
Abstract
Introduction
Tofacitinib is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis (RA). We evaluated the effect of concomitant methotrexate (MTX) or glucocorticoid (GC) use on tofacitinib clinical efficacy.
Methods
Data were pooled from two open-label, long-term extension studies of tofacitinib 5 or 10 mg twice daily in patients with RA. Response according to Clinical Disease Activity Index (CDAI) was assessed separately in patients who discontinued (no MTX/GC use within 30 days prior to year-3 visit; assessment at month 3/year 3) or initiated (on/before year 3; assessment at initiation and year 3) MTX/GC.
Results
By year 3, among patients receiving background MTX at baseline, 186/1608 (11.6%) discontinued MTX, and 319/1434 (22.2%) patients receiving GC at baseline discontinued GC. Overall, 70.4/69.1% of patients who discontinued/continued MTX and 72.7/65.9% who discontinued/continued GC achieved CDAI remission or low disease activity (LDA) at year 3. Month 3 remission/LDA rates were maintained at year 3 in the majority of patients, irrespective of MTX/GC discontinuation/continuation. By year 3, 6.2% of patients receiving tofacitinib without MTX at baseline had initiated concomitant MTX, and 25.1% receiving tofacitinib without GC initiated GC; 69.0% and 45.4% initiating MTX or GC, respectively, had a CDAI-defined incomplete response prior to initiation. RA signs/symptoms improved following MTX initiation; only modest improvement was observed with GC initiation.
Conclusions
Patients achieving remission/LDA with tofacitinib may discontinue MTX or GC and maintain treatment response. Patients with an incomplete response may benefit from adding concomitant MTX.
Funding
Pfizer Inc.
Trial registration
Study A3921024 [NCT00413699] and Study A3921041 [NCT00661661].
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Treatment options for rheumatoid arthritis (RA) include multiple classes of agents, and patients often receive background or concomitant treatment [1, 2] such as methotrexate (MTX) or glucocorticoid (GC), to improve or expedite clinical response. However, many patients experience adverse events with MTX or GC [3,4,5], and GCs should be used at the lowest possible dose for a minimum duration to minimize unwanted reactions [1, 2]. Therefore, it is important to understand the potential impact of discontinuing concomitant MTX or GC on the clinical efficacy of RA treatments.
Tofacitinib is an oral Janus kinase inhibitor approved for the treatment of RA. The efficacy and safety of tofacitinib 5 and 10 mg twice daily (BID) administered as monotherapy or in combination with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), mainly MTX, in patients with active RA have been demonstrated in phase II [6,7,8,9,10], phase III [11,12,13,14,15,16], and phase IIIb/IV [17] studies of up to 24 months’ duration and in long-term extension (LTE) studies with up to 114 months of observation [18,19,20].
In this study, post hoc analyses evaluated: (1) the possible effect of discontinuing MTX or GC on clinical responses in patients with RA receiving tofacitinib in two LTE studies, and (2) the potential effect of initiating MTX or GC in patients originally receiving tofacitinib without MTX or without GC, respectively, at LTE baseline.
Methods
Study Design
These were post hoc, pooled analyses from two multicenter, open-label LTE studies (ORAL Sequel [A3921024; NCT00413699] and Study A3921041 [NCT00661661]). ORAL Sequel was a global study that was ongoing at the time of the analysis (March 2015 data cut-off; database not locked; some values may change for the final locked database). Study A3921041 was conducted in patients in Japan. Full details of the LTE studies were reported previously [18, 19]. Both LTE studies were conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines. The final protocols were reviewed and approved by the Institutional Review Boards and/or Independent Ethics Committee at each study center, and all patients provided written, informed consent.
Patients
Full inclusion and exclusion criteria for the two LTE studies have been previously reported [18, 19]. In summary, patients aged ≥ 18 years (ORAL Sequel) or ≥ 20 years (A3921041) with a diagnosis of RA based on the American College of Rheumatology (ACR) 1987 Revised Criteria [21] were eligible to enroll in the LTE studies if they had completed a prior qualifying tofacitinib phase I (ORAL Sequel only), phase II, or phase III index study [6,7,8,9,10,11,12,13,14,15,16, 22,23,24,25,26], and patients must have received tofacitinib for at least 3 years in the LTE studies to be included in this analysis.
Patients with any serious medical condition (e.g., severe, progressive, or uncontrolled renal, hepatic, hematologic, gastrointestinal, metabolic, endocrine, pulmonary, cardiac, neurologic or cerebral disease, or history of lymphoproliferative disease) that would make treatment with tofacitinib potentially unsafe were excluded from the LTE studies.
Study Treatment
Patients received tofacitinib 5 or 10 mg BID in the LTE studies. Patients from phase II index studies initiated LTE study treatment with tofacitinib 5 mg BID, whereas patients from phase III index studies initiated LTE study treatment with tofacitinib 10 mg BID, except for patients from China and Japan who all initiated treatment with tofacitinib 5 mg BID per protocol. During the LTE studies, tofacitinib dose could be reduced from 10 to 5 mg BID for reasons of safety, and could be increased from 5 to 10 mg BID for reasons of inadequate response, at the discretion of the investigator.
The tofacitinib dose groups in the analysis were based on the total daily dose (TDD), defined as 5 mg BID for TDD < 15 mg and 10 mg BID for TDD ≥ 15 mg, and calculated by adding all doses received by each patient and dividing by the number of days a dose of tofacitinib was received in the study. Patients receiving tofacitinib 5 or 10 mg BID in the LTE studies were pooled to form a combined all-tofacitinib group.
Patients in the LTE studies were permitted to receive background RA therapy. Dose adjustments to permitted concomitant RA medications (including MTX, leflunomide, sulfasalazine, antimalarials, auranofin, injectable gold preparations, nonsteroidal anti-inflammatory drugs, and/or GCs at approved doses) were at the discretion of the investigator for inadequate efficacy or safety reasons, or could be tapered or discontinued in response to adequate RA disease control.
Analyses
Data from patients receiving tofacitinib 5 or 10 mg BID were pooled into a combined tofacitinib dose group for these analyses.
In patients who began LTE study treatment within 14 days of receiving the last tofacitinib dose in the qualifying index study, baseline values were taken from the index study, otherwise baseline values from the LTE studies were used. LTE baseline values were used for MTX and GC use/dose for all patients.
Discontinuation of MTX or GC was assessed separately in patients who received MTX or GC at baseline, and was defined as no MTX or GC use within the last 30 days prior to the year-3 visit. Initiation was assessed separately in patients who did not receive MTX or GC at baseline, and was defined as initiating MTX or GC on or before the year-3 visit.
Clinical efficacy was assessed at year 3 using the Clinical Disease Activity Index (CDAI). Remission was defined as CDAI ≤ 2.8, low disease activity (LDA) as CDAI > 2.8 to ≤ 10, and incomplete response as CDAI > 10. For maintenance analyses, patients receiving MTX or GC at LTE baseline were stratified by CDAI clinical response 3 months after LTE study entry and evaluated for maintenance of response through year 3.
For patients who initiated treatment with MTX or GC during the LTE studies, the proportion of patients with CDAI response (remission, LDA, or incomplete response) and RA disease parameters [tender joint count (TJC), swollen joint count (SJC), Patient Assessment of Arthritis Pain, assessed by visual analog scale (Pain VAS), Patient’s Global Assessment of disease activity (PtGA) and Physician’s Global Assessment of disease activity (PGA) assessed by VAS, Functional Assessment of Chronic Illness Therapy (FACIT)–Fatigue Scale, and C-reactive protein (CRP) levels] were evaluated. Assessments were recorded at baseline, last post-baseline visit prior to initiating MTX or GC, and at the year-3 visit.
Descriptive and summary statistics are presented.
Results
Patients Discontinuing MTX or GC
Of 4867 tofacitinib-treated patients, 2796 (57.4%) were receiving MTX at baseline and 1608/2796 (57.5%) had efficacy measurements at year 3. Of these patients, 186/1608 (11.6%) had discontinued MTX by year 3 and were evaluated in the analysis: 49/186 (26.3%) patients receiving tofacitinib 5 mg BID and 137/186 (73.7%) receiving tofacitinib 10 mg BID. Among all tofacitinib-treated patients who discontinued MTX, the mean (median) number of days spent off MTX prior to the year-3 visit was 577 (563) days.
Of 4867 tofacitinib-treated patients, 2553 (52.5%) were receiving GC at baseline and 1434/2553 (56.2%) had efficacy measurements through year 3. Of these patients, 319/1434 (22.2%) discontinued GC by year 3 and were evaluated in the analysis: 114/319 (35.7%) receiving tofacitinib 5 mg BID and 205/319 (64.3%) receiving tofacitinib 10 mg BID. Among all tofacitinib-treated patients who discontinued GC, the mean (median) number of days after GC discontinuation was 568 (547) days.
Patient demographics and disease characteristics were generally similar irrespective of whether patients discontinued or continued MTX or GC (Table 1).
MTX Discontinuation and CDAI Response
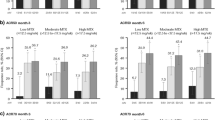
At year 3, 63/186 (33.9%) patients receiving tofacitinib who discontinued MTX achieved CDAI remission (CDAI ≤ 2.8), compared with 355/1422 (25.0%) who continued background MTX (Fig. 1a). Overall, 131/186 patients (70.4%) who discontinued MTX and 983/1422 patients (69.1%) who continued MTX were either in CDAI remission or LDA at year 3.

a CDAI response at year 3 in patients who were receiving tofacitinib with background MTX at baseline and subsequently discontinued or continued MTX; b maintenance of month-3 CDAI response at year 3 among patients who were receiving tofacitinib with background MTX at baseline and at the month-3 visit, who discontinued or continued MTX prior to the year-3 visit; c CDAI response at the last post-baseline visit prior to and following initiation of MTX at year 3 among patients who were receiving tofacitinib without MTX at baseline and subsequently initiated MTX. CDAI remission: ≤ 2.8; CDAI LDA: CDAI > 2.8 to ≤ 10; CDAI incomplete response as CDAI > 10. Year-3 data were missing from one patient who discontinued MTX, two patients who continued MTX, and one patient after initiation of MTX. CDAI Clinical Disease Activity Index, LDA low disease activity, MTX methotrexate
In the maintenance of efficacy analysis, of 41 patients who achieved CDAI remission at month 3 and later discontinued MTX (from month 3 to year 3), 28 (68.3%) maintained CDAI remission at year 3 (Fig. 1b); compared with 171/284 (60.2%) who continued MTX. A similar trend was observed for CDAI LDA or incomplete response, with the majority of patients maintaining month-3 CDAI response at year 3, regardless of MTX discontinuation.
Glucocorticoid Discontinuation and CDAI Response
At year 3, 109/319 patients (34.2%) who discontinued GC were in CDAI remission, while 226/1115 (20.3%) who continued GC were in CDAI remission (Fig. 2a). Overall, 232/319 patients (72.7%) who discontinued GC and 735/1115 (65.9%) who continued GC achieved either CDAI remission or LDA at year 3.

a CDAI response at year 3 in patients who were receiving tofacitinib with GC at baseline and subsequently discontinued or continued GC; b maintenance of month-3 CDAI response at year 3 among patients who were receiving tofacitinib with GC at baseline and at the month 3 visit, who discontinued or continued GC prior to the year-3 visit; c CDAI response at the last post-baseline visit prior to and following initiation of GC at year 3 among patients who were receiving tofacitinib without GC at baseline and subsequently initiated GC. CDAI remission: ≤ 2.8; CDAI LDA: CDAI > 2.8 to ≤ 10; CDAI incomplete response as CDAI > 10. Year-3 data were missing from one patient who discontinued GC, two patients who continued GC, four patients prior to initiation of GC, and three patients after initiation of GC. CDAI Clinical Disease Activity Index, GC glucocorticoid, LDA low disease activity
Of 54 patients who achieved CDAI remission at month 3 and later discontinued GC (from month 3 to year 3), 38 (70.4%) maintained CDAI remission at year 3, compared with 98/187 (52.4%) who continued GC (Fig. 2b). A similar trend was observed for CDAI LDA or incomplete response; the majority of patients maintained month-3 CDAI response at year 3, regardless of GC discontinuation.
Initiation of MTX or GC During the LTE Studies
Of 1044 patients in the LTE studies who were not receiving MTX at LTE baseline and had any efficacy measurement at year 3, 65 (6.2%) initiated MTX prior to/at year 3: 22/423 (5.2%) patients receiving tofacitinib 5 mg BID and 43/621 (6.9%) receiving tofacitinib 10 mg BID.
Of 1218 patients in the LTE studies who were not receiving GC at LTE baseline and had any efficacy measurement at year 3, 306 (25.1%) patients initiated GC prior to/at year 3: 82/417 (19.7%) patients receiving tofacitinib 5 mg BID and 224/801 (28.0%) receiving tofacitinib 10 mg BID.
Patient demographics and disease characteristics at baseline for those who initiated MTX or GC during the LTEs prior to the year-3 visit, and had a post-baseline visit prior to initiation, are presented in Table 2. Prior to initiation of MTX or GC, 40/58 (69.0%) and 128/282 (45.4%) patients, respectively, had a CDAI incomplete response (Figs. 1c, 2c). In patients who initiated MTX, CDAI remission or LDA was achieved in 18/58 (31.0%) patients at last post-baseline visit prior to initiation and in 29/58 (50.0%) after initiation at year 3 (Fig. 1c). In patients who initiated GC, CDAI remission or LDA was achieved in 150/282 (53.2%) patients at last post-baseline visit prior to initiation and in 155/282 (55.0%) after initiation at year 3 (Fig. 2c).
Overall, some improvements in CDAI remission/LDA rates and other RA disease parameters (TJC, SJC, Pain, PtGA, PGA, FACIT–Fatigue Scale Score, and CRP) were observed in patients receiving tofacitinib who initiated MTX; only modest improvement was observed in patients who initiated GC (Figs. 1c, 2c; Table 3).
Discussion
This post hoc, pooled analysis from two LTE studies evaluated how discontinuation of concomitant MTX or GC may impact the maintenance of clinical efficacy with tofacitinib 5 or 10 mg BID. In addition, the potential effect of MTX or GC initiation was investigated.
Previous analyses of these LTE studies demonstrated consistent efficacy and safety of tofacitinib over time (up to 114 months), when administered as monotherapy or in combination with csDMARDs [18–20]. In addition, in a vaccine sub-study of patients enrolled in the ORAL Sequel LTE, discontinuation of tofacitinib treatment for 2 weeks resulted in worsening of disease activity and physical function, which then showed improvement back to baseline levels approximately 1 month after reinitiation of tofacitinib [27]. However, this is the first analysis of the effect of discontinuation or initiation of concomitant treatment of either MTX or GC.
Overall, 11.6% of patients receiving tofacitinib with background MTX at LTE baseline discontinued MTX treatment prior to year 3; 22.2% who were receiving GC at LTE baseline discontinued GC. In patients not receiving MTX at baseline, 6.2% initiated MTX before year 3; 25.1% of patients not receiving GC at baseline initiated GC.
Similar CDAI response levels at year 3 between patients who discontinued MTX and those who continued use (among patients receiving background MTX at baseline) suggested that discontinuing concomitant MTX may not have a detrimental effect on response to tofacitinib in the majority of patients. Furthermore, at year 3, patients receiving tofacitinib generally maintained the same response to treatment achieved at month 3, irrespective of whether they discontinued MTX or not. Similarly, discontinuation of concomitant GC did not negatively impact CDAI response at year 3 or maintenance of month-3 CDAI response at year 3.
A phase IIIb/IV study (ORAL Strategy, NCT02187055) compared the efficacy and safety of tofacitinib monotherapy with tofacitinib in combination with MTX (and also adalimumab in combination with MTX), and showed that while both treatments provided improvement in clinical and functional responses, tofacitinib monotherapy was not non-inferior to tofacitinib combination therapy based on the primary endpoint of ACR50 response rate [17]. Although non-inferiority of monotherapy to combination therapy was not shown in this previous study based on ACR50 response rate, the results presented here suggest that in most patients receiving combination therapy who achieve remission, response can be maintained if MTX is then discontinued.
Further aims of this post hoc analysis were to investigate the proportions of patients who initiated treatment with MTX or GC during the two LTE studies, and the potential impact of MTX or GC initiation on the clinical efficacy of tofacitinib. For patients not receiving MTX at LTE baseline, the majority of those who initiated MTX were classified as CDAI incomplete responders at last post-baseline visit prior to initiating MTX. Although this suggests that the addition of another agent was likely due to limited efficacy of previous tofacitinib-based therapy, specific reasons for initiation were not captured during the LTE studies. In general, a numerical improvement in disease activity was observed after MTX initiation, suggesting that patients who experience an initial improvement with tofacitinib yet remain as CDAI-incomplete responders may derive some clinical benefit from the addition of MTX. However, the small number of patients who initiated MTX may limit the value of this finding. For patients not receiving GC at LTE baseline, prior to initiating GC, just under half were CDAI-incomplete responders. Overall, initiation of GC resulted in modest improvement in disease parameters.
This was a post hoc analysis of data from two open-label studies, which were not specifically designed to study the impact of concomitant MTX or GC discontinuation or initiation on tofacitinib; therefore, conclusions may be limited and should be interpreted with caution. In particular, the very small patient numbers in some analysis groups (e.g., patients used to investigate the impact of initiating MTX) limit this analysis to being exploratory in nature. It should be noted that patients had additional/differing exposure to tofacitinib in qualifying phase I–III studies prior to enrollment into the LTE studies evaluated in this analysis. The analysis is also limited by the definition of discontinuation and initiation used, which grouped together patients with different durations of MTX or GC treatment over 3 years. Also, patients receiving concomitant MTX and GC were not analyzed separately, which may confound results; however, similar proportions were observed in the continued vs. discontinued group for MTX or GC use separately among MTX or GC users at LTE baseline. A controlled study that includes a larger number of patients and is specifically designed to examine concomitant MTX/GC discontinuation and initiation with tofacitinib is needed; a prospective study investigating the effect of MTX discontinuation in LDA-responders initiated on tofacitinib in combination with MTX is ongoing (NCT02831855).
Conclusions
Patients achieving CDAI remission or LDA with tofacitinib may be able to discontinue MTX or GC and maintain their response to treatment. Moreover, patients who have an initial improvement with tofacitinib-based therapy but remain CDAI-incomplete responders (CDAI > 10) may benefit from the addition of MTX. Additional studies are required to support these findings.
References
Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68:1–26.
Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960–77.
Salliot C, van der Heijde D. Long-term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann Rheum Dis. 2009;68:1100–4.
Kavanaugh A, Wells AF. Benefits and risks of low-dose glucocorticoid treatment in the patient with rheumatoid arthritis. Rheumatology (Oxford). 2014;53:1742–51.
Rasch LA, Bultink IE, van Tuyl LH, Lems WF. Glucocorticoid safety for treating rheumatoid arthritis. Expert Opin Drug Saf. 2015;14:839–44.
Fleischmann R, Cutolo M, Genovese MC, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64:617–29.
Kremer JM, Bloom BJ, Breedveld FC, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60:1895–905.
Kremer JM, Cohen S, Wilkinson BE, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64:970–81.
Tanaka Y, Suzuki M, Nakamura H, et al. Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res (Hoboken). 2011;63:1150–8.
Tanaka Y, Takeuchi T, Yamanaka H, et al. Efficacy and safety of tofacitinib as monotherapy in Japanese patients with active rheumatoid arthritis: a 12-week, randomized, phase 2 study. Mod Rheumatol. 2015;25:514–21.
Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–60.
Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507.
Kremer J, Li Z-G, Hall S, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159:253–61.
Lee EB, Fleischmann R, Hall S, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377–86.
van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65:559–70.
van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19.
Fleischmann R, Mysler E, Hall S, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. 2017;390:457–68.
Wollenhaupt J, Silverfield J, Lee EB, et al. Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, long-term extension studies. J Rheumatol. 2014;41:837–52.
Yamanaka H, Tanaka Y, Takeuchi T, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open-label, long-term extension study. Arthritis Res Ther. 2016;18:34.
Wollenhaupt J, Silverfield J, Lee EB et al. Tofacitinib, an oral Janus kinase inhibitor, in the treatment of rheumatoid arthritis: safety and efficacy in open-label, long-term extension studies over 9 years. Arthritis & Rheumatology 69 [suppl 10], 683-4. 18-9-2017.
Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24.
Boyle DL, Soma K, Hodge J, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis. 2015;74:1311–6.
Conaghan PG, Østergaard M, Bowes MA, et al. Comparing the effects of tofacitinib, methotrexate and the combination, on bone marrow oedema, synovitis and bone erosion in methotrexate-naive, early active rheumatoid arthritis: results of an exploratory randomised MRI study incorporating semiquantitative and quantitative techniques. Ann Rheum Dis. 2016;75:1024–33.
Kremer JM, Kivitz AJ, Simon-Campos JA, et al. Evaluation of the effect of tofacitinib on measured glomerular filtration rate in patients with active rheumatoid arthritis: results from a randomised controlled trial. Arthritis Res Ther. 2015;17:95.
McInnes IB, Kim HY, Lee SH, et al. Open-label tofacitinib and double-blind atorvastatin in rheumatoid arthritis patients: a randomised study. Ann Rheum Dis. 2014;73:124–31.
Winthrop KL, Silverfield J, Racewicz A, et al. The effect of tofacitinib on pneumococcal and influenza vaccine responses in rheumatoid arthritis. Ann Rheum Dis. 2016;75:687–95.
Kaine J, Tesser J, DeMasi R, et al. Re-establishment of efficacy of tofacitinib, an oral Janus kinase inhibitor, in rheumatoid arthritis patients after temporary discontinuation. Ann Rheum Dis. 2017;76(S2):275–6.
Acknowledgements
Funding
This study and editorial support were sponsored by Pfizer Inc, New York, NY, USA. Pfizer Inc funded the article processing charges. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published.
Medical Writing and/or Editorial Assistance
Medical writing support under the guidance of the authors was provided by Nicole Jones BSc and Paul Scutt PhD on behalf of Complete Medical Communications, Macclesfield, UK, and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015; 163: 461–464).
Prior Presentation
Some of the data in this manuscript have been previously presented at the American College of Rheumatology Annual Scientific Meeting 2016 (November 11–16, 2017; Washington, DC, USA), and the Association of Women in Rheumatology 2017 National Conference (August 17–20; Hilton Head Island, SC, USA).
Disclosures
R. Fleischmann has received research grants and consulting fees from Pfizer Inc. J. Wollenhaupt is a shareholder and member of the speaker’s bureau for Pfizer Inc. S. Cohen has received research grants and consulting fees from Pfizer Inc. M.E. Weinblatt has received research grants from Amgen, BMS, Crescendo Bioscience, and UCB; and consulting fees from AbbVie, Amgen, BMS, Crescendo Bioscience, Eli Lilly, Gilead, Pfizer Inc, UCB, and Vertex. V. Bandi has provided consultant services to Pfizer Inc through Eliassen Group Inc. L. Wang is an employee and shareholder of Pfizer Inc. H. Fan is an employee and shareholder of Pfizer Inc. J. Andrews is an employee and shareholder of Pfizer Inc. L. Takiya is an employee and shareholder of Pfizer Inc. E. Bananis is an employee and shareholder of Pfizer Inc.
Compliance with Ethics Guidelines
Both LTE studies were conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines. The final protocols were reviewed and approved by the Institutional Review Boards and/or Independent Ethics Committee at each study center, and all patients provided written, informed consent.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to www.medengine.com/Redeem/8F3B4F6077797C60.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fleischmann, R., Wollenhaupt, J., Cohen, S. et al. Effect of Discontinuation or Initiation of Methotrexate or Glucocorticoids on Tofacitinib Efficacy in Patients with Rheumatoid Arthritis: A Post Hoc Analysis. Rheumatol Ther 5, 203–214 (2018). https://doi.org/10.1007/s40744-018-0093-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-018-0093-7