
Abstract
Background
Sickle cell disease (SCD) is a complex genetic disorder that manifests in infancy and progresses throughout life in the form of acute and chronic complications. As the upfront costs of potentially curative, genetic therapies will likely be high, an assessment and comprehensive characterization of the medical and non-medical cost burden will inform future decision making.
Objective
We sought to systematically summarize the existing literature surrounding SCD medical and non-medical costs.
Methods
We searched MEDLINE and EMBASE (2008–2020) and identified US-based studies that detailed medical or non-medical costs. Eligible studies provided empirical estimates about any aspect of cost or SCD individuals of all ages and their caregivers. Study quality was assessed using the Newcastle–Ottawa Scale, and costs were adjusted to 2019 US$.
Results
Search queries returned 479 studies, with 342 from medical burden searches and 137 from non-medical burden searches, respectively. Herein, we report the results of the 40 studies that contained relevant cost information: 39 detailed medical costs and 1 detailed non-medical costs. Costs were higher for SCD patients when compared with non-SCD individuals (cost difference range: $6636–$63,436 annually). The highest medical cost component for SCD patients was inpatient ($11,978–$59,851 annually), followed by outpatient and then pharmacy. No studies characterized the cost burden throughout the lifetime disease trajectory of an SCD individual, and no studies captured caregiver or productivity costs.
Conclusion
Our results reveal an incomplete characterization of medical and non-medical costs within SCD. A deeper understanding of the medical and non-medical cost burden requires completion of additional studies that capture the burden across the patient’s lifetime, in addition to expression of the impact of existing and emergent health technologies on disease trajectory.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This review highlights several gaps within the existing literature for both medical and non-medical costs. Medical costs should include a lifetime horizon and explore insurance type in greater detail. Non-medical cost literature is completely lacking outside of a caregiver burden proxy (lifetime income). |
The available cost literature for sickle cell disease (SCD) is heterogenous in study design, data sources, population, and analysis methods. These differences all contribute to difficulty in conducting meta-analyses for further synthesis beyond what we present. |
Future US cost research should give more detailed attention to medical and non-medical costs to fairly evaluate new and existing health technologies for SCD patients, referring to the Second Panel on Cost Effectiveness in Health and Medicine to gain insight on important cost measurements necessary for value considerations. |
1 Introduction
Sickle cell disease (SCD) refers to a group of single-gene, autosomal recessive disorders characterized by the presence of at least one β-globin allele with a sickle mutation, and a second, pathogenic variant permissive for abnormal hemoglobin polymerization. Excessive polymerization leads to red blood cell (RBC) membrane changes and fragility, which trigger a complex array of pathophysiologic pathways, resulting in occlusion of the microvasculature, dysregulation of nitric oxide, and inflammation, all of which contribute to pain and progressive organ damage over the individual’s lifetime. As such, SCD is associated with significant morbidity and an overall increased risk of mortality, as seen by up to 50% of individuals surviving past their fifth decade of life [1,2,3], translating to a reduced lifetime income of approximately $695,000 [4]. An estimated 100,000 individuals in the US are affected, with prevalence highest in African Americans [5].
Due to the progressive damage leading to both acute and chronic complications, individuals frequently visit acute and ambulatory healthcare settings to receive treatment, both of which contribute to the cost of care. Complications include acute events such as vaso-occlusive crises (VOC), infarctions, and acute chest syndrome. Similarly, chronic complications include cardiovascular disease, renal disease, neurodegenerative decline, and bone destruction. The armamentarium of treatment options is limited. Hydroxyurea is recommended for all patients with the most severe forms and has proven efficacy for the reduction in VOC and hospitalizations, but the drug is underprescribed and often not taken regularly [6, 7]. Blood transfusions have also documented efficacy [8], however they can lead to alloimmunization and result in iron overload over time [9, 10]. Three new agents have recently received US FDA approval: l-glutamine, voxelotor, and crizanlizumab, although their optimal use is still being established [11]. As these agents impact different pathophysiologic pathways, they offer the potential for multidrug therapy. Curative therapies such as hematopoietic stem cell transplant (HSCT) are available, although graft-versus-host disease may pose a significant concern, depending on the genetic match between donor and recipient as well as the source of donor cells. Lastly, the efficacy and safety of genetic therapies, aimed at modifying a patient’s own red cells so they are no longer sickle, are currently being investigated [12].
Taken together, ongoing vascular and organ damage, underutilization of hydroxyurea, and known adverse effects of effective therapies contribute to healthcare resource use (HCRU). HCRU increases are reflected by increases in economic burden, which we define as the combination of both medical and non-medical costs. Medical costs are those that originate in the formal healthcare sector as outlined by the Second Panel on Cost-Effectiveness in Health and Medicine, including areas such as drug, diagnostic, laboratory, and inpatient costs, to name a few [13]. Non-medical costs are those that then occur outside of the formal healthcare sector, including productivity, patient time, and education costs. One published systematic literature review captures the impact of SCD on health-related quality of life and HCRU [14]. This study reports on hospital length of stay, emergency department (ED) visits, rehospitalizations, and several other outcomes related to resource use; however, the authors do not extend resource use to a cost calculation and thus did not explicitly evaluate the economic burden. Given the recent expansion in treatment options within the US and the emergence of gene therapies, the need arises to properly assess their value in terms of what healthcare spending can expect to gain in health outcomes; therefore, a comprehensive and up-to-date summarization of medical and non-medical costs is warranted [15]. Our objective was to summarize the existing literature that describes both medical and non-medical costs associated with SCD, categorizing these costs into those that describe general burden, acute or chronic complications, treatment, and treatment complications. We undertook this work as part of the National Heart Lung and Blood Institute Cure Sickle Cell Initiative. We are members of the Clinical and Economic Impact Analysis group (https://curesickle.org).
2 Methods
2.1 Search Methods and Sources
We conducted a systematic review and landscape analysis following methods of the Cochrane Collaboration and the Agency for Healthcare Research and Quality (AHRQ) guides for systematic reviews, and adopted the population, intervention, comparator, outcomes, timing and setting/study design (PICOTS) framework to establish eligibility criteria (Appendix Table A1) [16, 17]. The adopted PICOTS framework reflects deliberations and decisions made over a 3-month period in late 2019 by an expert panel that included molecular biologists, clinicians who care for patients with SCD, health economists, evidence synthesis scientists, librarians, and patients with SCD. These stakeholders represent academia, clinical practice, the federal government, and the SCD population. This framework was then executed as multiple search strategies in the MEDLINE and EMBASE databases by an experienced health sciences librarian with expertise in evidence synthesis; see the electronic supplementary material for details on terms specifically related to the individual searches (Appendix List A1). These searches were run again in October 2020 to update the body of literature. Duplicates were removed and returned studies were screened for eligibility. For those that met the inclusion criteria, relevant data were extracted and synthesized. The content of this report aligns with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement for reporting of systematic reviews (see Appendix C for PRISMA checklists) [18].
2.2 Eligibility Criteria
We included US-based, English-language articles published in peer-reviewed journals from January 2008 to September 2020 and white papers from January 2018 to September 2020, reasoning that information provided in older white papers would not reflect current research findings. Eligible studies included those from the medical and non-medical cost searches that reported any costs related to SCD generally, acute or chronic complications, treatments, or treatment complications (Appendix List A1). Studies that simulated costs due to SCD were outside the scope of this work because we sought to summarize empirical cost estimates for our synthesis. Costs could be either the primary or secondary outcome. Included treatments were standard care therapies such as hydroxyurea and RBC transfusion, curative therapies (HSCT) with all matching types (matched unrelated donor, matched sibling, human leukocyte antigen identical sibling, or allogeneic) under both myeloablative and non-myeloablative regimens, and newer disease-modifying therapies (l-glutamine, voxelotor, and crizanlizumab). Individuals with sickle cell trait were excluded from our analysis as their β-hemoglobin gene carries one allele for the sickled cell mutation and one normal allele; these individuals commonly do not express the symptoms of SCD [19] and therefore have different implications for resource use.
2.3 Study Selection
Records identified through the databases and in consultation with experts were merged into the systematic review software—Distiller SR™ (Evidence Partners, V2.30.6). After duplicates were removed, one reviewer (ZB for the medical search, BJ for the non-medical search) independently screened the titles and abstracts of all the references, excluded those that did not meet the defined criteria, and assessed the full text of the remaining studies for eligibility.
2.4 Data Collection
One reviewer (ZB for the medical search, BJ for the non-medical search) extracted the main characteristics of studies into Microsoft® Excel (Microsoft Corporation, Redmond, WA, USA), including primary author, publication date, study design, sample size, population, age, data source, cost components (defined as inpatient, outpatient, pharmacy, ED, productivity, caregiver, patient time, and other), unit of analysis (per patient or per admission), cost year, adjusted and unadjusted dollar costs, and study-specific adjustment factors (such as age, sex, disease status, and medication use status). For studies that included figure-based costs without an accompanying table, WebPlotDigitizer© (automeris.io/WebPlotDigitizer) was used. A second reviewer (EC) then randomly sampled 50% of the included texts and validated the data extracted. Discrepancies were resolved between reviewers, and a third reviewer arbitrated (BJ or BD), when necessary. Studies were separated into four groups based on the economic burden reported: burden between SCD and non-SCD patients; and, within SCD, the burden associated with acute or chronic complications, treatments, and treatment complications. We did not preferentially extract selected dollar amounts but rather we reported all costs reported in the studies.
2.5 Quality Assessment
One reviewer (ZB) assessed the quality of included studies using the Newcastle–Ottawa Scale (NOS), as it was the most applicable scale given that the study sample consisted solely of non-randomized studies [20]. The NOS is adaptable to cross-sectional, cohort, and case-control studies. Assessment dimensions fall into three categories: selection, comparability, and outcome. These three categories address factors that can lead to biased study results, which would in turn affect study quality. We then weighted the scores within each dimension and applied the AHRQ standards of good, fair, or poor for assessment of overall study quality. We report both NOS dimension-specific and AHRQ ratings, as converted. As costs were sometimes a secondary outcome within a study, study authors did not always adjust their estimated costs by relevant covariates; we downrated these quality scores. A second reviewer (EC) quality-rated a random sample of 50% of the included articles. Discrepancies were resolved between reviewers and brought to a third reviewer (BJ or BD) to arbitrate, if necessary.
2.6 Synthesis of Results
Medical cost results were sorted by unit of analysis, either per patient or per admission. Patient-related costs were annualized when possible and adjusted to 2019 US$ using the Consumer Price Index (CPI) (https://www.usinflationcalculator.com/). The CPI was chosen for the adjustment index as our search sought non-medical costs in addition to medical costs, therefore purely adjusting off a medical-specific index would improperly adjust non-medical costs. Per admission costs were also adjusted for inflation, although these were not annualized as they represent a more transient economic burden to the healthcare system. Hospital charges were adjusted to reflect actual costs using a nationalized cost-to-charge ratio (CCR) of 0.37, which was derived from the 2017 Medicare Hospital Cost Report [21]. Due to high methodological heterogeneity among the included studies, it was not feasible to conduct a meta-analysis on costs from either a categorical or unit of analysis perspective; rather, we qualitatively synthesized the evidence by clinically meaningful categories and present these as the ranges (minimums and maximums) of the costs reported, further breaking them down by unit of analysis (per patient and per admission). We present these ranges across studies and most often report mean costs, unless otherwise specified. Incremental costs represent the difference associated with specific comparisons within studies, as expressed by the different population arms. We collapsed the underutilizing population (defined as medication possession ratio < 80% [22, 23] or by lowest tertile [24]) with the population that did not use any medication, to better capture the impact of interventions on resource utilization under ideal circumstances. We made no adjustments beyond those reported by the authors of individual studies. The first category for reported results is for studies that compare costs for patients with SCD versus those without SCD. For studies comparing costs within SCD patients, we report costs across the four original categories, with one additional category: general disease burden, acute or chronic complications, treatments, and treatment complications, and payer type. The additional category of payer type was agreed upon based on clinical importance after discussion among the authors and expert panel. Detailed descriptions of the initial reported dollar amounts are included in the evidence tables in Appendix B.
3 Results
3.1 Study Demographics
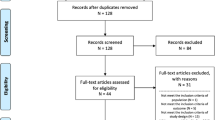
In total, we retrieved 479 sources from the literature; 342 for medical costs and 137 for non-medical costs. For medical costs, after removal of duplicates, we screened 313 abstracts; a full-text assessment was conducted on 151 articles, and 39 studies that met our inclusion criteria were evaluated. After removal of the 137 results returned from the non-medical cost search, we screened 108 abstracts and conducted a full-text assessment for 10 of these. One of these studies met our inclusion criteria and was included in our analysis. Herein, we report on the 40 studies (Fig. 1) [22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61]. The majority of studies (80%) were retrospective cohort studies [22,23,24, 26,27,28,29, 31,32,33,34,35, 37,38,39,40,41,42,43,44, 46,47,48,49,50, 53, 54, 56, 58,59,60,61], with a similar proportion of studies that explored patients of all ages (40%) [22, 23, 31, 32, 34, 35, 37, 38, 41, 43, 43, 52, 58,59,60,61] and under 18 years of age (37%) [33, 36, 39, 40, 44,45,46, 49,50,51, 53,54,55, 57]. Data sources were varied. Thirty percent of studies used information from Medicaid administrative claims data [22,23,24, 29, 31, 32, 35, 44, 47, 48, 59, 61]. Data for 25% of the studies were sourced from the Healthcare Cost and Utilization (HCUP) databases hosted by the AHRQ [25,26,27,28, 34, 36,37,38, 40, 50]. The HCUP databases provide the largest collection of longitudinal hospital care data in the US. Medical records [39, 51,52,53, 56, 57] and commercial claims databases [41,42,43, 47, 58, 60] each contributed data to 15% of the studies. Smaller proportions of studies were conducted using medical registries (7%) [30, 54, 55], the Pediatric Health Information System (7%) [33, 46, 55], and post hoc analysis of RCT data (5%) [45, 49].

PRISMA flow diagram. PRISMA Preferred Reporting Items for Systematic Reviews and Meta-Analyses
There was an equal split in the pattern for reported units of analysis, with per patient (47%) [22,23,24, 29, 31, 32, 35, 42, 43, 47,48,49, 54,55,56, 58,59,60,61] and per hospital admission (50%) [25,26,27,28, 30, 33, 34, 36,37,38,39,40,41,42, 44, 46, 50, 51, 53, 57] as the most frequently reported units. Inpatient costs [22, 23, 25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43, 46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61] were the most frequently reported cost component, followed by outpatient [23, 24, 31, 32, 35, 39, 42,43,44, 47, 48, 51, 54, 56, 58,59,60,61], pharmacy [22,23,24, 31, 32, 35, 42, 43, 47, 48, 56, 58,59,60,61], and ED costs [22, 23, 29, 31, 35, 36, 43, 44, 47, 48, 59,60,61]. The remaining cost components seen were classified as other and consisted of home health [39, 60], hospice [48], laboratory [48, 56, 60], surgery [48], physician [22], chelation [60], transfusion [60], office [48, 60], and other [35, 48, 60]. Only one study captured the income difference between SCD households [45]. No studies reported on productivity or caregiver burden associated with SCD individuals (Table 1).
The number of studies that we rated as good (n = 21) or poor (n = 18) quality were similar, with the remaining study given a fair rating (Table 2, Appendix Table B5). The low ratings in all studies that were rated poor were driven by a low NOS score in the comparability domain due to a lack of rigorous statistical adjustment for confounders on the cost result reported (Appendix Table B5).
3.2 Sickle Cell Disease (SCD) Versus Non-SCD
Five studies compared costs for those with versus those without SCD (Table 3, Appendix Table B1). Inpatient costs were the major driver for total medical cost burden in both cohorts, with 84–96% of total costs for SCD due to inpatient costs, compared with 81–84% for non-SCD (Appendix Table B1) [29]. Mean differences in general inpatient costs between SCD and non-SCD individuals ranged from $6636 to $63,436 per patient annually (Table 3). With respect to specific admission procedures, SCD individuals experienced a higher economic burden than non-SCD individuals during post-partum recovery [26], appendectomy, cholecystectomy, and hysterectomy [27]. In a study that explored cholecystectomy, authors reported that during admission for either an elective or acute procedure, the incremental costs due to SCD were $406 and $6510, respectively [28]. There was no significant difference in mean admission costs for ischemic bowel disease, although the magnitude of inpatient costs was $1072 greater for SCD individuals (Appendix Table B1) [25]. Cost differences for complications cited with high incidence in SCD populations (for example, stroke or acute kidney injury) were not explored in comparison with the general non-SCD population. No studies explored the differences in non-medical costs between SCD and non-SCD individuals.
3.3 SCD Burden
3.3.1 General SCD Burden
Eight studies explored costs within a general SCD setting (Table 4, Appendix Table B2). The annual aggregate economic burden for SCD across all ages in the US was cited as $864 million for inpatient admissions [34], while aggregate costs for SCD individuals under 18 years of age ranged from $164 million to $189 million (Appendix Table B2) [34, 36]. Within the general SCD population, total annual costs per patient ranged from $14,012 to $80,842 [31, 35, 47, 48]; inpatient costs were again frequently cited as the highest contributor to overall economic burden (Table 4). Per admission inpatient costs ranged from $5956 to $17,024 [30, 33, 37, 48]. One study found that mental health, when categorized by Short Form-12 scores, was not associated with significant differences in admission costs [30].
For studies that captured the general economic burden of SCD, age over or under 18 years was a frequently noticed cut-point among reported costs when using three different units of analysis: per individual admission, aggregate admission, and per patient [31, 33,34,35]. Across those three units of analysis, individuals over 18 years of age incurred higher costs. Inpatient costs led for both age groups, followed by pharmacy and outpatient costs. Cumulative estimates of lifetime healthcare costs for a 45-year-old with SCD were $1.2 million undiscounted and $602,000 when discounted 3% annually [35]. Age-specific annual contributions to this total rose after 18 years of age and peaked at approximately 30–39 years of age [34, 35]. The magnitude of medical cost burden was greater for patients ensured by Medicaid when compared with those insured by Medicare or commercial plans [47]. No studies explored non-medical costs in the general SCD population.
3.3.2 SCD Burden Associated with Acute or Chronic Complications
Eleven studies assessed the cost burden of either acute or chronic complications within SCD and compared those who experienced each with those who did not (Table 5, Appendix Table B2). The impact associated with specific complications varied widely over organ system involved (Table 5). Medical costs for acute complications ranged from $2616 [44] to $93,868 [42], represented by depression- and stroke-related admissions, respectively. Stroke cost estimates are reported with a wide range, between $56,316 [43] and $93,868 [42] per admission. Myocardial infarction admission costs were slightly lower than stroke, at $53,458 per visit [42]. VOC was the most frequently reported complication, with estimates ranging between $5335 [48] and $13,944 [39] per admission. Chronic complication costs ranged from $4398 [42] to $230,066 [43] per year, representing fatigue and stroke-year burden, respectively. Chronic VOC-related complication costs per patient were subject to variability as they ranged widely from $4609 [48] to $45,155 [47] annually per patient, due to cited higher frequencies in some individuals, which lead to increased hospital visits and healthcare utilization. Other complications were infrequently represented, many with one or two studies that met the inclusion criteria. Numerous complications expressed in our predefined PICOTS framework, and on which we searched, were entirely absent from the literature (Appendix B). One study found that the household income for SCD families with a patient with silent cerebral infarct history was $41,469, compared with $54,036 without silent cerebral infarcts; this was the only empirical estimate on a proxy outcome for caregiver burden and was also the only estimate found related to non-medical costs in our search [45].
3.4 SCD Burden Associated with Treatments
Twelve studies assessed the medical economic burden associated with SCD treatments (Table 6, Appendix Table B3). Long-term treatment of SCD can decrease total medical costs, with the magnitude of decrease varying across the treatment options (Table 6). The results of the studies suggested that hydroxyurea may decrease medical costs by up to $41,000 annually [24] in patients who are adherent to therapy [22,23,24, 49]. The magnitude of this cost savings was greatest with inpatient costs, although some the savings were reduced by costs incurred in the outpatient and pharmacy settings due to necessary patient monitoring and medication resource use [22, 23]. Still, the body of literature suggested that any expected costs from hydroxyurea utilization and monitoring were offset by the savings in emergency and acute healthcare utilization. Under a therapeutic program initiation in a small cohort, chronic transfusion (once monthly) recipients experienced decreased costs of $25,470 during the year of treatment compared with hydroxyurea alone, although these patients did not receive concurrent chelation therapy [51]. This savings was suggested to also arise from decreased inpatient costs. HSCT can lead to decreased medical costs in the years following treatment, although long-term data on this treatment are lacking [54,55,56]. One study reported costs that were unable to be annualized due to the unit of analysis chosen [55]. Initial procedure year costs ranged from $135,568 to $785,299, driven by different characteristics in procedure protocol for conditioning regimens (myeloablative vs. non-myeloablative), cell donor source (bone marrow vs. umbilical cord blood), and extent of the haplotype match (haploidentical vs. sibling vs. unrelated). None of the studies that met the inclusion criteria captured economic burden surrounding L-glutamine, crizanlizumab, and voxelotor. No studies explored the impacts that treatment would have on non-medical costs.
3.4.1 SCD Burden Associated with Treatment Complications
Iron overload treatment was the only treatment complication with economic data, although several other treatment complications were included in our initial search queries for hydroxyurea and HSCT (Appendix Table A1). Four studies assessed total medical costs when using iron chelation therapy (ICT) and reported conflicting results (Table 7, Appendix Table B4) [58,59,60,61]. Total incremental costs associated with ICT for prophylactic treatment of transfusion complications ranged from − $4688 to $21,723. These studies suggested that ICT results in higher medication costs, with the possible benefit of reduced costs in the inpatient or outpatient settings [58,59,60,61]. No studies explored how treatment complications may impact non-medical costs.
3.5 SCD Burden Associated with Insurance Type
Only one study directly characterized the influence that payer types (Medicaid, Medicare, and commercial) can have on the economic costs incurred due to SCD. Shah et al. reported that individuals on Medicaid incurred the highest average total costs per year, followed by Medicare and then commercial insurance ($42,843, $39,339, and $28,223, respectively) [47]. When these costs were stratified based on the annual VOC rate (0, 1, or 2), commercial patients had higher outpatient costs than both Medicaid and Medicare patients, while also generally having lower inpatient costs (Appendix Table B2). Indirect comparisons on the impact of payer type across the body of literature were only made within the category for SCD burden associated with ICT (Sect. 4.5), as comparisons across the other aforementioned groups (Sects. 4.2, 4.3, 4.4) were unreasonable due to vastly different study designs and objectives. Within the SCD burden associated with ICT, meaningful differences for total, pharmacy, and ED-related medical costs were not seen between Medicaid and private payers (Table 8). Medicaid inpatient costs trended higher than private payer costs ($20,705 [59] to $49,510 [58] vs. $3696 to $25,516 [60], respectively). Outpatient costs were also higher for the Medicaid population over the private population.
4 Discussion
This systematic review captured costs from studies that used varying study designs, data sources, populations, and time horizons. All of these factors can contribute to confounding, and thus the body of literature we characterized is heterogeneous, which may explain some of the wide ranges for cost estimates captured. Nonetheless, the results indicated that total annual medical costs for SCD individuals are considerably higher than in the general non-SCD population, with inpatient costs as the largest contribution to total costs when compared with other treatment settings. The magnitude of the difference in medical costs between SCD and non-SCD individuals varied widely dependent on the reason for admission, and are subject to limited generalizability with few estimates available to compare. Many complications frequently seen in SCD, such as stroke, myocardial infarction, and end-stage renal disease, were not characterized from the results of our search comparing these two populations. Among studies focused within the SCD population, total medical costs ranged widely from $21,819 to $80,843 per patient per year, with the magnitude of this variance reportedly driven by the age of the individual, i.e. either less than or greater than 18 years of age. Although collected evidence suggested that costs peak later in life, other studies on healthcare utilization pointed toward the highest use of medical services right after the transition of care (18 years of age) [62, 63], therefore further exploration is needed to clarify this distinction. Similarly, cost research that highlighted the relationship between mental illness and SCD’s economic burden suggest there is no association, yet this is in contrast to work performed previously that had shown significant uptake in HCRU for those with mental illness [64]. Results from studies on acute and chronic complications associated with SCD suggest that the extent of their impact varied widely dependent on the organ system involved; from fatigue ($4398) [41] to stroke ($230,066) [42] per patient per year. The quality ratings for literature surrounding complications were equally mixed between good and poor, and represent significant room to conduct studies that will improve how we understand these cost burdens. Studies that detailed treatments in SCD suggest that they may be cost-saving; however, these options need further exploration for a better understanding of their impact to economic burden. The economic consequences for complications from treatment with hydroxyurea have not yet been explored, and chronic transfusion therapy eventually requires iron-chelation therapy, for which there was conflicting evidence surrounding its overall impact on medical costs. HSCT is a promising option, with a significant cost reduction reported in the short-term [54,55,56,57]. However, these data contained no information beyond 2 years of receiving HSCT and represent a significant unknown for the future of HSCT and whether its promise for efficacy and safety is sustained. HCRU reduction in SCD may be achieved through healthcare system innovation [65], although formal cost analyses that evaluate the impact of those innovations are unavailable.
There exist substantial methodological differences in terms of study population, objectives, and analysis methods, making it difficult to conduct any meaningful meta-analysis across this body of literature. All medical costs are reported from a payer or institutional perspective, therefore there is no representation of the out-of-pocket costs or additional costs that patients experience as a result of the impact of SCD on their lives (e.g. ability to work). Many studies that evaluate treatment-specific interventions do not capture the impact on cost from specific complications. Across all SCD cost categories discussed (non-SCD vs. SCD, general SCD, complications, treatments, treatment complications, payer type), their respective impacts are largely explored within more common medical cost components such as inpatient, outpatient, pharmacy, and ED care. Thus, representation of SCD’s relative impact on economic costs outside of the formal healthcare sector is lacking for well-documented non-medical cost burdens of interest, such as patient [66] and caregiver time [67], transportation [66], productivity [68], education [69], and supportive/social services [70]. Therefore, the available economic estimates within SCD are incomplete and are not generalizable, as there exists significant gaps in the available medical cost literature as well as a lack of non-medical cost information. Thus, we endorse that future research describing costs in SCD should follow the Second Panel’s recommendations for capturing and categorizing costs [13], which are listed as (1) changes in HCRU; (2) changes in non-HCRU; (3) changes in use of caregiver time; (4) changes in use of patient time; and (5) productivity gains due to changes in health status.
Our review has several strengths, primarily being that it is the first of its kind within SCD to systematically summarize and evaluate the literature surrounding SCD costs within the US. We provide a comprehensive synthesis of medical costs in this systematic review and are able to highlight the relevant gaps in the literature surrounding informal aspects of healthcare. Future work should focus on the capture of non-medical costs, especially productivity and patient time impacts, to enhance how we understand SCD’s societal burden. A potential way to capture both medical and non-medical costs of SCD would be a national surveillance system that follows the individual throughout their lifetime, which would enable longitudinal explorations on their disease burden. Lastly, this work is part of a larger effort championed by National Heart, Lung, and Blood Institute (NHLBI) to bring greater attention to SCD [71], which we support through landscape characterization on the economic burden of SCD [72].
Our study also has limitations. The primary objective of this review was to assess cost data. If a study did not carry out a statistical adjustment on dollar costs reported, we assessed that evidence as lower quality; therefore, aggregate quality assessment may not reflect true study quality, but rather applicability to our research aims. A second limitation is that adjusted and unadjusted source costs were pooled together and thus may contribute to the potential for imprecise ranges of estimates. Adjusted costs with statistical methods to control for confounders are the gold standard for cost estimates, although if we restricted our analysis to only include adjusted costs we would have had much less data to explore. Methods for the collection of information on individuals living with SCD in large datasets has well-documented challenges with respect to sensitivity and specificity [73]. Furthermore, we did not appraise the included studies on their SCD sampling method, and thus this may add more uncertainty around the range of estimates reported on. Lastly, when we conducted the synthesis for excess costs associated with treatment use, we collapsed underutilizing patients and those with no medication use, which could underestimate the cost difference that should be seen. This underestimation would arise if patients with imperfect adherence could still derive some modest clinical benefit, and therefore experience lower medical costs. Overall, interpretation of the costs synthesized in this report should occur alongside appreciation of methodological heterogeneity among the studies included.
Future directions around cost literature generation in SCD should be devoted to several things: medical and non-medical costs with layering comorbidities, complications, and treatment effects; longitudinal age-related burden and payer status on all costs; long-term cost data on curative therapies; and exploration in aspects outside of direct medical care. With these facets of economic burden explored in greater detail, the value of future medical technologies can be better characterized from both a healthcare sector and a societal perspective. A better understanding of the value a treatment can provide will enable healthcare decision makers to better allocate resources to individuals significantly impacted by this disease, optimize their therapy, and improve patient and caregiver quality of life. Finally, with greater societal consideration of SCD’s economic burden, policy development will be more informed moving forward and will drive progress toward better health equity for this seriously burdened population.
5 Conclusion
Although the studies summarized in this review partially characterize the economic toll SCD exerts on healthcare resources, gaps in the evidence remain. The literature suggests that inpatient costs contribute the greatest to overall medical costs, followed by outpatient and emergency costs; these medical costs vary substantially among acute and chronic complications. Little evidence exists to describe non-medical costs such as productivity and caregiver burden. Additional cost characterizations by payer type and age of the individual will help to increase understanding of the burden of SCD on individuals, healthcare systems, and society. To fairly estimate the value of emerging therapies, better estimates will be needed for both medical and non-medical cost dimensions to comprehensively capture costs across the lifetime disease trajectory, as some treatment impacts are not fully captured in the current body of literature.
References
Panepinto JA, Bonner M. Health-related quality of life in sickle cell disease: past, present, and future. Pediatr Blood Cancer. 2012;59:377–85.
Centers for Disease Control and Prevention. Data and statistics on sickle cell disease. Centers for Disease Control and Prevention; 2020 [cited 26 Apr 2021]. Available at: https://www.cdc.gov/ncbddd/sicklecell/data.html.
Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44.
Lubeck D, Agodoa I, Bhakta N, Danese M, Pappu K, Howard R, et al. Estimated life expectancy and income of patients with sickle cell disease compared with those without sickle cell disease. JAMA Netw Open. 2019;2:e1915374.
Hassell KL. Population estimates of sickle cell disease in the US. Am J Prev Med. 2010;38:S512–21.
Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–22.
Zhou J, Han J, Nutescu EA, Gordeuk VR, Saraf SL, Calip GS. Hydroxycarbamide adherence and cumulative dose associated with hospital readmission in sickle cell disease: a 6-year population-based cohort study. Br J Haematol. 2018;182:259–70.
Evidence-based management of sickle cell disease: expert panel report, 2014. NHLBI, NIH [cited 26 Apr 2021]. Available at: https://www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease.
Miller ST, Kim H-Y, Weiner DL, Wager CG, Gallagher D, Styles LA, et al. Red blood cell alloimmunization in sickle cell disease: prevalence in 2010. Transfusion (Paris). 2013;53:704–9.
Meloni A, Puliyel M, Pepe A, Berdoukas V, Coates TD, Wood JC. Cardiac iron overload in sickle-cell disease. Am J Hematol. 2014;89:678–83.
Steinberg MH. Treating sickle cell anemia: a new era dawns. Am J Hematol. 2020;95:338–42.
Nardo-Marino A, Brousse V, Rees D. Emerging therapies in sickle cell disease. Br J Haematol. 2020;190:149–72.
Basu A. Estimating costs and valuations of non-health benefits in cost-effectiveness analysis. Cost-effectiveness in health and medicine. Oxford University Press [cited 14 Dec 2021]. Available at: https://oxford-universitypressscholarship-com.offcampus.lib.washington.edu/view/10.1093/acprof:oso/9780190492939.001.0001/acprof-9780190492939-chapter-8
Lee S, Vania DK, Bhor M, Revicki D, Abogunrin S, Sarri G. Patient-reported outcomes and economic burden of adults with sickle cell disease in the United States: a systematic review. Int J Gen Med. 2020;13:361–77.
Meltzer DO, Basu A, Sculpher MJ. Theoretical foundations of cost-effectiveness analysis in health and medicine. In: Cost-effectiveness in health and medicine. 2nd edn. New York: Oxford University Press; 2016 [cited 14 Dec 2021]. Available at: https://oxford.universitypressscholarship.com/10.1093/acprof:oso/9780190492939.001.0001/acprof-9780190492939-chapter-2
Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al. Cochrane handbook for systematic reviews of interventions version 6.0 (updated July 2019). The Cochrane Collaboration; 2019.
Trikalinos TA, Dahabreh IJ, Wallace BC, Schmid CH, Lau J. Towards a framework for communicating confidence in methodological recommendations for systematic reviews and meta-analyses. Rockville: Agency for Healthcare Research and Quality (US); 2013 [cited 2 Mar 2020]. Available at: http://www.ncbi.nlm.nih.gov/books/NBK164506/.
Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097.
Centers for Disease Control and Prevention. What is sickle cell trait? Centers for Disease Control and Prevention; 2017 [cited 23 Jan 2022]. Available at: https://www.cdc.gov/ncbddd/sicklecell/traits.html.
Wells G, Shea B, O’Connell D, Peterson J, Welch V, Losos M, et al. The Newcastle–Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp.
Medicare Hospital Cost Report PUF 2017. Data.CMS.gov [cited 26 Apr 2021]. Available at: https://data.cms.gov/Medicare-Inpatient/Medicare-Hospital-Cost-Report-PUF-2017/4sfm-kqny.
Candrilli SD, O’Brien SH, Ware RE, Nahata MC, Seiber EE, Balkrishnan R. Hydroxyurea adherence and associated outcomes among Medicaid enrollees with sickle cell disease. Am J Hematol. 2011;86:273–7.
Shah N, Bhor M, Xie L, Halloway R, Arcona S, Paulose J, et al. Treatment patterns and economic burden of sickle-cell disease patients prescribed hydroxyurea: a retrospective claims-based study. Health Qual Life Outcomes. 2019;17:155.
Lanzkron S, Haywood C, Fagan PJ, Rand CS. Examining the effectiveness of hydroxyurea in people with sickle cell disease. J Health Care Poor Underserved. 2010;21:277–86.
Akanbi O, Adejumo AC, Saleem N, Francisque F, Soliman M, Ogunbayo GO. Sickle cell disease is associated with higher mortality among patients hospitalized with ischemic bowel disease. Eur J Gastroenterol Hepatol. 2018;30:1027–32.
Bae E, Tangel V, Liu N, Abramovitz SE, White RS. Inpatient mortality and postpartum readmission rates in sickle cell disease pregnancies: a multistate analysis, 2007–2014. J Matern Fetal Neonatal Med. 2021;34(17):2783–92.
Brumm J, White RS, Arroyo NS, Gaber-Baylis LK, Gupta S, Turnbull ZA, et al. Sickle cell disease is associated with increased morbidity, resource utilization, and readmissions after common abdominal surgeries: a multistate analysis, 2007–2014. J Natl Med Assoc. 2020;112:198–208.
Ramdath A, Zeineddin A, Nizam W, Kearse L, Olufajo OA, Williams M. Outcomes after cholecystectomy in patients with sickle cell disease: does acuity of presentation play a role? J Am Coll Surg. 2020;230:1020–4.
Raphael JL, Dietrich CL, Whitmire D, Mahoney DH, Mueller BU, Giardino AP. Healthcare utilization and expenditures for low income children with sickle cell disease. Pediatr Blood Cancer. 2009;52:263–7.
Artz N, Zhang J, Meltzer D. Physical and mental health in adults hospitalized with sickle cell disease: impact on resource use. J Natl Med Assoc. 2009;101:139–44.
Blinder MA, Duh MS, Sasane M, Trahey A, Paley C, Vekeman F. Age-related emergency department reliance in patients with sickle cell disease. J Emerg Med. 2015;49:513-522.e1.
Carroll CP, Haywood C, Fagan P, Lanzkron S. The course and correlates of high hospital utilization in sickle cell disease: evidence from a large, urban Medicaid Managed Care Organization. Am J Hematol. 2009;84:666–70.
Dickerson AK, Klima J, Rhodes MM, O’Brien SH. Young adults with SCD in US children’s hospitals: are they different from adolescents? Pediatr Blood Cancer. 2012;58:741–5.
Fingar KR, Owens PL, Reid LD, Mistry KB, Barrett ML. Characteristics of inpatient hospital stays involving sickle cell disease, 2000–2016: Statistical Brief #251. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville: Agency for Healthcare Research and Quality (US); 2006 [cited 9 Apr 2021]. Available at: http://www.ncbi.nlm.nih.gov/books/NBK547764/.
Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84:323–7.
Peterson EE, Salemi JL, Dongarwar D, Salihu HM. Acute care utilization in pediatric sickle cell disease and sickle cell trait in the USA: prevalence, temporal trends, and cost. Eur J Pediatr. 2020;179(11):1701–10.
Thi Nhat Ho A, Shmelev A, Joshi A, Ho N. Trends in hospitalizations for sickle cell disease related-complications in USA 2004–2012. J Hematol. 2019;8:11–6.
Kumar V, Chaudhary N, Achebe MM. Epidemiology and predictors of all-cause 30-day readmission in patients with sickle cell crisis. Sci Rep. 2020;10:2082.
Raphael JL, Kamdar A, Wang T, Liu H, Mahoney DH, Mueller BU. Day hospital versus inpatient management of uncomplicated vaso-occlusive crises in children with sickle cell disease. Pediatr Blood Cancer. 2008;51:398–401.
Raphael JL, Mei M, Mueller BU, Giordano T. High resource hospitalizations among children with vaso-occlusive crises in sickle cell disease. Pediatr Blood Cancer. 2012;58:584–90.
Yeruva SLH, Paul Y, Oneal P, Nouraie M. Renal failure in sickle cell disease: prevalence, predictors of disease, mortality and effect on length of hospital stay. Hemoglobin. 2016;40:295–9.
Bradt P, Synnott P, Chapman R, Beinfeld M, Rind D, Pearson S, et al. Crizanlizumab, voxelotor, and l-glutamine for sickle cell disease: effectiveness and value. Institute for Clinical and Economic Review. 2020. Available at: https://icer-review.org/material/sickle-cell-disease-draft-evidence-report/.
Campbell A, Cong Z, Agodoa I, Song X, Martinez DJ, Black D, et al. The economic burden of end-organ damage among Medicaid patients with sickle cell disease in the United States: a population-based longitudinal claims study. J Manag Care Spec Pharm. 2020;26(9):1121–9.
Jerrell JM, Tripathi A, McIntyre RS. Prevalence and treatment of depression in children and adolescents with sickle cell disease: a retrospective cohort study. Prim Care Companion CNS Disord. 2011;13(2):e1–e7. https://doi.org/10.4088/PCC.10m01063.
King AA, Strouse JJ, Rodeghier MJ, Compas BE, Casella JF, McKinstry RC, et al. Parent education and biologic factors influence on cognition in sickle cell anemia. Am J Hematol. 2014;89:162–7.
McCormick M, Richardson T, Warady BA, Novelli EM, Kalpatthi R. Acute kidney injury in paediatric patients with sickle cell disease is associated with increased morbidity and resource utilization. Br J Haematol. 2020;189:559–65.
Shah NR, Bhor M, Latremouille-Viau D, Kumar Sharma V, Puckrein GA, Gagnon-Sanschagrin P, et al. Vaso-occlusive crises and costs of sickle cell disease in patients with commercial, Medicaid, and Medicare insurance—the perspective of private and public payers. J Med Econ. 2020;23(11):1345–55.
Shah N, Bhor M, Xie L, Paulose J, Yuce H. Medical resource use and costs of treating sickle cell-related vaso-occlusive crisis episodes: a retrospective claims study. J Health Econ Outcomes Res. 2020;7(1):52–60.
Wang WC, Oyeku SO, Luo Z, Boulet SL, Miller ST, Casella JF, et al. Hydroxyurea is associated with lower costs of care of young children with sickle cell anemia. Pediatrics. 2013;132:677–83.
Atwood CM, Gnagi SH, Teufel RJ, Nguyen SA, White DR. Blood transfusion in children with sickle cell disease undergoing tonsillectomy. Int J Pediatr Otorhinolaryngol. 2017;103:117–20.
Hilliard LM, Kulkarni V, Sen B, Caldwell C, Bemrich-Stolz C, Howard TH, et al. Red blood cell transfusion therapy for sickle cell patients with frequent painful events. Pediatr Blood Cancer. 2018;65:e27423.
Sarode R, Matevosyan K, Rogers ZR, Burner JD, Rutherford C. Advantages of isovolemic hemodilution-red cell exchange therapy to prevent recurrent stroke in sickle cell anemia patients. J Clin Apheresis. 2011;26:200–7.
Saylors RL, Watkins B, Saccente S, Tang X. Comparison of automated red cell exchange transfusion and simple transfusion for the treatment of children with sickle cell disease acute chest syndrome. Pediatr Blood Cancer. 2013;60:1952–6.
Arnold SD, Jin Z, Sands S, Bhatia M, Kung AL, Satwani P. Allogeneic hematopoietic cell transplantation for children with sickle cell disease is beneficial and cost-effective: a single-center analysis. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant. 2015;21:1258–65.
Arnold SD, Brazauskas R, He N, Li Y, Aplenc R, Jin Z, et al. Clinical risks and healthcare utilization of hematopoietic cell transplantation for sickle cell disease in the USA using merged databases. Haematologica. 2017;102:1823–32.
Saraf SL, Ghimire K, Patel P, Sweiss K, Gowhari M, Molokie RE, et al. Improved health care utilization and costs in transplanted versus non-transplanted adults with sickle cell disease. PLoS ONE. 2020;15:e0229710.
Soni S, Gross TG, Rangarajan H, Baker KS, Sturm M, Rhodes M. Outcomes of matched sibling donor hematopoietic stem cell transplantation for severe sickle cell disease with myeloablative conditioning and intermediate-dose of rabbit anti-thymocyte globulin. Pediatr Blood Cancer. 2014;61:1685–9.
Armstrong EP, Skrepnek GH, Sasane M, Snodgrass SM, Ballas SK. Long-term persistency and costs associated with the use of iron chelation therapies in the treatment of sickle cell disease within Medicaid programs. J Med Econ. 2013;16:10–8.
Blinder MA, Vekeman F, Sasane M, Trahey A, Paley C, Duh MS. Age-related treatment patterns in sickle cell disease patients and the associated sickle cell complications and healthcare costs. Pediatr Blood Cancer. 2013;60:828–35.
Delea TE, Hagiwara M, Thomas SK, Baladi J-F, Phatak PD, Coates TD. Outcomes, utilization, and costs among thalassemia and sickle cell disease patients receiving deferoxamine therapy in the United States. Am J Hematol. 2008;83:263–70.
Vekeman F, Sasane M, Cheng WY, Ramanakumar AV, Fortier J, Qiu Y, et al. Adherence to iron chelation therapy and associated healthcare resource utilization and costs in Medicaid patients with sickle cell disease and thalassemia. J Med Econ. 2016;19:292–303.
Crego N, Douglas C, Bonnabeau E, Earls M, Eason K, Merwin E, et al. Sickle-cell disease co-management, health care utilization, and hydroxyurea use. J Am Board Fam Med. 2020;33(1):91–105.
Hemker BG, Brousseau DC, Yan K, Hoffmann RG, Panepinto JA. When children with sickle-cell disease become adults: lack of outpatient care leads to increased use of the emergency department. Am J Hematol. 2011;86:863–5.
Leuche VT, Cutler GJ, Nelson SC, Jin J, Bergmann KR. Emergency Department Health Care Utilization and Opioid Administration among pediatric patients with sickle cell vasoocclusive pain crisis and coexisting mental health illness. Pediatr Emerg Care. 2020;38(2):e664–9.
Lanzkron S, Little J, Wang H, Field JJ, Shows JR, Haywood C, et al. Treatment of acute pain in adults with sickle cell disease in an Infusion Center versus the Emergency Department. Ann Intern Med. 2021;174(9):1207–13.
Callaway D, Chawla A, Sprinz P. Physical therapy in pediatric and young adult patients with sickle cell disease: assessing potential benefits and barriers. J Pediatr Hematol Oncol. 2020;42:463–6.
Moskowitz JT, Butensky E, Harmatz P, Vichinsky E, Heyman MB, Acree M, et al. Caregiving time in sickle cell disease: psychological effects in maternal caregivers. Pediatr Blood Cancer. 2007;48:64–71.
Rizio AA, Bhor M, Lin X, McCausland KL, White MK, Paulose J, et al. The relationship between frequency and severity of vaso-occlusive crises and health-related quality of life and work productivity in adults with sickle cell disease. Qual Life Res. 2020;29(6):1533–47.
Prussien KV, Jordan LC, DeBaun MR, Compas BE. Cognitive function in sickle cell disease across domains, cerebral infarct status, and the lifespan: a meta-analysis. J Pediatr Psychol. 2019;44:948–58.
Hsu LL, Green NS, Donnell Ivy E, Neunert CE, Smaldone A, Johnson S, et al. Community health workers as support for sickle cell care. Am J Prev Med. 2016;51:S87-98.
Cure Sickle Cell Initiative. NHLBI, NIH [cited 29 Jan 2022]. Available at: https://www.nhlbi.nih.gov/science/cure-sickle-cell-initiative.
Jiao B, Basu A, Roth J, Bender M, Rovira I, Clemons T, et al. The use of cost-effectiveness analysis in sickle cell disease: a critical review of the literature. Pharmacoeconomics. 2021;39:1225–41.
Reeves S, Garcia E, Kleyn M, Housey M, Stottlemyer R, Lyon-Callo S, et al. Identifying sickle cell disease cases using administrative claims. Acad Pediatr. 2014;14:S61-67.
Acknowledgements
The authors gratefully acknowledge the following collaborators: N. DiFronzo (NHLBI), and C. Henry, W. Wright, D. Louden, and J. Rich. The authors also appreciate the valuable insights and suggestions provided by the members of the Clinical and Economic Analysis Initiative Expert Panel.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Registration and Protocol
This study was conducted under the auspices of NHLBI and included a protocol and PICOTS criteria developed and approved by NHLBI.
Funding
This research was funded in part by the National Institutes of Health (NIH) Agreement OTA OT3HL152448, OT3HL151434. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the NIH.
Conflict of interest
Zachary Baldwin, Boshen Jiao, Anirban Basu, Joshua Roth, M.A. Bender, Zizi Elsisi, Kate M. Johnson, Emma Cousin, Scott D. Ramsey, and Beth Devine have no conflicts of interest to declare.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Ethics approval
Not applicable.
Author contributions
BJ, AB, JR, MAB, and BD designed the research. ZB, BJ, and BD performed the research. ZB and BJ collected the data. ZB, BJ, ZE, KMJ, and BD analyzed and interpreted the data. ZB wrote the first draft of the manuscript. ZB, BJ, AB, JR, MAB, EC, SDR, and BD reviewed and revised the manuscript. All the authors approved the final version for submission.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Baldwin, Z., Jiao, B., Basu, A. et al. Medical and Non-medical Costs of Sickle Cell Disease and Treatments from a US Perspective: A Systematic Review and Landscape Analysis. PharmacoEconomics Open 6, 469–481 (2022). https://doi.org/10.1007/s41669-022-00330-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41669-022-00330-w