
Abstract
Purpose of Review
Mendelian randomization (MR) is a strategy for evaluating causality in observational epidemiological studies. MR exploits the fact that genotypes are not generally susceptible to reverse causation and confounding, due to their fixed nature and Mendel’s First and Second Laws of Inheritance. MR has the potential to provide information on causality in many situations where randomized controlled trials are not possible, but the results of MR studies must be interpreted carefully to avoid drawing erroneous conclusions.
Recent Findings
In this review, we outline the principles behind MR, as well as assumptions and limitations of the method. Extensions to the basic approach are discussed, including two-sample MR, bidirectional MR, two-step MR, multivariable MR, and factorial MR. We also consider some new applications and recent developments in the methodology, including its ability to inform drug development, automation of the method using tools such as MR-Base, and phenome-wide and hypothesis-free MR.
Summary
In conjunction with the growing availability of large-scale genomic databases, higher level of automation and increased robustness of the methods, MR promises to be a valuable strategy to examine causality in complex biological/omics networks, inform drug development and prioritize intervention targets for disease prevention in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
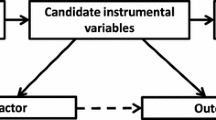
Causal inference in traditional observational epidemiological studies is hampered by the possibility of confounding and reserve causation [1]. Mendelian randomization (MR) is a method that can be used to uncover casual relationships between an exposure and outcome in the presence of such limitations. MR is a form of instrumental variable analysis, where genetic variants are used as proxies for the exposure of interest [2]. As Mendel’s Laws of Inheritance dictate, alleles segregate randomly from parents to offspring. Thus, offspring genotypes are unlikely to be associated with confounders in the population. In addition, germ-line genotypes are fixed at conception, and therefore, temporally precede the variables under observation, avoiding issues of reverse causation. The MR method involves finding genetic variants which are associated with an exposure, and then testing the association between these variants and the outcome. The causal “de-confounded” relationship between exposure and outcome can then be estimated when the necessary conditions are satisfied (Fig. 1a).


Design strategies for Mendelian randomization. a Standard MR: The causal relationship between an exposure variable (X) and an outcome (Y) is estimated using genetic variants (Z) as an instrument, regardless of the presence of variables (C) that may confound the observational association between the exposure and outcome. One method of estimation involves calculation of the Wald Ratio, [see Burgess review paper for description of the various instrumental variable (IV) estimators available] [3], where the causal estimate (\( {\widehat{\beta}}_{IV} \)) is derived by dividing the estimated regression coefficient of the outcome on the single nucleotide polymorphism (SNP) (\( {\widehat{\beta}}_{YZ} \)) by the estimated regression coefficient of the exposure on the SNP (\( {\widehat{\beta}}_{XZ} \)). b Two-sample MR. c Bidirectional MR. d Mediation and two-step MR. e Multivariable MR. f Factorial MR
In understanding how MR works, it can be useful to think of an MR study as being analogous to a randomized controlled trial (RCT), except that genotypes are used to randomize participants into different levels of the exposure/treatment. However, it is important to realize that this analogy is not perfect e.g., RCTs typically involve treatments over a short duration, whereas an individual’s genetics influences their biology from conception, meaning that many causal estimates from MR studies might reflect life-long exposures as well as developmental compensation that may arise from inheriting these mutations [4••].
Although initial applications of MR mostly focused on estimating the causal effect of environmental exposures on medically relevant outcomes, in recent years MR has found utility across a wide range of domains including the development of pharmaceutical agents (i.e., drug target validation, drug target repurposing, and side effect identification) and in the interpretation of high-dimensional omics studies. Table 1 lists several recent studies illustrating how MR has been used successfully across a wide variety of different contexts [5–37•, 106].
Core Assumptions Underlying Causal Inference in Mendelian Randomization Studies
In order for a genetic variant to qualify as a valid instrument for causal inference in a MR study, it must satisfy three core assumptions:
-
Assumption 1: The genetic variant must be truly associated with the exposure (NB the SNP need not be the functional variant responsible for the SNP-exposure association). Typically, SNPs which pass genome-wide significance (P < 5 × 10−8) and have been replicated in an independent sample are used as instruments in MR studies. The use of weak instruments can bias MR estimates towards the confounded observational estimate in one-sample MR settings and towards the null in two-sample MR settings (with non-overlapping samples). As common genetic variants frequently explain a small proportion of a trait’s variance, it may be useful to combine the effects of many SNPs together in an allelic score and use this as an instrument in MR studies.
-
Assumption 2: The genetic variant should not be associated with confounders of the exposure-outcome relationship. Although it is technically impossible to prove that this assumption holds in a MR study, it may be possible to disprove it by examining the association between the variant and known confounders of the exposure-outcome relationship.
-
Assumption 3: The genetic variant should only be related to the outcome of interest through the exposure under study. This is commonly referred to as the “no pleiotropy” assumption or the exclusion restriction criterion. Horizontal pleiotropy, where a SNP is associated with multiple traits independently of the exposure of interest, potentially violates this assumption. While it is not possible to prove that this assumption holds in an MR study, various extensions of the basic MR design can be used to detect its presence, and estimate the causal effect of the exposure even in the presence of such violation of the assumption (see below).
Even when these core assumptions have been met, MR has a number of limitations which need to be considered (summarized in Table 2), and which have been discussed at length elsewhere [2, 45,47,48,49,49, 50•, 51•, 52•].
Design of Mendelian Randomization Studies
The term MR covers a variety of approaches that use genetic variants to make inferences about the causal relationship between traits of interest [45, 52•]. Figure 1 illustrates some extensions to the basic MR design which are described in more detail in the paragraphs below.
Two-Sample Mendelian Randomization
Prior to 2011, most MR analyses were conducted using genetic instruments, exposure, and outcome of interest from individuals measured in the same sample (this is termed one-sample MR or single-sample MR). In such a scenario, the causal effect of the exposure on the outcome was typically estimated using 2-stage least-squares (2SLS) regression [53] (Fig. 1a). However, it is also possible to use MR to estimate causal effects where data on the exposure and outcome have been measured in different (or only partially overlapping) samples. This is known as two-sample MR [54•] (Fig. 1b). There are many advantages of using two-sample MR including in situations where it is difficult and/or expensive to measure the exposure and outcome in the same set of individuals (e.g., studies involving molecular gene expression data). Two-sample MR greatly increases the scope of MR analysis and continues to grow in popularity. For example, two-sample MR analyses can be performed on publicly available genome-wide association study (GWAS) summary data, a fact that has been taken advantage of by web software (and R packages) like MR-Base [55••]. Two-sample MR is understandably becoming increasingly popular in the research community. The percentage of all MR studies that used the two-sample design framework rose from close to 0% in 2011 to around 40% in 2016 [56].
Bidirectional Mendelian Randomization
In bidirectional MR, instruments for both exposure and outcome are used to evaluate whether the “exposure” variable causes the “outcome” or whether the “outcome” variable causes the “exposure” (Fig. 1c) [57•]. For example, in explaining the observational relationship between low levels of LDL cholesterol and risk of cancer, it may not be clear whether low levels of LDL cholesterol are causal for cancer, whether the presence of (undetected) cancer has a negative effect on LDL cholesterol, or whether the correlation between the two is due to latent confounding [58]. Bidirectional MR can help tease apart these relationships. MR analysis is first performed in one direction (i.e., “exposure” to “outcome”), and then performed in the opposite direction (i.e., “outcome” to “exposure”) using the SNPs robustly associated with each trait in the separate GWASs. The approach assumes that the causal association works through an underlying mechanism where it is possible to determine a single causal temporal direction. However, the complexity of biological systems, such as the existence of feedback loops between exposure and outcome variables, may make interpretation of the results of such analyses difficult [52•]. In these situations, it may be possible to use structural equation modeling to estimate feedback loops, although the properties of such approaches have yet to be examined thoroughly [51•].
Two-Step Mendelian Randomization
Two-step MR is used to assess whether an intermediate trait acts as a causal mediator between an exposure and an outcome [59•]. As shown in Fig. 1d, in the first step of the procedure, genetic instruments for the exposure are used to estimate the causal effect of the exposure variable on the potential mediator. In the second step of the procedure, genetic instruments for the potential mediator are used to assess the causal effect of the mediator on the outcome. Evidence of association in both steps implies some degree of mediation of the association between the exposure and the outcome by the intermediate variable. The magnitude of the direct effect (which is the effect of exposure on the outcome independent of the mediator) and indirect effect (which is the effect of the exposure on the outcome via the mediator) can be estimated separately by this method [60•]. However, this does require the assumptions of linearity and homogeneity for both the exposure-mediator and exposure-outcome relationships and no statistical interaction between exposure and mediator [60•]. Two-step MR and two-sample MR can be combined to facilitate the investigation of causal mediation in very large samples of individuals [50•].
Multivariable Mendelian Randomization
In some situations, genetic variants are pleiotropically associated with multiple correlated phenotypes. For example, genetic variants associated with lipoprotein metabolism rarely correlate with only one specific lipid fraction [61, 62•]. Single variable MR is likely to result in misleading conclusions regarding causality due to the presence of this horizontal pleiotropy. Multivariable MR is able to overcome this problem by using instruments associated with multiple exposures to jointly estimate the independent causal effect of each of the risk factors on the outcome (Fig. 1e) [63•, 64, 65]. For example, multivariable MR has recently been successfully employed in examining the relationship between high-density lipoprotein cholesterol and coronary heart disease. Univariate MR analyses, which ignore potential pleiotropic effects from other lipid fractions, suggest that increasing HDL levels lowers the risk of coronary heart disease. However, multivariable MR, which is able to account for SNPs’ pleiotropic effects through low-density lipoprotein and triglyceride levels [21•, 22], indicates that HDL is not causal for coronary heart disease, consistent with much of the evidence from randomized controlled trials [66,68,68].
Factorial Mendelian Randomization
The manner by which causes of disease act together to increase disease risk can have important public health implications, as above-additive effects act together to generate a greater burden of disease in the population [69]. Factorial MR can be used to determine the combined causal effects of the co-occurrence of two or more risk factors for disease [6•, 45] (Fig. 1f). In order to conduct factorial MR, individual level genotype data are required. For example, Ference et al. conducted a factorial MR study in order to investigate the effects of HMGCR and PCSK9 inhibition on CHD risk. In this study, a weighted genetic score for PCKS9 inhibition was constructed (with the weighting based on each SNP’s effect on LDL cholesterol levels) and participants were allocated into either a high or low inhibition group based on the median value of the PCSK9 score. The genetic score for HMGCR inhibition was constructed and the individuals were further allocated into groups based on the median value of the HMGCR score (Fig. 1f). The causal estimates for PCSK9 and HMGCR inhibition, and the combined effect of the two on CHD could then be determined. Results from this factorial analysis suggested that HMGCR and PCSK9 inhibition have independent effects on CHD, and act together in an additive manner to reduce CHD risk [16]. Another example of a factorial MR suggested that CETP inhibitors and statins were associated with decreased LDL-C and apoB levels and reduced risk of cardiovascular events. The reduction in CVD risk was proportional to the apoB reduction but less than expected for the LDL-C reduction [70].
Recent Developments
Resources for Performing Mendelian Randomization Analyses
MR, and in particular two-sample MR, provides a powerful, cost-efficient, and simple way to investigate potential causal relationships between many different human traits. Usefully, many GWAS consortia have made the results of their meta-analyses publicly available, greatly facilitating the running of such analyses [71,73,73]. For example, as a centralized GWAS data resource, Phenoscanner [74], can be used to search for genetic association across a large number of phenotypes. In addition, Ben Neale’s group have recently provided GWAS results of more than 2400 human traits based on up to 337,000 individuals from the latest UK Biobank release enabling two-sample MR analyses on a very large number of individuals (data can be downloaded from http://www.nealelab.is/blog/2017/7/19/rapid-gwas-of-thousands-of-phenotypes-for-337000-samples-in-the-uk-biobank). Several large-scale biobanks, such as the UK Biobank [75], the China Kadoorie biobank [76], and the HUNT study [77] allow researchers to apply for (a certain level of) genotype and phenotype information on large numbers of participants. These data sources can be used in one- or two-sample MR analyses when combined with other datasets. This idea led to the development of MR-Base [55••], which retrospectively collected, harmonized, and centralized complete GWAS summary datasets from the public domain. The curated summary data corresponds to 135 diseases, almost 2000 phenotypes in 1.5 million individuals and up to 4 billion SNP-trait associations, which is integrated with a software infrastructure (web interface, R package and API) for automating MR analyses. Therefore, MR-Base greatly increases the accessibility of GWAS summary results to other researchers, accelerates identification (discovery strand), prioritization (evidence synthesis strand), and evaluation (translational strand) of intervention targets.
Hypothesis-Free Investigations and “Mining the Phenome”
While there is obvious value in using MR to investigate the relationship between phenotypes for which causality has already been hypothesized, there is also an interest in detecting novel causal relationships. Hypothesis-free study designs such as genome-wide association studies (GWAS) and epigenome-wide association studies (EWAS) have shown tremendous success in recent years, and there are some instances where this strategy has shown promise in detecting putative causal relationships between phenotypes [38•, 51•, 78].
In a recent “one exposure to many outcomes” MR application, Haycock et al. systematically examined the association between telomere length and 22 cancers and 32 primary non-neoplastic diseases. The results suggested that longer telomeres were generally associated with increased risk for site-specific cancers but reduced risk for some non-neoplastic diseases, including cardiovascular diseases. This study highlighted the power of hypothesis-free MR in building a phenome-wide picture of traits of interest as opposed to the traditional “one exposure to one outcome” MR approach [28•].
Automation and data repositories provide solutions to some of the challenges involved in hypothesis-free MR. They trivialize the process of performing the analysis itself, and go some way towards improving reliability by (a) reducing human error [56] and (b) promoting the use of appropriate sensitivity analyses [3, 42••, 43•, 79, 80•, 81•]. However, many challenges still remain. Statistical power in MR is an issue even in the hypothesis-driven case, but hypothesis-free MR comes with a multiple testing burden that may be highly problematic. The nature of the data used in hypothesis-free MR is quite different from other hypothesis-driven study designs. There is often only a single consortium providing summary data for any one disease or trait which means that replication of a putative association in independent samples can be impossible. The emergence of large biobanks [75] may go some way to avoid this problem for many complex traits, but specific diseases for which cases need to be ascertained will still pose a challenge.
Another practical issue surrounds selecting those results from a hypothesis-free scan that are worthy of follow-up. Horizontal pleiotropy can manifest in many different patterns, which means that knowing the appropriate MR method to use for any particular pair of traits is difficult. Relying on a single method could lead to missed associations through of being overly conservative when there is no pleiotropy, or result in too many false positives because of miss-specifying the pleiotropic model. One method that has been developed recently to address this issue is MR-MoE (MR mixture of experts), which seeks to predict the most appropriate model based on the characteristics of the summary data [82•].
Another potential analytical strategy to mine the phenome would be to screen large publicly available disease and multi-omic GWAS summary results for evidence of genetic correlation using LD score regression via LD hub [83••, 84•]. Here, if traits are causally related and have non-zero heritability then there should be non-zero genetic correlations. However, genetic correlations can arise due to genetic confounding and horizontal pleiotropy and do not provide evidence on the direction of causality. Those disease-omic pairs showing evidence of genetic correlation could be followed up by conducting formal MR analyses [85]. One potential drawback of this approach is that the statistical efficiency of LD score regression may not be as high as that of MR in many cases, so selecting the appropriate scenarios in which to apply this as a screening method is important and warrants a priori power calculations.
The Role of MR in Disease Progression and Treatment
To date, the large majority of GWAS identify genetic variants (SNPs) associated with incidence or risk of disease. Such variants are informative for disease prevention, but not necessarily for treatment aimed at influencing disease progression [86, 87•]. For example, only ~ 8% of genetic association hits in the GWAS Catalog (p < 1 × 10−5) were reported by studies that have attempted to identify variants associated with disease progression or severity, and most of these GWAS have limited statistical power owing to small sample sizes (90% have N < 5000) [87•]. In a systematic search of the literature, Paternoster et al. were able to identify only 27 genetic studies that have used MR to identify risk factors influencing disease progression [87•], which leaves massive scope to extend MR methodologies and applications in this area. The introduction of collider bias when studying a selected (e.g., case only) group of individuals [87•] is a particular challenge when studying disease progression (more details of collider bias are given in Table 3 and 4) [44•, 104].
The Development of Approaches to Detect and Correct for Horizontal Pleiotropy in MR Analysis
The possibility of horizontal pleiotropy and the consequent violation of the exclusion restriction criterion are widely seen as the greatest threat to the validity of MR studies. Over the last few years, investigators have developed a suite of approaches that relax the strict requirement that genetic instruments exhibit no horizontal pleiotropy yet still produce causal effect estimates that are asymptotically consistent. These approaches often rely on different sets of assumptions to each other, meaning that if the results from all of these different analyses are largely consistent, then the investigator can be more confident in drawing conclusions regarding causality.
MR-Egger regression [42••] is one such approach where given a set of genetic variants that proxy an exposure variable of interest, estimates of the SNP-outcome association are regressed on estimates of the SNP-exposure association (this can be done in a one or two-sample MR framework), where each data point is weighted by the precision of the SNP-outcome coefficients. The slope of the weighted regression is an estimate of the causal effect of the exposure on the outcome. The intercept in this regression is free to vary, and the degree to which it departs from zero, is a function of the degree of directional pleiotropy present in the data.
The MR-Egger approach relaxes the requirement of no horizontal pleiotropy among the SNPs. Instead it assumes that there is no correlation between the gene-exposure association and the direct effect of the genetic variants on the outcome. This is referred to as the InSIDE assumption (Instrument Strength Independent of Direct Effect) and is a weaker requirement than the stricter exclusion restriction criterion. A drawback of the MR-Egger method is that it tends to suffer from low statistical power and is particularly susceptible to bias from weak instruments.
The weighted median estimator [94•] is a complementary method that permits up to 50% of the information in the MR analysis to come from SNPs that are invalid instruments. The mode-based estimate (MBE) further relaxes the assumption required for the weighted median approach and can estimate the causal effect when the most common pleiotropy value across instruments is zero [80•]. In addition, Bayesian modeling alternatives, such as Bayesian model averaging [81•], are under development, and may provide a framework to model pleiotropic effects and further relax MR assumptions, extending the scope of MR analysis.
In some circumstances, effect estimates are not consistent across independent instruments (e.g., with some genetic instruments showing unexpectedly large or small effects on the outcome, given the magnitude of their exposure effect), which could be indicative of horizontal pleiotropy. Formal statistical tests for heterogeneity can be used to assess this, such as Cochran’s Q statistic (for IVW) and the Rucker’s Q (for MR-Egger) [98, 103].
In addition to the above-mentioned methods, visual inspection can be helpful to identify pleiotropic variants (e.g., outlier detection). For example, funnel plots are used to display the MR estimate of individual genetic variants against their precision. Asymmetry in the funnel plot may arise due to some genetic variants having unusually strong effects on the outcome, which is indicative of directional pleiotropy [42••]. In addition, heterogeneous effects can be visualized by scatterplots of the gene-outcome and gene-exposure associations [94•] and forest plots of Wald ratios for each independent genetic instrument. In the leave-one-out plot, one SNP is removed at a time and the overall effect estimate is recalculated so that influential individual SNPs can be identified. As sensitivity analyses, all the above visualization methods are implemented in the MR-Base R package [55••]. Other graphical approaches have been proposed recently, such as the radial plot [99] and Q-contribution plots [103], which can further help to assess heterogeneity across genetic variants and detection of pleiotropic variants.
The Development of Approaches to Assess Instrument Strength
It is important to assess the instrument strength in order to avoid weak instrument bias in MR analysis. When weak instruments are estimated in GWAS with small sample sizes, MR approaches can violate the “NO Measurement Error” (NOME) assumption, which assume that the SNP-exposure associations (weights of the regression) are estimated without measurement error [43•]. For IVW, weak instruments that violate the NOME assumption can be reliably detected using the mean F-statistic [102]. For MR-Egger, the degree of violation of the NOME assumption can be quantified using the I2 statistic (IGX 2), a number ranging between 0 and 1, with higher values indicating less dilution of the causal effect estimate [43•].
Conclusion
MR is a flexible and robust statistical method which uses genetic variants as instrumental variables to detect and quantify causal relationships in observational epidemiological studies. In this review, we have endeavored to illustrate promising new findings and potential pitfalls of MR. The design strategies, assumptions, limitations, and potential of MR have been discussed. Given the growing availability of large-scale genetic resources and automated toolkits for implementing these methods, such as MR-Base and LD hub, we are now able to analyze all pairwise relationships within large multidimensional data sets in a hypothesis-free manner, producing evidence that can then be followed up in subsequent in-depth investigations.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Davey SG, Ebrahim S. Data dredging, bias, or confounding. BMJ. 2002;325(7378):1437–8.
Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res [Internet]. 2015 ; Available from: https://doi.org/10.1177/0962280215597579.
•• Holmes MV, Ala-Korpela M, Davey SG. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol. 2017;14(10):577–90. Reviews Mendelian randomization in the context of cardio-metabolic diseases. Describes the challenges in interpreting MR analyses such as assessing causality of multiple traits and evaluating pleiotropic effects.
• Würtz P, Wang Q, Soininen P, Kangas AJ, Fatemifar G, Tynkkynen T, et al. Metabolomic profiling of statin use and genetic inhibition of HMG-CoA reductase. J Am Coll Cardiol. 2016;67(10):1200–10. Highlights how genetic polymorphisms within the HMGCR gene can be used to mimic the effect of statins on a wide range of metabolic traits.
• Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol. 2015;65(15):1552–61. Applied factorial Mendelian randomization to estimate the combined effect of multiple exposures.
Myocardial Infarction Genetics Consortium Investigators, Stitziel NO, Won HH, Morrison AC, Peloso GM, Do R, Lange LA, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371(22):2072–82.
• Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72. First study to suggest that polymorphisms in the drug target gene, PCSK9, are strongly related to LDL cholesterol levels and risk of CHD.
Navarese EP, Kolodziejczak M, Schulze V, Gurbel PA, Tantry U, Lin Y, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med. 2015;163(1):40–51.
C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC), Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, et al. Association between C reactive protein and coronary heart disease: Mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548.
Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359(18):1897–908.
Polfus LM, Gibbs RA, Boerwinkle E. Coronary heart disease and genetic variants with low phospholipase A2 activity. N Engl J Med. 2015;372(3):295–6.
Millwood IY, Bennett DA, Walters RG, Clarke R, Waterworth D, Johnson T, et al. Lipoprotein-associated phospholipase A2 loss-of-function variant and risk of vascular diseases in 90,000 Chinese adults. J Am Coll Cardiol. 2016;67(2):230–1.
• Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JEL, Shah T, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet. 2012;379(9822):1214–24. Mendelian randomization study which suggested repurposing blockade of the interleukin-6 receptor (tocilizumab) as therapeutic approach to prevention of coronary heart disease.
• Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JEL, Shah T, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385(9965):351–61. Uses Mendelian randomization to explore whether the relation between statins and type 2 diabetes is a result of on-target or off-target effects.
Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med. 2016;375(22):2144–53.
Schmidt AF, Swerdlow DI, Holmes MV, Patel RS, Fairhurst-Hunter Z, Lyall DM, et al. PCSK9 genetic variants and risk of type 2 diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5(2):97–105.
Hägg S, Fall T, Ploner A, Mägi R, Fischer K, Draisma HHM, et al. Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int J Epidemiol. 2015;44(2):578–86.
Nordestgaard BG, Palmer TM, Benn M, Zacho J, Tybjaerg-Hansen A, Davey Smith G, et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 2012;9(5):e1001212.
White J, Swerdlow DI, Preiss D, Fairhurst-Hunter Z, Keating BJ, Asselbergs FW, et al. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol. 2016;1(6):692–9.
• Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45(11):1345–52. Suggests that lowering triglycerides will reduce risk of coronary heart disease independent from effect of HDL-C and LDL-C using a multivariable Mendelian randomization techinque.
Holmes MV, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36(9):539–50.
Tillmann T, Vaucher J, Okbay A, Pikhart H, Peasey A, Kubinova R, et al. Education and coronary heart disease: Mendelian randomisation study. BMJ. 2017;358:j3542.
Mokry LE, Ross S, Ahmad OS, Forgetta V, Davey SG, Goltzman D, et al. Vitamin D and risk of multiple sclerosis: a Mendelian randomization study. PLoS Med. 2015;12(8):e1001866.
Rhead B, Bäärnhielm M, Gianfrancesco M, Mok A, Shao X, Quach H, et al. Mendelian randomization shows a causal effect of low vitamin D on multiple sclerosis risk. Neurol Genet. 2016;2(5):e97.
Holmes MV, Dale CE, Zuccolo L, Silverwood RJ, Guo Y, Ye Z, et al. Association between alcohol and cardiovascular disease: Mendelian randomisation analysis based on individual participant data. BMJ. 2014;349:g4164.
Chen L, Davey SG, Harbord RM, Lewis SJ. Alcohol intake and blood pressure: a systematic review implementing a Mendelian randomization approach. PLoS Med. 2008;5(3):e52.
• Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, et al. Association between telomere length and risk of cancer and non-neoplastic diseases: a Mendelian randomization study. JAMA Oncol. 2017;3(5):636–51. Highlights the utility of Mendelian randomization at a phenome wide scale (one exposure to many outcomes), suggesting that longer telomeres increase risk for some cancers but reduce risk for some non-neoplastic diseases, including cardiovascular diseases.
Carreras-Torres R, Johansson M, Gaborieau V, Haycock PC, Wade KH, Relton CL, et al. The role of obesity, type 2 diabetes, and metabolic factors in pancreatic cancer: a Mendelian randomization study. J Natl Cancer Inst. 2017;109(9).
Bull CJ, Bonilla C, Holly JMP, Perks CM, Davies N, Haycock P, et al. Blood lipids and prostate cancer: a Mendelian randomization analysis. Cancer Med. 2016;5(6):1125–36.
Dimitrakopoulou VI, Tsilidis KK, Haycock PC, Dimou NL, Al-Dabhani K, Martin RM, et al. Circulating vitamin D concentration and risk of seven cancers: Mendelian randomisation study. BMJ. 2017;359:4761.
Censin JC, Nowak C, Cooper N, Bergsten P, Todd JA, Fall T. Childhood adiposity and risk of type 1 diabetes: a Mendelian randomization study. PLoS Med. 2017;14(8):e1002362.
Brion MJA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 2013;42:1497–01.
• Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167(5):1415–29.e19. Conducted GWAS on blood cell traits with Mendelian randomization as follow-up analysis. Detected causal relationship between blood cell indices and autoimmune diseases, schizophrenia, and coronary heart disease.
Richardson TG, Zheng J, Davey SG, Timpson NJ, Gaunt TR, Relton CL, et al. Causal epigenome-wide association study identifies CpG sites that influence cardiovascular disease risk. Am. J. Hum. Genet..2017;101(4):590–02.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–7.
• Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Consequences of natural perturbations in the human plasma proteome. BioRxiv. 2017; Available from: https://doi.org/10.1101/134551. Identifies causal roles for protein biomarkers in disease, which suggests a causal relationship for IL1RL1-IL18R1 loci on atopic dermatitis as well as MMP-12 on CHD.
• Evans DM, Brion MJA, Paternoster L, Kemp JP, McMahon G, Munafò M, et al. Mining the human phenome using allelic scores that index biological intermediates. PLoS Genet. 2013;9(10):e1003919. Uses a “genome-wide” allelic scores of biological phenotypes and subsequently use these scores to data mine GWAS.
Evans DM, Visscher PM, Wray NR. Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Hum Mol Genet. 2009;18(18):3525–31.
Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013;9(3):e1003348.
Richmond RC, Davey Smith G, Ness AR, den Hoed M, McMahon G, Timpson NJ. Assessing causality in the association between child adiposity and physical activity levels: a Mendelian randomization analysis. PLoS Med. 2014;11(3):e1001618.
•• Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25. Applied Egger regression (MR-Egger) to relax some violations of the standard Mendelian randomization assumptions, and provides an effect estimate which is not subject to directional pleiotropy.
• Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistics. Int J Epidemiol. 2016;45(6):1961–74. Proposes an adaptation of the I2 statistic from the field of meta-analysis to quantify the strength of NOME violation for MR-Egger.
• Munafò MR, Tilling K, Taylor AE, Evans DM, Davey Smith G. Collider scope: when selection bias can substantially influence observed associations. Int J Epidemiol [Internet]. 2017 Sep 27 [cited 2017 Oct 5]; Available from: https://academic.oup.com/ije/article/4259077. Discusses the impact of collider bias given the influence of selection and attrition.
Davey Smith G, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008;5(8):e177.
Bochud M, Rousson V. Usefulness of Mendelian randomization in observational epidemiology. Int J Env Res Public Health. 2010;7(3):711–28.
Davey Smith G. Random allocation in observational data: how small but robust effects could facilitate hypothesis-free causal inference. Epidemiology. 2011;22(4):460–3. discussion 467–8
VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in Mendelian randomization. Epidemiology. 2014;25(3):427–35.
• Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98. Describes new developments in Mendelian randomization including two-sample MR, bidirectional MR, network MR, two-step MR, factorial MR, and multiphenotype MR.
• Evans DM, Davey Smith G. Mendelian randomization: new applications in the coming age of hypothesis-free causality. Annu Rev Genomics Hum Genet. 2015;16:327–50. Details the potential of hypothesis-free Mendelian randomization and mining the phenome approaches.
• Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr. 2016;103(4):965–78. Discusses good practices and highlights the importance of the two-sample Mendelian randomization approaches in the post-GWAS era.
Angrist JD, Imbens GW. Two-stage least squares estimation of average causal effects in models with variable treatment intensity. J Am Stat Assoc. 1995;90(430):431–42.
• Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, EPIC-InterAct Consortium. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–52. Outlines the necessary steps for conducting Mendelian randomization investigations using published data.
•• Hemani G, Zheng J, Wade KH, Laurin C, Elsworth B, Burgess S, et al. MR-Base: a platform for systematic causal inference across the phenome using billions of genetic associations [Internet]. 2016 [cited 2017 Oct 5]. Available from: http://biorxiv.org/content/early/2016/12/16/078972. Describes the MR-Base and provides examples of applications in blood lipids.
Hartwig FP, Davies NM, Hemani G, Davey Smith G. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol. 2016;45(6):1717–26.
• Timpson NJ, Nordestgaard BG, Harbord RM, Zacho J, Frayling TM, Tybjærg-Hansen A, et al. C-reactive protein levels and body mass index: elucidating direction of causation through reciprocal Mendelian randomization. Int J Obes. 2011;35(2):300–8. Applies bidirectional Mendelian randomization approach to tease apart the direction of the relationship between C-reactive protein and body mass index.
Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet Lond Engl. 1986;1(8479):507–8.
• Relton CL, Davey Smith G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol. 2012;41(1):161–76. Reviews the development of two-step Mendelian randomization for mediation, and highlights its value in epigenetics research.
• Burgess S, Daniel RM, Butterworth AS, Thompson SG, EPIC-InterAct Consortium. Network Mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int J Epidemiol. 2015;44(2):484–95. Discusses the application of Mendelian randomization to investigate more complex networks between multiple interrelated exposures and disease outcomes.
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–83.
• Würtz P, Kangas AJ, Soininen P, Lehtimäki T, Kähönen M, Viikari JS, et al. Lipoprotein subclass profiling reveals pleiotropy in the genetic variants of lipid risk factors for coronary heart disease: a note on Mendelian randomization studies. J Am Coll Cardiol. 2013;62(20):1906–8. Illustrates the correlation between lipoprotein subclasses and shows the importance of considering pleiotropy to examine causality of lipid related traits.
• Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251–60. Discusses the multivariable Mendelian randomization approach, which allows causality of highly correlated phenotypes to be examined.
Burgess S, Dudbridge F, Thompson SG. Re: “multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects.”. Am J Epidemiol. 2015;181(4):290–1.
Kemp JP, Sayers A, Davey SG, Tobias JH, Evans DM. Using Mendelian randomization to investigate a possible causal relationship between adiposity and increased bone mineral density at different skeletal sites in children. Int J Epidemiol. 2016;45(5):1560–72.
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99.
Lilly E. Others. Lilly to discontinue development of evacetrapib for high-risk atherosclerotic cardiovascular disease. 2015.
Montgomery AA, Peters TJ, Little P. Design, analysis and presentation of factorial randomised controlled trials. BMC Med Res Methodol. 2003;3:26.
Ference BA, Kastelein JJP, Ginsberg HN, Chapman MJ, Nicholls SJ, Ray KK, et al. Association of genetic variants related to CETP inhibitors and statins with lipoprotein levels and cardiovascular risk. JAMA. 2017;318(10):947–56.
Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47(12):1449–56.
Horikoshi M, Beaumont RN, Day FR, Warrington NM, Kooijman MN, Fernandez-Tajes J, et al. Genome-wide associations for birth weight and correlations with adult disease. Nature. 2016;538(7624):248–52.
Kemp JP, Morris JA, Medina-Gomez C, Forgetta V, Warrington NM, Youlten SE, et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat Genet. 2017;49(10):1468–75.
Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016;32(20):3207–9.
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779.
Chen Z, Chen J, Collins R, Guo Y, Peto R, Wu F, et al. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int J Epidemiol. 2011;40(6):1652–66.
Krokstad S, Langhammer A, Hveem K, Holmen TL, Midthjell K, Stene TR, et al. Cohort profile: the HUNT study, Norway. Int J Epidemiol. 2013;42(4):968–77.
Blair DR, Lyttle CS, Mortensen JM, Bearden CF, Jensen AB, Khiabanian H, et al. A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell. 2013;155(1):70–80.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65.
• Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero-modal pleiotropy assumption. Int J Epidemiol [Internet]. 2017 Jul 12 [cited 2017 Oct 5]; Available from: https://academic.oup.com/ije/article-abstract/3957932/Robust-inference-in-summary-data-Mendelian. Describes the mode based estimate method, which can be used to infer the causal effect even if the majority of instruments are invalid.
• Thompson JR, Minelli C, Bowden J, Del Greco FM, Gill D, Jones EM, et al. Mendelian randomization incorporating uncertainty about pleiotropy. Stat Med. 2017; https://doi.org/10.1002/sim.7442. Discusses the advantages of using Bayesian model averaging when there is uncertainty about the nature of the pleiotropy.
• Hemani G, Bowden J, Haycock PC, Zheng J, Davis O, Flach P, et al. Automating Mendelian randomization through machine learning to construct a putative causal map of the human phenome [Internet]. 2017 [cited 2017 Oct 5]. Available from: https://www.biorxiv.org/content/early/2017/08/23/173682.abstract. Describes a machine learning approach, the MR-MoE (MR mixture of experts), which seeks to select the optimal Mendelian randomization model for different phenotypic characteristics.
•• Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–41. Introduces the use of LD Score regression for estimating genetic correlation between traits using GWAS summary data
• Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017;33(2):272–9. Describes LD Hub database, which collated and harmonized full GWAS results of more than 200 traits and explains the web interface for automated SNP heritability and genetic correlation analyses using LD score regression approach.
Zheng J, Haycock P, Hemani G, Elsworth B, Shihab H, Laurin C, et al. LD hub and MR-base: online platforms for preforming LD score regression and Mendelian randomization analysis using GWAS summary data. Behav Genet. 2016;46(6):815.
Davey Smith G, Paternoster L, Relton C. When will Mendelian randomization become relevant for clinical practice and public health? JAMA. 2017;317(6):589–91.
• Paternoster L, Tilling KM, Davey SG. Genetic epidemiology and Mendelian randomization for informing disease therapeutics: conceptual and methodological challenges PLoS Genet.. 2017;13(10):e1006944. Discusses the potential and pitfalls of Mendelian randomization studies of disease progression to identify potential new treatments.
• Verbanck M, Chen C-Y, Neale B, Do R. Widespread pleiotropy confounds causal relationships between complex traits and diseases inferred from Mendelian randomization [Internet]. 2017 [cited 2017 Oct 5]. Available from: https://www.biorxiv.org/content/early/2017/06/30/157552.abstract. Mendelian randomization approach that allows for the evaluation of pleiotropy in multi-instrument Mendelian randomization.
Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017.
Spiller W, Slichter D, Bowden J, Davey SG. Detecting and correcting for bias in Mendelian randomization analyses using gene-by-environment interactions [Internet]. 2017 [cited 2017 Oct 5]. Available from: https://www.biorxiv.org/content/early/2017/09/15/187849.abstract
• Millard L, Davies NM, Gaunt T, Davey SG, Tilling K. PHESANT: a tool for performing automated phenome scans in UK Biobank International Journal of Epidemiology. 2017 dyx204. Novel method for performing phenome scans in UK Biobank, which can be applied for MR phenome-wide association studies (MR-pheWAS).
Millard LA, Davies NM, Timpson NJ, Tilling K, Flach PA, Smith G. MR-PheWAS: hypothesis prioritization among potential causal effects of body mass index on many outcomes, using Mendelian randomization. Sci Rep. 2015;5:16645.
Zheng J, Richardson T, Millard L, Hemani G, Raistrick C, Vilhjalmsson B, Haycock P, Gaunt T. PhenoSpD: an integrated toolkit for phenotypic correlation estimation and multiple testing correction using GWAS summary statistics. BioRxiv. 2017; Available from: https://doi.org/10.1101/148627.
• Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14. Describes the weighted median approach, which permits up to 50% of the information in the MR analysis to come from SNPs that are invalid instruments.
van Kippersluis H, Rietveld CA. Pleiotropy-robust Mendelian randomization. Int J Epidemiol. 2017; https://doi.org/10.1093/ije/dyx002.
Cho Y, Shin S-Y, Won S, Relton CL, Davey Smith G, Shin M-J. Alcohol intake and cardiovascular risk factors: a Mendelian randomisation study. Sci Rep. 2015;5:18422.
Slichter D. Testing instrument validity and identification with invalid instruments. SOLE. 2014. http://www.sole-jole.org/14436.pdf
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. 2017;36(11):1783–802.
Bowden J, Spiller, W, Del Greco, F, Sheehan, N, Thompson, J, Minelli, C, et al. Improving the visualisation, interpretation and analysis of two-sample summary data Mendelian randomization via the radial plot and radial regression. BioRxiv. 2017. https://doi.org/10.1101/200378
Greco MFD, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34(21):2926–40.
Corbin LJ, Richmond RC, Wade KH, Burgess S, Bowden J, Davey SG, et al. BMI as a modifiable risk factor for type 2 diabetes: refining and understanding causal estimates using Mendelian randomization. Diabetes. 2016;65(10):3002–7.
Burgess S, Thompson SG. Bias in causal estimates from Mendelian randomization studies with weak instruments. Stat Med. 2011;30(11):1312–23.
Bowden J, Del Greco M F, Minelli C, Lawlor D, Sheehan N, Thompson J, et al. Improving the accuracy of two-sample summary data Mendelian randomization: moving beyond the NOME assumption [Internet]. 2017 [cited 2017 Oct 5]. Available from: https://www.biorxiv.org/content/early/2017/07/05/159442.abstract
Taylor AE, Munafò MR, CARTA consortium. Commentary: does mortality from smoking have implications for future Mendelian randomization studies? Int J Epidemiol. 2014;43(5):1483–6.
Spiller W, Davies NM, Palmer TM. Software Application Profile: mrrobust - A Tool For Performing Two-Sample Summary Mendelian Randomization Analyses. BioRxiv. 2017. https://www.biorxiv.org/content/early/2017/07/05/142125
Vaucher J, Keating BJ, Lasserre AM, Gan W, Lyall DM, Ward J, et al. Cannabis use and risk of schizophrenia: a Mendelian randomization study. Mol Psychiatry. 2017;
Funding
This work was supported by the MRC Integrative Epidemiology Unit with grants MC_UU_12013/1 and MC_UU_12013/4. Dr. Borges is supported by a Skills Development Fellowship from the UK Medical Research Council [grant number MR/P014054/1]. Dr. Bowden is supported by a Methodology Research Fellowship from the UK Medical Research Council (Grant Number MR/N501906/1). Dr. Haycock is supported by CRUK Population Research Postdoctoral Fellowship C52724/A20138. Prof. Evans is supported by the Australian Research Council Future Fellowship (Grant Number FT130101709). Dr. Baird was funded by the Arthritis Research UK project grant (ref 20,244).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jie Zheng, Denis Baird, Maria-Carolina Borges, Jack Bowden, Gibran Hemani, Philip Haycock, David M. Evans and George Davey Smith each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article contains no studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Genetic Epidemiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zheng, J., Baird, D., Borges, MC. et al. Recent Developments in Mendelian Randomization Studies. Curr Epidemiol Rep 4, 330–345 (2017). https://doi.org/10.1007/s40471-017-0128-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40471-017-0128-6