
Abstract
Today’s biodiversity was strongly impacted by glacial cycles during the Pleistocene. They generated species diversity and population structuring, which can easily remain undetected, if populations differentiation is not accompanied by divergence in external morphological traits. This phenomenon, described as cryptic diversity, has been intensively researched in bats, which are known to harbour cryptic phylogenetic lineages. Many studies however have a regional focus or suffer from geographical gaps. Widely understudied areas include Central and Western Asia, although they connect the western and eastern Palearctic fauna. These areas are characterized by topographic heterogeneity and therefore high ecosystem diversity. In this study we investigated the phylogeography of the Savii’s pipistrelle (Hypsugo savii), a bat species assumed to be widely distributed across the Palaearctic. We compiled published sequences from four mitochondrial genes (ND1, CytB, COI and 16 S), added new sequences especially from the Asian part of the distribution range and performed phylogenetic and phylogeographic analyses. They indicate that H. savii is a taxonomic unit with extensive cryptic diversity, comprising at least four major mitochondrial lineages with allopatric or parapatric distribution ranges. Divergence time dating reveals the impact of Pleistocene glaciations on shaping highly structured populations of a highly mobile mammal across the Palearctic. Our study also revealed several zones of secondary contact among populations where hybridization and gene flow likely occur. This study highlights the necessity for studying biodiversity in Western and Central Asia in order to understand biogeographic patterns, evolutionary processes and conservation needs in the area that connects eastern and western Palearctic faunas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Palearctic extends for more than 15,000 km from West to East comprising a continuum of temperate zone ecosystems between the Iberian and the Kamchatka Peninsula. Many taxa expanded their range throughout the whole Palearctic during climatically favourable conditions as similar ecosystems and ecological niches exist across the Eurasian continent. Subsequently, they may have diversified independently in different geographic regions, interrupted by periods of secondary contact (Michaux et al. 2005; Pentzold et al. 2013; Çoraman et al. 2019; Pârâu and Wink 2021). The evolutionary consequences such as phylogeographic patterns, hybridization, and introgression have been intensively studied in western Palearctic taxa (e.g., Michaux et al. 2005; Sperling and Nazari 2007; Lebarbenchon et al. 2010) and to a lesser extent in Eastern Asia (e.g., Ikeda and Motokawa 2021). In contrast, Central Asia has remained understudied (Zhang et al. 2008), even though the Caucasus and Central Asia might have played a central role in the evolution of Palearctic taxa (Mittermeier et al. 2004). Western and Central Asia are characterized by a strong heterogeneity in topography and ecosystems, which has a strong impact on diversity patterns and evolutionary processes such as adaptation. These regions not only represent expansion routes but also harbour biodiversity hotspots with high rates of endemism (e.g., Tojibaev et al. 2020; Zhang et al. 2020). As genetic data accumulate and sampling efforts concentrate on under-sampled geographic regions, studies with a pan-Palearctic perspective are now becoming much more feasible (e.g. Dufresnes et al. 2019).
Pleistocene glaciations have profoundly impacted the evolutionary histories and the genetic diversity of Palearctic species in the relatively recent geological past (Hewitt 2004). The advancing ice forced then-extant taxa to retreat into different southern regions. They served as geographically separated glacial refugia and drove allopatric divergence. After the ice receded following the glacial maximum, the newly emerged lineages spread towards the north where they may have met. Such secondary contact facilitated gene flow and the establishment of a genetically unique population of individuals. Furthermore, these glacial cycles repeated themselves several times, generating complex taxon-specific evolutionary histories and relationships. Many animal groups, covering all ecosystems including sea (Virgilio et al. 2009), freshwaters (Sworobowicz et al. 2015), land (Jablonski et al. 2019) and their combinations (Wielstra et al. 2013), have diversified by these climatic oscillations. However, some species lack genetic differentiation, as a result of expanding from single glacial refugium (Bernard et al. 2011).
Bats (Mammalia: Chiroptera) recently came into focus of taxonomic and phylogeographic studies for several reasons. First, they are characterized by high levels of cryptic diversity, newly discovered even in well studied areas such as the Western Palearctic (Mayer et al. 2007; Çoraman et al. 2019). Second, as top-predators of nocturnal insects, they play a central role in ecosystem-functioning and therefore became a target taxon in conservation management (Voigt and Kingston 2016). Finally, bats turned out as particularly rich natural reservoirs for viruses including those which occasionally spread to other mammalian species including humans. Understanding the distribution and population structure of pathogens’ hosts is therefore of crucial importance for risk assessments of infectious diseases (Letko et al. 2020).
Among Palearctic bats, Savii’s bat Hypsugo savii (Bonaparte, 1837) has an unusual wide distribution throughout most of the Palearctic temperate zone ranging from Western Europe/North-Western Africa to Central Asia (Juste and Paunović 2016). The wide distribution range and its occurrence from sea level up to 3000 m ASL indicates high ecological plasticity, with the species also being highly thermotolerant (Ancillotto et al. 2018). This is also illustrated by its current range expansion in Central Europe towards the north, where this species is established in urban areas and occurs now in close proximity to humans (Kipson et al. 2020). Individuals may vary in few morphological traits (e.g., Benda et al. 2006). Most conspicuous is the variation of pelt colouration of individuals from different populations but individuals within populations can also differ in their colouration (Arlettaz et al. 1993; Benda et al. 2006; Dolch et al. 2021). The degree of the hairiness of the tail membrane is another reported trait that differs between some populations (Dolch et al. 2021).
In contrast to the lack of apparent geographical morphological differentiation and ecological plasticity, several divergent lineages of the mitochondrial genome were found in different regions of the Palearctic (Mayer and Helversen 2001; Ibáñez et al. 2006; Mayer et al. 2007; García-Mudarra et al. 2009; Veith et al. 2011; Galimberti et al. 2012; Çoraman et al. 2013; Bogdanowicz et al. 2015; Batsleer et al. 2019; Mi̇fsud and Vella 2019; Borloti et al. 2020; Dolch et al. 2021). All studies had a regional focus in respect of the large Palearctic distribution of this species. Therefore, the larger extent and distribution of these lineages remained unclear, including larger geographic gaps of knowledge. So far, three mitochondrial lineages had been detected: A lineage from South-Western Europe (Pestano et al. 2003; Ibáñez et al. 2006; García-Mudarra et al. 2009; Veith et al. 2011), a wide-spread lineage ranging from Macaronesia to Italy (Pestano et al. 2003; Ibáñez et al. 2006; Mayer et al. 2007; García-Mudarra et al. 2009; Veith et al. 2011; Bogdanowicz et al. 2015; Batsleer et al. 2019; Mi̇fsud and Vella 2019; Borloti et al. 2020), and a South-Eastern European and Middle Eastern lineage (Ibáñez et al. 2006; Mayer et al. 2007; Çoraman et al. 2013; Mi̇fsud and Vella 2019; Dolch et al. 2021).
In this study, we investigate the genetic diversity of Savi’s pipistrelle bat Hypsugo savii throughout its whole Palearctic distribution range by compiling published sequence data and adding new sequences from unstudied areas. As previous studies involved different mitochondrial markers, we analysed each of the four markers separately and a concatenated data set. Phylogenetic and population genetic methods were used to delineate genetic lineages, infer phylogeographic patterns, identify regions of potential glacial refugia and to estimate evolutionary time scales for the divergence of genetic lineages. Finally, we discuss evolutionary scenarios and taxonomic implications for Palearctic bats of the genus Hypsugo based on this comprehensive analysis.
Materials and methods
Data collection
For the purpose of this study, we gathered all available sequences of H. savii (including those stored as Hypsugo sp. or H. darwinii) from previous studies from GenBank (NCBI 2022) and Barcode of Life (BOLD 2022) online depositories for four mitochondrial markers – ND1, CytB, COI, and 16 S rRNA genes. We also supplemented our dataset with additional sequences of ND1 mitochondrial gene, which had been sequenced following protocols described by Mayer et al. (2007) and Dietz et al. (2016), and deposited in the collection of Museum für Naturkunde, Berlin. As neighbouring congener species, all available sequences of H. alaschanicus, H. ariel, H. arabicus and the newly proposed H. stubbei (Dolch et al. 2021) were also included in the analysis. We refer to H. savii s.l. by following the recent taxonomy (Simmons 2005; Burgin et al. 2019; Kipson et al. 2020), which considers H. alaschanicus, H. ariel and H. arabicus as separate species. An alignment of all available sequences was constructed, using ClustalW algorithm, in MEGA-X programme (Kumar et al. 2018). Information on the sequences and their use in various segments of our analysis is given in Online Resource 1.
Analysis of genetic distances
Uncorrected p-distances were chosen as a measure of genetic distance and to allow continuity with previous studies (e.g., Mayer et al. 2007; Çoraman et al. 2013; Bogdanowicz et al. 2015; Mi̇fsud and Vella 2019). To obtain reliable results, we manually reduced each alignment by excluding sequences that (1) did not overlap with the majority or (2) were of lesser quality. Uncorrected p-distances were then calculated, and sequences were clustered into lineages according to their p-distance scores, with lineages themselves being visualised using Neighbour-Joining algorithm (Saitou and Nei 1987) under pairwise deletion. Sequences which were excluded due to their inadequate length and/or quality, were assigned to clades using some of the newly classified sequences with additional NJ phylograms, under a K2P substitution model (Kimura 1980), again using pairwise deletion. Statistical support for all NJ trees was obtained using bootstrap phylogeny test (Felsenstein 1985) with 1000 replicates.
Bayesian inference
To discern evolutionary relationships and to provide additional support for the lineages in individual marker datasets, we resolved to use Bayesian Inference (BI) reconstruction method. This method was also coupled with a molecularly calibrated clock to obtain estimates of divergence times. A few best sequences (i.e., longest and with least ambiguous sites) from each detected lineage were selected and retained in the alignment. Alongside H. savii, H. arabicus, H. alaschanicus, H. ariel and H. stubbei, H. cadornae was included as an outlying Hypsugo species, while Pipistrellus nathusii and Vespertilio murinus served as outgroups. A concatenated dataset was also constructed. Each lineage was represented by an artificial mitochondrial sequence constructed from selected marker sequences. When selecting marker sequences, we sought to choose those from same individuals or those from closest geographical locations. Most lineages were represented by a single concatenated sequence, however in cases of larger geographical distribution and potential additional internal splits indicated either by p-distance analysis or previous studies (e.g., Borloti et al. 2020), multiple concatenated sequences were constructed. Concatenated sequences were built using SequenceMatrix v.1.8 (Vaidya et al. 2011).
For conducting the Bayesian analysis, we used BEAST 2.5 programme package (Bouckaert et al. 2019). All data was treated as homochromous and ran under a Strict Clock model. Clock rates for ND1 and CytB were set as half of the pairwise value given by Ruedi and Mayer (2001) (0.024), with rates for COI following Russell et al. (2015) and Bairrd et al. (2017) (0.02). Rates for 16 S marker combined the reports of Mucedda et al. (2002) and Spitzenberger et al. (2006) with those by Ruedi and Mayer (2001), resulting in a value of 0.011225. Variations of substitution rate was set for each marker according to the lowest scoring BIC substitution model as proposed by jModelTest v. 2.1.10 (Darriba et al. 2012): HKY (Hasegawa et al. 1985) for ND1 and CytB markers and TrN (Tamura and Nei 1993) for COI and 16 S. If necessary, a gamma distribution shape parameter was also added (see Online Resource 2: Sect. 3 for more details). As a tree prior, a simplistic Yule Model was used in all cases and linked between partitions in the concatenated dataset. Individual marker datasets were run for 10 million generations, whilst the chain for concatenated dataset was ran for 5 million since good convergence was observed earlier when running individual markers. Each dataset was repeated with two additional independent reconstructions. Convergence of the MCMC was inspected with Tracer v.1.7.1 (Rambaut et al. 2018) (especially focusing on the ESS value) and a Maximum Clade Credibility (MCC) tree was then constructed using TreeAnnotator with 10% burn-in. Obtained trees from the three runs were then visually inspected in FigTree v.1.4.4 (Rambaut 2007). Since the results yielded identical topologies, the trees were not pooled. However, we did pool estimated divergence times from all reruns.
Haplotype network analysis
Additionally, in order to visualise lineage relationships further, we used PopART v. 1.7 software (Leigh and Bryant 2015) to draw Median-joining haplotype networks for each marker dataset. The sequences used in drawing these networks were extracted from the alignments for BI, omitting all sequences beyond what was determined to be H. savii and its nearest neighbour in the phylogenetic chronogram. These sequences were trimmed to provide complete deletion of uninformative sites. Epsilon value was left at default (zero).
Geographic visualization
Once all sequences were assigned to their respective group/lineage and good support for lineages was established, we geographically annotated them and visualised the results with QGIS v.3.4.14 (Madeira) programme. H. savii distribution map, used as a visual basis, was downloaded from IUCN Red List (IUCN 2016). For all data presentation purposes, we used a slightly modified colour scheme friendly to the colour-blind developed by Okabe and Ito (2002).
Results
We analysed 138 ND1, 57 CytB, 48 COI, and 42 16 S rRNA sequences, which represented Hypsugo savii or previously described taxa stemming from this species. Together with four other selected species of the genus Hypsugo, this meant a total number of 154 sequences for ND1, 66 for CytB, 68 for COI, and 47 for 16 S rRNA. According to sources listing the accessed sequences, they represent 250 H. savii s.l., 15 H. ariel, 15 H. alaschanicus, 1 H. stubbei, 4 H. cadornae and 5 H. arabicus individuals (Online Resource 1).
Within H. savii s.l., we detected six divergent mitochondrial lineages (Figs. 1c and 2), dubbed A, B, C, D, E and X. Genetic p-distances between well-defined lineages (A-E) were always larger than 6% for pairwise comparisons in protein coding genes. Lineage D clustered together with the newly described H. stubbei (Dolch et al. 2021). Within each lineage, p-distances did not exceed 2% and were therefore notably lower than those between lineages (Table 1). Accordingly, the five main lineages were separated by long and well-supported branches in phylogenetic analyses (Fig. 1c, Online Resource 2: Sects. 1, 2). In addition, we revealed a highly divergent lineage X, represented by a single individual (Figs. 1c and 2).

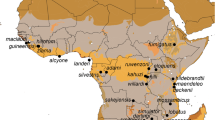
(a) Geographic origin of assigned Hyspugo savii sequences. For some sequences, only vague localities were available, and the placement therefore might be unprecise (see Online Resource 1). + marks the type locality of H. savii (Pisa, Italy). Projected lineage occurrence areas (coloured polygons) were created using buffer zones of approximately 200 km around each data point following anecdotal citations of migration distance (Juste and Paunović 2016; Dietz and Kiefer 2016) in QGIS v. 3.4.14. Question marks denote areas, where lineage distribution is at present unknown. (b) A H. savii individual photographed near Dragonja River, SW Slovenia, in 2018 by Jan Gojznikar. (c) MCC tree obtained by using the concatenated dataset. Posterior probabilities are shown next to their respective nodes. Value of the scale bar refers to million years before present. Squared tips indicate a concatenated sequence of a single individual, whilst triangles denote multiple concatenated sequences
The mitochondrial lineages show allopatric and parapatric distribution patterns including multiple contact zones (Fig. 1a). Lineage A is present in the Iberian Peninsula and apparently extends over the Pyrenees into Southern France. Its eastern border of distribution seems to be located along the Alps. This mitochondrial lineage was confirmed to exist on Sardinia. Lineage B is more widespread and appears in Macaronesia (Cape Verde and Canary Islands) throughout North-Western Africa into Malta and Italy, where it reaches its currently known northern limit in the Alps. A single sample was supposedly collected from central Asia Minor where lineage C is widely distributed. This record of lineage B so far east seems questionable, since it originates from a faecal sample and was processed in the lab together with individuals of lineage B from Italy (Veith et al. 2011). Therefore, it could originate from sequencing a contamination from a fresh sample or procedural error. In southern Spain (and possibly Portugal), Sardinia and Switzerland/Southern France/Western Italy, lineage B is in contact or even in sympatry with lineage (A). Lineage C occupies most of the Balkans and Middle East, reaching North into Caucasus, south into Israel and as far East as North-Eastern Iran. It was also found on Cyprus. Sequences from the Balkans clustered separate of those from further East (Online Resource 2). At the western distribution range, lineage C is likely in contact with lineage (B). The eastern border of lineage C’s distribution range is at present unknown, likely located in Central Asia. Lineage D (H. stubbei) was identified in Kyrgyzstan and Mongolia, suggesting a Central Asian distribution, where it can be found in sympatry with H. alaschanicus. Further to the south, lineage E was found in Pakistan and is represented by three samples from an unspecified location. Finally, the single individual representing the lineage X came from one locality in Israel.
The Bayesian inference for all mitochondrial markers yielded trees with good posterior probability support (above 0.95) for grouping sequences into the major mitochondrial lineages A to E. Lineages A to D formed a well-supported monophyletic clade. Surprisingly, lineage E, for which only the 16 S rRNA was available, was placed as an outgroup to all other Hypsugo sequences (Fig. 2). The phylogenetic position of lineage X represented by a single individual remains unclear. For the 16 S RNA gene we had rather restricted comparative data and the ND1 sequence insufficiently aligned for phylogenetic analysis. Lineage X grouped with H. savii in the 16 S RNA tree while a poorly supported sister-relationship with H. arabicus was suggested by the concatenated data set.

Chronograms, resulting from BI analysis of the four selected Hypsugo marker sequences ND1 (a), CytB (b), COI (c), and 16 S RNA (d). Only lineage A and B were detected in all four marker datasets. The dots denote nodes with posterior probability value equal or above 0.95. Other nodes were below the value we designated as reliable to successfully infer phylogenetic relations between detected lineages (0.95). The scale bar indicates approximate age in millions of years
Relationships between the lineages were mostly poorly resolved, with posterior probabilities frequently bellow 0.95 (Fig. 2). However, the trees usually suggested a sister relationship between lineages A and C as well B and D. Posterior probability values of more than 0.95 for some gene trees (Fig. 2) and the tree based on the concatenated data set (Fig. 1c) suggested H. alaschanicus as the sister species to H. savii s.l. In both 16 S and concatenated dataset, lineage E appeared to be basal to all other Hypsugo sequences covered by this study.
Accordingly, median-joining networks of mitochondrial haplotypes support the four lineages (A-D) as connections between them carry many more mutations than connections within each lineage (Fig. 3). The relatedness among lineages remained far less conclusive. However, a tendency to place lineage A closer to C was found, a result concordant with BI (Fig. 2).

Median-joining haplotype networks for the four marker sequences ND1 (a), CytB (b), COI (c), and 16 S RNA (d). Smaller coloured circles represent a single sequence, while the larger represent two or three. Hala refers to H. alaschanicus and Hstu to H. stubbei
Lineages A to D had an estimated node divergence height (age in highest posterior density (HPD) intervals, Table 2) between 2.1 and 2.6 million years ago (MYa) based on the data sets of protein coding genes, while the split from nearest neighbouring species (H. alaschanicus) was indicated to be between 3.1 and 3.7 MYa. Lineage E, represented only in the 16 S rRNA data set, was estimated to be significantly older (11.2 MYa, Table 2). Estimated divergence heights were a bit higher when it came to concatenated dataset, with lineage E still appearing to be noticeably older. Time estimate for divergence of lineage X in the concatenated dataset is likely not reliable due to missing sequence overlap with proposed sister taxon of H. arabicus (see Fig. 1c).
Discussion
The wide geographic sampling of bats traditionally assigned to Hypsugo savii revealed a total of six highly divergent mitochondrial lineages. The lineages differed among each other by p-distances between 6 and 11% for protein-coding mitochondrial markers. Lineages were found in different geographic regions resulting in widely allopatric distribution patterns. However, multiple contact zones exist among geographically neighbouring lineages.
Lineage diversity and distribution
Lineage A has its distribution range in South-Western Europe. It had been variously referred to as Western Mediterranean or Iberian lineage and was taxonomically designated as Hypsugo savii s.s. or Hypsugo savii ochromixtus (Veith et al. 2011; Batsleer et al. 2019; Dolch et al. 2021). Lineage B has a larger distribution range than lineage A, ranging from Macaronesian Archipelagos through North-Africa and Apennine Peninsula to the Alps. In the past, lineage B was referred to as Hypsugo cf. darwinii (Mayer et al. 2007; Veith et al. 2011; Datzmann et al. 2012; Dondini et al. 2016; Batsleer et al. 2019; Dolch et al. 2021), based from a description of the species from the Canary Islands (see Mayer et al. 2007). However, the type locality of H. savii is also within the range of lineage B, which indicates that H. cf. darwinii is synonymous to the nominate subspecies H. s. savii and therefore not a valid taxon. Lineage C has the largest distribution range, encompassing the Balkan Peninsula, Anatolia, the Caucasus, and parts of Middle East. Two subclades separated individuals from the Balkans from all the other regions. Lineage C likely represents the taxon H. s. caucasicus (Benda et al. 2006; Dolch et al. 2021). Lineage D was found in Kyrgyzstan and in Mongolia and likely represents the same taxon as the recently proposed H. stubbei from Mongolia (Dolch et al. 2021). However, animals from Kyrgyzstan and Mongolia appear to differ slightly in morphology, as Dolch et al. (2021) assigned animals from Kyrgyzstan as H. savii caucasicus. This contrasts with our analysis, which shows a split between H. s. caucasicus in Western Central Asia and H. (s.) stubbei in Kyrgyzstan and Mongolia. Finally, three individuals from Pakistan, labelled ‘H. savii’, and a single individual from Mt. Hermon in Israel turned out to represent two separate lineages respectively (respectively named E and X). Both lineages are highly divergent from other lineages within H. savii s.l. and thus likely represent other species.
Despite their divergence in the mitochondrial genome, individuals of H. savii from Europe to Eastern Asia closely resemble each other in morphology as variation in the most conspicuous trait (colouration) is high within and among populations (Arlettaz et al. 1993; Benda et al. 2006, 2012; Dolch et al. 2021).
Phylogenetic relationships
Although the detected mitochondrial lineages themselves are clearly defined, evolutionary relationships among them remain partially unresolved. The East-Asian Hypsugo alaschanicus appears to be the closest relative of H. savii s.l. This suggests an ancestral split about four MYa into H. savii in the West and H. alaschanicus in the East. Within H. savii, the divergence of multiple lineages within a narrow period about 2 to 3 MYa hindered the resolution of phylogenetic relationships. Nevertheless, Bayesian trees place lineages A and C in a monophyletic group, representing a sister clade to lineages B and D. This grouping is surprising, since it does not reflect geographic proximity of lineages. The Western European lineage A groups with the Eastern European and Caucasian lineage C, while the Central European lineage B is the sister lineage of eastern Asian lineage D. An explanation for this biogeographic pattern could be an early differentiation between a western European (lineage A), a southern lineage occupying northern Africa and the Apennine Peninsula (lineage B) and Asian lineage (D). Accordingly, eastern Europe was colonized by the western lineage north of the Alps, which established lineage C east of lineage B. Rigorously testing such conclusions would require genome-based analyses.
Lineage E is surprisingly different as it is placed basal to all other Hypsugo taxa covered by this study. A taxonomic assignment of lineage E remains open since we do not have morphological information of the individuals as only DNA sequences were available from GenBank. However, individuals of this lineage originate from the Indo-Malayan region where the purported Hypsugo taxon austenianus occurs. It is currently regarded as a subspecies within H. savii (H. savii austenianus according to Simmons (2005) and Simmons and Cirranello (2023). Our phylogenetic analysis now questions this and indicates a so far unrecognized species within the genus Hypsugo, which diverged from other Hypsugo taxa about 10 MYa.
The phylogenetic position of lineage X as a sister group to H. arabicus is intriguing. It may suggest that the distribution range of H. arabicus might extend further north of the Arabian Peninsula. However, this scenario should be treated with caution unless further records confirm the presence of this lineage in the Levant.
Biogeography
In the western Palearctic, H. savii shows a highly structured phylogeographic pattern that resembles those of many other terrestrial animal taxa (reviewed in Hewitt 2004). It is characterized by several glacial refugia in the Mediterranean region, resulting in a separation of populations between the Iberian, Italian and Balkan Peninsulas, and Asia Minor. This illustrates the effective dispersal barriers of the Mediterranean Sea as well the high mountain ranges of the Pyrenees and the Alps even for a highly mobile vertebrate as the Savi’s bat. A rather similar pattern of geographic structuring was recently found in among European Natterer’s bats. The three peninsulas are populated by three different lineages (Ibáñez et al. 2006; Çoraman et al. 2019; Juste et al. 2019).
Within Central Asia, two lineages of H. savii were found. The western parts were colonized by eastern Mediterranean lineage C, likely by an expansion to the east from Asia Minor or the Near East south of the Caspian Sea. An eastern glacial refugium is represented by lineage D, which was found from the Inner Asian Mountain corridor to Mongolia. So far, we know rather little about the number and location of glacial refugia in and postglacial colonization of Central Asia as this area was rarely sampled for genetic studies so far. In many taxa, Central Asian population show little to no geographic structuring due to rather recent colonization from the western Palearctic (e.g., Wielstra et al. 2013; Lombardo et al. 2022) or eastern (Kozyra et al. 2021), likely due to the lack of long-term glacial refugia in the area. In contrast, taxa adapted to cold climates can show complex patterns of populations structures (e.g., Dufresnes et al. 2019) reflecting the topographic heterogeneity in the region.
Since H. savii has a continuous distribution range from Western Europe and Northern Africa to Central Asia, multiple contact zones between lineages exist and were now located by sequencing mitochondrial DNA. Only on the island of Sardinia, two lineages occur in sympatry. This could be caused by the coexistence of two reproductively isolated populations and thus different biological species, but it can also reflect a single admixed population of individuals representing the two mitochondrial lineages A and B. Only the analysis of the nuclear genome can distinguish between both scenarios. The continuous distribution of H. savii s.l. allows the spread of parasites and diseases throughout the Palearctic distribution range of H. savii s.l. spanning more than 10,000 km and therefore connecting Western Europe with Eastern Asia. This is facilitated by the enormous ecological plasticity of H. savii, as indicated by its wide geographic distribution range, its occurrence from sea level up to 3000 m above mean sea level and even its settlement in urban areas (Uhrin et al. 2016; Kipson et al. 2020).
In contrast to bats, birds, the only other species-rich group of actively flying vertebrates, frequently show less geographic structuring of their mitochondrial genome (Carneiro de Melo Moura et al. 2019; Pârâu and Wink 2021; Lombardo et al. 2022). This apparent difference between mitochondrial population structures of birds and bats may be shaped by general behavioural differences in natal dispersal and in migration between both groups of animals. Bats of temperate zones frequently show natal philopatry by females (Kerth 2008) restricting the spread of the maternally inherited genome. Among birds, males tend to be the philopatric sex, while females disperse from their natal areas (Greenwood and Harvey 1982). This behaviour of female birds contributes to the dispersal of their mitochondrial genome across wider geographic areas as they settle for breeding distant from their place of hatching. In birds, mitochondrial dispersal is even further facilitated, as the proportion of migratory species is larger among birds than among bats and migratory distances are frequently often much larger in birds than in bats.
Conclusions
The allopatric and parapatric distribution pattern of mitochondrial lineages suggest a lack of effective hybridization barriers at least among neighbouring lineages. This biogeographic pattern of morphological homogeneity and mitochondrial parapatry is best described by distinguishing four sub-species within H. savii, namely H. s. ochromixtus (lineage A), H. s. savii (lineage B), H. s. caucasicus (lineage C) and H. s. stubbei (lineage D). However, we have to await the analysis of nuclear genome to see to which degree the genetic differentiation of the mitochondrial genome matches population structures in the nuclear genome and whether populations interbreed in contact zones.
References
Ancillotto L, Budinski I, Nardone V et al (2018) What is driving range expansion in a common bat? Hints from thermoregulation and habitat selection. Behav Processes 157:540–546. https://doi.org/10.1016/j.beproc.2018.06.002
Arlettaz R, Guibert E, Lugon A et al (1993) Variation of fur coloration in Savi’s bat Hypsugo savii (Bonaparte, 1837). Bonn Zool Beitr 44:293–297
Baird AB, Braun JK, Engstrom MD et al (2017) Nuclear and mtDNA phylogenetic analyses clarify the evolutionary history of two species of native hawaiian bats and the taxonomy of Lasiurini (Mammalia: Chiroptera). PLoS ONE 12:e0186085. https://doi.org/10.1371/journal.pone.0186085
Batsleer F, Portelli E, Borg JJ et al (2019) Maltese bats show phylogeographic affiliation with North-Africa: implications for conservation. Hystrix It J Mamm 30:172–177. https://doi.org/10.4404/hystrix-00237-2019
Benda P, Andreas M, Kock D et al (2006) Bats (Mammalia: Chrioptera) of the Eastern Mediterranean. Part 4. Bat fauna of Syria: distribution, systemantics, ecology. Acta Soc Zool Bohem 70:1–329
Benda P, Faiolâhi K, Andreas M et al (2012) Bats (Mammalia: Chrioptera) of the Eastern Mediterranean. Part 10. Bat fauna of Iran. Acta Soc Zool Bohem 76:163–582
Bernard R, Heiser M, Hochkirch A, Schmitt T (2011) Genetic homogeneity of the sedgling Nehalennia speciosa (Odonata: Coenagrionidae) indicates a single Würm glacial refugium and trans-Palaearctic postglacial expansion. J Zool Syst Evol Res 49:292–297. https://doi.org/10.1111/j.1439-0469.2011.00630.x
Bogdanowicz W, Hulva P, Černá Bolfíková B et al (2015) Cryptic diversity of Italian bats and the role of the Apennine refugium in the phylogeography of the western Palaearctic. Zool J Linn Soc 174:635–648. https://doi.org/10.1111/zoj.12248
BOLD (2022) Public Data Portal - Record List | BOLDSYSTEMS. https://www.boldsystems.org/index.php/Public_BINSearch?searchtype=records
Borloti I, Dinis H, Vasconcelos R (2020) Bats out of Africa: disentangling the systematic position and biogeography of bats in Cabo Verde. Genes 11:877. https://doi.org/10.3390/genes11080877
Bouckaert R, Vaughan TG, Barido-Sottani J et al (2019) BEAST 2.5: an advanced software platform for bayesian evolutionary analysis. PLoS Comput Biol 15:e1006650. https://doi.org/10.1371/journal.pcbi.1006650
Burgin CJ, Moratelli R, Cláudio VC et al (2019) Species accounts of Vespertilionidae. In: Wilson DE, Mittermeier RA (eds) Handbook of the mammals of the world – Volume 9: Bats: Lynx Edicions, pp 716–982
Carneiro de Melo Moura C, Bastian H-V, Bastian A et al (2019) Pliocene origin, ice ages and postglacial population expansion have influenced a panmictic phylogeography of the European bee-eater Merops apiaster. Diversity 11:12. https://doi.org/10.3390/d11010012
Çoraman E, Furman A, Karataş A, Bilgin R (2013) Phylogeographic analysis of Anatolian bats highlights the importance of the region for preserving the Chiropteran mitochondrial genetic diversity in the Western Palaearctic. Conserv Genet 14:1205–1216. https://doi.org/10.1007/s10592-013-0509-4
Çoraman E, Dietz C, Hempel E et al (2019) Reticulate evolutionary history of a western palaearctic bat complex explained by multiple mtDNA introgressions in secondary contacts. J Biogeogr 46:343–354. https://doi.org/10.1111/jbi.13509
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772–772. https://doi.org/10.1038/nmeth.2109
Datzmann T, Dolch D, Batsaikhan N et al (2012) Cryptic diversity in Mongolian Vespertilionid bats (Vespertilionidae, Chiroptera, Mammalia). Acta Chiropt 14:243–264. https://doi.org/10.3161/150811012X661594
Dietz C, Kiefer A (2016) Bats of Britain and Europe. Bloomsbury Publishing, London
Dietz C, Gazaryan A, Papov G et al (2016) Myotis hajastanicus is a local vicariant of a widespread species rather than a critically endangered endemic of the Sevan lake basin (Armenia). Mamm Biol 81:518–522. https://doi.org/10.1016/j.mambio.2016.06.005
Dolch D, Batsaikhan N, Stubbe A, Steinhauser D (2021) Hypsugo stubbei sp. nov., a novel cryptic bat species of the genus Hypsugo (Vespertilionidae, Chiroptera, Mammalia) from Mongolia. Erforsch Biol Ress Mongolei (Halle/Saale) 14:187–206
Dondini G, Vergari S, Fichera G, Kiefer A (2016) First record of Hypsugo cf. darwinii (tomes, 1859) in Tuscany, Italy. https://doi.org/10.14709/BarbJ.9.1.2016.01. Barbastella 9:
Dufresnes C, Mazepa G, Jablonski D et al (2019) Fifteen shades of green: the evolution of Bufotes toads revisited. Mol Phylogenet Evol 141:106615. https://doi.org/10.1016/j.ympev.2019.106615
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. https://doi.org/10.2307/2408678
Galimberti A, Spada M, Russo D et al (2012) Integrated operational taxonomic units (IOTUs) in echolocating bats: a bridge between molecular and traditional taxonomy. PLoS ONE 7:e40122. https://doi.org/10.1371/journal.pone.0040122
García-Mudarra JL, Ibáñez C, Juste J (2009) The Straits of Gibraltar: barrier or bridge to ibero-moroccan bat diversity? Biol J Linn Soc Lond 96:434–450. https://doi.org/10.1111/j.1095-8312.2008.01128.x
Greenwood PJ, Harvey PH (1982) The natal and breeding dispersal of birds. Ann Rev Ecol Syst 13:1–21
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174. https://doi.org/10.1007/BF02101694
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci 359:183–195. https://doi.org/10.1098/rstb.2003.1388
Ibáñez C, García-Mudarra JL, Ruedi M et al (2006) The Iberian contribution to cryptic diversity in European bats. Acta Chiropt 8:277–297. https://doi.org/10.3161/1733-5329(2006)8[277:TICTCD]2.0.CO;2
Ikeda Y, Motokawa M (2021) Phylogeography of the Japanese greater horseshoe bat Rhinolophus nippon (Mammalia: Chiroptera) in Northeast Asia: New insight into the monophyly of the Japanese populations. Ecol Evol 11:18181–18195. https://doi.org/10.1002/ece3.8414
IUCN (2016) The IUCN Red List of Threatened Species. In: IUCN Red List of Threatened Species. https://www.iucnredlist.org/en. Accessed 5 Sep 2022
Jablonski D, Nagy ZT, Avcı A et al (2019) Cryptic diversity in the smooth snake (Coronella austriaca). Amphib Reptil 40:179–192. https://doi.org/10.1163/15685381-20181025
Juste J, Paunović M (2016) Savi’s pipistrelle Hypsugo savii. The IUCN Red List of Threatened Species. In: IUCN Red List of Threatened Species. https://www.iucnredlist.org/species/44856/22072380. Accessed 3 May 2023
Juste J, Ruedi M, Puechmaille SJ et al (2019) Two new cryptic bat species within the Myotis nattereri species complex (Vespertilionidae, Chiroptera) from the Western Palaearctic. Acta Chiropt 20:285–300. https://doi.org/10.3161/15081109ACC2018.20.2.001
Kerth G (2008) Causes and consequences of sociality in bats. Bioscience 58:737–746. https://doi.org/10.1641/B580810
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/BF01731581
Kipson M, Gazaryan S, Horáček I (2020) Savi’s pipistrelle Hypsugo savii (Bonaparte, 1837). In: Hackländer K, Zachos FE (eds) Handbook of the mammals of Europe. Springer International Publishing, Cham, pp 1–18
Kozyra K, Zając TM, Ansorge H et al (2021) Late pleistocene expansion of small murid rodents across the Palearctic in relation to the past environmental changes. Genes 12:642. https://doi.org/10.3390/genes12050642
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lebarbenchon C, Poitevin F, Arnal V, Montgelard C (2010) Phylogeography of the weasel (Mustela nivalis) in the western-palaearctic region: combined effects of glacial events and human movements. Heredity 105:449–462. https://doi.org/10.1038/hdy.2009.186
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Letko M, Seifert SN, Olival KJ et al (2020) Bat-borne virus diversity, spillover and emergence. Nat Rev Microbiol 18:461–471. https://doi.org/10.1038/s41579-020-0394-z
Lombardo G, Rambaldi Migliore N, Colombo G et al (2022) The mitogenome relationships and phylogeography of barn swallows (Hirundo rustica). Mol Biol Evol 39:msac113. https://doi.org/10.1093/molbev/msac113
Mayer F, von Helversen O (2001) Cryptic diversity in European bats. Proc Royal Soc B 268:1825–1832. https://doi.org/10.1098/rspb.2001.1744
Mayer F, Dietz C, Kiefer A (2007) Molecular species identification boosts bat diversity. Front Zool 4:4. https://doi.org/10.1186/1742-9994-4-4
Michaux JR, Libois R, Filippucci M-G (2005) So close and so different: comparative phylogeography of two small mammal species, the yellow-necked fieldmouse (Apodemus flavicollis) and the woodmouse (Apodemus sylvaticus) in the Western Palearctic region. Heredity 94:52–63. https://doi.org/10.1038/sj.hdy.6800561
Mi̇fsud CM, Vella A (2019) Mitochondrial genetic diversity of bat species from the Maltese islands and applications for their conservation. NESciences 4:276–292. https://doi.org/10.28978/nesciences.646348
Mittermeier RA, Robles Gil P, Hoffman M et al (2004) Hotspots revisited, 1st English ed. Cemex, Mexico City, Mexico
Mucedda M, Kiefer A, Pidinchedda E, Veith M (2002) A new species of long-eared bat (Chiroptera, Vespertilionidae) from Sardinia (Italy). Acta Chiropt 4:121–135
NCBI (2022) Home - Nucleotide - NCBI. https://www.ncbi.nlm.nih.gov/nuccore
Okabe M, Ito K (2002) Color Universal Design (CUD) / Colorblind Barrier Free. In: Color Universal Design (CUD) - How to make figures and presentations that are friendly to colorblind people -. https://jfly.uni-koeln.de/color/. Accessed 3 May 2023
Pârâu LG, Wink M (2021) Common patterns in the molecular phylogeography of western palearctic birds: a comprehensive review. J Ornithol 162:937–959. https://doi.org/10.1007/s10336-021-01893-x
Pentzold S, Tritsch C, Martens J et al (2013) Where is the line? Phylogeography and secondary contact of western Palearctic coal tits (Periparus ater: Aves, Passeriformes. Paridae) Zoologischer Anzeiger - J Comp Zool 252:367–382. https://doi.org/10.1016/j.jcz.2012.10.003
Pestano J, Brown RP, Suárez NM, Fajardo S (2003) Phylogeography of pipistrelle-like bats within the Canary Islands, based on mtDNA sequences. Mol Phylogenet Evol 26:56–63. https://doi.org/10.1016/S1055-7903(02)00307-X
Rambaut A (2007) FigTree. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 5 Sep 2022
Rambaut A, Drummond AJ, Xie D et al (2018) Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032
Ruedi M, Mayer F (2001) Molecular systematics of bats of the genus Myotis (Vespertilionidae) suggests deterministic ecomorphological convergences. Mol Phylogenet Evol 21:436–448. https://doi.org/10.1006/mpev.2001.1017
Russell AL, Pinzari CA, Vonhof MJ et al (2015) Two tickets to paradise: multiple dispersal events in the founding of hoary bat populations in Hawai’i. PLoS ONE 10:e0127912. https://doi.org/10.1371/journal.pone.0127912
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Simmons N (2005) Order Chiroptera. In: Wilson DE, Reeder DM (eds) Mammal species of the world: a taxonomic and geographic reference. JHU, pp 312–529
Simmons NB, Cirranello AL (2023) Bat Species of the World: A taxonomic and geographic database. Version 1.3. https://batnames.org/. Accessed 6 Mar 2023
Sperling F, Nazari V (2007) Mitochondrial DNA divergence and phylogeography in western Palaearctic Parnassiinae (Lepidoptera: Papilionidae): how many species are there? Insect Syst Evol 38:121–138. https://doi.org/10.1163/187631207788783996
Spitzenberger F, Strelkov PP, Winkler H, Haring E (2006) A preliminary revision of the genus Plecotus (Chiroptera, Vespertilionidae) based on genetic and morphological results. Zool Scr 35:187–230. https://doi.org/10.1111/j.1463-6409.2006.00224.x
Sworobowicz L, Grabowski M, Mamos T et al (2015) Revisiting the phylogeography of Asellus aquaticus in Europe: insights into cryptic diversity and spatiotemporal diversification. Freshw Biol 60:1824–1840. https://doi.org/10.1111/fwb.12613
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023
Tojibaev KS, Jang CG, Lazkov GA et al (2020) An annotated checklist of endemic vascular plants of the Tian-Shan mountains in central Asian countries. Phytotaxa 464:117–158. https://doi.org/10.11646/phytotaxa.464.2.1
Uhrin M, Hüttmeir U, Kipson M et al (2016) Status of Savi’s pipistrelle Hypsugo savii (Chiroptera) and range expansion in Central and south-eastern Europe: a review. Mammal Rev 46:1–16. https://doi.org/10.1111/mam.12050
Vaidya G, Lohman DJ, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27:171–180. https://doi.org/10.1111/j.1096-0031.2010.00329.x
Veith M, Mucedda M, Kiefer A, Pidinchedda E (2011) On the presence of pipistrelle bats (Pipistrellus and Hypsugo; Chiroptera: Vespertilionidae) in Sardinia. Acta Chiropt 13:89–99. https://doi.org/10.3161/150811011X578642
Virgilio M, Fauvelot C, Costantini F et al (2009) Phylogeography of the common ragworm Hediste diversicolor (Polychaeta: Nereididae) reveals cryptic diversity and multiple colonization events across its distribution. Mol Ecol 18:1980–1994. https://doi.org/10.1111/j.1365-294X.2009.04170.x
Voigt CC, Kingston T (eds) (2016) Bats in the Anthropocene: conservation of bats in a changing world. Springer International Publishing, Cham
Wielstra B, Crnobrnja-Isailović J, Litvinchuk SN et al (2013) Tracing glacial refugia of Triturus newts based on mitochondrial DNA phylogeography and species distribution modeling. Front Zool 10:13. https://doi.org/10.1186/1742-9994-10-13
Zhang Y-J, Stöck M, Zhang P et al (2008) Phylogeography of a widespread terrestrial vertebrate in a barely-studied Palearctic region: green toads (Bufo viridis subgroup) indicate glacial refugia in Eastern Central Asia. Genetica 134:353–365. https://doi.org/10.1007/s10709-008-9243-0
Zhang Y, Zhang D, Li W et al (2020) Characteristics and utilization of plant diversity and resources in Central Asia. Reg Sustain 1:1–10. https://doi.org/10.1016/j.regsus.2020.08.001
Acknowledgements
We wish to thank Peter Trontelj for supporting first author’s master thesis, upon which this contribution is based. Genetic samples were collected in last decades by Eran Amichai, Christian Dietz, Heliana Dundarova, Astghik Ghazaryan, Suren Ghazaryan, Vanuhi Hambardzumyan, Darija Josić, Eran Levin, Tali Magory, Pirimkul Mamatkalykov, Eva Pavlovič, Begaim Sultanbekkyzy, and Otto von Helversen. We wish to thank Isabelle Waurick for DNA sequencing.
Bat tissue, collected from Slovenia, was handled under permits no. 35601-22/2015-5, 35601-65/2018-4, and 35601-59/2020-6 by Slovene Environmental Agency.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
JG collected some of the samples, compiled the sequences, ran the analysis, polished the results, and drafted the manuscript. FM designed the study, provided some of the samples and enabled their processing, supervised the study progress, and polished the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Handling editor: Patrick Arnold
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gojznikar, J., Mayer, F. Mitochondrial DNA reveals the impact of Pleistocene glaciations on a widespread palearctic bat species. Mamm Biol (2024). https://doi.org/10.1007/s42991-024-00449-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42991-024-00449-9