
Abstract
Background
Patients with persistent positive antiphospholipid antibodies (aPLs) and immune thrombocytopenia (ITP) hardly develop thrombosis but share many similar characteristics with antiphospholipid syndrome (APS).
Methods
This is a prospective cohort study consecutively enrolling thrombocytopenic patients with continuous positive aPLs. Patients developing thrombotic events are classified as the APS group. Then we compare the clinical characteristics and prognosis between aPLs carriers and patients with APS.
Results
This cohort included 47 thrombocytopenic patients with continuous positive aPLs and 55 with diagnosed primary APS. The proportion of smoking and hypertension are higher in the APS group (p = 0.03, 0.04, 0.03, respectively). The platelet count of aPLs carriers at admission was lower than APS patients [26 × 109/l (9 × 109/l, 46 × 109/l) vs. 64 × 109/l (24 × 109/l, 89 × 109/l), p = 0.0002]. Triple aPLs positivity is more common in primary APS patients with thrombocytopenia [24 (51.1%) vs. 40 (72.7%), p = 0.04]. Regarding the treatment response, the complete response (CR) rate is similar between aPLs carriers and primary APS patients with thrombocytopenia (p = 0.2). Nonetheless, the proportion of response, no response, and relapse differed significantly between the two groups [13 (27.7%) vs. 4 (7.3%), p < 0.0001; 5 (10.6%) vs. 8 (14.5%), p < 0.0001; 5 (10.6%) vs. 8 (14.5%), p < 0.0001, respectively]. In Kaplan–Meier analysis, primary APS patients had significantly more thrombotic events than aPLs carriers (p = 0.0006).
Conclusions
In the absence of other high-risk factors for thrombosis, thrombocytopenia could be an independent and long-lasting clinical phenotype of APS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
ITP patients with persistent aPLs hardly develop thrombosis. |
We aim to summarize the characteristics of patients with thrombocytopenia and persistent aPLs. |
Patients with thrombocytopenia and persistent positive aPLs (aPLs carriers) could be a phenotype of APS. |
aPLs carriers with thrombotic risk factors should still be ware of thromboembolism. |
Introduction
Antiphospholipid syndrome (APS) is an autoimmune disorder characterized as thrombosis, pregnancy morbidity, and thrombocytopenia with a distinct serological feature, which is positive antiphospholipid antibodies (aPLs) [1]. These aPLs are also frequently detected in immune thrombocytopenic purpura (ITP), a bleeding disorder resulting from antibody-mediated platelet destruction [2]. Harris et al. first discovered that aPLs were present in 30% of ITP patients at the time of diagnosis [3]. Multiple studies followed ITP patients with positive aPLs and found that most of them did not have thromboses like APS patients during years of follow-up [4,5,6].
The association between ITP and the occurrence of thrombosis has been controversial. A recent study analyzed risk factors for thrombosis, treatment, and platelet count of ITP patients, and concluded that the 5-year cumulative incidence rate of thrombosis in ITP is well below the predefined thresholds [4]. Yet, several studies have found that ITP patients with positive aPLs tend to develop thromboses compared to those without aPLs, and their clinical and laboratory characteristics differ [7, 8].
Thrombocytopenia is a common manifestation of APS [9]. There is a group of patients characterized as thrombocytopenia and persistent positive aPLs who do not develop thrombosis after a rather long period of follow-up. We believe this group of patients might be a clinical phenotype of APS. In this study, we compared aPLs carriers with thrombocytopenia and primary APS patients with thrombocytopenia from different aspects to further understand their characteristics.
Methods
This is a prospective cohort study. The inclusion criteria are: confirmed positive aPLs separated by a minimum of 12 weeks according to the 2006 Sydney classification criteria, thrombocytopenia, and continuous follow-up. Thrombocytopenia was defined as a platelet count < 100 × 109/l following the criterion used for the diagnosis of immune thrombocytopenia [10]. Patients with other underlying autoimmune diseases (e.g., systemic lupus erythematosus, rheumatoid arthritis, etc.) and other causes of thrombocytopenia were excluded. Bone marrow punctures indicated impairment of megakaryocyte maturation. Data of 112 patients were extracted from the Chinese Rheumatism Data Center (CRDC) at Peking Union Medical College Hospital from August 2015 to September 2021. Eight were lost during follow-up, two had incomplete data. Among the 102 patients, 55 (53.9%) who had thrombosis or pregnancy morbidity according to the 2006 Sydney revised classification of APS were classified as the APS group. Forty-seven (46.1%) patients who presented as thrombocytopenia and persistent positive aPLs but could not meet the classification criteria of APS were referred to as aPLs carriers. This study was conducted in accordance with Declaration of Helsinki principles and followed the International Conference on Harmonization Good Clinical Practice Guidelines. Written informed consent was obtained from all patients before enrollment. This study was approved by the medical ethics committee of Peking Union Medical College Hospital (approval number: JS-2038).
Patients were seen at 3- to 6-month intervals depending on their clinical status for evaluation of complete blood count, urine analysis, renal and liver biochemistry, complement levels, coagulation tests, aPLs and clinical events. Endpoint events include thrombotic events as confirmed by vascular ultrasound or other objective imaging modalities. Treatment response of thrombocytopenia was defined according to the guidelines of immune thrombocytopenia of the American Society of Hematology [10].
Levels of anticardiolipin (aCL) and anti-β2-glycoprotein (GP) I antibody isotypes were quantified by QUANTA Lite™ ELISA kits provided by INOVA diagnostics, Inc (San Diego, CA, USA). The defined cut-off values were > 40GPL/MPL. LA was measured by dilute Russel viper venom time (dRVVT), with the ratio above 1.20 considered positive. The manufacturer’s recommendations were followed carefully.
The Shapiro–Wilk test was performed to assess normality. Values are expressed as median and 25th and 75th percentiles [quantile 1 (Q1) and Q3, respectively] for continuous variables and numbers and percentages for categorical variable. Mann–Whitney U test was used to compare data not following a normal distribution. Chi-square (χ2) and Fisher’s exact tests were used to compare categorical variables. Cox regression analysis was used to detect predictors of events expressed as hazard ratio (HR) and 95% confidence interval (CI). Survival curves were determined by Kaplan–Meier survival analysis. A p value < 0.05 (two-sided) was considered statistically significant. Statistical analyses were performed using R statistical software, version 3.6.2 (http://www.R-project.org/).
Results
This cohort consecutively enrolled 102 patients with continuous positive aPLs and thrombocytopenia, 55 (53.9%) of which fulfilled the classification criteria for APS (Fig. 1). Patients were followed up for 826 patient-years. The overall median follow-up time is 5 years [quantile 1 (Q1) 2 years, Q3 9 years]. The minimum follow-up duration in this cohort was 1 year. The demographic and clinical characteristics on baseline are presented in Table 1. The range of age, gender, and disease duration is similar between aPLs carriers and primary APS patients (p = 0.2, 0.1, 0.7, respectively). Remarkably, the proportion of multiple risk factors for thrombosis was significantly higher in primary APS patients than aPLs carriers [6 (12.8%) vs. 19 (34.5%), p = 0.03 for smoking; 6 (12.8%) vs. 18 (32.7%), p = 0.04 for hypertension; 11.9 (10.3, 15) vs. 14.6 (11.3, 17.4), p = 0.03 for homocysteine (μmol/l)]. The prevalence of other APS-related clinical complications is generally low, although more primary APS patients had nephropathy than aPLs carriers [7 (12.7%) vs. 0, p = 0.01]. The treatment of studied patients is also summarized in Table 1. The proportion of anticoagulant therapy was higher in primary APS patients (76.4 vs. 12.8%, p < 0.001). Immunosuppressants (mainly including tacrolimus, sirolimus, cyclosporine A, azathioprine, mycophenolate mofetil, and cyclophosphamide, rituximab treatment was used rarely) were used for thrombocytopenia and other extra-criteria manifestations. The platelet count at admission was significantly lower in aPLs carriers [26 (× 109/l) (9, 46) vs. 64 (24, 89), p = 0.0002] (Table 2). Antibody profiles are summarized in Table 2. Triple aPLs positivity is more common in primary APS patients with thrombocytopenia [24 (51.1%) vs. 40 (72.7%), p = 0.04], although the proportion of positive single aPL is similar between the two groups (Table 2).

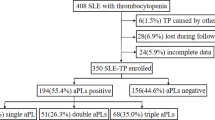
Flow chart of patient selection. A total of 189 patients at Peking Union Medical College Hospital had thrombocytopenia and positive aPLs separated by a minimum of 12 weeks. Seventy-seven were secondary APS. Of the 112 patients, two had incomplete data and eight were lost during follow-up. One hundred and two patients with thrombocytopenia were included in this cohort, of whom 55 were diagnosed with primary APS; 47 were aPLs carriers. aPLs antiphospholipid antibodies, APS antiphospholipid syndrome
We compared the treatment response of the studied group in Table 3 and found that only the rate of CR was similar between aPLs carriers and patients with APS (p = 0.2). More patients had response to treatment in aPLs carriers and less had NR and relapse (all p < 0.0001). Multivariate Cox regression was performed to distinguish factors associated with occurrences of thrombosis. Age, gender diagnosis of APS, smoking, hypertension, and three aPLs were analyzed, and diagnosis of APS was the only independent risk factor of thrombosis (HR 4.4, 95% CI 1.5–13.2, p = 0.008). In the course of at least 16 years, aPLs carriers hardly developed thrombosis while 67% of the thrombotic APS patients had recurrence of thrombosis. The Kaplan–Meier analysis is presented in Fig. 2. The profiles of four aPLs carriers who had thrombosis during follow-up were recorded in Table 4.

Kaplan–Meier curve of thrombotic events between aPLs carriers and primary APS patients with thrombocytopenia. p < 0.05, statistically significant. aPLs antiphospholipid antibodies, PAPS primary antiphospholipid syndrome
Discussion
This study compared aPLs carriers and primary APS patients both with thrombocytopenia. Differences in traditional cardiovascular risk factors were evident in terms of smoking and hypertension. Previous studies have illustrated that smoking and hypertension could induce thrombotic events, especially arterial thrombosis [11,12,13,14]. Homocysteine has been shown to trigger thrombosis [15]. Kassis et al. [16] reported an increased risk of venous and arterial thrombosis in patients with aPLs and hyperhomocysteinemia. While noticeable differences were not discovered in most clinical manifestations, which is likely due to the low incidence rates, aPLs-associated nephropathy was more frequent in APS patients. In our cohort, no clinical signs of nephropathy were found in aPLs carriers with thrombocytopenia. Lande et al. [17] and Nishizawa et al. [18] reported cases of membranous nephropathy complicated by ITP, but the aPLs profile was not further elucidated. The proportion of each separate kind of aPLs was similar between the two groups, except that LA was more frequent in thrombocytopenic APS patients but lacked statistical significance. Diz et al. [19] analyzed aCL and LA positivity of ITP patients with aPLs and APS patients, and found that the patients positive for LA was significantly higher in APS. Meanwhile, another research found mean titers of aCL and anti-β2GPI comparable between patients with ITP and APS [5]. Triple aPLs positivity has been suggested to be an independent risk factor for thrombosis in aPLs carriers and patients diagnosed with APS [20, 21].
Overall, aPLs carriers responded better to treatment than APS patients with thrombocytopenia. In multiple studies that compared ITP patients with and without aPLs, their treatment response did not seem to differ [7, 8, 22]. One particular study directly compared treatment response of ITP patients with positive aPLs and confirmed APS patients but without thrombocytopenia and also found no significant difference [19]. These discrepancies might be explained by the different patient selection. The reason why the two groups in our study manifest such evident difference in treatment response needs to be further discovered.
During follow-up, approximately half of thrombocytopenic patients with primary APS had a recurrence of thrombosis. In previous studies, the recurrence rate of thrombosis ranged from 16.6 to 45% among patients with APS during a 5-year period [23, 24]. As for ITP patients with positive aPLs, the occurrence rate of a first thrombotic event was 4.1–21.7% during a 3–5-year follow-up time [6,7,8, 22]. In our study, the incidence rate of thrombosis of aPLs carriers in over 16 years was 8.5% (4/47). This could indicate that the rate might not grow after a rather long period of follow-up. Information on the four aPLs carriers that developed thrombosis during follow-up is summarized in Table 4.
The differences of demographic characteristics and prognoses between the two groups could be explained by the second hit theory. This hypothesis has been suggested to explain the clinical observation that aPLs (first hit) might be persistently present but that thrombotic events only occur occasionally [25]. Inflammation, infection, and complement activation has been proposed as the possible second hit [25,26,27]. In this case, the cardiovascular risk factors could be serving as a second hit to cause reoccurrence of thrombosis in APS patients and the first thrombosis of ITP patients.
Although the role of thrombocytopenia in APS has raised much attention, it has not been formally included in the classification criteria of APS. The definition of other categories such as “possible” and “probable” APS was proposed [28]. Barbhaiya et al. [29] developed new classification criteria for APS including hematologic manifestation, which is a low platelet count < 150 × 109/l. A new version of classification criteria for APS with high sensitivity and specificity is expected, and we believe thrombocytopenia should be included.
There are some limitations in our study. The relatively small sample size might statistically widen the confidence interval and underestimate the differences in the clinical and biochemical findings. How the second hit serves as a trigger to promote thromboembolism should be further explored in future research.
Conclusions
In our cohort, aPLs carriers with thrombocytopenia share many similar manifestations with primary APS patients but present less thrombotic risk factors, and hardly develop thrombotic events. The distinct antibody profile indicated that aPLs carriers are not simply a manifestation of ITP. The differences between them indicate that thrombocytopenia could be a long-lasting clinical phenotype of APS in the absence of a second hit. For this group of patients, thrombotic risk factors should particularly be avoided. Anticoagulant therapy could be applied during the perioperative period and other high-risk events for thrombosis.
References
Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, de Groot PG, Koike T, Meroni PL, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295–306.
Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, Chong BH, Cines DB, Gernsheimer TB, Godeau B, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115(2):168–86.
Harris EN, Gharavi AE, Hegde U, Derue G, Morgan SH, Englert H, Chan JK, Asherson RA, Hughes GR. Anticardiolipin antibodies in autoimmune thrombocytopenic purpura. Br J Haematol. 1985;59(2):231–4.
Ruggeri M, Tosetto A, Palandri F, Polverelli N, Mazzucconi MG, Santoro C, Gaidano G, Lunghi M, Zaja F, De Stefano V, et al. Thrombotic risk in patients with primary immune thrombocytopenia is only mildly increased and explained by personal and treatment-related risk factors. J Thromb Haemost. 2014;12(8):1266–73.
Moulis G, Delavigne K, Huguet F, Fortenfant F, Beyne-Rauzy O, Adoue D. Antiphospholipid antibodies and the risk of thrombosis: a comparative survey between chronic immune thrombocytopenia and primary antiphospholipid syndrome. Rev Med Interne. 2011;32(12):724–9.
Cruz ACL, Colella MP, De Paula EV, Annichinno-Bizzachi J, Orsi FA. Clinical course of primary immune thrombocytopenia with positive antiphospholipid antibodies. Eur J Intern Med. 2019;69:e6–7.
Yang YJ, Yun GW, Song IC, Baek SW, Lee KS, Ryu HW, Lee MW, Lee HJ, Yun HJ, Kim S, et al. Clinical implications of elevated antiphospholipid antibodies in adult patients with primary immune thrombocytopenia. Korean J Intern Med. 2011;26(4):449–54.
Kim KJ, Baek IW, Yoon CH, Kim WU, Cho CS. Thrombotic risk in patients with immune thrombocytopenia and its association with antiphospholipid antibodies. Br J Haematol. 2013;161(5):706–14.
Sciascia S, Amigo MC, Roccatello D, Khamashta M. Diagnosing antiphospholipid syndrome: “extra-criteria” manifestations and technical advances. Nat Rev Rheumatol. 2017;13(9):548–60.
Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190–207.
de Souza AW, Silva NP, de Carvalho JF, D’Almeida V, Noguti MA, Sato EI. Impact of hypertension and hyperhomocysteinemia on arterial thrombosis in primary antiphospholipid syndrome. Lupus. 2007;16(10):782–7.
Erkan D, Yazici Y, Peterson MG, Sammaritano L, Lockshin MD. A cross-sectional study of clinical thrombotic risk factors and preventive treatments in antiphospholipid syndrome. Rheumatology (Oxford). 2002;41(8):924–9.
Li R, Zhou YS, Jia Y, Li ZG. Analysis of risk factors in development of thrombosis in patients with antiphospholipid syndrome. Beijing Da Xue Xue Bao Yi Xue Ban. 2012;44(5):788–91.
Girón-González JA, García del Río E, Rodríguez C, Rodríguez-Martorell J, Serrano A. Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals. J Rheumatol. 2004;31(8):1560–7.
Di Minno MN, Tremoli E, Coppola A, Lupoli R, Di Minno G. Homocysteine and arterial thrombosis: Challenge and opportunity. Thromb Haemost. 2010;103(5):942–61.
Kassis J, Neville C, Rauch J, Busque L, Chang ER, Joseph L, Le Comte M, Subang R, Fortin PR. Antiphospholipid antibodies and thrombosis: association with acquired activated protein C resistance in venous thrombosis and with hyperhomocysteinemia in arterial thrombosis. Thromb Haemost. 2004;92(6):1312–9.
Lande MB, Thomas GA, Houghton DC. Membranous nephropathy associated with chronic immune thrombocytopenic purpura in childhood. Am J Kidney Dis. 2001;37(5):E40.
Nishizawa K, Yamashita T, Ogawa Y, Kobayashi H. Membranous nephropathy complicated by immune thrombocytopenia treated with low-density lipoprotein apheresis: a case report and literature review. CEN Case Rep. 2022;11(1):43–9.
Diz-Küçükkaya R, Hacihanefioğlu A, Yenerel M, Turgut M, Keskin H, Nalçaci M, Inanç M. Antiphospholipid antibodies and antiphospholipid syndrome in patients presenting with immune thrombocytopenic purpura: a prospective cohort study. Blood. 2001;98(6):1760–4.
Frison L, Lombardi A, Caputo I, Semenzato G, Fabris F, Vianello F. Relevance of antiphospholipid antibody profile in the clinical outcome of ITP: a single-centre study. Hematology. 2019;24(1):134–8.
Hernández-Molina G, Espericueta-Arriola G, Cabral AR. The role of lupus anticoagulant and triple marker positivity as risk factors for rethrombosis in patients with primary antiphospholipid syndrome. Clin Exp Rheumatol. 2013;31(3):382–8.
Pierrot-DeseillignyDespujol C, Michel M, Khellaf M, Gouault M, Intrator L, Bierling P, Godeau B. Antiphospholipid antibodies in adults with immune thrombocytopenic purpura. Br J Haematol. 2008;142(4):638–43.
Bazzan M, Vaccarino A, Stella S, Sciascia S, Montaruli B, Bertero MT, Carignola R, Roccatello D. Patients with antiphospholipid syndrome and thrombotic recurrences: a real-world observation (the Piedmont cohort study). Lupus. 2016;25(5):479–85.
Cervera R, Khamashta MA, Shoenfeld Y, Camps MT, Jacobsen S, Kiss E, Zeher MM, Tincani A, Kontopoulou-Griva I, Galeazzi M, et al. Morbidity and mortality in the antiphospholipid syndrome during a 5-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. 2009;68(9):1428–32.
Shoenfeld Y, Blank M, Cervera R, Font J, Raschi E, Meroni PL. Infectious origin of the antiphospholipid syndrome. Ann Rheum Dis. 2006;65(1):2–6.
De Angelis V, Scurati S, Raschi E, Liutkus A, Belot A, Borghi MO, Meroni PL, Cimaz R. Pro-inflammatory genotype as a risk factor for aPL-associated thrombosis: report of a family with multiple anti-phospholipid positive members. J Autoimmun. 2009;32(1):60–3.
Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, Gavriilaki E, Alluri R, Streiff MB, Petri M, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. 2020;135(4):239–51.
Oztürk MA, Haznedaroğlu IC, Turgut M, Göker H. Current debates in antiphospholipid syndrome: the acquired antibody-mediated thrombophilia. Clin Appl Thromb Hemost. 2004;10(2):89–126.
Barbhaiya MZS, Ahmadzadeh Y, Costenbader K, Naden R, Erkan D. Development of new international classification criteria for antiphospholipid syndrome: phase III case collection results [abstract]. Arthritis Rheumatol. 2020;72 (suppl 10). https://acrabstracts.org/abstract/development-of-new-international-classification-criteria-for-antiphospholipidsyndrome-phase-iii-case-collection-results/
Acknowledgements
The authors would like to thank MD, Ph.D. Bruce L. Davidson for the review and kind suggestions of our manuscript. The authors would also like to thank the study participants for their involvement in the study.
Funding
This work and journal’s Rapid Service Fee was supported by the Chinese National Key Technology R&D Program, Ministry of Science and Technology (2021YFC2501301-5, 2017YFC0907601-3); Beijing Municipal Science & Technology Commission (Z201100005520022, 23, 25-27); CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-005) National High Level Hospital Clinical Research Funding (2022-PUMCH-A-008).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contribution
Xiaofeng Zeng and Jiuliang Zhao: conceptualization and funding acquisition. Yu Shi: data curation and analysis, writing. Hui Jiang: data curation. Yongqiang Zhao: review of the original draft. Mengtao Li: methodology.
Disclosures
Yu Shi, Hui Jiang, Yongqiang Zhao, Jiuliang Zhao, Mengtao Li, Xiaofeng Zeng have nothing to disclose.
Compliance with Ethics Guidelines
This study was approved by the medical ethics committee of Peking Union Medical College Hospital (approval number: JS-2038). This study was conducted in accordance with Declaration of Helsinki principles and followed the International Conference on Harmonization Good Clinical Practice Guidelines. Written informed consent was obtained from all patients before enrollment.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding authors
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shi, Y., Jiang, H., Zhao, Y. et al. Immune Thrombocytopenia Could be an Independent Clinical Phenotype of Antiphospholipid Syndrome: A Prospective Cohort Study. Rheumatol Ther 10, 649–658 (2023). https://doi.org/10.1007/s40744-023-00538-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-023-00538-0