
Abstract
Erwinia amylovora, the causative agent of fire blight of pome fruits and other rosaceous plants belongs to the group of regulated quarantine pests. The aim of this work was to characterize the populations of E. amylovora in Norway and their geographical distribution. A total of 238 E. amylovora isolates recovered from symptomatic host plants in Norway between 1986 and 2004 were genotyped by means of a short sequence repeat (SSR) marker (ATTACAGA) on plasmid pEa29. The SSR region was amplified and amplicon size determined using fluorescent labelling and rapid, automated capillary gel electrophoresis. All isolates contained the pEa29 plasmid harbouring the investigated marker. In total, ten genotypes were identified, of which two were detected only once. The number of repeats varied from 3 to 13, with 43% of the isolates containing five repeats. Of 17 isolates collected between 1986 and 1991, all but one contained five repeats, whereas more variation was observed in isolates from the period 2000 to 2004. Most of the isolates (80%) originated from Cotoneaster bullatus, hence no relationship between genotype of the isolate and host species that it was isolated from could be detected. This historic data suggests multiple introductions of E. amylovora to Norway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fire blight, caused by the bacterium Erwinia amylovora, is regarded as one of the most destructive diseases of pome fruits (Pedroncelli and Puopolo 2023). It rapidly kills blossoms and shoots, and may spread further into branches and the trunk, which usually leads to death of the tree (Thomson 2000). The most commonly affected hosts are pear (Pyrus communis), apple (Malus domestica), hawthorn (Crataegus spp.), Cotoneaster spp. (particularly the larger species), Sorbus aria and Pyracantha spp. (Momol and Aldwinckle 2000). Fire blight has been known in North America since 1780, whereas it appeared in New Zealand in 1919, and in England in 1957 (Bonn and van der Zwet 2000). Since then, the disease has spread to most of the European countries where susceptible hosts are growing (Kurz et al. 2021).
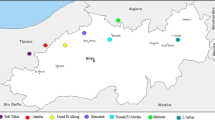
Fire blight was detected for the first time in Norway in 1986, when there was a limited outbreak in ornamental plants, particularly in Cotoneaster. Fire blight was not detected in the years 1993–1999, but after an unusually warm spring and summer with ample rain a major outbreak appeared in 2000 (Sletten and Melboe 2004). As a result of immediate and strict control measurements, the disease was restricted to a region along the southern West Coast for two decades (Fig. 1). However, E. amylovora has since spread further north and southeast along the coastline in southern Norway. According to annual surveillance programs, the bacterium has not yet been found in the major districts of commercial production of P. communis, M. domestica and stone fruits along the southern west coast and the Oslo-fjord. Uncontrolled movement of beehives is assumed to have been a main cause of the historic spread of fire blight in Norway, so it has been prohibited to move beehives in and out of infested areas between May and October, since 2004 (Sletten et al. 2017).

Map of Norway with sampling area in western Norway enlarged. Sampling sites in the counties Rogaland and Vestland where host plant material with fire blight symptoms were collected are indicated with dots. In total 238 isolates were obtained from 31 localities in the years 1986–2004
At the time of this study, fire blight was diagnosed on the basis of symptoms, sometimes followed by isolation of the bacterium and subsequent identification by nutritional and enzymatic tests, fatty acid profiling, serological tests, and molecular methods (Anonymous 2022). Today’s routine diagnostics entail qPCR screening of symptomatic or asymptomatic samples, followed by confirmation using a second qPCR assay targeting a different genomic region, although isolation is still attempted for symptomatic samples for identification purposes (Gottsberger 2010; Anonymous 2022). Based mainly on the results of biochemical and serological characterization, E. amylovora had previously been considered to be a species of great homogeneity (Momol and Aldwinckle 2000). Since then, molecular tools have provided improved means of identifying and genotyping E. amylovora, as well as tracking long- or short- range dispersal of the pathogen and for identifying possible sources of infection (Kurz et al. 2021). A range of methods has been developed ranging in scope from single loci with length or sequence polymorphisms to whole genome comparisons (McGhee and Jones 2000; Rezzonico et al. 2011; Smits et al. 2011; Mann et al. 2013; Bühlmann et al. 2014; Parcey et al. 2020; Kurz et al. 2021; Mendes et al. 2021). In addition, phenotypical variation has been observed in a set of Portuguese E. amylovora isolates that were characterized as clonal based on an MLSA using four housekeeping genes (Mendes et al. 2022). This emphasizes the limits of molecular tools, especially when focusing on selected loci, when trying to capture all facets of biological potential and malleability in E. amylovora.
For a long time, all virulent strains of E. amylovora had been assumed to carry a low copy number plasmid of 29 kb (pEa29) (Falkenstein et al. 1988; McGhee and Jones 2000). This plasmid has been used as a target in several PCR-based detection methods, two of which have been validated and approved by the European and Mediterranean Plant Protection Organization (EPPO). The validated primers and protocols were based on a conventional protocol developed by Bereswill et al. (1992) and a more sensitive nested protocol described by Llop et al. (2000), both targeting the same region of pEA29. Previously, using the Bereswill protocol Lecomte et al. (1997) had observed variation between isolates in the length of the amplified 1 kb fragment, and used this variation to group E. amylovora isolates from Central Europe into three types. This size variation was shown to be caused by a short sequence DNA repeat (SSR) of eight nucleotides (ATTACAGA), which is repeated 3–15 times, depending on the isolate (Kim and Geider 1999). The function of the SSR in pEA29 is unknown. Originally, the SSRs in pEa29 were thought to be unstable regions with frequent changes in length (Schnabel and Jones 1998; van Belkum et al. 1998; Jock et al. 2003). However, other studies have demonstrated that the number of repeats remains relatively stable, both under normal growth in laboratory as well as during stress conditions, establishing their usefulness as genetic markers (Kim and Geider 1999; Ruppitsch et al. 2004; Bühlmann et al. 2014). The SSR marker investigated here spans the same repeat region as the VNTR-A (variable number of tandem repeats A) typing region applied by Bühlmann et al. (2014). Multiple studies including Brennan et al. (2002) Llop et al. (2006) and Mendes et al. (2021) have described virulent isolates of E. amylovora without pEa29 that, consequently, gave negative results in diagnostic PCR reactions. To avoid false negatives as a result of the absence of the plasmid, the current EPPO diagnostic protocol contains several qPCR assays targeting chromosomal target regions in addition to the original PCR assays targeting the pEa29 plasmid (Anonymous 2022).
The main aim of this study was to characterize the diversity of Norwegian E. amylovora isolates from the first known outbreaks by using sequence length variability caused by SSRs in a region of the plasmid pEA29. Associations of SSR genotypes with spatial and temporal distributions as well as host plant species were investigated to help elucidate the epidemiology of E. amylovora in Norway.
Materials and methods
Collection and identification of isolates
Shoots of host plants with characteristic fire blight symptoms were collected on the West Coast of Norway (Fig. 1; Table 1). Sample extracts in 10 mM phosphate-buffered saline (PBS) at pH 7.2 were prepared from the margin of lesions and streaked onto plates with Difco nutrient agar supplemented with 5% sucrose. Levan-type colonies were picked and identified with biochemical methods described by Billing et al. (1961) and/or by immunofluorescence microscopy in the beginning of the outbreak, and later routinely by fatty acid analysis. Consequently, most strains have been analysed by fatty acid profiling as described in Persson and Sletten (1995). Of the 238 strains isolated, all originated from host plants within the apple subfamily (Maloideae): 190 from Cotoneaster bullatus, 15 from Cotoneaster divaricatus, 15 from Cotoneaster salicifolius, eleven from Cotoneaster horizontalis, four from Cotoneaster lucidus, two from M. domestica and one from Sorbus aria. Strain NCPPB595 (National Collection of Plant Pathogenic Bacteria, Central Science Laboratory, York, GB) and strain NCPPB683 (Collection Française de Bacteriés Phytopathogènes) were included in the tests as positive controls. All strains were stored on commercially available cryobeads (‘Protect’ microorganism cryopreservation system, Technical Service Consultants Ltd. England) at −80 °C according to the manufacturer’s instructions.
DNA preparation
Crude bacterial genomic DNA used for the PCR analyses was obtained by resuspending bacterial colonies into 40 µl TE (1 mM Tris-HCl (pH 8.0), 0.1 mM EDTA). The suspensions were used directly in the PCR reactions or frozen for storage until later use. For some symptomatic samples, plant genomic DNA was isolated using DNeasy Plant Mini Kit (QIAGEN) according to the manufacturer’s instructions. This was done to assess the sensitivity of the primers developed in this work in a diagnostic setting.
PCR reactions
We designed new PCR primers covering flanking regions of the SSR of pEa29, yielding a product that is shorter than the product obtained with comparable approaches (Bereswill et al. 1992; Llop et al. 2000). The SSR region amplified by this primer pair is also referred to as VNTR-A (Bühlmann et al. 2014). The SSR region was amplified with the following primers: SSR1f (5′- 6FAM (6-carboxyfluorescein)—AACAGGAAGAACGACGTATT-3′) and SSR1r—(5′-AGCCGGATGTCTTTCCTTT-3′). Amplifications were performed in 10-µl reaction volume with 1.0 u Taq DNA polymerase (Applied Biosystems), 200 mM dNTP, 10 pmol each of forward and reverse primer, 1.5 mM MgCl2, and 1 µl of 10x AmpliTaq buffer (500 mM KCl, 100 mM Tris-HCl pH 8.3, 15 mM MgCl2). A total of 1 µl from the bacterial suspension was used as a template. The thermal cycling was carried out as follows: 95 °C for 4 min, followed by 35 cycles of 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, and a final extension of 7 min at 72 °C, before cooling to 4 °C. To obtain insight in the occurrence of plasmid-free strains, which may have escaped detection by pEA29-based PCR, and compare sensitivity, isolates were screened using a second PCR that targets chromosomal DNA (Llop et al. 2000). The PCR was performed as described in the original publication, except that the volume of the PCR reaction was halved. All PCR reactions were performed with a GenAmp PCR System 9700 (PE Applied Biosystems).
Fragment analysis
The FAM-labelled PCR products were analysed using an ABI3730 DNA Analyzer with 48 capillaries × 36 cm using Performance Optimized Polymer for 3730 DNA Analyzers (POP-7; Applied Biosystems). For each PCR reaction, 1 µl of 50-fold-diluted PCR products was added to a loading buffer containing 8.8 µl Hi-Di™ formamide (Applied Biosystems), and 0.2 µl of GeneScan 500 LIZ size standard (Applied Biosystems). Electrophoresis of each sample was carried out at 66 °C and at 15 kV, for 20 min. The data was collected using the software Data Collection v 2.0 (AB), while GeneMapper v 3.0 (AB) was used to derive the fragment length of the FAM-labelled DNA-fragments using the known fragment lengths of the LIZ-labelled marker peaks.
Sequencing
For sequencing, SSR regions were amplified with the primers described by Llop et al. (2000). The amplicons were purified using the GenElute PCR Clean-up Kit (Sigma). Cycle sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction Kit v1.1 (Applied Biosystems) according to the manufacturer’s instructions. Analysis of sequence products was done on the ABI Prism 310 capillary (Applied Biosystems). Sequence analysis was performed using the Lasergene software package (DNAStar Inc.).
Genetic diversity calculations
Genotypic diversity was calculated by a normalized Shannon’s diversity index (Hs): HS = −∑ Pi ln Pi/ ln N, where Pi is the frequency of the ith genotype and N is the sample size. This diversity index corrects for differences in sample size (Sheldon 1969). Values for HS range from 0 (single genotype present) to 1 (each isolate in the sample has a different genotype).
Results
SSR analysis
The PCR primers SSR1f and SSR1r were designed to amplify the SSR region of E. amylovora pEa29 (ATTACAGA) based on publicly available nucleotide sequences from GenBank (accession numbers AF264948, NG005706, U19254) and the E. amylovora genome sequencing project (Sebaihia et al. 2010). The primers were designed to be E. amylovora specific and to amplify only a relatively small region (approximately 300 bp). The latter was necessary to ensure efficient amplification as well as to permit accurate fragment size determination in the ABI3730 capillary system. This SSR region was amplified from all 238 Norwegian isolates (Table 1) and from a British and a French control isolate. Fragment sizes were determined using an AB3730 automated fragment analysis system (Fig. 2). All 238 isolates gave fragments in the expected size range, indicating the presence of pEA29 in all tested isolates. All fragments were automatically assigned to the bins set up (8 bp intervals). After fragment analysis each isolate was assigned a number corresponding to the number of repeats, i.e. SSR3-SSR13. To verify fragment sizing by the automated fragment analysis system, one fragment from all variant genotypes was amplified (Fig. 3) and sequenced (data not shown). The number of repeats found in the sequenced samples confirmed the results from the automated fragment analysis. This show that capillary gel electrophoresis offers an automated, high speed, high resolution analytical method for determining the number of SSR repeats.

Example of electropherograms from capillary electrophoresis of FAM-labelled PCR products from the SSR loci of three different E. amylovora isolates. Grey peaks are size markers and black peaks are FAM-labelled SSR fragments. The vertical scale is relative fluorescence. The horizontal scale is size in base pairs. Isolate Ea595 (a) has one repeat unit (equivalent to 8 bp) fewer than isolate Ea0046 (b), and two repeat units (equivalent to 16 bp) fewer than isolate Ea0047 (c)

Example of PCR amplification products using primer pair SSR1f/r directed to the SSR region of pEa29. Lane 1, 100 bp ladder; lane 2–10, E. amylovora strains with three to eleven repeats respectively; lane 11, E. amylovora strain with thirteen repeats. Repeat numbers were determined by capillary electrophoresis and sequencing
SSR diversity
A total of ten different genotypes were identified among the 238 isolates in the collection and 43% of the isolates contained five repeats (Fig. 4). One British and one French isolate included in this study, also fell into this category. The British isolate (Ea595) was included in a previous study (Kim and Geider 1999), which gave the same results, confirming the reproducibility of the method used here. The other most common genotypes contained eight (23% of the isolates) and nine repeats (16%). The minimum number of repeats found in an isolate was three and this was found in one out of two isolates from M. domestica (Fig. 4). The maximum number of repeats found in an isolate was 13, which was only found in a single isolate from C. salicifolius. The genotypes with three and 13 repeats were the only genotypes occurring only once in the collection of 238 isolates. No isolates with twelve repeats were found.

Distribution of number of (ATTACAGA) repeats in pEA29 of Norwegian E. amylovora isolates collected over ten years from 30 locations in 13 municipalities, organized by location from north (top) to south, and years within each location. The percentage of isolates with a given number of repeats is shown for each region (top) as well as summarized across regions (bottom). For each bar, the number (n) of isolates that comprises 100% is shown
The largest number of genotypes in a single geographic location (seven) was found on the island Karmøy, which also was the origin of most isolates overall, 127 in total (Fig. 1; Table 2). In Eigersund, four genotypes were found, but from all the other locations only one or two genotypes were recovered (Table 2). The genotypic diversities, quantified by a normalized Shannon’s diversity index (Hs) were 0.29 for all isolates investigated in this study, and 0.22 for Karmøy. For all other locations Hs values were below 0.05 (Table 2).
SSR distribution in different host plant species
Most of the isolates (80%) originated from C. bullatus, and the remaining 48 isolates were from C. divaricatus (15), C. salicifolius (15), C. horizontalis (11), C. lucidus (4), M. domestica (2) and from Sorbus aria (1). The isolates from C. bullatus contained all genotypes except SSR3 and SSR13 which both were found only once. The 15 isolates from C. divaricatus were from four widely separated geographic locations, but they were all of genotype SSR5 except one (SSR6). C. salicifolius isolates comprised four genotypes, and ten of the 15 isolates were of genotype SSR5. Five of the eleven isolates from C. horizontalis were of genotype SSR5. The most common genotype (SSR5) was isolated from all the seven host species found in Norway.
SSR distribution over time
In the years 1986–1991 only 18 isolates were obtained, because of limited outbreaks. All, but one of these isolates were of genotype SSR5 and they were from a limited geographic region (Stavanger region). The isolate differing from this genotype was identified as SSR13, and was the only isolate from 1991. The other isolates were from 2000 (115), 2001 (32), 2002 (23), 2003 (4) and 2004 (46). Approximately 50% of all the isolates were collected in year 2000, and 30% of these isolates contained five repeats. In 2004, the last year of study and the year with second most isolates collected, 78% of the isolates were of genotype SSR5.
SSR primers for detection of fire blight
Although designed for genotyping and not particularly for detection, the sensitivity of the primers was tested by amplifying 10-fold dilution series of bacterial extracts and analysing amplification products on agarose gels (Fig. 5). The results were compared to the sensitivity of one of the routine PCR detection protocols for E. amylovora, which is a nested PCR protocol (Llop et al. 2000) (Fig. 5). Figure 5 shows that both protocols give positive signals when using a 10−5 dilution of the bacterial extract, corresponding to approx. 1000 cells per ml or 1 cell per reaction, and that the nested protocol is more sensitive. The designed primers were also tested on DNA extracted directly from infected leaves and fruits of host plants (Cotoneaster spp.) showing symptoms of fire blight. The primers detected E. amylovora in all tested cases, which was consistent with results (not shown) using the protocol developed by Llop et al. (2000).

PCR products from a dilution series (10-fold steps to 10−5) of E. amylovora extract (NCPPB595) equivalent to 108–103 CFU/ml of which 1 µl was used in the PCR reaction. Lanes 1–6, primer pair SSR1f/r (this study); Lanes 7–12, nested PCR protocol (Llop et al. 2000); Lane 13, negative control
Discussion
The most common genotype observed in this study, was SSR5, which occurred in 43% of the isolates. SSR5 was also the only genotype detected in the first five years of the fire blight outbreak in Norway (1986–1991). However, the total number of isolates from this period was only seventeen. The British and the French control isolates included in the study also belonged to the SSR5 genotype. Other frequent genotypes in this study were SSR8 (23%) and SSR9 (16%) (Fig. 4). This is somewhat different from frequencies of SSR genotypes observed in previous studies with isolates from other countries. Kim and Geider (1999) studied 130 isolates from all over the world and found that most of the isolates belonged to the three genotypes SSR6 (33%), SSR5 (25%) and SSR7 (17%). Moreover, they found only 5.5% of each of the genotypes SSR8 and SSR9. Later, the same group studied more than 100 isolates from multiple countries in western and central Europe, north America as well as Japan, and found, as in our study, SSR5 (38%) to be the most frequent genotype (Jock et al. 2003). SSR6 (30%) and SSR7 (15%) were also among the dominating genotypes in this second study (Jock et al. 2003). In a study of more than hundred Austrian E. amylovora isolates Ruppitsch et al. (2004) found the most frequent genotypes to be SSR7 (20%), SSR6 (17%), SSR5 (14%) and SSR4 (14%). They found few SSR8 (5%) and SSR9 (5%) genotypes. It should be noted that the numbers from Ruppitsch et al. (2004) given here have been corrected for differences resulting from the fact that they have counted GAATTACA repeats instead of ATTACAGA repeats, which leads to “loss” of one perfect repeat; for example: cgattaca-(GAATTACA)1-gaattatca (according to Ruppitsch et al. 2004) versus cg-(ATTACAGA)1 (ATTACAGA)2-attatca as used here and by Kim and Geider (1999), Jock et al. (2003) and a number of more recent publications (Taghdareh et al. 2014; Végh et al. 2017; Doolotkeldieva et al. 2019). Among the Norwegian isolates investigated here, only a few contained more than nine repeats (Fig. 4) and only one isolate contained as many as 13 repeats. In the studies of Kim and Geider (1999) and Jock et al. (2003) the frequencies of isolates with more than nine repeats were also low. In contrast, the Austrian study found that 25% of the isolates had more than nine repeats and the maximum number of repeats found was 15 (4% of the isolates) (Ruppitsch et al. 2004). Bühlmann et al. (2014), who targeted the same region under the name VNTR-A included 833 Erwinia amylovora isolates from North America, Northwestern and Southeastern Europe, and Oceania. In this dataset, SSR6 (25%), SSR5 (20%) and SSR7 (15%) were the largest observed groups with a notable regional difference being the higher prevalence of SSR7 and lower prevalence of SSR5 in North America compared to Northwestern and Southeastern Europe (Bühlmann et al. 2014). In summary, the high observed frequency of SSR5 in the Norwegian isolate collection investigated here is comparable to other studies of European isolates. The most apparent differences between the Norwegian fire blight isolates and the isolates from other parts of the world, based on SSR genotyping at the time this work was conducted, were the relatively high frequencies of SSR8 (23%) and SSR9 (16%) genotypes and the relatively low frequencies of SSR6 (3%) and SSR7 (8%) genotypes in the Norwegian isolates (Kim and Geider 1999; Jock et al. 2003; Ruppitsch et al. 2004; Bühlmann et al. 2014).
Genetic differentiation often occurs between geographically separated populations. Determination of whether different populations are genetically differentiated is useful for an understanding of the epidemiology of plant diseases. In most of the locations in this study only one or two genotypes could be detected (Table 2), which indicates that it is possible to follow the spread of E. amylovora to close neighbour areas with this simple genotyping tool.
The district Karmøy was clearly the location with most genetic variation (Table 2), 127 of the 238 isolates originated from Karmøy and they were all isolated in the period 2000 to 2004. The isolates from this district were from the island Karmøy (108 isolates) and from Nordheim (19 isolates), a small place at the mainland connected with a bridge of 200 meter to the island Karmøy. This high genetic variation in Karmøy compared to the rest of the region is intriguing since the area of Karmøy (178 km2) represents a small proportion of the total area (1500 km2) where the isolates originated from. All the seven genotypes found in Karmøy (Table 2) were isolated already in the first year of detection (year 2000). One possible explanation is that the number of repeats in pEa29 may not be constant under all conditions and that changes may happen more rapidly than generally assumed. Indeed, changes in SSR numbers have previously been found at low frequencies in laboratory studies after different types of cultivation or stress treatments (Schnabel and Jones 1998; Kim and Geider 1999; Jock et al. 2003). Alternatively, the results may be taken to indicate that E. amylovora was present before the year 2000 and that the diversity observed in 2000 is the result of prior evolution. Previously, different SSR genotypes have been found on plants with only very weak fire blight symptoms in a small area (Kim and Geider 1999). In principle, the observed difference in genetic variation between the various regions could be due to differences in selective pressure; however, there are no indications that the environmental conditions on Karmøy differ significantly from those in the rest of the regions. A more likely explanation for differences in genetic variation between the regions could be repeated introductions of the pathogen (from various sources) in some of these regions, e.g., via transport of beehives from infected areas (such transport was permitted until 2004).
Since the majority of the isolates (80%) originated from C. bullatus, and the remaining 48 isolates were from six different host species it was not feasible to draw any conclusion about relationships between genotype of the isolate and host species. From one of the host plant species, C. divaricatus, 14 of 15 isolates were of genotype SSR5 even though they were from four widely separated geographic locations. However, since SSR5 was the most prevalent genotype in this study, this was likely a coincidence. In other studies of E. amylovora isolated from Maloideae, using RAPD analysis (Taylor and Hale 1998; Brennan et al. 2002) or PCR-RFLP analysis of the SSR region (Lecomte et al. 1997), no significant relationship was found between genotype and host species.
The present study strongly suggests that the first fire blight outbreak in Norway started with a strain of the SSR5 genotype since this was the only genotype recovered during the first five years after detection of the disease in 1986. In this period all the isolates were also from a relatively limited geographic region (the Stavanger district). The relatively high frequency of this genotype in many European countries (Kim and Geider 1999; Jock et al. 2003; Ruppitsch et al. 2004) also makes initial infection by an SSR5 genotype quite probable. In the last five years of the study, SSR5 was also the dominating genotype, except in 2001, when the genotypes SSR8 and SSR9 together made up 91% of the 32 isolates. All the SSR8 and SSR9 isolates from 2001 were however from one location on Karmøy. Whether the isolates that were recovered in 2000, from many locations in a large geographic region originated from the first outbreak detected 10 years earlier or whether there has been more than one introduction of E. amylovora to Norway is uncertain. The appearance of more and other SSR genotypes could be caused by both new introduction and/or evolution of the pathogen in what must have been a latent undetected phase.
The primer set used in this study was designed for genotyping and not for detection. It was less sensitive than the protocol from Llop et al. (2000), a nested PCR recommended by EPPO (Fig. 5). Regardless, the assay detected E. amylovora in symptomatic plant material as well as in highly diluted pure cultures. Compared to a nested PCR, the method described here requires fewer reagents and saves over one hour per PCR run. Nesting PCRs increases sensitivity often at the cost of specificity, and for the nested protocols by Llop et al. (2000) and McManus and Jones (1995) a propensity towards false positives was shown (Powney et al. 2011).
Since this work was conducted, the development of an alternative genotyping approach based on the CRISPR region has proven powerful in reconstructing the dissemination of Erwinia amylovora over time (Kurz et al. 2021). That work was performed on isolates that had previously been genotyped by other methods, including SSR analysis of VNTR-A, making it possible to infer a relationship between results obtained from the two methods. The CRISPR archetypal genotype A correlated with a higher number of repeats in the SSR marker used in this work, while genotype D correlated with a lower number of repeats for isolates from Austria, but this relationship was not observed for other geographic regions (Rezzonico et al. 2011; Kurz et al. 2021). Single nucleotide polymorphism (SNP) analysis based on whole genome sequencing of isolates has become affordable enough to emerge as an obtainable gold standard for genotyping, surpassing both microsatellite and CRISPR-based approaches. Future findings on the correlation between SNPs on a genome scale and the two latter approaches will provide additional clarity on the implications of the SSR variations shown in this work.
Data availability
The data presented in the study are included in the manuscript.
References
Anonymous (2022) PM 7/20 (3) Erwinia amylovora. EPPO Bull 52:198–224
Bereswill S, Pahl A, Bellemann P, Zeller W, Geider K (1992) Sensitive and species-specific detection of Erwinia amylovora by polymerase chain reaction analysis. Appl Environ Microbiol 58:3522–3526
Billing E, Baker LAE, Crosse JE, Garrett CME (1961) Characteristics of English isolates of Erwinia amylovora (Burrill) Winslow et al. J Appl Bacteriol 24:195–211
Bonn V, van der Zwet T (2000) Distribution and economic importance of fire blight. In: Vanneste JL (ed) Fire blight. The disease and its causative agent, Erwinia amylovora. CABI, Wallingford, pp 37–53
Brennan JM, Doohan FM, Egan D, Scanlan H, Hayes D (2002) Characterization and differentiation of Irish Erwinia amylovora isolates. J Phytopathol 150:414–422
Bühlmann A, Dreo T, Rezzonico F, Pothier JF, Smits THM, Ravnikar M, Frey JE, Duffy B (2014) Phylogeography and population structure of the biologically invasive phytopathogen Erwinia amylovora inferred using minisatellites. Environ Microbiol 16:2112–2125
Doolotkeldieva T, Bobushova S, Schuster C, Konurbaeva M, Leclerque A (2019) Isolation and genetic characterization of Erwinia amylovora bacteria from Kyrgyzstan. Eur J Plant Pathol 155:677–686
Falkenstein H, Bellemann P, Walter S, Zeller W, Geider K (1988) Identification of Erwinia amylovora, the fireblight pathogen, by colony hybridization with DNA from Plasmid pEA29. Appl Environ Microbiol 54:2798–2802
Gottsberger RA (2010) Development and evaluation of a real-time PCR assay targeting chromosomal DNA of Erwinia amylovora. Lett Appl Microbiol 51:285–292
Jock S, Jacob T, Kim WS, Hildebrand M, Vosberg HP, Geider K (2003) Instability of short-sequence DNA repeats of pear pathogenic Erwinia strains from Japan and Erwinia amylovora fruit tree and raspberry strains. Mol Genet Genomics 268:739–749
Kim WS, Geider K (1999) Analysis of variable short-sequence DNA repeats on the 29 kb plasmid of Erwinia amylovora strains. Eur J Plant Pathol 105:703–713
Kurz M, Carnal S, Dafny-Yelin M, Mairesse O, Gottsberger RA, Ivanović M, Grahovac M, Lagonenko AL, Drenova N, Zharmukhamedova G, Doolotkeldieva T, Smits THM, Rezzonico F (2021) Tracking the dissemination of Erwinia amylovora in the Eurasian continent using a PCR targeted on the duplication of a single CRISPR spacer. Phytopathol Res 3:18
Lecomte P, Manceau C, Paulin JP, Keck M (1997) Identification by PCR analysis on plasmid pEA29 of isolates of Erwinia amylovora responsible of an outbreak in Central Europe. Eur J Plant Pathol 103:91–98
Llop P, Bonaterra A, Peñalver J, López MM (2000) Development of a highly sensitive nested-PCR procedure using a single closed tube for detection of Erwinia amylovora in asymptomatic plant material. Appl Environ Microbiol 66:2071–2078
Llop P, Donat V, Rodriguez M, Cabrefiga J, Ruz L, Palomo JL, Montesinos E, Lopez MM (2006) An indigenous virulent strain of Erwinia amylovora lacking the ubiquitous plasmid pEA29. Phytopathol 96:900–907
Mann RA, Smits TH, Bühlmann A, Blom J, Goesmann A, Frey JE, Plummer KM, Beer SV, Luck J, Duffy B (2013) Comparative genomics of 12 strains of Erwinia amylovora identifies a pan-genome with a large conserved core. PLoS One 8:e55644
McGhee GC, Jones AL (2000) Complete nucleotide sequence of ubiquitous plasmid pEA29 from Erwinia amylovora strain Ea88: gene organization and intraspecies variation. Appl Environ Microbiol 66:4897–4907
McManus P, Jones A, (1995) Detection of Erwinia amylovora by nested PCR and PCR-dot-blot and reverse blot hybridizations. In: Proceedings of the VII international workshop on fire blight, pp 87–90
Mendes RJ, Amaro C, Luz JP, Tavares F, Santos C (2022) Variability within a clonal population of Erwinia amylovora disclosed by phenotypic analysis. PeerJ 10:e13695
Mendes RJ, Luz JP, Santos C, Tavares F (2021) CRISPR genotyping as complementary tool for epidemiological surveillance of Erwinia amylovora outbreaks. PLoS One 16:e0250280
Momol M, Aldwinckle H (2000) Genetic diversity and host range of Erwinia amylovora. In: Vanneste JL (ed) Fire blight. The disease and its causative agent, Erwinia amylovora. CABI, Wallingford, pp 55–72
Parcey M, Gayder S, Morley-Senkler V, Bakkeren G, Úrbez-Torres JR, Ali S, Castle AJ, Svircev AM (2020) Comparative genomic analysis of Erwinia amylovora reveals novel insights in phylogenetic arrangement, plasmid diversity, and streptomycin resistance. Genomics 112:3762–3772
Pedroncelli A, Puopolo G (2023) This tree is on fire: a review on the ecology of Erwinia amylovora, the causal agent of fire blight disease. J Plant Pathol. https://doi.org/10.1007/s42161-023-01397-y
Persson P, Sletten A (1995) Fatty acid analysis for the identification of Erwinia carotovora subsp. atroseptica and E. carotovora subsp. carotovora. EPPO Bull 25:151–156
Powney R, Beer SV, Plummer K, Luck J, Rodoni B (2011) The specificity of PCR-based protocols for detection of Erwinia amylovora. Australas Plant Pathol 40:87–97
Rezzonico F, Smits TH, Duffy B (2011) Diversity, evolution, and functionality of clustered regularly interspaced short palindromic repeat (CRISPR) regions in the fire blight pathogen Erwinia amylovora. Appl Environ Microbiol 77:3819–3829
Ruppitsch W, Stöger AR, Keck M (2004) Stability of short sequence repeats and their application for the characterization of Erwinia amylovora strains. FEMS Microbiol Lett 234:1–8
Schnabel EL, Jones AL (1998) Instability of a pEA29 marker in Erwinia amylovora previously used for strain classification. Plant Dis 82:1334–1336
Sebaihia M, Bocsanczy AM, Biehl BS, Quail MA, Perna NT, Glasner JD, DeClerck GA, Cartinhour S, Schneider DJ, Bentley SD, Parkhill J, Beer SV (2010) Complete genome sequence of the plant pathogen Erwinia amylovora strain ATCC 49946. J Bacteriol 192:2020–2021
Sheldon AL (1969) Equitability indices - dependence on species count. Ecology 50:466–467
Sletten A, Melboe NS (2004) Experiences with the control of fireblight in Norway during 1986/2003. EPPO Bull 34:361–363
Sletten A, Talgo V, Rafoss T, Melboe NS (2017) Fire blight in Norway: a review of strategies and control measures from 1986 to 2016. J Plant Pathol 99:137–139
Smits TH, Rezzonico F, Duffy B (2011) Evolutionary insights from Erwinia amylovora genomics. J Biotechnol 155:34–39
Taghdareh G, Baghaee-Ravari S, Moslemkhani C, Mahdikhani-Moghaddam E (2014) Evaluation of repeat sequences on plasmid pEA29 of Erwinia amylovora from Iran. Eur J Plant Pathol 140:735–744
Taylor RK, Hale CN (1998) Identification and characterisation of isolates of Erwinia amylovora from cotoneaster in Australia. Australas Biotechnol 8:353–356
Thomson SV (2000) Epidemiology of fire blight. In: Vanneste JL (ed) Fire blight. The disease and its causative agent, Erwinia amylovora. CABI, Wallingford, pp 9–36
van Belkum A, Scherer S, van Alphen L, Verbrugh H (1998) Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev 62:275–293
Végh A, Hevesi M, Pájtli É, Palkovics L (2017) Characterization of Erwinia amylovora strains from Hungary. Eur J Plant Pathol 147:455–461
Acknowledgements
This research was supported by the NIBIO basic funding. Isolates were collected through fire blight surveys commissioned by the Norwegian Food Safety Authority. We thank Anne B. Nilsen for construction of the Norwegian map indicating the collected fire blight samples. The fragment analysis was performed at the FUGE technology platform Centre for Integrative Genetics, Ås, Norway.
Funding
Open access funding provided by Norwegian Institute of Bioeconomy Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brurberg, M., Elameen, A., Sletten, A. et al. Genetic characterization of historic Norwegian Erwinia amylovora isolates by SSR-genotyping. J Plant Pathol (2024). https://doi.org/10.1007/s42161-024-01596-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42161-024-01596-1