
Abstract
Introduction
While dose escalation of golimumab has been used for patients with rheumatoid arthritis who demonstrate an inadequate response to the standard dose, its effectiveness has not been fully evaluated. The aim of this study was to assess the clinical outcome observed by dose escalation of golimumab for patients with rheumatoid arthritis in the daily clinical setting.
Methods
A post hoc analysis was performed of data from the 24-week post-marketing surveillance conducted in Japan (n = 5154). A total of 301 patients with moderate or high disease activity at baseline who underwent dose escalation of golimumab were assessed for effectiveness at 24 weeks based on several variables, such as DAS28-CRP, SDAI, and CDAI, as well as for medication persistence through 24 weeks. In addition, the study population was stratified by the time to dose escalation, and effectiveness was likewise evaluated. Logistic regression analysis was performed to identify factors associated with a moderate/good EULAR response to golimumab at 24 weeks.
Results
Patients with golimumab dose escalation showed significant improvement of the clinical signs and symptoms of rheumatoid arthritis at 24 weeks, as indicated by reduction of the DAS28-CRP (∆0.89), SDAI (∆8.64), and CDAI (∆8.28) scores. This result was relatively consistent across the subgroups stratified by the timing of dose escalation. According to Kaplan-Meier analysis, 78.1% of the patients continued to receive golimumab at 24 weeks, and this was also similar among the subgroups stratified by the time to dose escalation. Multivariate analysis identified male sex and previous biologic therapy as factors that were significantly associated with the clinical response at 24 weeks.
Conclusion
In real-world clinical practice, improvement of disease activity was observed after uptitration of golimumab from 50 to 100 mg regardless of the timing. Male patients and biologic-naive patients were more likely to respond to dose escalation of golimumab.
Trial Registration
UMIN-CTR, Identifier: UMIN000015895.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Golimumab (GLM), a fully human monoclonal antibody targeting TNF, is approved at 50 mg or 100 mg every 4 weeks for the treatment of rheumatoid arthritis (RA) in Japan |
In real-world practice, dose escalation of GLM from 50 to 100 mg seems to achieve better control in RA patients with higher disease activity, but there have been only few reports supporting a clinical benefit of this strategy |
The aim of this study is to assess the clinical outcome of GLM dose escalation in a real-world clinical practice using the data from post-marketing surveillance of GLM conducted in Japan |
What was learned from the study? |
In a real-world clinical setting, improvement of disease activity was observed after uptitration of golimumab from 50 to 100 mg regardless of the timing |
Male patients and biologic-naive patients were more likely to respond to dose escalation of golimumab, but further investigation will be required to identify useful predictors at the time of dose escalation |
Introduction
Rheumatoid arthritis (RA) is a chronic debilitating systemic inflammatory disease that leads to joint destruction and functional disability. It is estimated that up to 1% of the Japanese population has RA [1]. The strategy for management of RA has been improved considerably since the introduction of biologic agents targeting key molecules implicated in the pathogenesis of this disease. Tumor necrosis factor (TNF) inhibitors were among the first biologic agents proven to be effective for suppression of disease activity and progression of joint damage [2].
Golimumab (GLM), a fully human monoclonal antibody targeting TNF, has been demonstrated to be an effective treatment for RA [3,4,5,6,7]. In Japan, subcutaneous GLM (50 mg or 100 mg every 4 weeks) is approved for adults with moderate to severe RA. In the phase 3 GO-FORTH study conducted in Japan, clinical efficacy was comparable between the two doses of GLM, but inhibition of radiographic progression was numerically better in patients treated with 100 mg of GLM + methotrexate (MTX) than in patients receiving 50 mg of GLM + MTX [4]. The GO-FORTH study was not designed to compare GLM dosages, but post hoc analysis suggested that a GLM dose of 100 mg was more likely to be effective than 50 mg for preventing structural damage in patients with high disease activity and patients with high CRP levels [8]. There have been few reports supporting a clinical benefit of GLM dose escalation. In the GO-FORTH study, 9 out of 86 subjects who initiated GLM at a dose of 50 mg underwent dose escalation to 100 mg as rescue treatment because of lack of efficacy, resulting in subsequent improvement of the signs and symptoms of RA [4]. In real-world daily practice, dose escalation of GLM seems to achieve better control in RA patients with higher disease activity [9]. However, dose escalation of biologic agents needs to be carefully appraised since this approach can lead to higher medical costs [10] that impose a greater burden on the healthcare system.
Therefore, the present study was designed to assess the clinical effect of GLM dose escalation in real-world clinical practice. We analyzed data from post-marketing surveillance (PMS) of GLM conducted in Japan, focusing on the population of patients who underwent dose escalation during the 24-week study period.
Methods
Study Design and Patients
As described previously [11], 5154 patients with RA who were treated with golimumab (GLM) in Japan between September 2011 and May 2013 were monitored for 24 weeks at selected medical facilities across Japan. The enrolled patients received subcutaneous GLM (50 mg or 100 mg) once every 4 weeks, with dose escalation (from 50 to 100 mg) or reduction (from 100 to 50 mg) at the discretion of the treating physician. Patients who underwent dose escalation of GLM were included in this study if they had moderate or high disease activity according to the DAS28-CRP score at study entry. Patients who underwent dose escalation of GLM multiple times and patients who started GLM at 100 mg were excluded.
This article is based on previously conducted PMS study and does not contain any new interventional studies with human participants or animal subjects performed by any of the authors. The PMS study protocol and ethical considerations were assessed by the internal review board members and approved by the Japanese PMDA. This PMS study was registered with the University Hospital Medical Information Network Clinical Trials Registry (UMIN-CTR, Identifier: UMIN000015895), was conducted in accordance with the Japanese regulations (Ministry of Health, Labor, and Welfare Ministerial Ordinance No. 171) on Good Post-marketing Study Practice (GPSP), and was carried out by Janssen Pharmaceutical K.K. after a contract with each study center.
Assessment of Effectiveness
The effectiveness of GLM was assessed by analyzing changes of the Disease Activity Score 28 based on C-reactive protein (DAS28-CRP), the Clinical Disease Activity Index (CDAI), and the Simplified Disease Activity Index (SDAI) at 24 weeks from baseline. The EULAR response was also evaluated from the absolute change of DAS28-CRP as well as the remission rates based on DAS28-CRP, CDAI, and SDAI scores at 24 weeks. Cutoff values for disease activity (high/moderate/low) were defined for the DAS28-CRP (5.1/3.2/2.6), SDAI (26/11/3.3), and CDAI (22/10/2.8) scores. Remission was defined as DAS28-CRP ≤ 2.6, SDAI ≤ 3.3, and CDAI ≤ 2.8. A good EULAR response was defined as a decrease of DAS28-CRP from baseline by > 1.2 if it was ≤ 3.2 at 24 weeks. A moderate EULAR response was defined as a decrease of DAS28-CRP from baseline by > 1.2 if the value was > 3.2 at 24 weeks or a decrease of DAS28-CRP from baseline by > 0.6 and ≤ 1.2 if it was ≤ 5.1 at 24 weeks.
Statistical Analysis
Data are presented as mean ± standard deviation (SD) for continuous variables and as proportions (%) for categorical valuables unless otherwise described. The paired t test was performed to compare changes of variables from baseline to week 24, and statistical significance was defined as p < 0.05 (two-tailed). P values were adjusted for multiplicity by the Bonferroni method. Kaplan-Meier analysis was conducted to estimate treatment persistence, which was defined as the period from initiation of GLM to its discontinuation for any reason. Survival curves were compared with the log-rank test, using the subgroup who underwent dose escalation at 4 weeks as a reference, and log-rank p values were corrected for multiple comparisons by the Bonferroni method.
Univariate logistic regression analysis was performed to examine the association between patient characteristics and a good or moderate EULAR response, followed by multivariate logistic regression analysis. Variables tested in the univariate logistic regression analysis were also evaluated in multivariate models. In all analyses, p < 0.05 was considered statistically significant. Associations were assessed by calculating the odds ratio (OR) with 95% confidence interval (CI).
Effectiveness was assessed in the modified intent-to-treat (ITT) population, which included patients who at least had baseline data. If the 24-week data were missing, baseline values were carried forward for imputation. Effectiveness was also assessed in the observed case (OC) population, which included patients who had data at both baseline and 24 weeks, as a sensitivity analysis to confirm the robustness of results obtained by ITT analysis.
All statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC).
Results
Patient Disposition and Baseline Characteristics
As reported previously, a total of 5154 patients were enrolled in the post-marketing surveillance (PMS) of golimumab (GLM), and 5137 patients were included in the safety analysis set, of which 4331 started GLM at 50 mg every 4 weeks [11]. Among these patients, 301 had high or moderate disease activity at study entry and underwent one dose escalation (to 100 mg every 4 weeks) during the 24-week treatment, fulfilling the study criteria for the ITT effectiveness analysis population (Fig. 1). In this population, dose escalation was more frequent at earlier time points (week 4, n = 85; week 8, n = 63; week 12, n = 62; week 16, n = 44; week 20, n = 47). The patients showed female predominance (81.7%). The mean age was 62.3 ± 13.4 years, and the mean disease duration was 10.4 ± 10.0 years. The mean disease activity scores were as follows: DAS28-ESR, 5.48 ± 1.10; DAS28-CRP, 4.81 ± 0.99; SDAI, 28.59 ± 12.14; CDAI, 26.18 ± 11.60. Approximately two-thirds of the patients (64.5%) had previously used biologic disease-modifying antirheumatic drugs (bDMARDs), and the majority were on concomitant glucocorticoids (62.2%, mean dose: 5.01 ± 3.13 mg) and MTX (78.8%, mean dose: 8.97 ± 3.61 mg) during the study period (Table 1). When stratified by the time to dose escalation (4, 8, 12, 16, or 20 weeks), overall demographic and baseline characteristics were generally comparable across all subgroups. However, the patients who underwent dose escalation at 8 weeks showed harder-to-treat baseline characteristics such as a higher frequency of comorbidities (82.5% vs. 75.7% in the total effectiveness population) and previous bDMARD exposure (79.4% vs. 64.5% in the total effectiveness population; Table 1).

Flow diagram of patient disposition demonstrating the number of patients included in each population of the present study. ITT intent to treat, GLM golimumab. *Patients could be allocated to more than one category for exclusion criteria
Persistence with GLM Stratified by the Time to Dose Escalation
Kaplan-Meier analysis revealed that persistence (i.e., average time to discontinuation) with GLM after dose escalation was as follows in each subgroup: week 4, 19.7 weeks; week 8, 20.3 weeks; week 12, 21.6 weeks; week 16, 22.7 weeks; week 20, 23.5 weeks. When the week 4 subgroup was compared with the other subgroups, only the week 20 subgroup was significantly different (log-rank p = 0.005), presumably because of the short follow-up period after dose escalation (Fig. 2). Overall, 21.9% of patients discontinued GLM treatment after dose escalation, and the primary reason was the progression of disease in all the subgroups (Supplementary Table 1).

Persistence with GLM treatment stratified by the timing of dose escalation. Kaplan-Meier analysis was performed to assess persistence with GLM treatment based on the time when the GLM dose was increased from 50 to 100 mg during the observation period of the 24-week post-marketing surveillance. Each of the Kaplan-Meier curves was compared with the week 4 curve by the log-rank test, and the p values were adjusted for multiple comparisons by using Bonferroni correction. Descriptive statistics are shown in the table. GLM golimumab
Effectiveness of GLM Stratified by the Time to Dose Escalation
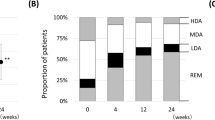
The mean DAS28-CRP of the total study population improved significantly from 4.81 to 3.92 at 24 weeks (p < 0.001). Similarly, subgroup analyses demonstrated a significant decrease of DAS28-CRP regardless of the timing of dose escalation (Fig. 3a: p < 0.001 with Bonferroni correction). Pairwise comparisons between subgroups failed to detect statistically significant differences for any combinations (Supplementary Table 2). Likewise, significant reduction of the CDAI and SDAI scores from baseline was observed in the overall study population (Fig. 3b, c: both p < 0.001). This was also true when stratified by the timing of dose escalation, with no significant differences among the subgroups being detected by pairwise comparisons (Supplementary Table 2). Overall, 46.1% of patients who underwent dose escalation achieved a good (24.4%) or moderate (21.7%) EULAR response at 24 weeks. Consistent with this result, approximately 50% of the patients in each dose escalation subgroup demonstrated a good or moderate EULAR response (Fig. 4a), with no statistical differences among the subgroups (Supplementary Table 3). Remission based on DAS28, SDAI, and CDAI was observed in 23.2%, 9.5%, and 9.1% of the overall study population, respectively (Fig. 4b), increasing to 30.8%, 27.7%, and 32.1%, respectively, when patients with low disease activity were included (Fig. 4c). Although numerical differences were observed, we did not detect any significant inter-subgroup differences (Supplementary Table 3). Across the overall effectiveness analysis, the week 8 subgroup tended to show less improvement, which was possibly related to its baseline characteristics. Overall, patients who received GLM dose escalation from 50 to 100 mg demonstrated significant improvement of their signs and symptoms irrespective of the timing of dose escalation. This conclusion was strengthened by the sensitivity analysis in the OC population, which revealed similar trends for all variables assessed (Supplementary Table 4).

Disease activity at baseline and week 24 in patients on GLM treatment. For each subgroup stratified by the timing of dose escalation, disease activity at baseline (0 week) and 24 weeks was evaluated from the DAS28-CRP (a), CDAI (b), or SDAI (c) score. DAS28-CRP, Disease Activity Score 28 based on C-reactive protein, CDAI Clinical Disease Activity Index, SDAI Simplified Disease Activity Index, GLM golimumab. ***p < 0.001 versus baseline by the paired t-test. †p < 0.0001 versus baseline by the paired t-test with Bonferroni correction for multiplicity

Disease activity measures at 24 weeks stratified by the timing of GLM dose escalation. a Percentage of patients achieving a EULAR response at 24 weeks stratified by GLM dose escalation at the time point indicated. The clinical response to golimumab at 24 weeks was evaluated using the EULAR response criteria based on DAS28-CRP. A good or moderate response was defined as improvement of DAS28–CRP by > 1.2 from any baseline score or improvement by 0.6–1.2 from a baseline score of ≤ 5.1. b Percentage of patients achieving remission as defined by the DA28-CRP (score < 2.6), SDAI (score ≤ 3.3), or CDAI (score ≤ 2.8). c Percentage of patients achieving low disease activity as defined by the DA28-CRP (score ≤ 3.2), SDAI (score ≤ 11), or CDAI (score ≤ 10). EULAR European League Against Rheumatism, DAS28-CRP Disease Activity Score 28 based on C-reactive protein, CDAI Clinical Disease Activity Index, SDAI Simplified Disease Activity Index, REM remission, LDA low disease activity, GLM golimumab
Logistic regression analysis suggested that the likelihood of achieving a moderate or good EULAR response at 24 weeks was significantly higher in males (p = 0.015) and in biologic-naïve patients (p = 0.013). No significant associations were identified for the other demographic and baseline clinical characteristics (Table 2).
Discussion
Although GLM is approved at two dosages (i.e., 50 mg and 100 mg) for the treatment of active RA in Japan, there has been little evidence as to whether dose escalation confers additional clinical benefit for control of disease activity. The present study demonstrated that RA patients with moderate to high disease activity who underwent GLM dose escalation significantly improved the clinical signs and symptoms as indicated by reduction of the DAS28-CRP, CDAI, and SDAI scores. When effectiveness was assessed by the EULAR response, about half of the patients exhibited a good or moderate response after dose escalation. In addition, approximately 30% improved to show low disease activity or remission after dose escalation. We also demonstrated that the clinical response at 24 weeks was similar regardless of the time to dose escalation of GLM, suggesting that emergence of the response was prompt and a plateau occurred within 4 weeks.
The effect of dose escalation observed in the current study of a real-world patient population is consistent with the results of the GO-FORTH randomized clinical trial conducted in Japanese RA patients with an inadequate response to MTX. In that study, nine patients received rescue treatment by GLM dose escalation from 50 to 100 mg due to lack of efficacy and demonstrated improvement of the ACR20 response rate [4]. Here, we found that the patients who underwent dose escalation of GLM demonstrated improvement of disease activity at 24 weeks. Since the current study only included patients with moderate or high disease activity at study entry, it is speculated that this population was likely to undergo dose escalation due to insufficient response to GLM at a dose of 50 mg. Our results therefore suggest that poor responders to 50 mg of GLM may be benefited from uptitration of GLM to 100 mg in controlling disease activity. On the other hand, a previous post hoc analysis of the GO-FORTH study, which suggested that initiating GLM therapy at 100 mg might be more beneficial than initiating at 50 mg for preventing joint destruction in patients with high disease activity or a high CRP [8]. Currently, there is no evidence directly comparing the efficacy between initiating GLM at 100 mg versus increasing the dose of GLM to 100 mg. Yet, given the fact that patients and physicians are required to consider economic implications when selecting treatment strategies, it would be valuable to provide more therapeutic options as suggested by the data presented in the current study. However, it should be noted that the current study did not analyze patient characteristics at the time of dose escalation because the PMS protocol did not require clinical evaluation at time points other than weeks 0 and 24. Thus, while our data clearly indicate the clinical effect of starting GLM at 50 mg and increasing the dose to 100 mg as needed, the net benefit of dose escalation itself cannot be determined.
The clinical benefit of dose escalation of biologic agents for RA has been best studied with infliximab, which is indicated for dose escalation or dose intensification if the response is insufficient. Previous studies have demonstrated that patients with an inadequate response to infliximab at a starting dose of 3 mg/kg generally had a low trough serum concentration [12,13,14]. Immunogenicity is strongly associated with low serum concentrations of infliximab and subsequent attenuation of the clinical response [15,16,17,18]. It has previously been suggested that dose escalation of infliximab is efficacious in patients with a low anti-infliximab antibody titer, while this approach may not be as effective for patients with a high titer because the anti-drug antibody cannot be overcome by the increased drug dose [19, 20]. For GLM, a recent study re-examined immunogenicity using banked blood samples from previous phase 3 clinical trials with a highly sensitive assay, revealing that 24.9% of patients in the GO-FORWARD study were anti-GLM antibody positive, which was far higher than detected by the original assay [15]. However, this study also showed that the anti-GLM antibody titers were predominantly low (< 1:100) and appeared to have no significant impact on the serum GLM concentration or the clinical response [15]. Therefore, it is hypothesized that dose escalation could be beneficial in most patients with an insufficient response to GLM at 50 mg, while a small group of patients with high anti-GLM antibody titers could possibly receive more benefit from switching to a different biologic agent. However, further studies are required to verify this hypothesis and to determine the predictors of clinically relevant reduction of the serum GLM concentration.
The present study revealed that 21.9% of patients discontinued GLM treatment even after dose escalation due primarily to the progression of disease, suggesting that dose escalation of GLM is not always effective. Therefore, it is crucial to identify characteristics of patients who would respond to GLM dose escalation after inadequate response to 50 mg of GLM. To this end, univariate and multivariate logistic regression analyses were employed. Our results suggested that dose escalation might be less effective in patients who have previously used biologics (OR 0.50, p = 0.013). It is widely believed that sequential switching to an alternative TNF inhibitor is effective for reducing disease activity, but the response is lower than with the first TNF inhibitor [21,22,23,24,25], probably because of channeling bias favoring patients with more severe disease [26]. Biologic-naïve patients are presumed to have lower disease activity than biologic-experienced patients, thereby being more responsive to GLM dose escalation. In addition, our analysis also suggested that male sex predicted a clinical response to dose escalation (OR 2.37, p = 0.015). The reason why male sex was a positive predictor of a clinical response is not clear. However, it is interesting that several studies have identified male sex as a positive predictor of the clinical response to MTX [27,28,29]. Because approximately 80% of our study population was on concomitant MTX therapy, it is possible that concomitant use of MTX might have contributed to a better clinical response to dose escalation of GLM. On the other hand, concomitant use of MTX was not identified as an independent predictive factor by the present analysis, so further investigation of a larger sample size will be required to clarify the association between male sex and the clinical response to dose escalation.
The present analysis had some limitations as a retrospective study. First, the lack of blinding and randomization may have caused selection bias as well as response bias. In particular, decisions about dose escalation were made at the physician’s discretion, potentially acting as a confounder. Second, the short study duration precluded us from conducting more thorough analyses of effectiveness and persistence. Third, we were unable to obtain data on patient characteristics at the time of dose escalation because of the design of the original PMS study, so we had to use the baseline patient characteristics. Thus, clinical improvement was evaluated by comparing disease activity at 24 weeks with that at baseline instead of activity at the time of dose escalation, preventing us from evaluating the improvement related to dose escalation per se. Furthermore, predictive factors were likewise determined by using baseline patient characteristics, but it would be more informative for daily clinical practice if we could identify useful predictors at the time of dose escalation. Finally, the effectiveness population of the current study included patients on GLM 50 mg monotherapy, which is not approved in Japan. This might have led to underestimation of effectiveness because those patients could have undergone dose escalation to 100 mg monotherapy, which is approved in Japan, for reasons other than an inadequate response such as cost, since Japanese national health insurance only covers approved indications [9].
Conclusions
We demonstrated that RA patients with moderate to high disease activity experienced clinical improvement after dose escalation of GLM to 100 mg in real-world clinical practice. Our findings provide a critical insight into the effect of GLM dose escalation, which will facilitate more effective use of GLM in daily practice in Japan. However, use of a higher dose of GLM incurs a higher cost, consequently imposing a greater economic burden on the healthcare system. Therefore, further studies are required to more precisely identify the characteristics of patients who would respond to GLM dose escalation and evaluate the balance between the clinical benefit versus economic burden of GLM dose escalation.
Change history
08 October 2020
Under Results section, heading: Effectiveness of GLM Stratified by the Time to Dose Escalation, the remission based on values of DAS28, SDAI, and CDAI was published incorrectly. The correct values are: 16.1%, 5.0% and 4.3%
References
Yamanaka H, Sugiyama N, Inoue E, Taniguchi A, Momohara S. Estimates of the prevalence of and current treatment practices for rheumatoid arthritis in Japan using reimbursement data from health insurance societies and the IORRA cohort (I). Mod Rheumatol. 2014;24(1):33–40.
Furst DE, Breedveld FC, Kalden JR, et al. Updated consensus statement on biological agents for the treatment of rheumatoid arthritis and other immune mediated inflammatory diseases (May 2003). Ann Rheum Dis. 2003;62(Suppl 2):2–9.
Takeuchi T, Harigai M, Tanaka Y, et al. Golimumab monotherapy in Japanese patients with active rheumatoid arthritis despite prior treatment with disease-modifying antirheumatic drugs: results of the phase 2/3, multicentre, randomised, double-blind, placebo-controlled GO-MONO study through 24 weeks. Ann Rheum Dis. 2013;72(9):1488–95.
Tanaka Y, Harigai M, Takeuchi T, et al. Golimumab in combination with methotrexate in Japanese patients with active rheumatoid arthritis: results of the GO-FORTH study. Ann Rheum Dis. 2012;71(6):817–24.
Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every 4 weeks in methotrexate-naive patients with active rheumatoid arthritis: 24-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60(8):2272–83.
Smolen JS, Kay J, Doyle MK, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374(9685):210–21.
Keystone EC, Genovese MC, Klareskog L, et al. Golimumab, a human antibody to tumour necrosis factor α given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann Rheum Dis. 2009;68(6):789–96.
Tanaka Y, Harigai M, Takeuchi T, et al. Prevention of joint destruction in patients with high disease activity or high C-reactive protein levels: post hoc analysis of the GO-FORTH study. Mod Rheumatol. 2016;26(3):323–30.
Okazaki M, Kobayashi H, Ishii Y, Kanbori M, Yajima T. Real-world treatment patterns for golimumab and concomitant medications in Japanese rheumatoid arthritis patients. Rheumatol Ther. 2018;5(1):185–201.
Sugiyama N, Kawahito Y, Fujii T, et al. Treatment patterns, direct cost of biologics, and direct medical costs for rheumatoid arthritis patients: a real-world analysis of nationwide Japanese claims data. Clin Ther. 2016;38(6):1359–75.
Kanbori M, Suzuka H, Yajima T, et al. Postmarketing surveillance evaluating the safety and effectiveness of golimumab in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2018;28(1):66–75.
Takeuchi T, Miyasaka N, Tatsuki Y, et al. Baseline tumour necrosis factor alpha levels predict the necessity for dose escalation of infliximab therapy in patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(7):1208–15.
Rahman MU, Strusberg I, Geusens P, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:1233–8.
van der Bijl AE, Goekoop-Ruiterman YP, de Vries-Bouwstra JK, et al. Infliximab and methotrexate as induction therapy in patients with early rheumatoid arthritis. Arthritis Rheum. 2007;56:2129–34.
Leu JH, Adedokun OJ, Gargano C, Hsia EC, Xu Z, Shankar G. Immunogenicity of golimumab and its clinical relevance in patients with rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. Rheumatology. 2019;58(3):441–6.
Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13(12):707–18.
Cludts I, Spinelli FR, Morello F, Hockley J, Valesini G, Wadhwa M. Anti-therapeutic antibodies and their clinical impact in patients treated with the TNF antagonist adalimumab. Cytokine. 2017;96:16–23.
Pascual-Salcedo D, Plasencia C, Ramiro S, et al. Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology. 2011;50(8):1445–52.
Ducourau E, Mulleman D, Paintaud G, et al. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. 2011;13(3):R105.
Wolbink GJ, Vis M, Lems W, et al. Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(3):711–5.
Chatzidionysiou K, Askling J, Eriksson J, Kristensen LE, van Vollenhoven R, ARTIS group. Effectiveness of TNF inhibitor switch in RA: results from the national Swedish register. Ann Rheum Dis. 2015;74(5):890–6.
Smolen JS, Kay J, Matteson EL, et al. Insights into the efficacy of golimumab plus methotrexate in patients with active rheumatoid arthritis who discontinued prior anti-tumour necrosis factor therapy: post hoc analyses from the GO-AFTER study. Ann Rheum Dis. 2014;73(10):1811–8.
Virkki LM, Valleala H, Takakubo Y, et al. Outcomes of switching anti-TNF drugs in rheumatoid arthritis—a study based on observational data from the Finnish Register of Biological Treatment (ROB-FIN). Clin Rheumatol. 2011;30(11):1447–54.
Hyrich KL, Lunt M, Watson KD, Symmons DP, Silman AJ, British Society for Rheumatology Biologics Register. Outcomes after switching from one anti-tumor necrosis factor alpha agent to a second anti-tumor necrosis factor alpha agent in patients with rheumatoid arthritis: results from a large UK national cohort study. Arthritis Rheum. 2007;56(1):13–20.
Gomez-Reino JJ, Carmona L, BIOBADASER Group. Switching TNF antagonists in patients with chronic arthritis: an observational study of 488 patients over a four-year period. Arthritis Res Ther. 2006;8(1):R29.
Rubbert-Roth A, Finckh A. Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res Ther. 2009;11(Suppl 1):S1.
Smolen JS, van Vollenhoven RF, Florentinus S, Chen S, Suboticki JL, Kavanaugh A. Predictors of disease activity and structural progression after treatment with adalimumab plus methotrexate or continued methotrexate monotherapy in patients with early rheumatoid arthritis and suboptimal response to methotrexate. Ann Rheum Dis. 2018;77(11):1566–72.
Saevarsdottir S, Wallin H, Seddighzadeh M, et al. Predictors of response to methotrexate in early DMARD naive rheumatoid arthritis: results from the initial open-label phase of the SWEFOT trial. Ann Rheum Dis. 2011;70:469–75.
Wessels JA, van der Kooij SM, le Cessie S, et al. A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 2007;56:1765–75.
Acknowledgements
The authors acknowledge the cooperation of all the patients and physicians who participated in this PMS study. The authors are also grateful to Toshiro Yano (Ikuyaku, Integrated Value Development Division, Mitsubishi Tanabe Pharma Corp.) for cooperation with study conception and design.
Funding
Sponsorship for this study and the Rapid Service Fee were jointly provided by Janssen Pharmaceutical K.K. (Tokyo, Japan) and Mitsubishi Tanabe Pharma Corporation (Osaka, Japan). All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and Other Assistance
The authors thank Hiroki Nakane (EPS Corp.) for performing the statistical analysis. Medical writing support, under the guidance of the authors, was provided by Dr. David Robert McQuire of Yamada Translation Bureau, Inc. Statistical analysis support and medical writing assistance were both funded by Janssen Pharmaceutical K.K. and Mitsubishi Tanabe Pharma Corporation.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Hirohito Shimizu is an employee of Janssen Pharmaceutical K.K., a wholly owned subsidiary of Johnson & Johnson. Hisanori Kobayashi, Masayoshi Kanbori, and Yutaka Ishii are employees of Janssen Pharmaceutical K.K., a wholly owned subsidiary of Johnson & Johnson, and may hold stock and/or stock options in the company.
Compliance with Ethics Guidelines
This article is based on previously conducted PMS study and does not contain any new interventional studies with human participants or animal subjects performed by any of the authors. The PMS study protocol and ethical considerations were assessed by the internal review board members and approved by the Japanese PMDA. This PMS study was registered with the University Hospital Medical Information Network Clinical Trials Registry (UMIN-CTR, Identifier: UMIN000015895), was conducted in accordance with the Japanese regulations (Ministry of Health, Labor and Welfare Ministerial Ordinance No. 171) on Good Post-marketing Study Practice (GPSP), and was carried out by Janssen Pharmaceutical K.K. after a contract with each study center.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available due confidentiality clauses signed with participating medical institutions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to: https://doi.org/10.6084/m9.figshare.11836053.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shimizu, H., Kobayashi, H., Kanbori, M. et al. Effect of Golimumab Dose Escalation in Japanese Patients With Rheumatoid Arthritis: Post-Hoc Analysis of Post-Marketing Surveillance Data. Rheumatol Ther 7, 311–325 (2020). https://doi.org/10.1007/s40744-020-00198-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-020-00198-4