
Abstract
South Africa is a semi-arid country in need of soil conservation methods for sustainable agroecosystem practices. Over time, researchers have worked extensively on the plant growth beneficial effects of fungi. However, archaeal groups in the soil and rhizosphere of different plants are under-studied. The impact of soil edaphic factors on the community structure of archaeal and fungal groups in the maize rhizosphere is the focus of this research. This will confirm the hypothesis that environmental factors have a significant impact on the plant–soil rhizobiome. To achieve this objective, a shotgun metagenomics approach was used to analyze maize rhizosphere and bulk soils collected from North West and Gauteng provinces of South Africa. Our findings revealed that both the maize rhizosphere and bulk soils have several archaeal and fungal phyla in common. Besides, Ascomycota (30‒51%), Thaumarchaeota (14‒26%), Euryarchaeota (19‒29%), and Crenarchaeota (8‒12%) have significantly dominated the maize rhizosphere and surrounding bulk soils. In comparison, genera such as Nitrosopumilus (Ls-18.32%, Lc-16.25%; Rs-15.45%, Rc-11.47%), Candidatus Nitrososphaera (Ls-15.29%, Lc-13.37%; Rs-10.53%, Rc-8.29%), Cenarchaeum (Ls-8.84%, Lc-8.16%; Rs-7.63%, Rc-5.38%), Chaetomium (Ls-25.15%, Lc-23.05%; Rs-14.83%, Rc-6.29%), and Neurospora (Ls-2.09%, Lc-1.95%; Rs-1.05%, Rc-0.99%) were significantly higher in maize rhizosphere soils compared to bulk soils (p < 0.05). Diversity indices showed that the archaeal and fungal composition were significantly different across samples with the presence of unclassified groups that could indicate the possibility of novel organisms in the sampling areas. The forward selection of soil edaphic factors predicted N–NH4, pH, and organic matter as the major contributing factors to the variation in the fungal and archaeal communities. To fully exploit the advantages possessed by these species, it is necessary to better understand the main environmental factors driving microbial assemblages of the soil microbiome. In addition, mapping out a culture-based method to unveil the unclassified groups could assist in the discovery of biotechnologically important archaeal and fungal groups for sustainable plant growth.
Article highlights
-
Important soil edaphic factors such as N–NH4, pH, and organic matter serves as the major contributing factors to the variation in archaeal and fungal community in maize rhizosphere.
-
Using meta’omics, important fungal and archaeal groups such as Ascomycota, Thaumarchaeota, Euryarchaeota, and Crenarchaeota significantly dominated the maize rhizosphere and surrounding bulk soils.
-
Diversity indices showed that the archaeal and fungal composition were significantly different across samples with the presence of unclassified groups that could indicate the possibility of novel organisms in the sampling areas.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Plant rhizosphere is an important interface that enhances the exchange of materials between plants and their proximal environment, most of which are influenced by either host plant exudates or pedological factors [1]. Microbes inhabiting the rhizosphere and phyllosphere of plants can be classified as epiphytic and endophytic organisms. An example of plant rhizobiome includes those organisms associated with the roots and heavily affected by plant root exudates, while phyllosphere organisms inhabit above-ground portion of plant [2,3,4].
Often times, the host plant protects its microbiome by providing energy sources, adjust pH of the soil, and reduces competition for beneficial organisms. The microorganisms, in turn, help in the biogeochemical cycling of important nutrients and induce indirect plant growth-promoting mechanisms that improve plant growth [5,6,7]. The composition of soil microorganisms in the rhizosphere of plant is affected by the genetic composition of the host plant across different plant species, and also within different genotypes of a single species [8]. In a study conducted by Gomes et al. [9] on the root-associated microbiome of maize genotypes with different phosphorus use efficiency, a considerable difference in fungal and bacterial diversity associated with the plant could be ascribed to factors, viz. host genotype, the phosphorus content of the soil, and the compartment the microbiome is associated with. The understanding of the host plant–microbial interplay and plant alleles involved in this interaction could help in the modification of plant breeds and other biotechnological applications [10].
Considering the main microbial groups inhabiting soil, bacteria have been the focal point of most studies, as they dominate soils with about 103–106 per gram of soil [11]. However, fungal and archaeal groups are also important, with most studies showing their ubiquity in the soil [12]. A study on Antarctic and crust soils showed that these two environments harbor 104 and 107 per gram of fungi propagules, respectively. The disparity in the ecology and physiology of these two habitats was referred to the temporal and spatial variations imposed by distinct edaphic factors, such as organic matter, phosphate, nitrate, and ammonia composition of the soil [13, 14].
Several studies have reported the enormous effect of soil physical and chemical factors on the community structure of microorganisms in the soil [15, 16]. A study by Banda et al. [15] showed the high abundance of archaeal phylum—Euryarchaeota in saline soil. The variation in Cladosporiaceae and Nectriaceae in salty environment serve as a marker to measure the salinity level of the saline soil. Consequently, the microbial variation and other soil composition could be used as a measure to predict the salinity level of an environment. Asides the effect of salinity, plant microcosm is also affected by abiotic factors viz. moisture content, ammonia, pH, and organic matter. As reported by Gao et al. [16] soil pH could increase due to saline ion exchange, which is a function of the microbial inhabitation. The variation in the K, Na, C/N, exchangeable sodium percentage and pH between sodic and saline soil were the determinant factors influencing the community structure and abundance of archaeal, fungal and bacterial communities in the sampling sites indicating the strong relationship between soil microbiota and environmental variables [16].
Microorganisms, such as bacteria, fungi, archaea, and viruses live in the rhizosphere of plants. The plant rhizosphere microbiome is a reservoir of essential phytochemicals and bioactive metabolites that aid in plant immunity to soil stressors as well as plant growth promotion [4, 17]. Rhizosphere fungi are either beneficial or pathogenic to their host depending on the growth stage and environmental factors [5, 18]. There has been extensive research on the plant beneficial traits possessed by fungi and their use in the agroecosystem [19,20,21]. However, the archaeal group has been under-explored, and they are indispensable components of the plant microbiome [22]. Recently, researchers are now interested in the metabolic potential, composition, and diversity of archaea in the plant rhizosphere. Meanwhile, studies have referred to archaeal groups as understudied tiny helpers of land plants with potential symbiosis in agroecosystem [22, 23]. While culture-based methods for isolating these organisms can be difficult, the advent of next-generation sequencing has enhanced metagenomic studies of plant-associated microbes [24]. Shotgun metagenomics give an extensive description of the structural and functional attributes of the entire microbial communities in an environment. Apart from the benefits derived from the use of shotgun metagenomics, a PCR-dependent metagenomics approach has also proven to be helpful in the study of community structure and functional diversity of each microbial group (i.e., fungi and archaea) associated with plant’s rhizosphere [25].
Maize (Zea mays L.) is a staple food that plays a critical role in global food security since it feeds more than a third of the world's population. North West-South Africa (NW-SA) is a well-known province for its high-yielding maize, accounting for more than 40% of total maize production in South Africa, compared to other provinces under similar cultivation, such as Mpumalanga, Free State, and Kwazulu-Natal [26, 27]. In developed countries, maize is consumed as a second-cycle meal. In contrast, it is consumed directly in developing countries, where it is a staple food for nearly 90% of the middle and lower classes [27].
Maize possesses important physiological properties that enable them to thrive in arid and semi-arid soils. These features include; it’s unique nutrient assimilation genes, as well as other agronomic traits that enable easy recruitment of soil microorganisms with reliable nitrogen metabolic quality [28]. Here, we hypothesized that our sampling sites serve as a reservoir for important fungi and archaea, with their distribution influenced by various pedological factors. We used a shotgun metagenomic method to investigate the community structure of these species in the maize rhizosphere to determine the composition of microbial groups proximal to the plant. This approach can help uncover unculturable species present in the sampling sites while reducing the bias associated with PCR-dependent DNA metagenomic analysis.
2 Materials and methods
2.1 Soil sampling and physicochemical analysis
Rhizosphere soil samples were collected from two maize farms and adjacent soil samples (control), namely, Randfontein (26° 11′ 52.0″ S 27° 33′ 18.3″ E) and Lichtenburg (25° 59′ 40.4″ S 26° 31′ 44.5″ E) towns in Gauteng and North West provinces of South Africa, respectively (Fig. 1). Both sampling sites in this study were formerly grassland for animal grazing, but recently converted to croplands. They witnessed yearly rainfall between April and November (mean depth of 365–mm). The temperature experienced in the sites were 3 to 21 °C and 2 to 23 °C in the winter, while the summer experiences 17 to 27 °C and 14 to 30 °C for the Randfontein and Lichtenburg sampling sites, respectively. Randfontein town is known for gold mining, while Lichtenburg is situated at the heart of the maize triangle, i.e., mainly known for maize farming. The varieties of maize cultivated on the sampling sites were WE5323 and WE3128 hybrids for Lichtenburg and Randfontein farms, respectively. The rhizosphere soils (combined composite samples from a plant) were collected from three different plants (triplicate-Ls and Rs) using a sterile scalpel blade to scrape soil off the roots of plant at a radius of 3 cm away from the plant, 20-cm depth with 2 × 4 m2 area for each sample, while the bulk soils (controls-Lc and Rc) were collected from an adjacent area 10-m apart from each sampling site [29]. All the samples were transported to the laboratory in a cooler box filled with ice and stored at − 20 °C.

Location of the sampling sites at Gauteng and North West Provinces
For soil physicochemical analyses, 20 g of each sample was mixed, air-dried, and sieved (2-mm sieve) to disaggregate larger particles. At a ratio of 1:2.5 (soil-deionized water), the pH was measured using an MS02 soil pH-meter (Sonkir, Australia). Available phosphorus (P) content was measured using a Spectrophotometer [30]. After which, Organic carbon (Org C) was determined using dichromate digestion [31]. Total carbon (Total C), Organic matter (OM), and Organic carbon (Org C) were determined using the method of Shi et al. [28] and Walkley and Black [32]. The ammonium (N–NH4) content of samples was measured using a modified manual calorimeter as described by Baethgen and Alley [33], while nitrate (N–NO3) content was determined using LAQUA twin nitrate ion meter (B-742). Exchangeable potassium (K) was obtained using a flame photometer [34]. Soil sulfate was measured using the method of Walker [35] with data collected from triplicate readings used for statistical analysis.
2.2 DNA extraction and illumina sequencing
2.2.1 Molecular analysis
The whole metagenomic DNA extraction was performed using a PowerMax soil kit (DNeasy MOBIO, Carlsbad, CA) according to the quick-start protocol containing the manufacturer’s instructions with little adjustment. Five grams of soil sample was used to elute 5 mL of DNA using the protocol. Afterward, 25–50 ng of the DNA of each sample was used for library preparation with the use of the Illumina Nextera Sample Preparation kit (New York, USA). During sequencing, the samples were fragmented, and adapter sequences were added. The concentration of the libraries was measured using the Qubit® dsDNA HS Assay Kit (Life Technologies, Jiangsu, China), and the Agilent 2100-Bioanalyzer (Agilent Technologies, Waldbronn, Germany) was used to determine the average library size. The libraries were pooled and mixed up to 0.6 nM with the Illumina NovaSeq system used to sequence the pair-end for 300-cycles.
2.2.2 Downstream analysis of metagenome sequences
The downstream analysis of the sample reads was performed using the default settings of the Metagenomics Rapid Annotations Subsystems Technology (MG-RAST server -v4). The MG-RAST pipeline involves the quality assessment of raw reads using SolexaQA to dereplicate the metagenome data and trimming of low-quality reads [36]. The Duplicate Reads Inferred Sequencing Error Estimation (DRISEE) was used to remove sample-sequencing error based on the measuring of artificial duplicate reads (ADRs), and the Bowtie aligner was used to screen reads related to model organisms such as human, fly, mouse, and other animals [37]. The BLAST-like alignment (BLAT-algorithm) was adopted to ensure the annotation of the sequences against the M5NR-database that provides non-redundant integration of several other databases [38]. Still on MG-RAST pipeline, the Ribosomal Database Project-RDP was used for the taxonomic classification of fungal and archaeal groups with other taxa screened-out.
To normalize high levels of systematic variation and reduce false positive results, this study adopted a trimmed mean of M-values (TMM). TMM was conducted using the edgeR Bioconductor package (v 3.10.5). The default settings of egdeR automatically trims 30% of log fold-change and 5% of mean abundance [39]. The mean values deduced from the merging of the taxon of fungi and archaea from the replicates were agglomerated, and unclassified reads related to these groups were also retained for statistical analysis. Also, the soil functional features were performed using SEED subsystem level 1 with a minimum identity of 60%, maximum alignment length of 15 base pairs, and an e-value of 1 × 10–5. Annotated functional tables extracted from MG-RAST were agglomerated based on functional level and unclassified functional reads also retained for statistical analysis.
2.3 Statistical analysis
After annotation on MG-RAST, the rarefaction curve was plotted using the SEED subsystem of MG-RAST. CANOCO 5 (Microcomputer-Power, Ithaca, NY) was used to depict the principal component analysis (PCA), principal coordinate analysis (PCoA), and canonical correspondence analysis (CCA) of analyzed domains. Alpha and beta-diversity were calculated by comparing the Pielou evenness and Shannon indices of each habitat using Kruskal–Wallis and analysis of similarity (ANOSIM) on PAST -v3.20 [40, 41]. The beta diversity was depicted using the principal coordinate analysis (PCoA) based on Euclidean distance matrix and the one-way analysis of similarities (ANOSIM) via 999 permutations was used to test for differences in community composition between the groups of samples. The physicochemical parameters of the samples were compared using analysis of variance (ANOVA) adopting Tukey HSD and Monte-Carlo permutation test and sketched with the aid of GraphPad Prism -v5.0 to ensure effective comparison within and between the sampling sites. In addition, the circos software (http://circos.ca/) was used to visualize different taxa of fungal and archaeal groups.
3 Results
3.1 Physical and chemical of maize rhizosphere and bulk soils
The physicochemical assessment of sampling sites showed that the pH (Rs-6.76, Rc-6.73), Org C (Rs-1.09, Rc-0.87) and P (Rs-257.14 mgkg−1, Rc-206.54 mgkg−1) of Randfontein samples were significantly higher than the pH (Ls-5.62, Lc-5.87), Org C (Ls-0.61, Lc-0.60) and P (Ls-50.98 mgkg−1, Lc-65.86 mgkg−1) of Lichtenburg samples and controls. However, the result was vice versa when considering the N–NO3 (Ls-16.29 mgkg−1, Lc-16.24 mgkg−1) and K (Ls-240.00 mgkg−1, Lc-243.00 mgkg−1), which were substantially higher than the N–NO3 (Rs-8.52 mgkg−1, Rc-7.38 mgkg−1) and K (Rs-167.00 mgkg−1, Rc-148.50 mgkg−1) of both sampling areas and their controls (p ≤ 0.05). Nevertheless, the mean values from environmental variables such as sulfate, Total C, and OM showed an insignificant difference between rhizosphere soils and their controls, while N–NH4 had a mixed result (Table 1).
3.2 Metagenome dataset of sampling sites
The metagenome reads uploaded on MG-RAST database were between the range of 17,000,000–22,000,000 (Ls), 14,000,000–15,000,000 (Lc), 13,000,000–17,000,000 (Rs), and 13,000,000–15,000,000 (Rc) for triplicates reads of the rhizosphere and bulk samples, respectively. The post quality-control assessment retained reads were between the range of 15,000,000–20,000,000 (Ls) and 12,000,000–15,000,000 (Rs) rhizosphere samples, while 13,000,000–14,000,000 (Lc) and 12,000,000–13,000,000 (Rc) were assigned to the bulk samples (Table S1). The taxonomic reads assigned to archaea were between the range of 28,000–35,000 (Ls), 18,000–23,000 (Rs), 23,000–26,000 (Lc), and 12,000–21,000 (Rc) for the rhizosphere and bulk samples. In the case of eukaryotes, the reads allocated to samples were between the range of 42,000–59,000 (Ls), 21,000–31,000 (Rs), 23,000–29,000 (Lc), and 24,000–35,000 (Rc) (Table S1). The rarefaction curve to determine the richness of the samples was evaluated using MG-RAST SEED subsystem (Figure S1).
The distribution of fungal and archaeal (phylum level) community at the rhizosphere and surrounding soils.
Our sorting from the RDP classification of MG-RAST showed a total number of thirteen (13) conspicuous phyla from the fungal and archaeal domain. These include; Ascomycota (32‒51%), Crenarchaeota (8‒10%), Euryarchaeota (19‒29%), Basidiomycota (1‒2%), Korarchaeota (0.2‒0.32%), Thaumarchaeota (14‒26%), Blastocladiomycota (0.01%), Nanoarchaeota (0.01%), Chytridiomycota (0.1%), Glomeromycota (0.01%), Microsporidia (0.03‒0.05%), and unclassified derived from fungi and archaea (1‒2%). The dominant phyla from the two groups include; Ascomycota (32‒51%), Basidiomycota (1‒2%), and Chytridiomycota (0.1%) from fungi, while Thaumarchaeota (14‒26%), Euryarchaeota (19‒29%), and Crenarchaeota (8‒10%) were the dominant archaeal phyla (Fig. 2). Phyla such as Ascomycota (p = 0.009), Thaumarchaeota (p = 0.046), and Euryarchaeota (p = 0.033) were significantly high in the bulk samples, while Crenarchaeota (p = 0.047), and unclassified derived from archaea (p = 0.007) were significantly high in the rhizosphere samples (Table S2).

The relative abundance of fungal and archaeal phyla in maize rhizosphere soils and the surrounding soils. Microbial phyla were represented as Ascomy (Ascomycota), Nanoarch (Nanoarchaeota), Basidiomy (Basidiomycota), Glomeromy (Glomeromycota), Crenarch (Crenarchaeota), Thaumarch (Thaumarchaeota), Chytridiomy (Chytridiomycota), Euryarch (Euryarchaeota), Korarch (Korarchaeota), Blastocladiomy (Blastocladiomycota). Ls lichtenburg rhizosphere sample, Lc lichtenburg bulk (control) sample, Rs randfontein rhizosphere sample, Rc randfontein bulk (control) sample. The percentages and color distribution of the phyla can be traced to their samples from right side of the circus to the left
3.3 Community structure of archaea across sampling sites
The metagenome study of our samples showed that Euryarchaeota (19.90–28.65%), Thaumarchaeota (14.51–22.20%), Crenarchaeota (8.22–11.46%), and unclassified derived from archaea were the major phyla derived from our sampling sites as revealed by the RDP database. Meanwhile, all the major phyla were significantly different across the sites (p < 0.05) as presented in Fig. 2; table S2.
At the order level, Halobacteriales (18.05–20.97%), unclassified derived from Thaumarchaeota (10.79–17.39%), unclassified Crenarchaeota (11.41–14.41%), Nitrosopumilales (6.53–9.90%), Methanosarcinales (7.37–10.22%), Methanomicrobiales (6.02–8.28%), Cenarchaeales (3.26–4.78%), Thermococcales (3.55–4.20%), unclassified Euryarchaeota (3.08–3.70%), Sulfolobales (2.12–2.61%), Desulfurocaccales (2.32–2.57%), Methanobacteriales (1.89–2.37%), Methanococcales (1.77–2.04%), and Methanocellales (1.65–1.95%) were the major order from our metadata (Fig. 3a). Meanwhile, Nitrosopumilales (Ls), unclassified Thaumarchaeota (Ls), and Desulfurococcales (Rs) were significantly high in the rhizosphere samples, while only Methanosarcinales (Rc) was higher in Randfontein bulk sample (p ≤ 0.05) (Table S3).

Heatmap of maize rhizosphere archaeal community: Order (a) and Genus (b). The color saturation (z-score) represents the relative abundance of maize rhizosphere archaea. Ls lichtenburg rhizosphere sample, Lc lichtenburg bulk (control) sample, Rs randfontein rhizosphere sample, Rc randfontein bulk (control) sample
Besides, considering major groups at the genus level, Nitrosopumilus (18.32%), Candidatus Nitrososhaera (15.29%), and Cenarcheum (8.84%) were the most abundant in Ls, Thermococcus (4.30%), Methanocella (3.70%) and Archaeoglobus (2.23%) were the most abundant in Rs. At the same time, Methanosarcina, Haloterrigina, Sulfolobus, Methanoshaerula, Halalkalicoccus, Haladaptatus, Methanoculleus, Thermoplasma, Halogeometricum, Aciduliprofundum and Methanospirillum were the most abundant in the control samples (Fig. 3b). High significant difference was observed in genera such as C. Nitrososphaera (p = 0.004), Nitrosopumilus (p = 0.014), Methanosarcina (p = 0.005), Cenarchaeum (p = 0.012), Halloterigena (p = 0.000), Sulfolobus (p = 0.002), Methanosphaerula (p = 0.001), and Methanospirillum (p = 0.018) when comparing archaeal genus across sampling sites (Table S5). PCA was adopted to depict the distribution of archaea between the sites with Lc-1-3, Rs-2, Rc-1, and Rc-2 sites having the most archaeal inhabitation (Fig. 4).

Principal component analysis (PCA) of archaeal metagenomes. The length of each vector represents the influence of the archaeal group metagenomes on the environment. Coordinate axis 1 (87.45%) and 2 (10.80%) explain variation in the sampling sites. Ls-1-3 triplicate rhizosphere soil from Lichtenburg site, Lc-1-3 triplicate bulk (control) soil from Lichtenburg site, Rs-1-3 triplicate rhizosphere soil from Randfontein site, Rc-1-3 triplicate bulk (control) soil from Randfontein site
3.4 Community structure of fungi across sampling sites
Our metadata showed four (4) major fungal phyla involved in the rhizosphere and bulk soil samples. These include; Ascomycota (Rc-50.85%), which was predominantly higher in the control samples, while Basidiomycota (Rs-1.70%) and Chytridiomycota (Ls-0.09%) were higher in the rhizosphere samples (Fig. 2). Out of the fungal phyla identified from our metadata, only Ascomycota was significantly high across sampling sites (p = 0.009) (Table S2).
The major fungal orders present in our metagenome study includes; Sordariales (12.15–36.72%), Pleosporales (15.86–46.45%), Eurotiales (11.99–16.10%), Hypocreales (11.66–16.17%), Helotiales (1.69–5.18%), Onygenales (3.46–5.89%), Agaricales (2.10–4.06%), Saccharomycetales (2.06–3.85%), Phyllachorales (0.98–1.65%), unclassified Sordariomycetes (0.94–1.54%), Magnaporthales (0.84–1.26%), Tremellales (0.50–1.10%), Pezizales (0.35–0.70%), Malasseziales (0.10–0.26%), Polyporales (0.12–0.36%), Schizosaccharomycetales (0.29–0.39%), and Puccinales (0.24–0.26%) (Fig. 5a). A significantly high difference was noticed in orders such as Sordariales (Ls-36.72%), Agaricales (Rs-4.06%), Saccharomycetales (Rs-3.85%), Ustilaginales (Rs-0.89%), Malasseziales (Rs-0.26%), Polyporales (Rs-0.36%), and Schizosaccharomycetales (Rs-0.39%) across the sites (p ≤ 0.05) (Table S4).

Heatmap of maize rhizosphere fungal community: Order (a) and Genus (b). The color saturation (z-score) represents the relative abundance of maize rhizosphere fungi. Ls lichtenburg rhizosphere sample, Lc lichtenburg bulk (control) sample, Rs randfontein rhizosphere sample, Rc randfontein bulk (control) sample
Nonetheless, a significantly high abundance was observed in major fungal genus, viz. Chaetomium-Ls (p = 0.014) and Neurospora-Ls (p = 0.050), while genera such as Sordaria, Aspergillus, Gibberella, Podospora, Nectria, Penicillium, Metarhizium, Pyrenophora, Verticillium, Neurospora, Magnaporthe, and Phaeosphaeria were insignificantly different across the sites (p ˃ 0.05) (Fig. 5b, Table S6). The distribution of fungi across the sites was depicted using PCA with Rs-1-3 having the highest fungi distribution, followed by both Lc-2 and Lc-3 (Fig. 6).

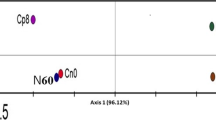
Principal component analysis (PCA) of fungal metagenomes. The length of each vector represents the influence of the fungal group metagenomes on the environment. Coordinate axis 1 (76.27%) and 2 (23.73%) explain variation in the sampling sites. Ls-1-3 triplicate rhizosphere soil from Lichtenburg site, Lc-1-3 triplicate bulk (control) soil from Lichtenburg site, Rs-1–3 triplicate rhizosphere soil from Randfontein site, Rc-1-3 triplicate bulk (control) soil from Randfontein site
3.5 Diversity indices of fungal and archaeal communities across the rhizosphere and bulk soils
The community variation of the sampling sites within samples (α-diversity) was assessed using pielou evenness and Shannon diversity. Alpha diversity of fungal and archaeal communities showed an insignificant difference between assessed taxonomical cadre (Fungal phylum = 0.97, order = 0.77, genus = 0.87; Archaeal phylum = 0.97, order = 0.99, genus = 0.50). The significant difference was confirmed using Kruskal–Wallis one-way analysis of variance (Table 2).
The β-diversity of assessed microbial communities (fungi and archaea) was depicted using principal coordinate analysis (PCoA) Bray–Curtis dissimilarity and confirmed using ANOSIM. The PCoA plot of rhizosphere fungi showed that samples are widely separated except Ls-1-3 and Lc-1-3 samples that were very close to each other (Fig. 7a). Using ANOSIM with R = 0.58 and p = 0.01, this shows that the samples have fungal communities of wide difference across the sampling sites. Similarly, β-diversity of archaeal communities was widely separated between the sampling sites (Fig. 7b) as depicted by PCoA and further explained using ANOSIM (R = 0.60, p = 0.01).

Principal coordinate analysis (PCoA) plot of rhizosphere fungal (a) and archaeal (b) community across sampling sites using Bray–Curtis dissimilarities. Coordinate axis 1 (82.50%), 2 (17.50%) and (91.48%), (06.53%) for fungi and archaea, respectively explain separation in the sampling sites. Ls-1-3 triplicate rhizosphere soil from Lichtenburg site, Lc-1-3 triplicate bulk (control) soil from Lichtenburg site, Rs-1-3 triplicate rhizosphere soil from Randfontein site, Rc-1-3 triplicate bulk (control) soil from Randfontein site
3.6 Impact of pedological variables on archaeal and fungal communities
The correlation between the analyzed physicochemical variables and the relative abundances of fungal and archaeal phyla was depicted using CCA. Three parameters such as OM, pH, and N–NH4 were selected based on a significant test and contribution of the variables to microbial disparity (Fig. 8).

Canonical correspondence analysis (CCA) of fungal and archaeal phyla explained using forward selection of environmental variables that best predict variation in the microbiome composition of each site
Inference deduced from the CCA showed that fungal and archaeal communities are greatly controlled by soil physicochemical properties with CCA permutation test = 0.03. Nanoarchaeota, Chytridiomycota, Blastocladiomycota, Thaumarchaeota, Glomeromycota, and unclassified derived from archaea positively correlated with OM and negatively with pH and N–NH4. Also, Korarchaeota, Ascomycota, Basidiomycota, and unclassified derived from fungi positively correlated with N–NH4 and negatively with pH and OM. Additionally, Euryarchaeota, Crenarchaeota, and Microsporidia positively correlated with pH but against N–NH4, and OM. To test environmental variables that best explain differences in fungi and archaea composition, the Monte-Carlo permutation test with 999 random sorting and forward selection of pedological factors was used. CCA showed that N–NH4 significantly (p = 0.048) contributed 50.4% of the variation, pH significantly (p = 0.050) contributed 37.2% variation, while OM insignificantly (p = 0.080) contributed 12.4% of the variation in archaeal and fungal communities of the sampling sites (Table 3).
3.7 Pathways involved in nutrient cycling and stress responses obtained from the sampling sites
The important nutrient pathways and stress responses associated with the dominant archaeal and fungal groups viz. Euryarchaeota, Thaumarchaeota, Crenarchaeota and Ascomycota revealed using the SEED subsystem technology of MG-RAST were showed in Table 4. Significantly high values were noticed in the nitrogen (Rs-6.55, Rc-6.38) and phosphorus (Rs-6.13, Rc-6.12) metabolism of Randfontein samples compared to nitrogen (Ls-5.58, Lc-5.83) and phosphorus (Ls–5.77, Lc-6.11) metabolism of Lichtenburg samples and controls. In the case of stress responses, rhizosphere samples of both sites showed considerably high values (Ls-13.90, Rs-13.81) compared to their controls (Lc-11.75, Rc-12.07). Besides, other nutrient pathways such as carbohydrate, potassium, sulfur, and iron acquisition and metabolism were insignificantly different across the rhizosphere samples and their controls.
4 Discussion
Plant microbiome acts as a major determinant of plant health and agricultural productivity [42]. The structural composition of a plant microbiome varies according to plant compartments and tissue networks, which help ease microbial inhabitation [43]. However, environmental variables have also proved to be an important evolutionary and ecological tool shaping the ecological complexity of rhizosphere organisms [44]. In this study, we surveyed the influence of environmental variables on the diversity of fungal and archaeal communities associated with the rhizosphere of maize using a shotgun metagenomics approach. Our metadata showed high fungal and archaeal diversity in the sampling sites, as depicted by the rarefaction curve. The rarefaction curve of our sampling site showed that the sampling efforts were fully covered, most especially the Ls site, because most of the reads reached saturation.
In this study, most of the assessed soil properties showed a spatial heterogeneity except for Organic matter, total carbon, and sulfate that were insignificantly different (p ≤ 0.05) between samples. The considerably high values of pH, P, N and Organic Carbon in Randfontein samples compared to Lichtenburg samples could be attributed to the proximity of Randfontein samples to the mining area. High values deduced from the aforementioned chemical properties were similar to the to the findings of Liu et al. [45]. Liu and colleagues showed that the chemical properties (available P, K, N, and pH) of most reclaimed mine soil were higher than natural soils with a significant change in the soil profile after mining and reclamation in an opencast coal mine [45]. Nevertheless, available K was also higher in the Lichtenburg samples and could be attributed to the low level of pH, because the pH of samples Lc (5.87) and Ls (5.62) were lower than the acceptable range (6–7.2), which indicates an equilibrium in the available soil nutrient. In addition, sample Rc (6.73) and Rs (6.76) were within the normal range for measuring available H+ and OH− concentration in plant’s rhizosphere [46]. According to Tale and Ingole [46], soil pH ensures the availability of important soil nutrients such as available K, Fe, Zn, Mn, and Cu. The lower the pH, the higher the soil micronutrient solubility and vice versa. Meanwhile, both extremes, i.e., an imbalance ionic strength may inhibit plant growth. Other measured chemical parameters such as N–NH4 (2.42–4.75), K (128–240), and N–NO3 (7–16) in this study were considerably normal for the growth of plants. The values deduced from this study were similar to the findings of Fadiji et al. [1] and Akinola et al. [4]. With a considerable level of nutrients in the soil, the microbial activities are usually high, especially in plant soils with high organic matter [4].
This study revealed three (3) major archaeal phyla namely: Thaumarchaeota, Euryarchaeota, and Crenarchaeota. The major phyla were significantly high in the rhizosphere samples compared to their controls. The presence of high major phyla in rhizosphere samples compared to the bulk is generally possessed by plant-soil rhizobiome as reported by Lundberg et al. [47], Zverev et al. [48], and Wei et al. [49] on Arabidopsis thaliana, gramineous plant and Chinese medicinal herb (Alisma), respectively. At the genus level, Nitrosopumilus, Candidatus Nitrososphaera and Sulfolobus were conspicuous in the Ls site. The inference from this study could be attributed to an elevated level of nitrite and slightly acidic nature of Ls site. According to Gubry-Rangin et al. [50], nitrification in an agricultural soil with these properties is usually controlled by archaea rather than bacteria. Our findings also echo the previous studies by Caldwell et al. [51], Zhang et al. [52] and Ayiti et al. [53]. Caldwell and co. discovered Candidatus Nitrososphaera as one of the most abundant genera in a prokaryotic study of the rhizosphere of coffee plants grown under organic, transitional, and intensive farm practices. Likewise, Zhang et al. [52] conducted a phylogenetic analysis and community structure of the archaeal amoA gene in rice rhizosphere with most clusters falling within Nitrosopumilus and Nitrososphaera-like clusters. The aforementioned genera belonging to the phyla Thaumarchaeota and Crenarchaeota are well known to be involved in the biogeochemical cycling of nutrients [54, 55]. Sulfur is another nutrient recycled by archaea to form hydrogen sulfide (H2S) through sulfidogenesis. H2S reacts directly with metals to form sulfides followed by oxidation through acidophilic archaea such as Sulfolobales, Euryarchaeota, and Thaumarchaeota, which are well represented in the sampling sites [56]. The presence of hyperthermophilic archaea such as Sulfolobales, Thermococcales, Thermoproteales, Thermoplasmatales, Desulfurococcales, Methanobacteriales, Archaeoglobales, Methanococcales, and Methanopyrales in the maize rhizosphere, and surrounding soils seems anomalous because organisms affiliated to these archaeal groups are exclusively endemic to hot springs, abyssal hot vents, and solfataric fields with an in situ temperature range between 80 and 115 °C [57, 58]. In addition, methanogenic archaea such as Methanosphaerula, Methanosarcina, Methanospirillum, Methanoregula, and Methanothermobacter significantly dominated Rs and Rc. This can be attributed to the closeness of these sites to a mining area. The proximity to active mines for several decades may have accumulated relatively large concentrations of hydrocarbons originating from the mine pits [59]. Methanogenic archaea thrive in an anoxic environment, and they are highly involved in the mineralization and assimilation of carbon from bicarbonate or carbon dioxide (CO2) [60]. However, the presence of unclassified organisms derived from classes, viz. Euryarchaeota, Crenarchaeota, and Thaumarchaeota showed the complexity (i.e., community structure) of the sampling sites.
To ascertain the influence and abundance of soil rhizobiomes on the sampling sites, we adopted PCA to depict archaeal structural diversity in the sampling sites. PCA showed that each sample has a predominant archaeal group with a combined variation of 98.25% between the rhizosphere samples and their controls. The vector arrows revealed which archaeal order most strongly influences the distribution. For example, Methanopyrales, unclassified archaea derived from Euryarchaeota, Cenarchaeales, Nitrosopumilales, unclassified derived from Crenarchaeota, Sulfolobales, and Thermoplasmatales predominated Ls and Lc samples, while Methanosarcinales, Methanomicrobiales, Methanococcales, Thermococcales, Halobacteriales, Acidilobales, and Methanobacteriales predominated Rs and Rc samples. The difference in archaeal dominance could also interfere with the function of these organisms in the proximal environment. The high abundance of Methanogenic archaea in Rc and Rs confirmed the aforementioned ecological functions, such as the assimilation and mineralization of carbon. The presence of Nitrosopumilales (ammonia-oxidizing archaea) and Methanopyrales (methanogenic archaea) in Ls and Lc showed their contribution to the biogeochemical cycling of nitrogen and carbon in the agroecosystem as reported by Nazaries et al. [61] and Qin et al. [62].
The metagenome study of the samples identified three (3) major fungal phyla (Ascomycota, Basidiomycota, and Chytridiomycota) with the exemption of the unclassified group. Significantly, Ascomycota was conspicuously dominant over all other phyla with abundance levels across all sampling sites represented in this order Rc ˃ Ls ˃ Rs ˃ Lc, while other dominant phyla, viz. Basidiomycota and Chytridiomycota were insignificantly higher in Rs and Ls sites than their controls (Table S2). These important phyla have been reported in different studies on pepper, black soil, maize, and tomato [63,64,65,66]. Several studies have also shown that Ascomycota is the most abundant and diverse group of fungi at the proximity of plants [19, 67]. Besides, Ascomycota also serves as a major decomposer in plant soil and its genera have been reported as highly influential plant growth-promoters [68]. Although phylum Basidiomycota may not be the most abundant in this study, they are also decomposers of lignin and cellulose [69]. Some of the rhizosphere fungal genera observed in this study, such as Chaetomium, Sordaria, Aspergillus, Gibberella, Podospora, Nectria, Penicllium, Metarhizium, Pyrenophora, Phaenosphaeria, Verticillium, Glomerella, Neurospora, Magnaporthe, and Leptosphaeria, have been reported as growth-promoters in similar studies on apple orchards, maize, rye, and wheat [48, 69, 70]. Additionally, a significant difference between the samples was noticed only in genera such as Chaetomium and Neurospora (Table S6). The high abundance level of some fungal communities noticed in the bulk samples compared to their rhizosphere soils was expected since maize produce metabolites, such as benzoxazinoids, that are capable of reducing the community assembly at the rhizosphere, roots, and shoots of the plant [71]. Studies on soil microbial co-occurrence networks showed that benzoxazinoids attract only important microbes and reduce unwanted organisms [71].
The genera Penicillium, Sordaria, and Aspergillus are some of the most abundant fungal genera in this study. Although some of these genera are parasitic, several studies have shown that these organisms are capable of producing several biological compounds that act as plant growth-promoters and also inhibit the growth of parasitic organisms [72]. According to a study by Franke-Whittle et al. [69] the aforementioned genera positively correlated with plant growth promotion and could be adopted to increase soil fertility.
Besides, beneficial fungi within the sphere of plants induce both direct and indirect mechanisms such as biogeochemical cycling of nutrients, trigger induced systemic resistance, activate plant genes beneficial to growth, and phytohormone production [17, 73,74,75]. The principal component analysis (PCA) was used to determine the influence and dominant fungal groups in the sampling sites. PCA showed that each site has a predominant fungal group with a combined variation of 100% (i.e., axis 1 and 2) between the maize rhizosphere samples and their controls. The position of each metagenome in Fig. 7 shows the frequency of the sequences correlated with each sample, and the vector arrows showed which group most strongly influences the distribution. Almost 80% of the fungal orders, viz. Eurotiales, Pezizales, Saccharomycetales, Malasseziales, Agaricales, Onygenales, Phyllachorales, and Ustilaginales predominated Rs-1-3. Ls and Lc were insignificantly different and both samples were predominated by Ophiostomatales, Helotiales, and Sordariales, while Pleosporales, Tremellales, and Pucciniales correlated with Rc-1-3 bulk samples. The different fungi dominance in each environment could influence the ecological role played by the microbial community.
Soil pedological variables determine the microbial turnover, as different microbiome interactions are favored by a change in soil physicochemical properties [41, 76]. The CCA was used to depict the correlation between the environmental variables and the relative abundance of fungal and archaeal communities. Considering the vector length of the evaluated physicochemical parameters (N–NH4, pH, and OM), the CCA plot showed that all parameters greatly influence the community structure of fungi and archaea. The N–NH4 content of the samples significantly (p = 0.048) contributed 50.4% of the variation, pH significantly (p = 0.050) contributed 37.2% variation, while OM insignificantly (p = 0.080) contributed 12.4% variation in fungal and archaeal communities. As depicted by CCA, most fungal communities were influenced by OM and N–NH4, while most of the archaeal communities were predicted by OM and pH of the sampling sites. The result deduced from this research is similar to the studies of Pereira e Silva et al. [76] and Boer et al. [77] that showed that OM and N–NH4 of the soil induce variation in archaeal and fungal structural diversity. In the study by Pereira e Silva et al. [76] OM had a positive influence on the community structure of archaeal groups in agricultural soils. This result showed that OM provides a substantial amount of carbon to archaeal communities associated with the sample. In a study by Hallam et al. [78] and Walker et al. [79] on genomic analyses of Nitrosopumilus maritimus and Crenarchaeum symbiosum, it was observed that these archaeal groups are capable of mixotrophy, and that the Crenarchaeota, Nanoarchaeota, and Thaumarchaeota groups are capable of growing on methane and methanol [80]. This concludes that archaea might not solely depend on ammonia oxidation [81]. Nevertheless, the abundance of fungal groups was significantly affected by OM and NH4 content of the sampling site. This result is in consistent with the saprotrophic nature of fungi [77]. The positive influence of OM content on the fungal distribution, as reflected on the CCA could also be attributed to the saprotrophic nature of fungi [77].
5 Conclusions
Using a shotgun metagenomics approach, this study has shown that Ascomycota, Thaumarchaeota, Euryarchaeota, and Crenarchaeota were significantly present in the maize rhizosphere and surrounding bulk soils in two South African sites. More so, at the genus level, diverse plant growth-promoting organisms were detected across the sampling sites with Nitrosopumilus, Candidatus Nitrososphaera, Cenarchaeum, Chaetomium, and Neurospora found significantly abundant in the maize rhizosphere soils compared to the bulk soils. The α-diversity showed indifferent community structure within the samples, while β-diversity revealed that the rhizobiome was significantly different across the maize rhizosphere soil samples and their controls. We also discovered a strong relationship between soil physicochemical properties and the structural diversity of archaeal and fungal communities with N–NH4, pH, and OM observed as the major contributing factor to the variation in the assessed microbial communities. This demonstrated that changes in the community structure of archaea and fungi could be altered by edaphic factors as predicted by the forward selection of important environmental variables. To understand the key pedological factors driving the microbial assemblage of soil rhizobiome, a more detailed understanding of the interplay controlling these important soil microbiomes is needed with microcosm experiments and fieldwork. Nevertheless, the presence of unclassified archaea in this study demonstrated the likelihood of detecting novel organisms of agricultural and industrial use. Therefore, we recommend the isolation and characterization of these unclassified groups to fully exploit the benefits possessed by these organisms.
Data availability
The raw sequences have been made publicly available on the NCBI SRA database under the Bioproject numbers PRJNA645371 and PRJNA645385 for the rhizosphere and bulk samples of Lichtenburg and Randfontein sites, respectively.
References
Fadiji AE, Ayangbenro AS, Babalola OO (2020) Metagenomic profiling of the community structure, diversity, and nutrient pathways of bacterial endophytes in maize plant. Antonie Van Leeuwenhoek 4:1–13
Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM (2006) The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol 57:233–266
Koopman MM, Fuselier DM, Hird S, Carstens BC (2010) The carnivorous pale pitcher plant harbors diverse, distinct, and time-dependent bacterial communities. Appl Environ Microbiol 76:1851–1860
Akinola SA, Ayangbenro AS, Babalola OO (2021) The diverse functional genes of maize rhizosphere microbiota assessed using shotgun metagenomics. J Sci Food Agric 101(8):3193–3201
Dlamini SP, Akanmu AO, Fadiji AE, Babalola OO (2023) Maize rhizosphere modulates the microbiome diversity and community structure to enhance plant health. Saudi J Biol Sci 30(1):103499
Yim B, Ibrahim Z, Rüger L, Ganther M, Maccario L, Sørensen SJ, Heintz-Buschart A, Tarkka MT, Vetterlein D, Bonkowski MS (2022) Soil texture is a stronger driver of the maize rhizosphere microbiome and extracellular enzyme activities than soil depth or the presence of root hairs. Plant Soil 478(1–2):229–251
Igiehon NO, Babalola OO (2018) Below-ground-above-ground plant-microbial interactions: focusing on soybean, rhizobacteria and mycorrhizal fungi. Open Microbiol 12:261
Bouffaud ML, Kyselková M, Gouesnard B, Grundmann G, Muller D, Moënne-Loccoz Y (2012) Is diversification history of maize influencing selection of soil bacteria by roots? Mol Ecol 21:195–206
Gomes EA, Lana UG, Quensen JF, de Sousa SM, Oliveira CA, Guo J, Guimarães LJ, Tiedje JM (2018) Root-associated microbiome of maize genotypes with contrasting phosphorus use efficiency. Phytobiomes 2:129–137
Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci 110:6548–6553
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Sci 309:1387–1390
Buckley DH, Graber JR, Schmidt TM (1998) Phylogenetic analysis of nontermophilic members of the kingdom Crenarchaeota and their diversity and abundance in soils. Appl Environ Microbiol 64:4333–4339
Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R et al (2011) Examining the global distribution of dominant archaeal populations in soil. ISME J 5:511–517
Jung J, Yeom J, Kim J, Han J, Lim HS et al (2011) Change in gene abundance in the nitrogen biogeochemical cycle with temperature and nitrogen addition in Antarctic soils. Res Microbiol 168:1028–1026
Banda JF, Zhang Q, Ma L, Pei L, Du Z, Hao C, Dong H (2021) Both pH and salinity shape the microbial communities of the lakes in Badain Jaran Desert. NW China Sci Total Environ 791:148108
Gao J, Zhao Q, Chang D, Ndayisenga F, Yu Z (2022) Assessing the effect of physicochemical properties of saline and sodic soil on soil microbial communities. Agriculture 12(6):782
Fadiji AE, Ayangbenro AS, Babalola OO (2020) Organic farming enhances the diversity and community structure of endophytic archaea and fungi in maize plant: a shotgun approach. J Soil Sci Plant Nutr 45:1–13
Jia M, Chen L, Xin H-L, Zheng C-J, Rahman K, Han T, Qin L-P (2016) A friendly relationship between endophytic fungi and medicinal plants: a systematic review. Front Microbiol 7:906
Fontaine S, Hénault C, Aamor A, Bdioui N, Bloor J, Maire V, Mary B, Revaillot S, Maron P-A (2011) Fungi mediate long term sequestration of carbon and nitrogen in soil through their priming effect. Soil Biol Biochem 43:86–96
Fuentes A, Herrera H, Charles TC, Arriagada CJM (2020) Fungal and bacterial microbiome associated with the rhizosphere of native plants from the Atacama desert. Microorganisms 8:209
Gómez-Muñoz B, Lekfeldt JD, Magid J, Jensen LS, de Neergaard A (2018) Seed treatment with Penicillium sp. or Mn/Zn can alleviate the negative effects of cold stress in maize grown in soils dependent on soil fertility. J Agron Crop Sci 204:603–612. https://doi.org/10.1111/jac.12288
Moissl-Eichinger C, Pausan M, Taffner J, Berg G, Bang C, Schmitz RA (2018) Archaea are interactive components of complex microbiomes. Trends Microbiol 26:70–85
Taffner J, Cernava T, Erlacher A, Berg G (2019) Novel insights into plant-associated archaea and their functioning in arugula (Eruca sativa Mill.). J Adv Res 19:39–48
Akinola SA, Babalola OO (2020) The importance of adverse soil microbiomes in the light of omics: Implications for food safety. Plant Soil Environ 66:421–430
Taffner J, Erlacher A, Bragina A, Berg C, Moissl-Eichinger C, Berg G (2018) What is the role of Archaea in plants? New insights from the vegetation of alpine bogs. MSphere 3:e00122-e218
Gravelet-Blondin R (2013) Prospectus on the South African maize industry. The Maize Trust, Pretoria, South Africa. https://sacota.co.za/wp-content/uploads/Prospectus-on-the-South-African-Maize-Industry.pdf.
Jéan du Plessis DdVB, Justinus M, Apie P, Thinus P (2003) Maize Production. The Department of agriculture and obtainable from resource centre directorate agricultural information services Private Bag X144, Pretoria, 0001 South Africa pp 1–35
Hirel B, Bertin P, Quilleré I, Bourdoncle W, Attagnant C, Dellay C, Gouy A, Cadiou S, Retailliau C, Falque M (2001) Towards a better understanding of the genetic and physiological basis for nitrogen use efficiency in maize. J Plant Physiol Biochem 125:1258–1270
Zhang J, Wang P, Tian H, Jiang H, Wang Y, Yan C (2018) Identification of interior salt-tolerant bacteria from ice plant Mesembryanthemum crystallinum and evaluation of their promoting effects. Symbiosis 76:243–252
Olsen SR, Cole CV, Watanabe FS, Dean LA (1954) Estimation of available phosphorus in soils by extraction with sodium bicarbonate. USDA Circular 939. U.S. Government Printing Office, Washington DC
Shi J-Y, Yuan X-F, Lin H-R, Yang Y-Q, Li Z-Y (2011) Differences in soil properties and bacterial communities between the rhizosphere and bulk soil and among different production areas of the medicinal plant Fritillaria thunbergii. Int J Mol Sci 12:3770–3785
Walkley A, Black IA (1934) An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. J Soil Sci 37:29–38
Baethgen W, Alley M (1989) A manual colorimetric procedure for measuring ammonium nitrogen in soil and plant Kjeldahl digests. Commun Soil Sci Plant Anal 20:961–969
Deke AL, Adugna WT, Fite AT (2016) Soil physico-chemical properties in termite mounds and adjacent control soil in Miyo and Yabello Districts of Borana zone. Southern Ethiopia Am J Agric Forest 4:69
Walker D (1972) Soil sulfate I. Extraction and measurement. Can J Soil Sci 52:253–260
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A (2008) The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform 9:1–8
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Gen Res 12:656–664
Wilke A, Harrison T, Wilkening J, Field D, Glass EM, Kyrpides N, Mavrommatis K, Meyer F (2012) The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform 13:141
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Hammer Ø, Harper DA, Ryan PD (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeontol Elect 4:9
Akinola SA, Ayangbenro AS, Babalola OO (2021) Metagenomic insight into the community structure of maize-rhizosphere bacteria as predicted by different environmental factors and their functioning within plant proximity. Microorganisms 9:1419
Lee SA, Kim Y, Kim JM, Chu B, Joa J-H, Sang MK, Song J, Weon H-Y (2019) A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci Rep 9:1–15
Omomowo OI, Babalola OO (2019) Bacterial and fungal endophytes: tiny giants with immense beneficial Potential for plant growth and sustainable agricultural productivity. Microorganisms 7:481
Wang N, Wang A, Xie J, He M (2019) Responses of soil fungal and archaeal communities to environmental factors in an ongoing antimony mine area. J Sci Total Environ 652:1030–1039
Liu X, Bai Z, Cao Y, Zhang G (2017) Changes in soil properties in the soil profile after mining and reclamation in an opencast coal mine on the Loess Plateau. China Ecol Engineer 98:228–239
Tale KS, Ingole S (2015) A review on the role of physico-chemical properties in soil quality. J Chem Sci Rev Lett 4:57–66
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Del Rio TG (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90
Zverev A, PershinaE SV, Kichko A, Mitrofanova O, Kobylyanskii V, Yuzikhin O, Belimov A, Andronov E (2020) Molecular analysis of the rhizosphere microbial communities from gramineous plants grown on contrasting soils. MicrobiologyOpen 89:231–241
Wei C, Gu W, Tian R, Xu F, Han Y, Ji Y, Li T, Zhu Y, Lang P, Wu W (2022) Comparative analysis of the structure and function of rhizosphere microbiome of the Chinese medicinal herb Alisma in diferent regions. Arch Microbiol 204:448
Gubry-Rangin C, Nicol GW, Prosser JI (2010) Archaea rather than bacteria control nitrification in two agricultural acidic soils. FEMS Microbiol Ecol 74(3):566–574
Caldwell AC, Silva LCF, da Silva CC, Ouverney CC (2015) Prokaryotic diversity in the rhizosphere of organic, intensive, and transitional coffee farms in Brazil. PLoS ONE 10(6):e0106355
Zhang J, Zhou X, Chen L, Chen Z, Chu J, Li Y (2016) Comparison of the abundance and community structure of ammonia oxidizing prokaryotes in rice rhizosphere under three different irrigation cultivation modes. World J Microbiol Biotechnol 32:85
Ayiti OE, Ayangbenro AS, Babalola OO (2022) 16S Amplicon sequencing of nitrifying bacteria and archaea inhabiting maize rhizosphere and the influencing environmental factors. Agriculture 12:1328. https://doi.org/10.3390/agriculture12091328
Weildler GW, Gerbl FW, Stan-Lotter H (2008) Crenarchaeota and their role in the nitrogen cycling in a subsurface radioactive thermal spring in the Austrian Central Alps. Appl Environ Microbiol 4:5934–5942
Kimble JC, Winter AS, Spilde MN, Sinsabaugh RL, Northup DE (2018) A potential central role of Thaumarchaeota in N-cycling in a semi-arid environment, Fort Stanton cave, Snowy River passage, New Mexico, USA. FEMS Microbiol Ecol 94:1–17
Islam W, Noman A, Naveed H, Huang Z, Chen HY (2020) Role of environmental factors in shaping the soil microbiome. Environ Sci Pollut Res 4:1–23
Stetter KO (1996) Hyperthermophilic procaryotes. FEMS Microbiol Rev 18:149–158
Sugai A, Uda I, Itoh YH, Itoh T (2004) The core lipid composition of the 17 strains of hyperthermophilic archaea. Thermococcales J Oleo Sci 53(1):41–44
Beckmann S, Lueders T, Krüger M, von Netzer F, Engelen B, Cypionka H (2011) Acetogens and acetoclastic Methanosarcinales govern methane formation in abandoned coal mines. Appl Environ Microbiol 77(10):3749–3756
Gong B, Cao H, Peng C, Perčulija V, Tong G, Fang H, Wei X, Ouyang S (2019) High-throughput sequencing and analysis of microbial communities in the mangrove swamps along the coast of Beibu Gulf in Guangxi. China Sci Rep 9:1–10
Nazaries L, Murrell JC, Millard P, Baggs L, Singh BK (2013) Methane, microbes and models: fundamental understanding of the soil methane cycle for future prediction. Env Microbiol 15(9):2395–2417
Qin W, Martens-Habbena W, Kobelt JN, Stahl DA (2016) Candidatus Nitrosopumilales—Bergey’s manual of systematics of archaea and bacteria. John Wiley & Sons, Inc. in association with Bergey’s Manual Trust. Doi:https://doi.org/10.1002/9781118960608.obm00122.
Al-Maliki S, AL-MammoryScullion HJ (2018) Interactions between humic substances and organic amendments affecting soil biological properties and growth of Zea mays L. in the arid land region. Arid Land Res Manage 32:455–470
Umadevi P, Anandaraj M, Srivastav V, Benjamin S (2018) Trichoderma harzianum MTCC 5179 impacts the population and functional dynamics of microbial community in the rhizosphere of black pepper (Piper nigrum L.). Braz J Microbiol 49:463–470
Zhou J, Jiang X, Zhou B, Zhao B, Ma M, Guan D, Li J, Chen S, Cao F, Shen D (2016) Thirty-four years of nitrogen fertilization decreases fungal diversity and alters fungal community composition in black soil in northeast China. Soil Biol Biochem 95:135–143
Adedayo AA, Fadiji AE, Babalola OO (2022) The effects of plant health status on the community structure and metabolic pathways of rhizosphere microbial communities associated with Solanumlycopersicum. Horticult 8:404. https://doi.org/10.3390/horticulturae8050404
Xiong J, Peng F, Sun H, Xue X, Chu H (2014) Divergent responses of soil fungi functional groups to short-term warming. Microbial Ecol 68:708–715
Wang J-T, Zheng Y-M, Hu H-W, Zhang L-M, Li J, He J-Z (2015) Soil pH determines the alpha diversity but not beta diversity of soil fungal community along altitude in a typical Tibetan forest ecosystem. J Soils Sediments 15:1224–1232
Franke-Whittle IH, Manici LM, Insam H, Stres B (2015) Rhizosphere bacteria and fungi associated with plant growth in soils of three replanted apple orchards. Plant Soil 395:317–333
Zhang R, Mu Y, Li X, Li S, Sang P, Wang X, Wu H, Xu N (2020) Response of the arbuscular mycorrhizal fungi diversity and community in maize and soybean rhizosphere soil and roots to intercropping systems with different nitrogen application rates. Sci Total Environ 740:139810
Kudjordjie EN, Sapkota R, Steffensen SK, Fomsgaard IS, Nicolaisen M (2019) Maize synthesized benzoxazinoids affect the host associated microbiome. Microbiome 7:1–17
Nicoletti R, de Stefano M (2012) Penicillium restrictum as an antagonist of plant pathogenic fungi. Dynamic Biochem Process Biotechnol Mol Biol 6:61–69
Guillot J, Bond R (2020) Malassezia yeasts in veterinary dermatology: an updated overview. Front Cell Infect Microbiol 10:79
Mendes JF, Von Groll A, Poester VR, Brasil CL, Brandolt TM, Klafke GB, Nogueira CEW, Meireles MCA, Xavier MO (2019) Paracoccidioides spp. in soil from the Pampa Biome in Southern Brazil. Curr Microbiol 76:258–262
Hossain MM, Sultana F, Islam S (2017) Plant growth-promoting fungi (PGPF): phytostimulation and induced systemic resistance Plant-microbe interactions in agro-ecological perspectives. Springer, pp 135–191.
Pereira e Silva MC, Dias ACF, van Elsas JD, Salles JF (2012) Spatial and temporal variation of archaeal, bacterial and fungal communities in agricultural soils. PLoS ONE 7:e51554
Boer WD, Folman LB, Summerbell RC, Boddy L (2005) Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev 29:795–811
Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe Y-I (2006) Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc Natl Acad Sci 103:18296–18301
Walker CB, de la Torre JR, Klotz MG, Urakawa H, Pinel N (2010) Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc Natl Acad Sci 107:8818–8823
Bomberg M, Timonen S (2007) Distribution of cren- and euryarchaeota in scots pine mycorrhizosphere and boreal forest humus. Microb Ecol 54:406–416
Jia Z, Conrad R (2009) Bacteria rather than Archaea dominate microbial ammonia oxidation in an agricultural soil. Environ Microbiol 11:1658–1671
Funding
Open access funding provided by North-West University. This research was funded by the National Research Foundation (ZA) grant (UID123634 and UID132595) awarded to OOB.
Author information
Authors and Affiliations
Contributions
SAA did the laboratory work as part of his PhD at NWU, analyzed and interpreted the results with the help of ASA, and wrote the first draft of the manuscript. OOB initiated the next-generation sequence research, supervised all the listed coauthors, helped shape the research, verified the analytical methods, edited and commented on the document at all stages.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Akinola, S.A., Ayangbenro, A.S. & Babalola, O.O. Pedological factors as drivers of archaeal and fungal communities in maize rhizosphere: a shotgun metagenomic sequencing approach. SN Appl. Sci. 5, 351 (2023). https://doi.org/10.1007/s42452-023-05603-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-023-05603-5