campbell A, Hudgens S, Jones L, King-Kallimanis BL, Kluetz P, et al. Broadening the definition of tolerability in cancer clinical trials to better measure the patient experience. Friends of Cancer Research. 2018. https://friendsofcancerresearch.org/wp-content/uploads/Comparative-Tolerability-Whitepaper_FINAL.pdf. Accessed 30 Jun 2023.
Mendoza TR, Dueck AC, Bennett AV, Mitchell SA, Reeve BB, Atkinson TM, et al. Evaluation of different recall periods for the US National Cancer Institute’s PRO-CTCAE. Clin Trials. 2017;14(3):255–63.
Basch E, Dueck AC, Mitchell SA, Mamon H, Weiser M, Saltz L, et al. Patient-reported outcomes during and after treatment for locally advanced rectal cancer in the PROSPECT Trial (Alliance N1048). J Clin Oncol. 2023;41(21):3724–34.
Botero JP, Thanarajasingam G, Warsame R. Capturing and incorporating patient-reported outcomes into clinical trials: practical considerations for clinicians. Curr Oncol Rep. 2016;18(10):1–6.
Kluetz PG, Chingos DT, Basch EM, Mitchell SA. Patient-reported outcomes in cancer clinical trials: measuring symptomatic adverse events with the National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). Am Soc Clin Oncol Educ Book. 2016;36(36):67–73.
Reckamp KL, Giaccone G, Camidge DR, Gadgeel SM, Khuri FR, Engelman JA, et al. A phase 2 trial of dacomitinib (PF-00299804), an oral, irreversible pan-HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non-small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer. 2014;120(8):1145–54.
Mita MM, LoRusso PM, Papadopoulos KP, Gordon MS, Mita AC, Ferraldeschi R, et al. A phase I study of ASTX660, an antagonist of inhibitors of apoptosis proteins, in adults with advanced cancers or lymphoma. Clin Cancer Res. 2020;26(12):2819–26.
Laing R, Uribe J, Uribe-Lewis S, Money-Kyrle J, Perna C, Chintzoglou S, et al. Low-dose-rate brachytherapy for the treatment of localised prostate cancer in men with a high risk of disease relapse. BJU Int. 2018;122(4):610–7.
Rose TL, Harrison MR, Deal AM, Ramalingam S, Whang YE, Brower B, et al. Phase II study of gemcitabine and split-dose cisplatin plus pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive bladder cancer. J Clin Oncol. 2021;39(28):3140–8.
Laughlin BS, Thorpe CS, DeWees TA, Voss MM, Chang JH, Hartsell WF, et al. Initial quality of life and toxicity analysis of a randomized phase 3 study of moderately hypofractionated radiation therapy with or without androgen suppression for intermediate-risk adenocarcinoma of the prostate: PCG GU003. Adv Radiat Oncol. 2023;8(3): 101142.
Tan DSW, Felip E, de Castro G, Solomon BJ, Greystoke A, Cho BC, et al. Canakinumab versus placebo in combination with first-line pembrolizumab plus chemotherapy for advanced non-small-cell lung cancer: results from the CANOPY-1 trial. J Clin Oncol. 2024;42(2):192–204.
Gotlib J, Reiter A, Radia DH, Deininger MW, George TI, Panse J, et al. Efficacy and safety of avapritinib in advanced systemic mastocytosis: interim analysis of the phase 2 PATHFINDER trial. Nat Med. 2021;27(12):2192–9.
Meixner E, Wark A, Forster T, Weykamp F, Lang K, König L, et al. Health-related quality of life and patient-reported symptoms after postoperative proton beam radiotherapy of cervical and endometrial cancer: 2-year results of the prospective phase II APROVE-trial. Radiat Oncol. 2023;18(1):5.
Sebastian M, Rydén A, Walding A, Papadimitrakopoulou V. Patient-reported symptoms possibly related to treatment with osimertinib or chemotherapy for advanced non-small cell lung cancer. Lung Cancer. 2018;122:100–6.
Sio TT, Atherton PJ, Birckhead BJ, Schwartz DJ, Sloan JA, Seisler DK, et al. Repeated measures analyses of dermatitis symptom evolution in breast cancer patients receiving radiotherapy in a phase 3 randomized trial of mometasone furoate vs placebo (N06C4 [alliance]). Support Care Cancer. 2016;24(9):3847–55.
Jagsi R, Griffith KA, Boike TP, Walker E, Nurushev T, Grills IS, et al. Differences in the acute toxic effects of breast radiotherapy by fractionation schedule: Comparative analysis of physician-assessed and patient-reported outcomes in a large multicenter cohort. JAMA Oncol. 2015;1(7):918–30.
Chera BS, Amdur RJ, Green R, Shen C, Gupta G, Tan X, et al. Phase II trial of de-intensified chemoradiotherapy for human papillomavirus-associated oropharyngeal squamous cell carcinoma. J Clin Oncol. 2019;37(29):2661–9.
Mavroidis P, Price A, Fried D, Kostich M, Amdur R, Mendenhall W, et al. Dose–volume toxicity modeling for de-intensified chemo-radiation therapy for HPV-positive oropharynx cancer. Radiother Oncol. 2017;124(2):240–7.
Nelson B, Meier T, Zhang Y, Wang K, Mascia AE, Paquette I, et al. Feasibility trial of intensity modulated proton therapy to reduce toxicity in anal cancer patients. Am J Clin Oncol. 2023;46(7):293–9.
Hopwood P, Haviland JS, Sumo G, Mills J, Bliss JM, Yarnold JR. Comparison of patient-reported breast, arm, and shoulder symptoms and body image after radiotherapy for early breast cancer: 5-year follow-up in the randomised Standardisation of Breast Radiotherapy (START) trials. Lancet Oncol. 2010;11(3):231–40.
Tane K, Egawa C, Takao S, Yamagami K, Miyashita M, Baba M, et al. Body mass index and menopausal disorders during menopause affect vasomotor symptoms of postmenopausal Japanese breast cancer patients treated with anastrozole: a prospective multicenter cohort study of patient-reported outcomes. Breast Cancer. 2017;24(4):528–34.
Smulian AG, Moore DM, Robertson JC, Kralovic SM. Phase I study demonstrates safety and tolerability of radiofrequency ablation (RFA) of the anal mucosa. HIV Clin Trials. 2014;15(1):36–44.
Landrum LM, Nugent EK, Zuna RE, Syzek E, Mannel RS, Moore KN, et al. Phase II trial of vaginal cuff brachytherapy followed by chemotherapy in early stage endometrial cancer patients with high-intermediate risk factors. Gynecol Oncol. 2014;132(1):50–4.
Wortel RC, Heemsbergen WD, Smeenk RJ, Witte MG, Krol SDG, Pos FJ, et al. Local protocol variations for image guided radiation therapy in the multicenter Dutch hypofractionation (HYPRO) trial: impact of rectal balloon and MRI delineation on anorectal dose and gastrointestinal toxicity levels. Int J Radiat Oncol Biol Phys. 2017;99(5):1243–52.
Herstein A, Wallner K, Merrick G, Mitsuyama H, Armstrong J, True L, et al. I-125 versus Pd-103 for low-risk prostate cancer. Cancer J. 2005;11(5):385–9.
Mansfield AS, Każarnowicz A, Karaseva N, Sánchez A, De Boer R, Andric Z, et al. Safety and patient-reported outcomes of atezolizumab, carboplatin, and etoposide in extensive-stage small-cell lung cancer (IMpower133): a randomized phase I/III trial. Ann Oncol. 2020;31(2):310–7.
Atallah E, Schiffer CA, Radich JP, Weinfurt KP, Zhang MJ, Pinilla-Ibarz J, et al. Assessment of outcomes after stopping tyrosine kinase inhibitors among patients with chronic myeloid leukemia. JAMA Oncol. 2021;7(1):42–50.
Nielsen LK, Klausen TW, Jarden M, Frederiksen H, Vangsted AJ, Do T, et al. Clarithromycin added to bortezomib-cyclophosphamide-dexamethasone impairs health-related quality of life in multiple myeloma patients. Eur J Haematol. 2019;102(1):70–8.
Paz-Ares L, Barlesi F, Siena S, Ahn MJ, Drilon A, Conley A, et al. Patient-reported outcomes from STARTRK-2: a global phase II basket study of entrectinib for ROS1 fusion-positive non-small-cell lung cancer and NTRK fusion-positive solid tumours. ESMO Open. 2021;6(3): 100113.
Hinchcliff EM, Knisely A, Adjei N, Fellman B, Yuan Y, Patel A, et al. Randomized phase 2 trial of tremelimumab and durvalumab in combination versus sequentially in recurrent platinum-resistant ovarian cancer. Cancer. 2024;130(7):1061–71.
Madariaga A, Mitchell SA, Pittman T, Wang L, Bowering V, Kavak N, et al. Patient self-reporting of tolerability using PRO-CTCAE in a randomized double-blind, placebo-controlled phase II trial comparing gemcitabine in combination with adavosertib or placebo in patients with platinum resistant or refractory epithelial ovarian carcinoma. Gynecol Oncol. 2022;167(2):226–33.
Gelderblom H, Jones RL, Blay JY, George S, von Mehren M, Zalcberg JR, et al. Patient-reported outcomes and tolerability in patients receiving ripretinib versus sunitinib after treatment with imatinib in INTRIGUE, a phase 3, open-label study. Eur J Cancer. 2023;192: 113245.
Sidana S, Dueck AC, Thanarajasingam G, Griffin JM, Thompson C, Durani U, et al. Longitudinal patient reported outcomes with CAR-T cell therapy versus autologous and allogeneic stem cell transplant. Transplant Cell Ther. 2022;28(8):473–82.
Bailey AJ, Parmar MK, Stephens RJ. Patient-reported short-term and long-term physical and psychologic symptoms: results of the continuous hyperfractionated accelerated [correction of acclerated] radiotherapy (CHART) randomized trial in non-small-cell lung cancer. CHART Steering Committee. J Clin Oncol. 1998;16(9):3082–93.
List MA, Siston A, Haraf D, Schumm P, Kies M, Stenson K, et al. Quality of life and performance in advanced head and neck cancer patients on concomitant chemoradiotherapy: a prospective examination. J Clin Oncol. 1999;17(3):1020–8.
Buron C, Le Vu B, Cosset JM, Pommier P, Peiffert D, Delannes M, et al. Brachytherapy versus prostatectomy in localized prostate cancer: results of a French multicenter prospective medico-economic study. Int J Radiat Oncol Biol Phys. 2007;67(3):812–22.
US Food and Drug Administration. Project patient voice. 2023. https://www.fda.gov/about-fda/oncology-center-excellence/project-patient-voice. Accessed 25 Nov 2023.
Basch E, Leahy AB. Reporting standards for patient-reported outcomes in clinical trial protocols and publications. J Natl Cancer Inst. 2019;111(11):1116–7.
Thomson DJ, Ho KF, Ashcroft L, Denton K, Betts G, Mais KL, et al. Dose intensified hypofractionated intensity-modulated radiotherapy with synchronous cetuximab for intermediate stage head and neck squamous cell carcinoma. Acta Oncol (Madr). 2015;54(1):88–98.
Yeung AR, Deshmukh S, Klopp AH, Gil KM, Wenzel L, Westin SN, et al. Intensity-modulated radiation therapy reduces patient-reported chronic toxicity compared with conventional pelvic radiation therapy: updated results of a phase III trial. J Clin Oncol. 2022;40(27):3115–9.
Schuurhuizen CSEW, Marino P, Braamse AMJ, Buffart LM, Joly F, Fizazi K, et al. Impact of patient- and clinician-reported cumulative toxicity on quality of life in patients with metastatic castration-naïve prostate cancer. J Natl Compr Canc Netw. 2018;16(12):1481–8.
Hall E, Cameron D, Waters R, Barrett-Lee P, Ellis P, Russell S, et al. Comparison of patient reported quality of life and impact of treatment side effects experienced with a taxane-containing regimen and standard anthracycline based chemotherapy for early breast cancer: 6year results from the UK TACT trial (CRUK/01/001). Eur J Cancer. 2014;50(14):2375–89.
Boons CCLM, Timmers L, Janssen JJWM, Westerweel PE, Blijlevens NMA, Smit WM, et al. Response and Adherence to Nilotinib in Daily practice (RAND study): an in-depth observational study of chronic myeloid leukemia patients treated with nilotinib. Eur J Clin Pharmacol. 2020;76(9):1213–26.
Post CCB, de Boer SM, Powell ME, Mileshkin L, Katsaros D, Bessette P, et al. Long-term toxicity and health-related quality of life after adjuvant chemoradiation therapy or radiation therapy alone for high-risk endometrial cancer in the randomized PORTEC-3 trial. Int J Radiat Oncol Biol Phys. 2021;109(4):975–86.
Duriseti S, Kavanaugh JA, Szymanski J, Huang Y, Basarabescu F, Chaudhuri A, et al. LITE SABR M1: a phase I trial of lattice stereotactic body radiotherapy for large tumors. Radiother Oncol. 2022;167:317–22.
Dueck AC, Mendoza TR, Mitchell SA, Reeve BB, Castro KM, Rogak LJ, et al. Validity and reliability of the US National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). JAMA Oncol. 2015;1(8):1051–9.
National Cancer Institute. Frequently asked questions. 2023. https://healthcaredelivery.cancer.gov/pro-ctcae/faqs.html. Accessed 5 May 2023.
National Cancer Institute. PRO-CTCAE instrument & form builder. 2023. https://healthcaredelivery.cancer.gov/pro-ctcae/instrument-pro.html. Accessed 5 May 2023.
HealthMeasures. PROMIS. 2023. https://www.healthmeasures.net/explore-measurement-systems/promis. Accessed 25 Nov 2023.
Basch E, Dueck AC, Rogak LJ, Minasian LM, Kelly WK, O’Mara AM, et al. Feasibility assessment of patient reporting of symptomatic adverse events in multicenter cancer clinical trials. JAMA Oncol. 2017;3(8):1043–50.
Basch E, Dueck AC, Rogak LJ, Mitchell SA, Minasian LM, Denicoff AM, et al. Feasibility of implementing the patient-reported outcomes version of the Common Terminology Criteria for Adverse Events in a multicenter trial: NCCTG N1048. J Clin Oncol. 2018;36(31):3120–5.
Shepshelovich D, McDonald K, Spreafico A, Razak ARA, Bedard PL, Siu LL, et al. Feasibility assessment of using the complete Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) Item Library. Oncologist. 2019;24(4):e146–8.
Basch E, Pugh SL, Dueck AC, Mitchell SA, Berk L, Fogh S, et al. Feasibility of patient reporting of symptomatic adverse events via the Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) in a Chemoradiotherapy Cooperative Group multicenter clinical trial. Int J Radiat Oncol Biol Phys. 2017;98(2):409–18.
Pietanza MC, Basch EM, Lash A, Schwartz LH, Ginsberg MS, Zhao B, et al. Harnessing technology to improve clinical trials: study of real-time informatics to collect data, toxicities, image response assessments, and patient-reported outcomes in a phase II clinical trial. J Clin Oncol. 2013;31(16):2004–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Conflicts of Interest
Minna Grahvendy, Bena Brown and Laurelie R. Wishart have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
All data generated or analysed during this study are included in this published article and its supplementary information files.
Code Availability
Not applicable.
Author Contributions
MG conceptualised the study, completed the title and abstract screening, completed full-text review, analysed the data and wrote the manuscript. BB developed the concept of the study, completed full-text review and revised the manuscript. LW developed the concept of the study and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
40271_2024_689_MOESM1_ESM.pdf
Supplementary file1 Online Resource 1. Preferred Reporting Items for Systematic reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) Checklist (PDF 190 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Grahvendy, M., Brown, B. & Wishart, L.R. Adverse Event Reporting in Cancer Clinical Trials: Incorporating Patient-Reported Methods. A Systematic Scoping Review. Patient 17, 335–347 (2024). https://doi.org/10.1007/s40271-024-00689-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40271-024-00689-4