
Abstract
The take-all disease is one of the most important maladies in cereals and grasses, being caused by the fungus Gaeumannomyces tritici. Secondary metabolites are known to perform critical functions during the infection process of various phytopathogens. However, the current understanding of the biosynthesis of secondary metabolites in G. tritici is limited. Similarly, comprehensive analyses of the expression, conservation, and evolution of these biosynthesis-related genes are crucial for enhancing our knowledge of the molecular mechanisms that drive the development of the take-all disease. Here we have performed a deep survey and description of secondary metabolite biosynthetic gene clusters in G. tritici, analyzed a previously published RNA-seq of a mimicked infection condition, and assessed the conservation among 10 different Magnaporthales order members. Notably, the majority of the 35 putative gene clusters identified were conserved among these species, with GtPKS1, GtPKS3, and GtTERP4 uniquely identified in G. tritici. In the mimicked infection condition, seven gene clusters, including the GtPKS1 cluster, exhibited upregulated expression. Through comparative genomic analysis, GtPKS1 was associated with the production of dichlorodiaporthin, a metabolite with cytotoxic and antifungal activity. In addition, GtPKS10 and GtPKSNRPS3 showed similarities to already characterized biosynthetic pathways involved in the synthesis of ACR-toxin (phytotoxic) and trichosetin (phytotoxic and antibiotic), respectively. These three gene clusters were further scrutinized through phylogenetic inference, which revealed the distribution of orthologous sequences across various plant-associated fungi. Finally, the detailed identification of several genes enrolled in secondary metabolite biosynthesis provides the foundation for future in-depth research, supporting the potential impact of several small molecules on G. tritici lifecycle and host interactions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Magnaporthales order encompass several economically important plant pathogenic fungi (Luo et al. 2015). Gaeumannomyces tritici is the causative agent of the take-all disease (TAD), one of the most devastating root diseases of wheat, being also able to infect other cereals and grasses, as triticale, barley, and rye (Keenan et al. 2015). Several studies reported the capability of this fungus to grow in a wide range of temperatures from 4 to 30 °C, and, although the disease is more common in wheat grown under moist conditions, it can occur under conditions of low precipitation, giving the pathogen a cosmopolitan status (Kwak and Weller 2013).
The TAD generally occurs when the practice of monoculture prevails (Kwak and Weller 2013). However, abiotic factors, such as soil pH and humidity can also influence the progression and severity of the disease (Smiley 1973; James Cook 2003). The primary infection occurs when young seedling roots meet fungus-carrying debris, which allows the fungus to invade and destroy the root tissues (Palma-Guerrero et al. 2021). Secondary infections spread through root-to-root contact, resulting in field patches of acutely infected plants (Palma-Guerrero et al. 2021). The cosmopolitan status of G. tritici leads to diminished crop yields and lower grain quality worldwide. In the United Kingdom, approximately 50% of wheat are impacted by the disease, resulting in average yield losses ranging from 5 to 20%, although severe epidemics can result in yield losses as high as 60% (McMillan et al. 2011; Agriculture and Horticulture Development Board (AHDB)).
Despite the substantial impact and importance of TAD pathogens, a knowledge gap persists regarding the molecular mechanisms involved in disease development, such as virulence determinants (Yang et al. 2015). To tentatively address this matter, the genome of G. tritici has been made available. Additionally, genomes from multiple Magnaporthales species with diverse lifestyles have been published, including Magnaporthiopsis poae, Pyricularia oryzae, Pyricularia pennisetigena, Pyricularia grisea, Magnaporthiopsis incrustans, Magnaporthiopsis rhizophila, Nakataea oryzae, Pseudohalonectria lignicola, Ophioceras dolichostomum, and Falciphora oryzae (Dean et al. 2005; Xu et al. 2015; Zhong et al. 2016; Gómez Luciano et al. 2019). These sequences can be exploited through comparative genomic analysis aiming to understand the evolutionary relationships between species, adaptation to environmental conditions, and to locate putative virulence determinants, involved in host-pathogen interactions.
Secondary metabolites (SMs) are small molecules with a myriad of biological activities. Several pathogenic fungi, including phytopathogens, employ toxic SMs to gain advantages while interacting with their hosts (Osbourn 2010; Gibson et al. 2014; Keller 2015). For example, the phytopathogen Fusarium graminearum, responsible for several diseases in wheat and barley (head blight, crown rot, and seedling blight), produces an unusual non-ribosomal octapeptide, fusaoctaxin A, that facilitates the invasion through a cell-to-cell penetration process (Jia et al. 2019). Pyrenophora tritici-repentis, which causes tan spot of wheat, produces necrosis-inducing toxins called triticones (Rawlinson et al. 2019), and Parastagonospora nodorum, another wheat pathogen, produces phomacins, compounds that disrupt the cytoskeletal rearrangements of the host, affecting positively the infection outcome (Li et al. 2020).
In filamentous fungi, the genes enrolled in the same SM biosynthetic pathway are usually in spatial proximity, arranged in a cluster-like manner in the genomes. These biosynthetic gene clusters (BGCs) are not only common in filamentous fungi as they are also found in bacteria (operons), oomycetes and, more recently, have been discovered in plant species (Osbourn 2010; Nützmann and Osbourn 2014). The physical linkage of SM genes potentially minimizes the number of regulatory steps in the biosynthetic machinery, thereby contributing to physiological optimization (Gacek and Strauss 2012). Moreover, the BGCs are frequently organized around backbone genes, as polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS) or hybrids (PKS-NRPS). Since the domains from these backbone genes have been largely explored, bioinformatic tools for the prediction of potential BGCs rely on these domains for accurate prediction (Medema et al. 2011; Wolf et al. 2015). Notably, the greater accessibility of gene sequencing techniques combined with BGS prediction tools, have located a vast array of unexplored SM gene clusters. Many of these BGCs remain silent under standard laboratory culture conditions. These compounds are only synthesized during certain stages of the complex fungal life cycle (e.g., miktospiromide A and kitrinomycin A are only produced in a coculture of Penicillium brasilianum and Aspergillus nomius) (Cowled et al. 2023). Noteworthy, the activation of silent BGCs and characterization of the producing compounds present significant scientific and commercial opportunities, considering the extensive range of biological activities of SMs (Brakhage 2013; Yaegashi et al. 2014; Rutledge and Challis 2015; Keller 2015).
Secondary metabolites may be valuable virulence determinants for G. tritici. Furthermore, these compounds can also exhibit roles in the environmental maintenance of the pathogen and resistance against mycopathogens. To investigate TAD-related BGCs, we explored the genome of G. tritici strain R3-111a-1. The conservation of the identified BGCs was assessed within other ten species of the Magnaporthales order to identify BGCs unique to G. tritici or conserved among several phytopathogens. Some selected gene clusters were deeply explored through comparative genomic analysis and phylogeny, and potential compounds were suggested. Furthermore, to identify BGCs that may play a vital role in host-pathogen interaction, we also analyzed previously published transcriptomic data comparing axenic culture with infected wheat roots.
Methods
Prediction of secondary metabolite biosynthetic gene clusters
All fungal genomes were obtained from the NCBI Genome Database. The Bioproject accession numbers are displayed in Online Resource 1. Genome assembly and completeness were inferred from analysis with BUSCO (Simão et al. 2015). The prediction of putative BGCs was conducted as described previously (Sbaraini et al. 2017), with few modifications. G. tritici R3-111a-1 BGCs were identified with antiSMASH 7.0 (using the genome assembly sequence as input) and SMIPS (using the predicted proteins as input) algorithms (Wolf et al. 2015; Blin et al. 2023). Furthermore, the Synthase Domain Parser Tool (Gilchrist and Chooi 2021a) was employed for a more accurate prediction of the domain architecture of backbone proteins.
Conservation of the predicted secondary metabolite biosynthetic gene clusters among species from the Magnaporthales order
The conservation of the predicted BGCs among species from the Magnaporthales order with annotated genome (M. poae ATCC 64411, P. oryzae 70 − 15, P. pennisetigena Br36 and P. grisea NI907) was assessed using MultiGeneBlast 1.1.14 (Medema et al. 2013), based, primarily, on backbone gene conservation (amino acid sequences; e-value < 1 × 10−5, query coverage ≥ 45%, and identity ≥ 45%). For species without annotated genome (M. incrustans M35, M. rhizophila M23, N. oryzae M69, P. lignicola M95, O. dolichostomum CBS 114926 and F. oryzae R5-6-1) the analysis was firstly conducted using BLASTN, based on the potential conservation of the backbone gene CDS region (nucleotide sequences; e-value < 1 × 10−5, query coverage ≥ 45%, and identity ≥ 45%). Subsequently, the nucleotide fragments (containing the potential backbone genes) were annotated using FGENESH (gene-finding parameters for the closest species presented in the algorithm) (Solovyev et al. 2006) and validated with BLASTP (employing the same cut-offs used in the MultiGeneBlast step). The BLASTN/FGENESH/BLASTP procedure was also used as a second line of verification for all BGCs from annotated genomes that did not show conservation in the MultiGeneBlast analyses.
Prediction of BGC boundaries and synteny comparison analyses
The cblaster tool was utilized for predicting the boundaries of all identified BGCs (Gilchrist et al. 2021; Blin et al. 2023). Based on the results obtained from cblaster, selected BGCs of interest were further analyzed for synteny comparison using the clinker tool (Gilchrist and Chooi 2021b). Both cblaster and clinker were operated using their default parameters.
Phylogenetic analyses
For phylogenetic analyses, the backbone genes of specific BGCs were chosen (GtPKS1, GtPKS10, and GtPKSNRPS3) and orthologous sequences were selected through routine BLASTP against the non-redundant protein sequences (nr) database. In addition, several fungal genome sequences have been deposited at NCBI as raw or incomplete assemblies. These projects do not have predicted proteins/genes/mRNAs deposited at NCBI and are, therefore, inaccessible through routine BLASTP/BLASTN. Thus, a screening using BLASTN against the WGS database was also performed to incorporate those putative orthologous sequences in the analyses.
When a putative positive match was found, the genome sequences were downloaded, and genes were predicted with FGENESH (gene-finding parameters for the closest species presented in the algorithm) (Solovyev et al. 2006). Genes that satisfied the previously fixed cutoffs (e-value < 1 × 10− 5, query coverage ≥ 50%, and identity ≥ 45%) were incorporated n the phylogeny. PRANK v.140603 was employed for sequence alignment (Löytynoja and Goldman 2010). The best-fit evolutionary model was estimated using Prottest 3.4 (Darriba et al. 2011). Phylogenetic reconstruction (Maximum Likelihood) was conducted using PhyML 3.1 (Guindon et al. 2010) with aLRT SH-like (approximate likelihood ratio test Shimodaira–Hasegawa) branch support estimation (Anisimova and Gascuel 2006; Anisimova et al. 2011).
The phylogeny of PKS and PKS-NRPS genes can help to determine if the collected entries are true orthologs or artifacts, however, this analysis can be problematic, particularly for ortholog definition. To address this issue, a previously established phylogenetic-based approach was employed (Sbaraini et al. 2016). In this phylogeny, we included the KS and AT domains of PKS and PKS-NRPS genes from G. tritrici, and the domains of all characterized PKS and PKS-NRPS genes stored in the MIBiG database (i.e., a database of characterized BGCs) (Medema et al. 2015). The amino acid alignment was built using PRANK v.100701, without manual curation. Phylogenetic reconstruction was conducted employing Maximum Likelihood as described above.
Transcriptome and RNA-Seq differential expression analysis
To validate the expression of the identified BGCs and gain insights into their regulation during plant infection, we analyzed a previously published sequencing data from G. graminis var. tritici strain Ggt-C2 cells cultured in PDA plates or inoculated in wheat roots with or without the biocontrol agent Bacillus velezensis (Kang et al. 2019). The raw RNA-seq libraries were downloaded from the NCBI SRA database under the BioProject accession numbers PRJNA485739 and PRJNA496308. Low quality (Q < 30) and adapter sequences were removed using Trim Galore! (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and the libraries were mapped to the latest annotation and assembly version of the G. tritici genome using HISAT2 (Kim et al. 2019) with default parameters. Aligned reads were counted with FeatureCounts and differential expression analysis was carried out using the DESeq2 pipeline (Liao et al. 2013; Love et al. 2014). Genes with an FDR-corrected p-value lower than 0.05 and absolute log fold-change expression higher than 1.5 were considered differentially expressed. The parameters were based on those used by Kang and coworkers (2019).
Results
Predictions of secondary metabolite biosynthetic gene clusters
Thirty-five putative BGCs were identified in G. tritici R3-111a-1 genome using two prediction tools: antiSMASH 7.0 and SMIPS. The 35 BGCs are from different classes, including 14 PKS (13 type I and 1 type III), 5 NRPS, 3 NRPS-like, 5 terpenes (TERP), 3 PKS-NRPS hybrids, and 5 indols (IND) (Fig. 1, Online Resource 2). Additionally, among the 13 type I PKS backbone genes identified, 2 are potential non-reducing (NR) PKS, 10 are potential highly-reducing (HR) PKS, while 1 is a potential partially reducing (PR) PKS (Online Resource 3).

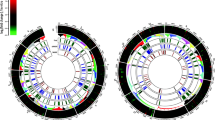
Conservation of the predicted BGCs of G. tritici in ten species of the order Magnaporthales. Conservation was inferred using the backbone genes amino acid sequences (e-value < 1 × 10−5, query coverage ≥ 45%, and identity ≥ 45%), in 10 species of the order Magnaporthales. a) Conservation of PKS BGCs. b) Conservation of terpene and indol BGCs. c) Conservation of NRPS, NRPS-like and PKS-NRPS BGCs. PPE (P. pennisetigena) POR (P. oryzae); PGR (P. grisea); MPO (M. poae); MRH (M. rhizophila); MIN (M. incrustans); NOR (N. oryzae); FOR (F. oryzae); PLI (P. lignicola); ODO (O. dolichostomum). The conservation of the predicted BGCs among species with annotated genome was assessed using MultiGeneBlast v1.1.14, based, primarily, on backbone gene conservation (amino acid sequences; e-value < 1 × 10−5, query coverage ≥ 45%, and identity ≥ 45%). For species without annotated genome, the analysis was firstly conducted using BLASTN, based on the potential conservation of the backbone gene CDS region (nucleotide sequences; e-value < 1 × 10−5, query coverage ≥ 45%, and identity ≥ 45%). Subsequently, the nucleotide fragments (containing the potential backbone genes) were annotated using FGENESH (gene-finding parameters for the closest species presented in the algorithm) and validated with BLASTP
Conservation of BGCs in the Magnaporthales order
To understand the distribution of the identified BGCs in close species with different infection traits we assessed the conservation of these genes in ten species of the Magnaporthales order, encompassing three families: Magnaporthaceae (5 evaluated species), Pyriculariaceae (3 evaluated species) and Ophioceraceae (2 evaluated species). The proximity of the selected species to G. tritici was confirmed by phylogeny (Online Resource 4). The majority of BGCs found in G. tritici were conserved in the phytopathogenic N. oryzae M69 (29 orthologous BGCs; ~ 83%) and in the endophytic F. oryzae (27 orthologous BGCs; ~ 77%). Additionally, more than 68.5% of identified BGCs were conserved in P. grisea NI907, P. oryzae 70 − 15, P. pennisetigena Br36, M. incrustans M35 and M. rhizophila M23 with respectively 28, 26, 24, 24 and 24 orthologous BGCs (Fig. 1, Online Resource 2). M. poae ATCC 64411, a phytopathogenic fungus closely related to G. tritici, harbors only 19 orthologous BGCs (~ 54%), showing less conservation than the other species of the Magnaporthaceae family evaluated (Fig. 1, Online Resource 2), as well as species from more distantly related family Pyriculariaceae. However, the BUSCO analysis indicated that M. poae ATCC 64411 genome has a low quality, which can potentially affect the BGC identification pipeline (Online Resource 5). Notably, only the non-pathogenic saprotrophic fungi O. dolichospermum CBS 114926 (17 orthologous BGCs) and P. lignicola M95 (16 orthologous BGCs), from the family Ophioceraceae, showed less than 54% of conserved BGCs (Fig. 1, Online Resource 2). Notably, GtPKS1, GtPKS3, and GtTERP4 were found only in G. tritici, while 11 BGCs were conserved in all ten species (Fig. 1, Online Resource 2).
Comparative genomic analyses and phylogeny of G. tritici BGCs
Considering that no putative BGCs have been functionally characterized in TAD pathogens to date, we employed comparative genomics to determine the potential final products of these BGCs. Furthermore, a phylogenetic analysis was also performed using all PKS/PKS-NRPS backbone enzymes available at MIBIG to support the suggested orthology (Online Resource 6). These comparisons revealed three interesting BGCs that exhibited similarity with gene clusters characterized in other species: GtPKS1, GtPKS10, and GtPKSNRPS3 putatively linked with the biosynthesis of dichlorodiaporthin, ACR-toxin, and trichosetin compounds, respectively. Moreover, GtPKS9 backbone enzyme (GGTG_00407; XP_009216417.1) is likely implicated in melanin biosynthesis, displaying 61% identity with the characterized PKS involved in Glarea lozoyensis melanin biosynthesis (pks1; AAN59953.1); GtNRPS2 backbone enzyme (GGTG_08621/ XP_009224727.1) is likely implicated in the biosynthesis of the siderophore ferricrocin, displaying 55% identity with SSM1 (i.e., the ferricrocin NRPS characterized in Pyricularia spp.; XP_003719607.1/AAX49357.1); and GtNRPS5 backbone enzyme (GGTG_02228; XP_009218263.1) is likely implicated in the biosynthesis of coprogen siderophores, displaying 60% identity with SSM2 (i.e., the coprogen NRPS characterized in Pyricularia spp.; XP_030985539.1). Additionally, GtNRPS1 backbone enzyme (GGTG_13228; XP_009229397.1) shows 55% with Abt1 implicated in aureobasidin A biosynthesis. However, Abt1 and the GtNRPS1 backbone enzyme have a different domain organization.
The GtPKS1 backbone enzyme (XP_009229208.1; GGTG_13042) exhibits 56% identity with the Aspergillus oryzae dichlorodiaporthin backbone enzyme (DiaA; BAE62229.1). For product release in A. oryzae, a β-lactamase-like enzyme (DiaB; BAE62228.1) is involved, and this enzyme is also conserved in G. tritici (54% of identity; XP_009229207.1). Additionally, a short-chain dehydrogenase/reductase (DiaC; BAE62225.1), and a flavin-dependent monooxygenase (DiaD; BAE62224.1) are also conserved in G. tritici (XP_009229202.1, and XP_009229201.1) showing 66% and 54% of identity, respectively (Fig. 2a). While an ortholog for AoiQ (BAE62227.1), which catalyzes the incorporation of the halogens observed in dichlorodiaporthin, was not initially identified in the G. tritici cluster, a reannotation of the region using FGENESH revealed a single gene that was initially predicted as two separate genes, XP_009229205.1 (GGTG_13039) and XP_009229206.1 (GGTG_13040). This newly defined gene shares a 63% identity with AoiQ (required for dichlorodiaporthin production, with its absence leading to citreoisocoumarin accumulation), and both genes possess methyltransferase and flavin-dependent-halogenase domains (Fig. 2a). Furthermore, the capability of G. tritici to produce dichlorodiaporthin is strengthened by two pieces of evidence. Firstly, cblaster analysis did not detect any supplementary genes as part of the putative BGC (Online Resource 7). Secondly, the transcriptomic analysis revealed that the genes XP_009229201.1-XP_009229208.1 (excluding XP_009229204.1) are the only ones within this locus that are upregulated under infection conditions (Online Resource 8). Based on these findings and the existing body of knowledge about dichlorodiaporthin, we constructed a potential biosynthetic pathway for GtPKS1 in G. tritici (Fig. 2b).

Putative dichlorodiaporthin gene cluster (GtPKS1), conservation and synteny. a) BGC organization and synteny comparison of GtPKS1. Backbone genes are identified with the full protein ID and accessory genes are identified with the last digits that differ from the backbone gene code. Homologous genes are indicated by color. (* indicates a reannotated gene) b) The proposed dichlorodiaporthin biosynthetic pathway
We also explored the distribution of dichlorodiaporthin PKS employing phylogenetic approaches. The inferred phylogenetic tree revealed that the GtPKS1 backbone enzyme is in close evolutionary proximity with PKSs found in the phytopathogens Colletotrichum sublineola and Grosmannia clavigera, as well as with the backbone enzyme of the saprobe/eventually pathogen Lachnellula occidentalis, and Neurospora sp. (which present endophytic, phytopathogenic and saprotrophic lifestyles) (Fig. 3).

Phylogenetic analysis of GtPKS1. The GtPKS1 backbone enzyme (XP_009229208.1; GGTG_13042; highlighted in bold) exhibits 56% identity with the A. oryzae dichlorodiaporthin backbone enzyme (DiaA; BAE62229.1; highlighted in bold), being also in close evolutionary proximity with PKSs found in Lachnellula occidentalis, Neurospora sp., and in the phytopathogens Colletotrichum sublineola and Grosmannia clavigera
The GtPKS10 backbone enzyme (XP_009218470.1; GGTG_02434) shows 53% of identity with ACRTS2 (BAN19720.1) (Fig. 4a), an A. alternata PKS responsible for the biosynthesis of ACR-toxin (Izumi et al. 2012). Both enzymes also share a common domain organization. ACRTS1, a putative hydrolase, theoretically involved in the release of the carbon chain from the PKS’ acyl carrier protein domain, is another essential enzyme in ACR-toxin biosynthesis. Notably, ACRTS1 (L8AXV5) has a predicted cytochrome P450 family domain, and a similar domain is found in XP_009218466.1 (GGTG_02430), located in the vicinities of the GtPKS10 backbone gene, although ACRTS1 and XP_009218466.1 are not orthologs. In this way, XP_009218466.1 can potentially release the carbon chain of the G. tritici PKS (Fig. 4). Interestingly, the gene XP_009218466.1 is conserved in more distantly related orthologous gene clusters, such as the gene cluster found in M. anisopliae (Fig. 4a). However, it is challenging to determine whether the entire gene cluster for ACR-toxin is conserved in the TAD pathogen, as other potential enzymes involved in compound biosynthesis have not been investigated. Moreover, XP_009218469.1 (GGTG_02433) is a potential transcription factor (harboring a Zn2Cys6 binuclear cluster DNA-binding domain and a fungal-specific transcription factor domain) which could regulate the expression of the entire cluster. This potential transcription factor is also present in BGCs of several phytopathogens (e.g., P. nodorum, P. lindquistii, P. teres f. teres and N. serpens) (Fig. 4a).

Putative ACR-toxin gene cluster (GtPKS10), conservation and synteny. a) BGC organization and synteny comparison of GtPKS10. Backbone genes are identified with the full protein ID and accessory genes are identified with the last digits that differ from the backbone gene code. Homologous genes are indicated by color. b) ACR-toxin 2D chemical structure
Noteworthy, the phylogenetic analysis, employing ACR-toxin backbone enzymes, revealed orthologs in several plant-associated species. Interestingly, GtPKS10 backbone enzyme is in close evolutionary proximity with orthologous sequences found in Mycosphaerella populi (plant pathogen), dead wood inhabiting saprobes from order Xylariales, and plant-associated fungi from order Hypocreales (entomopathogens, endophytes, and phytopathogens) (Fig. 5) Moreover, orthologs for the GtPKS10 BGC were found in the three evaluated species from the Magnaporthiopsis genus and in P. pennisetigena (Fig. 1a, Online Resource 2).

Phylogenetic analysis of GtPKS10. The GtPKS10 backbone enzyme (XP_009218470.1; GGTG_02434; highlighted in bold) shows 53% of identity with ACRTS2 (BAN19720.1; highlighted in bold), an A. alternata PKS responsible for the biosynthesis of ACR-toxin. GtPKS10 presented similarity with PKSs found in several plant-associated fungi
The predicted backbone enzyme for GtPKSNRPS3 BGC (XP_009223648.1; GGTG_07560) displays 54% identity with the backbone enzyme EqxS (AGO86662.1), derived from F. heterosporum (Fig. 6a), which is responsible for the biosynthesis of equisetin. Like the lovastatin biosynthesis pathway, a trans-ER (EqxC; AGO86659.1) is mandatory for equisetin biosynthesis, and a predicted trans-ER was also found conserved in the GtPKSNRPS3 (XP_009223650.1 (GGTG_07562); 53% of the identity with EqxC). Noteworthy, other genes found in the equisetin BGC, and not explored through knockout mutants, are also conserved in the GtPKSNRPS3 BGC: XP_009223649.1 (GGTG_07561) displays 36% of identity with Eqx3 (AGO86663.1; hypothetical protein); XP_009223647.1 (GGTG_07559) displays 49% of identity with EqxF (AGO86660.1; transcription factor/regulator); XP_009223646.1 (GGTG_07558) displays 64% of identity with EqxG (AGO86666; MFS transporter) (Fig. 6a). However, an ortholog for EqxD (AGO86665.1) was not found in the GtPKSNRPS3 BGC. EqxD is a N-methyltransferase and knockout mutants for this gene in F. heterosporum led to abolition of equisetin production and accumulation of trichosetin, a phytotoxic metabolite. In this way, we predict that G. tritici can, potentially, produce trichosetin or trichosetin-like compounds. These results are further corroborated by the cblaster analysis, which did not predict additional genes in this gene cluster (i.e., the BGC comprises XP_009223646.1- XP_009223650.1). Drawing on these findings and the existing knowledge about trichosetin (Fisch 2013), we constructed a potential biosynthetic pathway for GtPKSNRPS3 in G. tritici (Fig. 6b).

Putative trichosetin gene cluster (GtPKSNRPS3), conservation and synteny. a) BGC organization and synteny comparison of GtPKSNRPS3. Backbone genes are identified with the full protein ID code and accessory genes are identified with the last digits that differ from the backbone gene code. Homologous genes are indicated by color. b) The proposed trichosetin biosynthetic pathway
Notably, the phylogenetic analysis, employing equisetin/trichosetin backbone enzymes, revealed orthologs in several plant-associated species, including plant-pathogens as Alternaria spp., Raffaelea spp., and Penicillium expansum, besides Fusarium spp. (Fig. 7). Among the species from Magnaporthales order evaluated, the GtPKSNRPS3 backbone gene exhibited orthologs in all three Pyricularia species and in M. incrustans (Fig. 1, Online Resource 2). However, as uncovered by the phylogenetic analysis, equisetin/trichosetin orthologs in the Magnaporthales order do not have a monophyletic origin (Fig. 7).

Phylogenetic analysis of GtPKSNRPS3. The predicted backbone enzyme for GtPKSNRPS3 BGC (XP_009223648.1; GGTG_07560; highlighted in bold) displays 54% identity with the backbone enzyme eqxS, derived from F. heterosporum (highlighted in bold), responsible for the biosynthesis of equisetin. GtPKSNRPS3 is in evolutionary proximity with PKSs found in several phytopathogens and plant-associated fungi
Expression profile of predicted BGCs in wheat root mimicked infection condition
To gain insights about the expression of predicted BGC backbone genes in wheat root conditions, we explored a previously published RNA-seq data (Kang et al. 2019). In this experiment, wheat roots infected with G. tritici (Wr_Gt), and infected with G. tritici in the presence of the biocontrol agent Bacillus velezensis (Wr_Gt_Bv) were compared with G. tritici grown on potato dextrose agar plates (Pda_Gt). Thus, three comparative analyses were performed (Wr_Gt x Pda_Gt; Wr_Gt_Bv x Pda_Gt; Wr_Gt x Wr_Gt_Bv). Moreover, it is important to notice that the detailed experimental procedure was previously described by Kang and coworkers (2019). The differential expression analysis showed that of the 35 backbone genes, 3 were downregulated when conditions Wr_Gt x Pda_Gt were compared (GtNRPS2, GtNRPS-like3, and GtPKSNRPS1); 6 backbone genes were upregulated when the conditions Wr_Gt x Pda_Gt were compared (in addition to the already described GtPKS1, the gene clusters GtPKS12, GtPKS13, GtNRPS5, GtNRPS-like1, and GtTERP2 were also differentially expressed), and three backbone genes were upregulated when the conditions Wr_Gt_Bv x Pda_Gt were compared (GtPKS1, GtPKS8, and GtPKS12) (Fig. 8). Noteworthy, gene clusters differentially expressed had the predicted boundaries adjusted following the expression profile results (Online Resource 9).

Heatmap of backbone genes differentially expressed in at least one infection conditions. Differential expression analysis of wheat roots infected with G. tritici compared with G. tritici grown on potato dextrose agar plates (Wr_Gt x Pda_Gt), and wheat roots infected with G. tritici in the presence of the biocontrol agent Bacillus velezensis compared with G. tritici grown on potato dextrose agar plates (Wr_Gt_Bv x Pda_Gt). Genes not differentially expressed are indicated in grey
Furthermore, 29 out of 35 backbone genes were expressed in at least one of the conditions explored (FPKM values ≥ 2). Although not upregulated, the backbone genes of the GtPKS10 BGC (i.e., putatively implicated in ACR-toxin-like compound biosynthesis) had FPKM values ≥ 2 in all three conditions evaluated; while the GtPKSNRPS3 backbone gene (i.e., putatively implicated in trichosetin biosynthesis) had FPKM values ≥ 2 in the Wr_Gt_Bv condition. Moreover, the BGCs implicated in melanin (GtPKS9), ferricrocin (GtNRPS2), and coprogens (GtNRPS5) biosynthesis also had FPKM values ≥ 2 in all three conditions explored. These results stress the functionality of the predicted backbone genes/BGCs, besides suggesting a potential activity of the produced metabolites in these conditions.
Discussion
Phytopathogenic fungi have a worldwide impact, being responsible for significant losses in the main cultivated crops. These pathogens have been the focus of intensive research, given the socioeconomic impact that such pathogens may cause (Dean et al. 2012). Comparative genomic studies have proven to be important to understand the evolution of fungal pathogens and highlight potential virulence determinants (Dean et al. 2012), but initiatives exploring G. tritici are still scarce. The availability of G. tritici genome, along with sequences of several other species of the order Magnaporthales, creates a platform for highlighting putative virulence determinants. In this context, secondary metabolites can be important tools for microorganisms, being produced to bypass the host’s defenses and guarantee the success of these organisms in the environment (Keller 2015). We explored the potential of the secondary metabolism of G. tritici and identified three interesting BGCs: GtPKS1, GtPKS10 and GtPKSNRPS3.
Our analysis showed that orthologs for the GtPKS1 backbone gene were absent in all evaluated Magnaporthales species. Furthermore, this cluster was differentially expressed under infection conditions and is similar to an A. oryzae BGC involved in dichlorodiaporthin biosynthesis. Through heterologous expression and in vitro reactions, the function of BAE62229.1 (DiaA), BAE62228.1 (DiaB), BAE62225.1 (DiaC), BAE62224.1 (DiaD), and BAE62227.1 (AoiQ) were explored in A. oryzae. These approaches led to the isolation of 8-methyldichlorodiaporthin, dichlorodiaporthin, citreoisocoumarin, and 20 other compounds (Chankhamjon et al. 2016; Liu et al. 2021). The heterologous expression of the complete BGC resulted in the production of 8-methyldichlorodiaporthin and dichlorodiaporthin, both of which have cytotoxic activity (Almeida et al. 2018; Cai et al. 2018). Moreover, dichlorodiaporthin also showed antifungal activity, against Colletotrichum musae and Rhizoctonia solani and α-glucosidase inhibitory activity (Li et al. 2016). Several structurally related isocoumarins exhibit phytotoxic activity, as well as a variety of potential biotechnological applications (Saeed 2016; Meepagala et al. 2018; Shabir et al. 2021). Given the production of dichlorodiaporthin and related compounds by G. tritici and the upregulation of several genes in the GtPKS1 BGC under infection conditions, we propose that this gene cluster may play a significant role in G. tritici infection process in wheat.
The GtPKS10 gene cluster was potentially associated with the biosynthesis of a molecule similar to the ACR-toxin. In A. alternata, ACR-toxin is a host-selective toxin that affects rough lemon (Citrus jambhiri) mitochondrion, uncoupling oxidative phosphorylation and causing leakage of cofactor NAD + from the Krebs cycle (Izumi et al. 2012). Furthermore, two genes in the GtPKS10 cluster had their putative function identified: XP_009218466.1 (GGTG_02430) being a cytochrome P450, harboring a potential role in the product release; and XP_009218469.1 (GGTG_02433), harboring a potential role as a transcription factor (Fig. 4). Notably, the GtPKS10 backbone gene XP_009218470.1 (GGTG_07560) was not differentially expressed under infection conditions, indicating that it may not be involved in the infection process.
The next BGC putatively linked with an important compound was GtPKSNRPS3. This gene cluster was related to eqx, a F. heterosporum BGC involved in the biosynthesis of equisetin, an N-methyl serine-derived acyl tetramic acid with antibiotic, cytotoxic, and HIV inhibitory activities (Vesonder et al. 1979; Singh et al. 1998; Kakule et al. 2013). However, the absence of an eqxD ortholog in G. tritici indicates that the most likely final product is trichosetin, an N-desmethyl precursor of equisetin, which has phytotoxic and antibiotic activity (Fig. 6) (Marfori et al. 2002, 2003). Multiple GtPKSNRPS3 accessory genes were conserved in F. heterosporum eqx BGC, reinforcing the functionality of this cluster in G. tritici. Moreover, other genes intertwined in GtPKSNRPS3 BGC can also help in compound biosynthesis potentially acting as a transporter or transcription factor/regulator. Notably, the phylogenetic analysis, employing equisetin/trichosetin backbone enzymes, revealed orthologs in several plant-associated species (Fig. 7).
Besides the three BGCs already discussed, other clusters also proved to be interesting targets for future studies, like GtPKS9, GtNRPS2 and GtNRPS5, which are likely implicated in melanin, ferricrocin and coprogen biosynthesis, respectively. Melanin has several activities such as photoprotection and thermoregulation, and in G. tritici is also an important virulence determinant, being necessary for the development of TAD (Henson et al. 1999; Cordero and Casadevall 2017). The siderophores ferricrocin and coprogen are low-molecular mass iron chelators, involved in the acquisition of iron, an essential element for the development of filamentous fungi (Haas et al. 2008; Khan et al. 2018). Furthermore, siderophores play a crucial role in infection processes across various host-pathogen interactions (Oide et al. 2006; Hof et al. 2007; Haas et al. 2008; Chen et al. 2013). In the phytopathogenic bacteria Erwinia chrysanthemi, the siderophore chrysobactin emerges as a key determinant influencing the infection outcome (Neema et al. 1993). This siderophore effectively diminishes the iron concentration in colonized tissues, thereby depriving plant cells of essential iron resources (Neema et al. 1993). Ferricrocin is an intracellular siderophore that facilitates iron storage, and Hof and coworkers (2007) demonstrated its involvement in the capability of P. grisea appressoria to penetrate the plant surface, where deletion of the gene responsible for its biosynthesis led to decreased virulence in rice. However, in our analysis of the transcriptomic data published by Kang and coworkers (2019), the GtNRPS2 backbone gene was downregulated under infection conditions (Wr_Gt x Pda_Gt and Wr_Gt_Bv x Pda_Gt), although other nearby genes were upregulated, and further studies are needed to elucidate the role of this BGC in the TAD. Moreover, coprogen is an iron(III) hydroxamate, involved in the acquisition of extracellular iron, mostly produced under iron deprivation, being also an important virulence factor for several pathogenic fungi (Miethke and Marahiel 2007; Hof et al. 2009; Voß et al. 2020). In the transcriptomic analysis, GtNRPS5 was upregulated under Wr_Gt x Pda_Gt condition, reinforcing the hypothesis that GtNRPS5 is involved in the infection of G. tritici in wheat. Surprisingly, in the presence of B. velezensis (Wr_Gt_Bv x Pda_Gt), the GtNRPS5 backbone gene was not differentially expressed, which may indicate a possible interference caused by the biocontrol agent. In summary, the presence of intra- and extracellular siderophores already identified as virulence determinants in pathogenic fungi is an excellent indicator for the putative involvement of ferricrocin and coprogen in the infection process of G. tritici.
Despite the limitations of the differential expression analysis, this method allows the identification of genes possibly involved in the infection process and, therefore, provides relevant information to be investigated in future works. Two BGCs with no orthologs characterized in other species, GtTERP2 and GtPKS13, were positively regulated in the mimicked infection condition and presented extensive conservation in the Magnaporthales order, being of great interest for future investigation.
Conclusions
The availability of the genome of the TAD pathogen G. tritici, in addition to several other species belonging to the Magnaporthales order, has enabled comparative analyses. These analyses facilitate the identification of putative BGCs, and provide insights into aspects of genome organization, gene expression, and their potential roles as determinants of virulence. Although the importance of GtPKS1, GtPKS9, GtPKS10, GtNRPS2, GtNRPS5, and GtPKSNRPS3 BGCs during G. tritici pathogenic infection to economically important cereals and grasses requires further confirmation, our findings are important for future research. Dichlorodiaporthin, trichosetin, ACR-toxin, and siderophores compounds can play significant roles in fungal-plant interactions in several models, including economically important phytopathogenic fungi.
References
Agriculture and Horticulture Development Board (AHDB) Take-all disease in cereals: prevent, detect, control. Accessed 6 Dec 2023 https://ahdb.org.uk/take-all
Almeida C, Pérez-Victoria I, González-Menéndez V, de Pedro N, Martín J, Crespo G, Mackenzie T, Cautain B, Reyes F, Vicente F, Genilloud O (2018) Non-geminal aliphatic dihalogenation pattern in Dichlorinated diaporthins from Hamigera fusca NRRL 35721. J Nat Prod 81:1488–1492. https://doi.org/10.1021/acs.jnatprod.8b00041
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: a fast, Accurate, and powerful alternative. Syst Biol 55:539–552. https://doi.org/10.1080/10635150600755453
Anisimova M, Gil M, Dufayard JF, Dessimoz C, Gascuel O (2011) Survey of Branch Support methods demonstrates Accuracy, Power, and robustness of fast likelihood-based approximation schemes. Syst Biol 60:685–699. https://doi.org/10.1093/sysbio/syr041
Blin K, Shaw S, Augustijn HE, Reitz ZL, Biermann F, Alanjary M, Fetter A, Terlouw BR, Metcalf WW, Helfrich EJN, van Wezel GP, Medema MH, Weber T (2023) antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res 51:W46–W50. https://doi.org/10.1093/nar/gkad344
Brakhage AA (2013) Regulation of fungal secondary metabolism. Nat Rev Microbiol 11:21–32. https://doi.org/10.1038/nrmicro2916
Cai R, Wu Y, Chen S, Cui H, Liu Z, Li C, She Z (2018) Peniisocoumarins A–J: isocoumarins from Penicillium commune QQF-3, an endophytic fungus of the Mangrove Plant Kandelia candel. J Nat Prod 81:1376–1383. https://doi.org/10.1021/acs.jnatprod.7b01018
Chankhamjon P, Tsunematsu Y, Ishida-Ito M, Sasa Y, Meyer F, Boettger‐Schmidt D, Urbansky B, Menzel K, Scherlach K, Watanabe K, Hertweck C (2016) Regioselective Dichlorination of a non‐activated Aliphatic Carbon Atom and Phenolic Bismethylation by a multifunctional fungal flavoenzyme. Angew Chem Int Ed 55:11955–11959. https://doi.org/10.1002/anie.201604516
Chen L-H, Lin C-H, Chung K-R (2013) A nonribosomal peptide synthetase mediates siderophore production and virulence in the citrus fungal pathogen Alternaria alternata. Mol Plant Pathol 14:497–505. https://doi.org/10.1111/mpp.12021
Cordero RJB, Casadevall A (2017) Functions of fungal melanin beyond virulence. Fungal Biol Rev 31:99–112. https://doi.org/10.1016/j.fbr.2016.12.003
Cowled MS, Kalaitzis JA, Crombie A, Chen R, Sbaraini N, Lacey E, Piggott AM (2023) Fungal duel between Penicillium brasilianum and aspergillus nomius results in dual induction of Miktospiromide A and Kitrinomycin A. J Nat Prod 86:2398–2406. https://doi.org/10.1021/acs.jnatprod.3c00593
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. https://doi.org/10.1093/bioinformatics/btr088
Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu JR, Pan HQ, Read ND, Lee YH, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li WX, Harding M, Kim S, Lebrun MH, Bohnert H, Coughlan S, Butler J, Calvo S, Ma LJ, Nicol R, Purcell S, Nusbaum C, Galagan JE, Birren BW (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434:980–986. https://doi.org/10.1038/nature03449
Dean R, Van Kan JAL, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, Foster GD (2012) The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13:414–430. https://doi.org/10.1111/j.1364-3703.2011.00783.x
Fisch KM (2013) Biosynthesis of natural products by microbial iterative hybrid PKS–NRPS. RSC Adv 3:18228–18247. https://doi.org/10.1039/C3RA42661K
Gacek A, Strauss J (2012) The chromatin code of fungal secondary metabolite gene clusters. Appl Microbiol Biotechnol 95:1389–1404. https://doi.org/10.1007/s00253-012-4208-8
Gibson DM, Donzelli BGG, Krasnoff SB, Keyhani NO (2014) Discovering the secondary metabolite potential encoded within entomopathogenic fungi. Nat Prod Rep 31:1287–1305. https://doi.org/10.1039/C4NP00054D
Gilchrist CLM, Chooi Y-H (2021a) Synthaser: a CD-Search enabled Python toolkit for analysing domain architecture of fungal secondary metabolite megasynth(et)ases. Fungal Biol Biotechnol 8:13. https://doi.org/10.1186/s40694-021-00120-9
Gilchrist CLM, Chooi Y-H (2021b) Clinker & clustermap.js: automatic generation of gene cluster comparison figures. Bioinformatics 37:2473–2475. https://doi.org/10.1093/bioinformatics/btab007
Gilchrist CLM, Booth TJ, van Wersch B, van Grieken L, Medema MH, Chooi Y-H (2021) Cblaster: a remote search tool for rapid identification and visualization of homologous gene clusters. Bioinf Adv 1. https://doi.org/10.1093/bioadv/vbab016
Gómez Luciano LB, Tsai IJ, Chuma I, Tosa Y, Chen Y-H, Li J-Y, Li M-Y, Lu M-YJ, Nakayashiki H, Li W-H (2019) Blast fungal genomes show frequent chromosomal changes, Gene gains and losses, and Effector Gene turnover. Mol Biol Evol 36:1148–1161. https://doi.org/10.1093/molbev/msz045
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to Estimate Maximum-Likelihood Phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. https://doi.org/10.1093/sysbio/syq010
Haas H, Eisendle M, Turgeon BG (2008) Siderophores in fungal physiology and virulence. Annu Rev Phytopathol 46:149–187. https://doi.org/10.1146/annurev.phyto.45.062806.094338
Henson JM, Butler MJ, Day AW, THE DARK SIDE OF THE MYCELIUM (1999) Melanins of Phytopathogenic Fungi. Annu Rev Phytopathol 37:447–471. https://doi.org/10.1146/annurev.phyto.37.1.447
Hof C, Eisfeld K, Welzel K, Antelo L, Foster AJ, Anke H (2007) Ferricrocin synthesis in Magnaporthe grisea and its role in pathogenicity in rice. Mol Plant Pathol 8:163–172. https://doi.org/10.1111/j.1364-3703.2007.00380.x
Hof C, Eisfeld K, Antelo L, Foster AJ, Anke H (2009) Siderophore synthesis in Magnaporthe grisea is essential for vegetative growth, conidiation and resistance to oxidative stress. Fungal Genet Biol 46:321–332. https://doi.org/10.1016/j.fgb.2008.12.004
Izumi Y, Ohtani K, Miyamoto Y, Masunaka A, Fukumoto T, Gomi K, Tada Y, Ichimura K, Peever TL, Akimitsu K (2012) A polyketide synthase gene, ACRTS2, is responsible for biosynthesis of host-selective ACR-toxin in the rough lemon pathotype of Alternaria alternata. Mol Plant Microbe Interact 25:1419–1429. https://doi.org/10.1094/MPMI-06-12-0155-R
James Cook R (2003) Take-all of wheat. Physiol Mol Plant Pathol 62:73–86. https://doi.org/10.1016/S0885-5765(03)00042-0
Jia L-J, Tang H-Y, Wang W-Q, Yuan T-L, Wei W-Q, Pang B, Gong X-M, Wang S-F, Li Y-J, Zhang D, Liu W, Tang W-H (2019) A linear nonribosomal octapeptide from Fusarium graminearum facilitates cell-to-cell invasion of wheat. Nat Commun 10:922. https://doi.org/10.1038/s41467-019-08726-9
Kakule TB, Sardar D, Lin Z, Schmidt EW (2013) Two related pyrrolidinedione synthetase loci in Fusarium heterosporum ATCC 74349 produce divergent metabolites. ACS Chem Biol 8:1549–1557. https://doi.org/10.1021/cb400159f
Kang X, Guo Y, Leng S, Xiao L, Wang L, Xue Y, Liu C (2019) Comparative transcriptome profiling of Gaeumannomyces graminis var. tritici in wheat roots in the absence and Presence of Biocontrol Bacillus velezensis CC09. Front Microbiol 10. https://doi.org/10.3389/fmicb.2019.01474
Keenan S, Cromey MG, Harrow SA, Bithell SL, Butler RC, Beard SS, Pitman AR (2015) Quantitative PCR to detect Gaeumannomyces graminis var. tritici in symptomatic and non-symptomatic wheat roots. Australas Plant Pathol 44:591–597. https://doi.org/10.1007/s13313-015-0379-y
Keller NP (2015) Translating biosynthetic gene clusters into fungal armor and weaponry. Nat Chem Biol 11:671–677. https://doi.org/10.1038/nchembio.1897
Khan A, Singh P, Srivastava A (2018) Synthesis, nature and utility of universal iron chelator– siderophore: a review. Microbiol Res 212–213:103–111. https://doi.org/10.1016/j.micres.2017.10.012
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915. https://doi.org/10.1038/s41587-019-0201-4
Kwak Y-S, Weller DM (2013) Take-all of wheat and Natural Disease suppression: a review. Plant Pathol J 29:125–135. https://doi.org/10.5423/PPJ.SI.07.2012.0112
Li W, Xu J, Li F, Xu L, Li C (2016) A New Antifungal Isocoumarin from the endophytic Fungus Trichoderma sp. 09 of Myoporum bontioides A. Gray. Pharmacogn Mag 12:259–261. https://doi.org/10.4103/0973-1296.192204
Li H, Wei H, Hu J, Lacey E, Sobolev AN, Stubbs KA, Solomon PS, Chooi Y-H (2020) Genomics-Driven Discovery of Phytotoxic cytochalasans involved in the virulence of the Wheat Pathogen Parastagonospora nodorum. ACS Chem Biol 15:226–233. https://doi.org/10.1021/acschembio.9b00791
Liao Y, Smyth GK, Shi W (2013) The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41:e108–e108. https://doi.org/10.1093/nar/gkt214
Liu M, Ohashi M, Hung Y-S, Scherlach K, Watanabe K, Hertweck C, Tang Y (2021) AoiQ catalyzes Geminal Dichlorination of 1,3-Diketone Natural products. J Am Chem Soc 143:7267–7271. https://doi.org/10.1021/jacs.1c02868
Love MI, Huber W, Anders S (2014) Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Löytynoja A, Goldman N (2010) webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics 11:579. https://doi.org/10.1186/1471-2105-11-579
Luo J, Qiu H, Cai G, Wagner NE, Bhattacharya D, Zhang N (2015) Phylogenomic analysis uncovers the evolutionary history of nutrition and infection mode in rice blast fungus and other Magnaporthales. Sci Rep 5:9448. https://doi.org/10.1038/srep09448
Marfori EC, Kajiyama S, Fukusaki E, Kobayashi A (2002) Trichosetin, a novel tetramic acid antibiotic produced in dual culture of Trichoderma harzianum and Catharanthus roseus Callus. Z Naturforsch C J Biosci 57:465–470. https://doi.org/10.1515/znc-2002-5-611
Marfori EC, Kajiyama S, ichiro, Fukusaki E, Kobayashi A (2003) Phytotoxicity of the tetramic acid metabolite trichosetin. Phytochemistry 62:715–721. https://doi.org/10.1016/s0031-9422(02)00629-5
McMillan VE, Hammond-Kosack KE, Gutteridge RJ (2011) Evidence that wheat cultivars differ in their ability to build up inoculum of the take-all fungus, Gaeumannomyces graminis var. tritici, under a first wheat crop. Plant Pathol 60:200–206. https://doi.org/10.1111/j.1365-3059.2010.02375.x
Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, de Bruijn I, Chooi YH, Claesen J, Coates RC, Cruz-Morales P, Duddela S, Düsterhus S, Edwards DJ, Fewer DP, Garg N, Geiger C, Gomez-Escribano JP, Greule A, Hadjithomas M, Haines AS, Helfrich EJN, Hillwig ML, Ishida K, Jones AC, Jones CS, Jungmann K, Kegler C, Kim HU, Kötter P, Krug D, Masschelein J, Melnik A v, Mantovani SM, Monroe EA, Moore M, Moss N, Nützmann H-W, Pan G, Pati A, Petras D, Reen FJ, Rosconi F, Rui Z, Tian Z, Tobias NJ, Tsunematsu Y, Wiemann P, Wyckoff E, Yan X, Yim G, Yu F, Xie Y, Aigle B, Apel AK, Balibar CJ,Balskus EP, Barona-Gómez F, Bechthold A, Bode HB, Borriss R, Brady SF, Brakhage AA,Caffrey P, Cheng Y-Q, Clardy J, Cox RJ, de Mot R, Donadio S, Donia MS, van der Donk WA, Dorrestein PC, Doyle S, Driessen AJM, Ehling-Schulz M, Entian K-D, Fischbach MA,Gerwick L, Gerwick WH, Gross H, Gust B, Hertweck C, Höfte M, Jensen SE, Ju J, Katz L, Kaysser L, Klassen JL, Keller NP, Kormanec J, Kuipers OP, Kuzuyama T, Kyrpides NC, Kwon H-J, Lautru S, Lavigne R, Lee CY, Linquan B, Liu X, Liu W, Luzhetskyy A,Mahmud T, Mast Y, Méndez C, Metsä-Ketelä M, Micklefield J, Mitchell DA, Moore BS,Moreira LM, Müller R, Neilan BA, Nett M, Nielsen J, O’Gara F, Oikawa H, Osbourn A,Osburne MS, Ostash B, Payne SM, Pernodet J-L, Petricek M, Piel J, Ploux O, Raaijmakers JM, Salas JA, Schmitt EK, Scott B, Seipke RF, Shen B, Sherman DH, Sivonen K, Smanski MJ, Sosio M, Stegmann E, Süssmuth RD, Tahlan K, Thomas CM, Tang Y, Truman AW, Viaud M, Walton JD, Walsh CT, Weber T, van Wezel GP, Wilkinson B, Willey JM, Wohlleben W,Wright GD, Ziemert N, Zhang C, Zotchev SB, Breitling R, Takano E, Glöckner FO (2015)Minimum Information about a Biosynthetic Gene cluster. Nat Chem Biol 11:625–631. https://doi.org/10.1038/nchembio.1890
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–W346. https://doi.org/10.1093/nar/gkr466
Medema MH, Takano E, Breitling R (2013) Detecting sequence homology at the Gene Cluster Level with MultiGeneBlast. Mol Biol Evol 30:1218–1223. https://doi.org/10.1093/molbev/mst025
Meepagala KM, Briscoe WE, Techen N, Johnson RD, Clausen BM, Duke SO (2018) Isolation of a phytotoxic isocoumarin from Diaporthe eres infected Hedera helix (English ivy) and synthesis of its phytotoxic analogs. Pest Manag Sci 74:37–45. https://doi.org/10.1002/ps.4712
Miethke M, Marahiel MA (2007) Siderophore-Based Iron Acquisition and Pathogen Control. Microbiol Mol Biol Rev 71:413–451. https://doi.org/10.1128/MMBR.00012-07
Neema C, Laulhere JP, Expert D (1993) Iron Deficiency Induced by Chrysobactin in Saintpaulia leaves inoculated with Erwinia chrysanthemi. Plant Physiol 102:967–973. https://doi.org/10.1104/pp.102.3.967
Nützmann HW, Osbourn A (2014) Gene clustering in plant specialized metabolism. Curr Opin Biotechnol 26:91–99
Oide S, Moeder W, Krasnoff S, Gibson D, Haas H, Yoshioka K, Turgeon BG (2006) NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell 18:2836–2853. https://doi.org/10.1105/tpc.106.045633
Osbourn A (2010) Secondary metabolic gene clusters: evolutionary toolkits for chemical innovation. Trends Genet 26:449–457. https://doi.org/10.1016/j.tig.2010.07.001
Palma-Guerrero J, Chancellor T, Spong J, Canning G, Hammond J, McMillan VE, Hammond-Kosack KE (2021) Take-all Disease: New insights into an important wheat Root Pathogen. Trends Plant Sci 26:836–848. https://doi.org/10.1016/j.tplants.2021.02.009
Rawlinson C, See PT, Moolhuijzen P, Li H, Moffat CS, Chooi Y, Oliver RP (2019) The identification and deletion of the polyketide synthase-nonribosomal peptide synthase gene responsible for the production of the phytotoxic triticone A/B in the wheat fungal pathogen pyrenophora tritici‐repentis. Environ Microbiol 21:4875–4886. https://doi.org/10.1111/1462-2920.14854
Rutledge PJ, Challis GL (2015) Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol 13:509–523. https://doi.org/10.1038/nrmicro3496
Saeed A (2016) Isocoumarins, miraculous natural products blessed with diverse pharmacological activities. Eur J Med Chem 116:290–317. https://doi.org/10.1016/j.ejmech.2016.03.025
Sbaraini N, Guedes RLM, Andreis FC, Junges Â, de Morais GL, Vainstein MH, de Vasconcelos ATR, Schrank A (2016) Secondary metabolite gene clusters in the entomopathogen fungus Metarhizium anisopliae: genome identification and patterns of expression in a cuticle infection model. BMC Genomics 17:736. https://doi.org/10.1186/s12864-016-3067-6
Sbaraini N, Andreis FC, Thompson CE, Guedes RL, Junges A, Campos T, Staats CC, Vainstein MH, de Vasconcelos ATR, Schrank A (2017) Genome-wide analysis of secondary metabolite gene clusters in Ophiostoma_ulmi and Ophiostoma novo-ulmi reveals a fujikurin-like gene cluster with a putative role in infection. Front Microbiol 8. https://doi.org/10.3389/fmicb.2017.01063
Shabir G, Saeed A, El-Seedi HR (2021) Natural isocoumarins: structural styles and biological activities, the revelations carry on…Phytochemistry 181:112568. https://doi.org/10.1016/j.phytochem.2020.112568
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva E v., Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Singh SB, Zink DL, Goetz MA, Dombrowski AW, Polishook JD, Hazuda DJ (1998) Equisetin and a novel opposite stereochemical homolog phomasetin, two fungal metabolites as inhibitors of HIV-1 integrase. Tetrahedron Lett 39:2243–2246. https://doi.org/10.1016/S0040-4039(98)00269-X
Smiley RW (1973) Relationship between take-all of wheat and Rhizosphere pH in Soils fertilized with ammonium vs. Nitrate-Nitrogen Phytopathol 63:882. https://doi.org/10.1094/Phyto-63-882
Solovyev V, Kosarev P, Seledsov I, Vorobyev D (2006) Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol 7:S10. https://doi.org/10.1186/gb-2006-7-s1-s10
Vesonder RF, Tjarks LW, Rohwedder WK, Burmeister HR, Laugal JA (1979) Equisetin, an antibiotic from Fusarium equiseti NRRL 5537, identified as a derivative of N-methyl-2,4-pyrollidone. J Antibiot (Tokyo) 32:759–761. https://doi.org/10.7164/antibiotics.32.759
Voß B, Kirschhöfer F, Brenner-Weiß G, Fischer R (2020) Alternaria alternata uses two siderophore systems for iron acquisition. Sci Rep 10:3587. https://doi.org/10.1038/s41598-020-60468-7
Wolf T, Shelest V, Nath N, Shelest E (2015) CASSIS and SMIPS: promoter-based prediction of secondary metabolite gene clusters in eukaryotic genomes. Bioinformatics. https://doi.org/10.1093/bioinformatics/btv713
Xu X-H, Su Z-Z, Wang C, Kubicek CP, Feng X-X, Mao L-J, Wang J-Y, Chen C, Lin F-C, Zhang C-L (2015) The rice endophyte Harpophora oryzae genome reveals evolution from a pathogen to a mutualistic endophyte. Sci Rep 4:5783. https://doi.org/10.1038/srep05783
Yaegashi J, Oakley BR, Wang CCC (2014) Recent advances in genome mining of secondary metabolite biosynthetic gene clusters and the development of heterologous expression systems in Aspergillus nidulans. J Ind Microbiol Biotechnol 41:433–442. https://doi.org/10.1007/s10295-013-1386-z
Yang L, Xie L, Xue B, Goodwin PH, Quan X, Zheng C, Liu T, Lei Z, Yang X, Chao Y, Wu C (2015) Comparative transcriptome profiling of the early infection of wheat roots by Gaeumannomyces graminis var. tritici. PLoS ONE 10:e0120691. https://doi.org/10.1371/journal.pone.0120691
Zhong Z, Norvienyeku J, Chen M, Bao J, Lin L, Chen L, Lin Y, Wu X, Cai Z, Zhang Q, Lin X, Hong Y, Huang J, Xu L, Zhang H, Chen L, Tang W, Zheng H, Chen X, Wang Y, Lian B, Zhang L, Tang H, Lu G, Ebbole DJ, Wang B, Wang Z (2016) Directional selection from Host Plants is a major force driving host specificity in Magnaporthe Species. Sci Rep 6:25591. https://doi.org/10.1038/srep25591
Acknowledgements
The authors would like to thank to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS). Some data here presented previously encompassed an oral presentation at GENÉTICA 2021 - Brazilian Congress of Genetics. The manuscript has been read and approved by all named authors.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
FG: Conceptualization, formal analysis, investigation, data curation, writing - original draft, visualization. IP: Formal analysis, data curation, writing - original draft. MSC: Formal analysis, writing - review & editing. AS: Writing - review & editing, supervision. NS: Conceptualization, formal analysis, investigation, writing - review & editing, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Geremia, F., Paim, I., da Silva Camargo, M. et al. Secondary metabolite gene clusters from the phytopathogenic fungus Gaeumannomyces tritici. J Plant Pathol 106, 657–669 (2024). https://doi.org/10.1007/s42161-024-01605-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-024-01605-3