
Abstract
Background and aim
Congenital disorders of glycosylation (CDG) are a large heterogeneous group of about 170 rare inherited metabolic disorders due to defective protein and lipid glycosylation. This study aimed to assemble and summarise available data on the epidemiology of CDG.
Methods
A set of keywords related to epidemiology and CDG was defined. The keywords were combined through a custom Python script, search through the MEDLINE database, using PubMed as the search engine. The script retrieved the correspondent MEDLINE data from each article, and the relevant information was exported. Next, inclusion and exclusion criteria were set and applied during the selection phase. Finally, epidemiology-related information was extracted and compiled.
Results
One hundred sixty-five papers on CDG epidemiology were included in this literature review. Most of them reported on the frequency of symptoms in CDG patients followed in cohort studies, on pathogenic variant allelic frequency, and on the prevalence of the disorder in populations. According to this review, the most reported CDG was phosphomannomutase-2 deficiency (PMM2-CDG) followed in descending order by FKTN-CDG, EXT1/EXT2-CDG, ALG6-CDG, and PIGA-CDG.
Conclusions
We provide an overview on epidemiological data regarding 93 CDG by compiling information from the literature. Generating epidemiological data on CDG is important to appropriately target resources for CDG research and drug development and to support public health decision-making.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A rare disease (RD) affects a small number of people [1]. According to the Rare Disease Act of 2002, a RD “affects fewer than 200,000 people in the USA” [2], whilst in Europe, a disease is considered to be rare when it affects no more than one person in 2000 [3]. An operational definition would consider the specific clinical and qualitative challenges associated with the low prevalence of rare diseases. Rare Diseases International is working with a multi-stakeholder group of experts to develop an international operational definition of rare diseases as part of its Memorandum of Understanding with the World Health Organization [4].
Close to 8000 RDs are known and about 80% have a genetic origin [5, 6]. Hence, when jointly considered, RDs are not so rare. A recent analysis yielded a conservative, evidence-based estimate for the population prevalence of rare diseases of 3.5–5.9%, corresponding to 263–446 million persons affected globally at any point in time [7]. One in 17 people will be affected by a RD at some point in their lives [8]. This amounts to 30 million people across Europe and 25 million in the United States of America (USA) [9]. Despite the many efforts of the medical, research, and patient communities, most RDs lack effective treatment [10].
This article focuses specifically on congenital disorders of glycosylation (CDG), first reported clinically in 1980 by Jaeken et al. [11]. These are rare genetic disorders caused by pathogenic variants in the genes that code for proteins needed for the glycosylation and deglycosylation processes (building glycan trees and attaching them to proteins and lipids). Approximately 170 different CDG have been described [12, 13]. They are named by the official gene symbol followed by the suffix “-CDG”. They can be classified according to the underlying affected glycosylation pathways: protein N-glycosylation, protein O-glycosylation, glycosphingolipid synthesis, glycosylphosphatidylinositol (GPI)-anchor synthesis, and other glycosylation pathways. In N-glycosylation, the assembling of the glycan occurs in the cytosol and the endoplasmic reticulum, and the remodelling in the Golgi apparatus. O-glycosylation mainly occurs in the Golgi apparatus. It has no processing and thus consists only of the assembly, allowing for a more variable set of O-glycans. In some CDG, there is a combined defect in N- and O-glycosylation. Glycosphingolipids consist of membrane lipids linked to a glycan. GPI-anchored proteins are localised in the plasma membrane [14,15,16]. N-glycosylation defects can be divided into CDG type I (CDG-I) and CDG type II (CDG-II). CDG-I defects impair the biosynthesis and attachment of the lipid-linked oligosaccharide to proteins, thus generating proteins which have some unoccupied glycosylation sites. CDG-II defects impair the glycan remodelling, thus generating defective glycoproteins [17].
According to the Centers for Disease Control and Prevention [18], epidemiology consists of the methods used to understand the causes and progression of diseases and health outcomes in populations. It is the study of the distribution of these determinants, and this can be used to control health problems and their implications for society.
Two important epidemiologic tools are prevalence and incidence: Prevalence refers to the ratio between the number of patients with a particular disease in a population and the number of people in that population at a given moment. Incidence refers to the number of individuals who develop a specific disease during a particular time period. The key difference between these two epidemiologic measures is that incidence only considers new cases, while prevalence includes new and pre-existing cases. These terms are often confused with “frequency”, which is the number of events in a certain time [18].
Therefore, generating epidemiological data on CDG, and other RDs, is fundamental to:
-
1)
Evaluate the burden of disease
-
2)
Identify unmet clinical needs. For instance, epidemiological data contributes to creating guidelines for patient management and follow-up. Clinical management guidelines are essential to ensure that data is systematically collected and that patients receive a uniform, high-quality care [7]
-
3)
Expedite drug approval, by supporting the designation of new drugs as “orphan drugs”: One criterium for a drug to obtain an orphan drug designation by the Food and Drug Administration (FDA) [19] and European Medicine Agency (EMA) [20] is adequate documentation on relevant epidemiological data (surveys, cohort studies, patient registries, databases, etc.) demonstrating that the intended condition is rare
-
4)
Identify eligible target populations for therapies prior to being marketed, since relevant epidemiological data is a limiting factor for clinical trial development
This review aimed to assemble and summarise published epidemiological data on CDG and to report the main obstacles to research in this area.
Methods
A set of keywords related to epidemiology and CDG were defined (Table 1S). A customised Python script (tj_articles_extraction - https://github.com/tatianarijoff/tjbioarticles) was used to combine the keywords and search the MEDLINE database, using PubMed as the search engine. The script retrieves the corresponding MEDLINE data (title, abstract, MeSH terms, etc.) from each article from the XML and exports them to a Pandas DataFrame. Using the LaTeX Python library, we generated PDF files containing the title, year of publication, and abstract of the articles selected by the script and eliminated duplicate entries.
We identified 628 article abstracts and 115 unique abstracts were extracted.
Inclusion and exclusion criteria selected: (1) only articles that included information regarding incidence, prevalence, number of patients, and epidemiology; (2) that were related/relevant to CDG; (3) written in English; and (4) no reviews, opinion articles, and other types of articles, such as short/brief communications and letters. The flowchart for study selection is shown in Fig. 1. Of the 115 unique records selected, sixty-three articles were reviewed. An additional 101 articles were identified through authors’ referrals and screening of the extracted reviews’ references.

PRISMA flowchart of study selection. *Some articles were excluded for more than one reason
We extracted information from the 165 included articles regarding their study aim, disease(s) addressed, incidence, prevalence, number of patients in the total population, and significant findings used in the results section of this article. In their cohort studies, several studies involving patients from different countries could not represent the total national population. As some patients may have been reported in different studies, only studies that explicitly stated that they report the total number of patients per country were considered. Seaborn (https://seaborn.pydata.org/, a Python data visualisation library) [21, 22] and Excel were used to generate the figures used in this manuscript.
Results
Characteristics of the included studies
The 165 articles reviewed fall apart into five groups: (i) 81 case reports; (ii) 43 patient cohort studies which reported on the frequency of CDG symptoms [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]; (iii) 11 papers [38, 39, 42, 64,65,66,67,68,69,70,71] on the frequency of carriers of specific pathogenic variants, most of European origin, and prevalence of CDG; (iv) 9 papers [65, 72,73,74,75,76,77,78,79] on pathogenic variant allelic frequency, mostly describing PMM2 pathogenic variants; and (v) 8 other original articles, which did not focus on epidemiological data, but on data (mechanisms of disease; 3D modelling of the proteins associated with CDG, etc.), reporting information relevant to the present work [62, 80,81,82,83,84,85,86]. Three questionnaires were also included in this review [25, 26, 69].
These 165 studies reported data from 38 different countries, with the largest number of publications (n = 31) from the USA, followed by Belgium (n = 15), France and the Netherlands (n = 12 for each), and Germany (n = 11) (Fig. 2). Only the most recent data was included in this literature revision.

The geographical distribution of the publications included in the literature review. Articles whose corresponding authors were from different countries were counted for each country
Of the 170 different described CDG, 93 were included in this review. The most used method to screen for/diagnose CDG was transferrin isoelectric focusing (TfIEF) (n=11) [20, 23, 31, 36,37,38,39, 42, 45, 49, 50], followed by polymerase chain reaction (PCR) (n=5) [64, 71, 72, 74, 76], whole exome/genome sequencing (WES/WGS) (n=6) [27, 35, 38, 39, 69, 75], Sanger sequencing (n=3) [24, 27, 35], sequence-specific primers (SSP) [64, 76], ELISA [71], single nucleotide polymorphisms (SNP) and short tandem repeat (STR) genotypic analysis [74], and/or other studies [30, 36, 67, 72, 78, 87]. CDG prevalence in the total population was calculated mostly by dividing the total number of reported patients by the number of the total population, whilst disease frequency based on allele frequency was mostly assessed using the Hardy-Weinberg equilibrium. Seven studies presented an estimated prevalence that was determined with 95% confidence interval (CI) [38, 42, 47, 68, 71, 72, 88]. Most of the studies covered data regarding CDG patients in Europe, as most reported patients had a European origin.
Summary of demographic and clinical characteristics
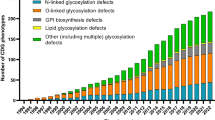
Table 1 summarises the total number of CDG patients in the included studies. A total of 3057 CDG patients were considered. Additionally, a comparison between the reported CDG patients and a systematic survey of literature carried out by Orphanet in January 2022 is presented [89]. In Fig. 1S, the data presented in Table 1 is organised in a pie-chart format.
Figure 3 shows the number of reported CDG patients in different countries by continent. Some case reports did not report the patients’ nationality but their ethnicity. Europe was the continent with the highest number of reported CDG patients (n=618), followed by Asia (n=416), America (n=243, approximately 190 from the USA), and Africa (n=22). Additionally, two XYLT2-CDG Australian patients [167] were considered as well. Japan had the highest number of reported patients, with approximately 215 (including 207 FKTN-CDG patients), followed by the USA (n=188), Italy (n=110), France (n=103), Spain (n=97), and Saudi Arabia (n=68).

Number of reported CDG patients by continent until October 2021. The patients are divided by country and ethnic groups. The USA axis includes patients of different ethnicities. Delgado et al. [48] did a cohort study on Argentinian and Chilean EXT1/EXT2-CDG patients but did not specify the patients’ nationality. Geis et al. [49] reported 35 POMT1-CDG patients from 27 independent families (16 families of Turkish origin; 8 of German origin; and, individually, 1 with Indonesian, Gipsy, and African origins). The authors did not report the nationality of the patient with African parents
The data from 521 patients collected from case reports is summarised in Table 2S. A total of 161 of them had their age at diagnosis available. It ranged from prenatal diagnosis [51] to 47 years [132]. The average age at diagnosis for these patients was 86.8 months (± 7.2 years).
In their patient cohort, Pérez-Cerdá et al. observed that the CDG patients born before 1996 had an average age at diagnosis of 13.4 ± 4.3 years, whilst in the patients born after 1995, it was 2.3 ± 2.4 years [41].
Four studies reported the mean or range of age at diagnosis [25, 29, 47, 93], showing a propensity for young patients. For instance, Francisco et al. [25] reported that 74% CDG patients were diagnosed before 5 years of age, and in 2000, 54% of diagnosed patients were below 10 years of age [168]. Whilst 37.9% of non-PMM2-CDG patients were diagnosed after six, only 17.2% of PMM2-CDG were diagnosed at the same age range [25]. Witters et al. [29] and van de Loo et al. [93] reported the same age range for the diagnosis of PMM2-CDG and other CDG, respectively: between 2 weeks and 21 years.
The mean age at diagnosis of EXT1-CDG was 3 years [47]. Except for X-linked CDG, no difference was observed between the incidence of CDG in male and female patients [43, 54, 157].
The mortality rate in infants with PMM2-CDG was 20% [25, 36]. A similar rate (18%) was observed in Portuguese PMM2-CDG patients aged 1 to 12 months [65].
Fourteen articles besides providing data on incidence also shared information about the clinical manifestations and disease progression [23,24,25,26, 28,29,30, 32, 33, 35, 36, 42, 93, 169]. Six of these articles reported symptoms and disease progression of PMM2-CDG patients.
Epidemiological data
From the overall number of reported CDG patients in Table 1, the five most frequently found CDG are PMM2-CDG (32.7% of the total number of CDG patients included in this revision), followed by FKTN-CDG (6.5%), EXT1/EXT2 (3.7%), ALG6-CDG (3.3%), and PIGA-CDG (2.9%).
The total number of diagnosed European CDG patients might exceed 2500, making the prevalence of CDG in Europe around 0.1–0.5:100,000 [87].
The birth incidence of PMM2-CDG ranges from 5:100,000 to 0.06:100,000 births worldwide [68, 71]. The frequency of PMM2-CDG in patients under 18 years old has been estimated in the Estonian population to be 1: 79,000 [38]. The estimated frequency of PMM2-CDG ranged from 50:1,000,000 [71] in the total Dutch, Flemish, and Danish populations to 0.4:1,000,000 in Poland [68]. The expected carrier frequency of PMM2-CDG in the Saudi Arabian and Turkish was considerably lower compared to European populations (1.4×10−5 and 3.5×10−6, respectively) [42, 69] (Table 2).
MPI-CDG, ALG9-CDG, COG5-CDG, COG6-CDG, and COG8-CDG are extremely rare in European Americans and were predicted to be considerably more prevalent in African Americans [169]. Despite that, a total of thirty-six MPI-CDG patients have been reported.
In the following subsections, we provide more detailed information on seventeen CDG: PMM2-CDG, FKTN-CDG, EXT1/EXT2-CDG, ALG6-CDG, PIGA-CDG, GALNT3-CDG, SLC35A2-CDG, ST3GAL5-CDG, DPAGT1-CDG, GMPPA-CDG, ALG12-CDG, ALG8-CDG, GALNT14-CDG, GALNT5-CDG, MPDUI-CDG, RFT1-CDG, and SRD5A3-CDG.
PMM2-CDG
Twenty-two publications exclusively reported epidemiologic data on PMM2-CDG [31, 35,36,37,38, 42, 46, 71, 74,75,76,77,78, 88, 105, 168, 189,190,191,192,193,194]. PMM2-CDG accounts for most of the published CDG patients, reaching 62% in 2018 [87]. There are at least 1000 reported PMM2-CDG patients, but the number of diagnosed patients is undoubtedly much higher [38].
The most common compound heterozygous pathogenic genotype is P113L/R419H [29], and the most frequent pathogenic variant is R141H, a missense variant [25, 29, 35, 39, 46, 71, 72, 74, 75, 77, 87, 168, 195] (Table 3).
The geographic distribution and frequency of the different pathogenic variants varied greatly in the European countries, except for the frequent R141H variant. The latter was reported from all countries and ranged from 20.5% (in Portugal, 2021, based on allelic frequency) [65] to 86.4% (in Denmark, 2000, estimation based on haplotype analysis) [71] of all pathogenic variants (Table 3). The high incidence of R141H in European individuals might be due to preferential transmission (transmission ratio distortion) and a geographic origin event [74]. Evidence for a transmission ratio distortion was indeed found: there is an increased recurrence risk in PMM2-CDG families ranging from 38% in Scandinavian families to 34% in other European families (2004) [77]. On the other hand, haplotype analysis of patients with this variant and from different geographic origins suggested that R141H is an ancestral variant in the Caucasian population, representing an old and single event derived from linkage disequilibrium with other alleles [71].
The R141H/R141H genotype has never been reported. This can be explained by its lethality, as the enzymatic activity of the recombinant R141H protein is null [77].
A founder effect for the missense F119L variant, the second most common pathogenic variant amongst the Scandinavian population, was found in Southern Scandinavia [72, 168]. Denmark, Sweden, the Netherlands, and Belgium, mentioned in descending order, reported the highest number of patients per number of inhabitants. In Denmark, it accounted for 48% of the disease alleles. Moving southward, this percentage gradually decreased: F119L varied between 17% in the Netherlands and 11% in Germany. This pathogenic variant has not been found in Southern European countries [168]. The preferential transmission was also observed for F119L amongst Scandinavian patients as this was suggested to be related to reproductive advantage at the stage of gametogenesis, fertilisation, implantation, or embryogenesis [77].
D65Y is an Iberian founder missense pathogenic variant [65]. Alongside R141H, it is the most prevalent pathogenic variant found in Portugal and Spain (19.3% and 18% of PMM2-CDG patients, respectively) [65, 74].
Additionally, V44A, a missense pathogenic variant, probably also originated in the Iberian Peninsula, as it has only been reported in Portuguese and Latin American patients [78].
By 2000, the E139K missense variant had been reported in four French patients, and it is likely a founder pathogenic variant of French origin [168].
The Italian population’s second most common pathogenic variant is the L32R missense pathogenic variant (16% of disease alleles). Besides L32R and R141H variants, 14 others were found [168].
The missense pathogenic variant V231M was commonly found amongst Polish and Estonian PMM2-CDG patients (21% and 23%, respectively) [38, 68].
Amongst the Turkish population, V231M was the most prevalent pathogenic variant (2020). The low prevalence of PMM2-CDG in the Turkish population can be explained by the fact that the R141H allele is rare in the Turkish population (3.81×10−4 vs 3.97×10−3 in Public DataBases). The total frequency of likely pathogenic alleles in the Turkish population was 1.91×10−3, and it was considerably lower than allele frequencies reported before in Public DataBases. It should be pointed out that the rate of consanguineous marriages in Turkey is high (23%), increasing the risk for homozygous variants [42].
Most of the pathogenic PMM2-CDG variants have been reported in Europe. This might be explained by its less intensive study in other continents.
FKTN-CDG (FCMD)
FKTN-CDG, also known as Fukuyama congenital muscular dystrophy (FCMD), has a high incidence in Japan (0.7–1.2 per 10,000 births) [60]. It is caused by a founder pathogenic variant in the 3′-UTR of the fukutin (FKTN gene) within the Japanese population [121, 196]. A Japanese national registry reported 207 patients by 2013 [55].
EXT1/EXT2-CDG
EXT1/EXT2-CDG is one of the few dominant CDG. They are also known as hereditary multiple exostoses (HME) or hereditary multiple osteochondromas (HMO); so reports prior to the new nomenclature proposal [154] would not be identified through our set of keywords.
More than 110 patients have been reported [48, 58]. The estimated incidence of EXT1/EXT2-CDG patients ranges between 5.56×10−5 and 2×10−5. The highest estimated incidence has been identified in Pauingassi, a Canadian Indian community (1:77) [180], followed by a closed sub-population of the Chamorros population from the island of Guam (1:1000) [57]. In Europe and North America, the estimated incidence is 1:50 000 (2×10−5) [114]. EXT1 variants account for 56 to 78 % of all EXT1/EXT2-CDG patients, whilst variants in the EXT2 gene account for 20.5 to 44% [58].
ALG6-CDG
Three publications exclusively reported epidemiologic data on ALG6-CDG [67, 197, 198]. Hundred one patients with ALG6-CDG have been reported, making it the second most frequent CDG-I, following PMM2-CDG [87]. The frequency and the prevalence of ALG6-CDG in the global population are not known. Almost all reported ALG6-CDG patients were found in Europe [197]. Some patients were diagnosed in South Africa as descendants from European colonists [198].
Half of the ALG6-CDG patients showed the homozygous A333V missense variant. Twenty different ALG6 pathogenic variants have been described, with the c.998T variant resulting in the A333V substitution making up most of the alleles [64]. Another frequent variant is the missense L453V variant, with an allelic frequency of 0.012 [73].
ALG6 missense variant Y131H occurs at a frequency of 0.021 in the general North American population. Based on the allelic frequency, the birth rate of homozygotes is predicted to be 4.55×10−4. It was suggested that if this homozygous variant causes CDG, it may not be detected by serum TfIEF and thus could be missed [67].
PIGA-CDG
PIGA-CDG, also known as multiple congenital anomalies-hypotonia-seizures syndrome type 2 (MCAHS2), is an X-linked recessive disease [199]; thus, symptoms have only been reported in male patients [43]. As of 2020, 88 patients have been reported [59].
GALNT3-CDG
GALNT3-CDG is also known as familial hyperphosphatemic tumoral calcinosis (HFTC). At least 66 patients from 42 different families have been reported [83, 125, 126].
The incidence is highest in patients of African origin, followed by white patients with Middle Eastern origin. One Chinese GALNT3-CDG patient has been reported [125].
SLC35A2-CDG
According to our literature search, 62 SLC35A2-CDG patients have been reported [54, 153]. It is an X-linked dominant disease, making the phenotype more aggressive in male patients (with a higher mortality rate in males than females) but a higher incidence in females than in males. The frequency and the prevalence of SLC35A2-CDG are not known. Seven per cent of the CDG-II patients have been diagnosed with SLC35A2-CDG [54].
ST3GAL5-CDG
Fifty ST3GAL5-CDG patients have been reported; 38 of them are Amish (the largest Amish populations are found in the states of Pennsylvania, Ohio, and Indiana), due to a founder pathogenic missense variant (c.862C > T, R288X), with a predicted incidence of 1:1200. The reported non-Amish patients have been found inside and outside the USA: 4 African American, 3 Pakistani, 2 French, 2 South Korean, and 1 Iranian [56].
DPAGT1-CDG
Forty-five DPAGT1-CDG patients have been reported and around 46% (13/28) are female (Table 2S). In Ng et al. cohort study, the mortality in infants was 30%, making DPAGT1-CDG more severe than PMM2-CDG, in which 20% of the patients with the most severe symptoms died before the age of 1 [63]. Most of the pathogenic variants associated with DPAGT1-CDG are missense (Table 2S). Pérez-Cerdá et al. have reported eight Spanish patients [41].
GMPPA-CDG
GMPPA-CDG has been identified in 21 patients. The homozygous GMPPA frameshift pathogenic variant L89fs is likely a founder variant originating from the Maya-Mam population (Guatemala, Central America) [132].
Other CDG
In Table 4, we clustered information on other CDG, regarding frequent pathogenic variants. It should be clarified that the pathogenic variants grouped in Table 4 do not represent all the reported pathogenic variants, but their allelic frequency was documented in the articles reviewed in this literature revision.
Discussion
We found 165 papers on epidemiology data in CDG, focusing mainly on incidence, prevalence, and allelic frequency.
The prevalence and carrier frequencies of most CDG are unknown. Few articles provided information on the incidence and prevalence of CDG, as there is only information regarding the prevalence of PMM2-CDG for nine countries (Denmark, Estonia, Flanders (Belgium), France, Netherlands, Poland, Saudi Arabia, Sweden, and Turkey) [38, 42, 68, 71, 72, 88]; EXT1/EXT2-CDG for Bulgaria, Pauingassi (Canadian indigenous people), Chamorros (indigenous people of Guam), the UK, and the USA [47, 57, 70, 114, 180]; CDG-I for Sweden [72]; CDG (non-specified) for Poland, Saudi Arabia, and the USA [68, 69, 188]; and the birth prevalence for FKTN-CDG in Japan [60]. Although 42 cohort studies were identified, none reported patients from sub-Saharan African countries, Russia, and China. As these are huge countries, a high underdiagnosis of CDG is expected.
Excluding Japan, with more than 210 patients, 207 with FKTN-CDG [55], the country with the highest number of reported CDG patients was the USA [188]. This may be due to the existence of research consortiums (such as Frontiers in Congenital Disorders of Glycosylation [200]) and internationally referenced clinics, facilitating both the diagnosis and a closer follow-up of these patients. Besides the size of the country, the USA is ethnically diverse. Therefore, a large portion of the population might originate from regions with a high prevalence of some CDG (either by a founder effect or a high carrier frequency of certain pathogenic variants).
Although some 160 CDG-causing genes associated with about 210 distinct clinically defined phenotypes have been reported [12, 13], we present epidemiological data on only 93 CDG. The number of patients of the remaining CDG is too small or the reported information is too scarce for an epidemiological study [44, 85, 86, 97, 112, 119, 120, 133, 155, 157]. This underlies CDG heterogeneity, patient geographical dispersion, and the likelihood that many patients are still unreported.
Regarding the most frequent CDG, PMM2-CDG, Denmark, Netherlands, and Flanders (Belgium) are the regions with the highest prevalence of PMM2-CDG described so far (1:20,000) [71]. The European country with the highest number of reported PMM2-CDG patients is France (n=96) [40], followed by Spain (n=71) [41], Portugal (n=39) [65], Italy (n=37) [46], Denmark (n=22) [71], the Netherlands (n=19) [71], Poland (n=17) [68], the UK (n=14) [50], Germany (n=12) [71], Belgium (n=9) [71], Estonia (n=5) [38], Ireland (n=2) [71], and Switzerland (n=1) [71].
Note that the number of reported Dutch, Belgium, and Danish patients and the prevalence of PMM2-CDG in those countries were estimated in 1998, and since then, there is no or insufficient update of the actual prevalence.
The prevalence of CDG in Asia is certainly underestimated due to a lack of data. Japan, Saudi Arabia, and Turkey have the most reported CDG patients, with 214, 68, and 54, respectively [33, 42, 55, 60, 69, 121, 122, 126, 151, 166]. The most reported CDG in Turkey is POMT1-CDG (n=18) [49] and PMM2-CDG (n=11) [42]. The expected carrier frequency of PMM2-CDG in the Saudi Arabian and Turkish populations was lower than in European populations. We did not find reported PMM2-CDG patients from the following countries: Bahrain, India, Iran, Iraq, Israel, Kuwait, Lebanon, Pakistan, Palestine, South Korea, UAE, and Vietnam. Additionally, no CDG had a particularly high incidence in these countries. Some Middle Eastern and West Asian countries such as Iran, Iraq, Pakistan, Saudi Arabia, and Turkey have a high incidence of consanguineous marriages [33, 42, 144, 145, 156, 160, 165, 166], increasing the rate of autosomal recessive diseases. Indeed, excluding Japan, these countries reported the highest number of CDG patients in Asia [49, 90, 99, 100, 102, 110, 112, 120, 121, 123, 127, 132, 133, 139, 144, 156, 158,159,160, 166, 167, 189, 192, 196, 197, 201,202,203,204,205].
The South American country with the highest number of reported CDG patients is Argentina [48, 97, 98]. There may be underdiagnosis in other South American countries, but the high influx of European immigrants (and thus, genetic import) in the last century could also have contributed to this observation in Argentina. This also explains also why the R141H PMM2 variant was prevalent amongst these patients [97]. Furthermore, the only Latin American centre specialising in CDG is found in Argentina (https://cemeco.fcm.unc.edu.ar/), which implies a bias towards the diagnosis of Argentinian patients.
ALG6-CDG is considered the second most common CDG [87], but in our review, it fell behind EXT1/EXT2-CDG [48, 58] and FKTN-CDG [55]. Regardless of the explanation, this may well correspond to the reality as EXT-CDG patients are often not considered as a CDG because they are monosymptomatic whilst most CDG are polysymptomatic. The incidence of FKTN-CDG in Japan is 0.7–1.2 per 10,000 births [60]. This high incidence is caused by a founder FKTN pathogenic variant. Traditionally, FKTN-CDG is classified as congenital muscular dystrophy (CMD). However, fukutin deficiency as a CDG surpasses ALG6-CDG as the second most common CDG.
In 2021, Pajusalu et al. [206] performed a statistical analysis to estimate the prevalence of 27 N-linked CDG across different populations. The estimation was based on allele frequencies disclosed in gnomAD (https://gnomad.broadinstitute.org/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). The expected prevalence differed from the one available in previously published literature. Regarding PMM2-CDG, the group reported that the Finnish population has the higher prevalence (1:18,745), followed by Ashkenazi-Jews (1:19,908) and non-Finnish European populations (1:27,465). ALG6-CDG was more prevalent amongst non-Finish Europeans (1:623,512). Amongst East Asian populations, MAN2B2-CDG and FUK-CDG have a high prevalence (1:11,323 and 1:12,248, respectively). ALG1-CDG is more prevalent amongst Ashkenazi-Jews (1:47,656), compared to other populations. The other N-linked CDG had a considerably lower prevalence amongst the different populations.
Concerning the diagnosis and screening of CDG, the most used method for screening congenital disorders of N-linked glycosylation is TfIEF. Since transferrin carries sialylated N-glycans, there is a cathodal shift caused by a partial deficiency of sialic acid in CDG. Normal serum transferrin is mainly constituted of tetrasialotransferrin with negligible small amounts of mono-, di-, tri-, penta-, and hexasialotransferrins. Abnormal TfIEF results, caused by the cathodal shift, can be divided into types 1 and 2—in type 1, there is an increase of both disialotransferrin and asialotransferrin and a decrease of tetrasialotransferrin, whilst type 2 is also characterised by an increase of triasialotransferrin or monosialotransferrin [14, 207].
Although this technique is one of the most applied whilst screening for CDG, a normal TfIEF result does not exclude CDG, as congenital disorders of O-glycosylation, (GPI)-anchor synthesis, and other glycosylation pathways are overlooked. In addition, other inborn errors of metabolism, like hereditary fructose intolerance (HFI) [208] and classic galactosemia [209], have also been associated with abnormal TfIEF results.
The identification of CDG and other RDs through genome sequencing aids in understanding the subjacent pathophysiologies, which in turn helps to target therapies [210].
NGS methods like WGS and WES applied in large population studies are an excellent tool for estimating a more real prevalence of CDG and other RDs. These methods remove the necessity to prioritise candidate genes for sequencing. Genome-wide association studies using data from thousands of individuals, Biobanks, and population samples aid to identify genetic variations associated with a particular disease.
Currently, WES is the major tool of CDG diagnosis. It is cost-effective, as it has a higher yield of relevant gene variants compared to WGS—although the human exome represents less than 2% of the genome, approximately 85% of known disease-related pathogenic variants occur in exons [211].
Limitations and strengths
The primary limitations faced whilst conducting our review were the diversity of the data, which was derived from (1) CDG multiplicity and heterogeneity; (2) the differences between studies (study population(s); information sources; study design; etc.) and the duplication of patients in different studies; and (3) a lack of uniform, accepted standards for the collection and organisation of CDG epidemiological data. Additionally, we did not identify studies from Africa, South-Eastern Asia, and Russia. That gap is also reflected in the fact that we only identified epidemiological data on 93 CDG of the approximately 170 types currently reported. Importantly, the number of new CDG has rapidly grown during recent years, from 128 in 2017 to approximately 170 in 2021 [12], hindering the follow-up of the real number of CDG patients and the actual prevalence of the diseases.
Also, published data inconsistencies were an important limitation. The inconsistent number of reported CDG patients in scientific publications and reports [89] underlines a low level of consistency between publications and registries/health institutes and agencies. This leads to a lack of clarity as for real numbers, impacting both data quality and quantity. In an attempt to centralise information and make it broadly available, we will create a virtual toolkit in the worldcdg.org platform, consisting in a web section dedicated to epidemiology, an ePoster about CDG epidemiology, and the recording of a talk on the subject presented at the 5th World CDG Conference.
Moreover, our search method was a limitation in our work, as it failed to extract a high number of case reports. Indeed, most of the additional included papers by authors’ referrals were case reports. Hence, a search method refinement, namely by improving keywords, should be applied in future studies. Still, our search methodology did not only allow us to identify 3057 CDG patients, but it also brought methodological advances as it promoted the automation of the search process. A literature research using a customised Python script is more time efficient since it retrieves and analyses information faster.
This study solely relied on data from the medical literature. No complementary data sources (such as patient registries and other databases) were used, which limited our analysis. However, the lack of centralised and open CDG data repositories also hindered the possibility of accessing and incorporating such information in this study.
Our literature review provides an overview of what is known regarding the epidemiology of CDG. It highlights the urgency of collecting and grouping epidemiological data with standardised parameters, to create a more realistic estimate of the real number of CDG patients.
The social and economic burden of CDG and other RDs ends up being overlooked, due to the limited evidence on the prevalence of these diseases. Advances in the epidemiological study of CDG and other RDs help to guide healthcare decision-makers in prioritising healthcare policies and clinical management guidelines related to them.
Our review brings awareness on the necessity of creating patients’ registries and databases of high quality to support the target of patients’ populations for clinical trials and the development of orphan drugs.
Conclusion
This is the first literature review compiling published data on the global epidemiology of CDG. This study identified several challenges and gaps regarding CDG epidemiology, with data scarcity, inconsistency, and CDG heterogeneity amongst them. Also, this study red flagged a remarkable lack of uniformity and accepted standards for the collection and organisation of CDG epidemiological data. Higher-quality epidemiological data and more realistic estimates of the actual number of people living with CDG are important in order to target resources for CDG research and drug development, to manage and support public health decision-making. To achieve this, future work should explore this issue further by assembling epidemiological data from databases (ClinVar; LOVD; etc.), patient registries, medical records biobanks, and international collaborations with patient and professional networks.
Availability of data and materials
The data generated and analysed in this study are included in the main text and supplementary information file.
Abbreviations
- AAMR:
-
Alacrima, achalasia, and mental retardation syndrome
- CDG:
-
Congenital disorders of glycosylation
- CDG-I:
-
CDG type I
- CDG-II:
-
CDG type II
- CI:
-
Confidence interval
- CMD:
-
Congenital muscular dystrophy
- EIEE36:
-
Early infantile epileptic encephalopathy-36
- EIEE55:
-
Early infantile epileptic encephalopathy-55
- EMA:
-
European Medicines Agency
- FCMD:
-
Fukuyama congenital muscular dystrophy
- FDA:
-
Food and Drug Administration
- GPI:
-
Glycosylphosphatidylinositol
- HFTC:
-
Familial hyperphosphatemic tumoral calcinosis
- HME:
-
Hereditary multiple exostoses
- HMO:
-
Hereditary multiple osteochondromas
- MCAHS2:
-
Multiple congenital anomalies-hypotonia-seizures syndrome 2
- MCAHS3:
-
Multiple congenital anomalies-hypotonia-seizures syndrome 3
- NGS:
-
Next-generation sequencing
- PCR:
-
Polymerase chain reaction
- RD:
-
Rare disease
- SNP:
-
Single nucleotide polymorphism(s)
- SSP:
-
Sequence-specific primers
- STR:
-
Short tandem repeat
- TfIEF:
-
Transferrin isoelectric focusing
- UK:
-
United Kingdom
- UAE:
-
United Arab Emirates
- USA:
-
United States of America
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
References
Richter T, Nestler-Parr S, Babela R, Khan ZM, Tesoro T, Molsen E, et al. Rare disease terminology and definitions—a systematic global review: report of the ISPOR Rare Disease Special Interest Group. Value Health. 2015;18(6):906–14.
Rare Diseases Act of 2002, Pub. L. No. 107-280, 116 Stat. 1988. https://www.congress.gov/107/plaws/publ280/PLAW-107publ280.pdf. 2002.
Baldovino S, Moliner AM, Taruscio D, Daina E, Roccatello D. Rare diseases in Europe: from a wide to a local perspective. Isr Med Assoc J. 2016;18(6):359–63.
Operational definition of rare diseases - Rare Diseases International. Available from: https://www.rarediseasesinternational.org/description-for-rd/. Cited 2021 Dec 20.
Ferreira CR. The burden of rare diseases. Am J Med Genet A. 2019;179(6):885–92.
Haendel M, Vasilevsky N, Unni D, Bologa C, Harris N, Rehm H, et al. How many rare diseases are there? Nat Rev Drug Discov. 2020;19(2):77–8.
Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020;28:165–73.
What is a rare disease? - Rare Disease UK. Available from: https://www.raredisease.org.uk/what-is-a-rare-disease/. Cited 2021 Dec 20.
Baekelandt E, de Haan J, de Vrueh R, Schuttelaar. Priority medicines for Europe and the world: 2013 update report; 2013. p. 148–51.
Kaufmann P, Pariser AR, Austin C. From scientific discovery to treatments for rare diseases – the view from the National Center for Advancing Translational Sciences – Office of Rare Diseases Research. Orphanet J Rare Dis. 2018;13(1):196.
Jaeken J, Vanderschueren-Lodeweyckx M, Casaer P, Snoeck L, Corbeel L, Eggermont E, et al. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG-deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome?: 90. Pediatr Res. 1980;14(2):179.
Blăniţă D, Boiciuc C, Morava E, Uşurelu N. Congenital disorders of glycosylation: a booming chapter in pediatric genetics. In: International congress of geneticists and breeders from the Republic of Moldova. 11th. ed. Chişinău: Institute of Genetics, Physiology and Plant Protection; 2021. p. 41.
Francisco R, Marques-da-Silva D, Brasil S, Pascoal C, Dos Reis Ferreira V, Morava E, et al. The challenge of CDG diagnosis. Mol Genet Metab. 2019;126(1):1–5.
Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–8.
Maeda Y, Ashida H, Kinoshita TBT-M in E. CHO glycosylation mutants: GPI anchor. In: Glycomics, vol. 416. Cambridge: Academic Press; 2006. p. 182–205. https://doi.org/10.1016/S0076-6879(06)16012-7.
Russo D, Parashuraman S, D’Angelo G. Glycosphingolipid–protein interaction in signal transduction. Int J Mol Sci. 2016;17(10):1732.
Freeze HH. Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med. 2007;7(4):389–96.
Department Health UO, Services H, for Disease Control C. Principles of epidemiology in public health practice, third edition: an introduction. 2006.
Developing products for rare diseases & conditions | FDA. Available from: https://www.fda.gov/industry/developing-products-rare-diseases-conditions. Cited 2021 Dec 20.
Medicines Agency E. Committee for Orphan Medicinal Products (COMP) points to consider on the estimation and reporting on the prevalence of a condition for the purpose of orphan designation COMP guideline. 2019.
Waskom ML. seaborn: statistical data visualization. J Open Source Softw. 2021;6(60):3021.
Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9(3):90–5.
Linssen M, Mohamed M, Wevers RA, Lefeber DJ, Morava E. Thrombotic complications in patients with PMM2-CDG. Mol Genet Metab. 2013;109(1):107–11.
Fiumara A, Barone R, Del Campo G, Striano P, Jaeken J. Electroclinical features of early-onset epileptic encephalopathies in congenital disorders of glycosylation (CDGs). In: Morava E, Baumgartner M, Patterson M, Rahman S, Zschocke J, Peters V, editors. JIMD reports, vol. 27. Berlin: Springer; 2016. p. 93–9. https://doi.org/10.1007/8904_2015_497.
Francisco R, Pascoal C, Marques-da-Silva D, Brasil S, Pimentel-Santos FM, Altassan R, et al. New insights into immunological involvement in congenital disorders of glycosylation (CDG) from a people-centric approach. J Clin Med. 2020;9(7):2092.
Marques-da-Silva D, Francisco R, dos Reis Ferreira V, Forbat L, Lagoa R, Videira PA, et al. An electronic questionnaire for liver assessment in congenital disorders of glycosylation (LeQCDG): a patient-centered study. In: Morava E, Baumgartner M, Patterson M, Rahman S, Zschocke J, Peters V, editors. JIMD reports, vol. 44. Berlin: Springer; 2019. p. 55–64. https://doi.org/10.1007/8904_2018_121.
Hall BD, Stevenson RE, Jones JR. Fatal hyperkeratosis syndrome in four siblings due to dolichol kinase deficiency. Am J Med Genet Part A. 2020;182(6):1421–5.
Björklund JEM, Stibler H, Kristiansson B, Johansson SGO, Magnusson CGM. Immunoglobulin levels in patients with carbohydrate-deficient glycoprotein syndrome type I. Int Arch Allergy Immunol. 1997;114(2):116–9.
Witters P, Honzik T, Bauchart E, Altassan R, Pascreau T, Bruneel A, et al. Long-term follow-up in PMM2-CDG: are we ready to start treatment trials? Genet Med. 2019;21(5):1181–8.
Damen G, de Klerk H, Huijmans J, den Hollander J, Sinaasappel M. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr. 2004;38(3):282–7.
Jamroz E, Adamek D, Paprocka J, Adamowicz M, Marszał E, Wevers RA. CDG type Ia and congenital cytomegalovirus infection: two coexisting conditions. J Child Neurol. 2009;24(1):13–8.
Brucker WJ, Croteau SE, Prensner JR, Cullion K, Heeney MM, Lo J, et al. An emerging role for endothelial barrier support therapy for congenital disorders of glycosylation. J Inherit Metab Dis. 2020;43(4):880–90.
Incecik F, Herguner O, Mungan N. Clinical features and molecular genetics of autosomal recessive ataxia in the Turkish population. J Pediatr Neurosci. 2020;15(2):86.
Haijes HA, Jaeken J, Foulquier F, Van Hasselt PM. Hypothesis: lobe A (COG1-4)-CDG causes a more severe phenotype than lobe B (COG5-8)-CDG. J Med Genet. 2018;55(2):137–42.
Chong M, Yoon G, Susan-Resiga D, Chamberland A, Cheillan D, Paré G, et al. Hypolipidaemia among patients with PMM2-CDG is associated with low circulating PCSK9 levels: a case report followed by observational and experimental studies. J Med Genet. 2020;57(1):11–7.
Monin M-LL, Mignot C, De Lonlay P, Héron B, Masurel A, Mathieu-Dramard M, et al. 29 French adult patients with PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J Rare Dis. 2014;9(1):207.
Pérez-Dueñas B, García-Cazorla A, Pineda M, Poo P, Campistol J, Cusí V, et al. Long-term evolution of eight Spanish patients with CDG type Ia: typical and atypical manifestations. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2009;13(5):444–51.
Vals MA, Pajusalu S, Kals M, Mägi R, Õunap K. The prevalence of PMM2-CDG in Estonia based on population carrier frequencies and diagnosed patients. In: Morava E, Baumgartner M, Patterson M, Rahman S, Zschocke J, Peters, editors. JIMD reports, vol. 39. Berlin: Springer; 2018. p. 13–7. https://doi.org/10.1007/8904_2017_41.
Al Teneiji A, Bruun TUJ, Sidky S, Cordeiro D, Cohn RD, Mendoza-Londono R, et al. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol Genet Metab. 2017;120(3):235–42.
Schiff M, Roda C, Monin M-L, Arion A, Barth M, Bednarek N, et al. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J Med Genet. 2017;54(12):843–51.
Pérez-Cerdá C, Girós ML, Serrano M, Ecay MJ, Gort L, Pérez Dueñas B, et al. A population-based study on congenital disorders of protein N- and combined with O-glycosylation experience in clinical and genetic diagnosis. J Pediatr. 2017;183:170–177.e1.
Yıldız Y, Arslan M, Çelik G, Kasapkara ÇS, Ceylaner S, Dursun A, et al. Genotypes and estimated prevalence of phosphomannomutase 2 deficiency in Turkey differ significantly from those in Europe. Am J Med Genet Part A. 2020;182(4):705–12.
Bayat A, Knaus A, Pendziwiat M, Afenjar A, Barakat TS, Bosch F, et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia. 2020;61(6):1142–55.
Funke S, Gardeitchik T, Kouwenberg D, Mohamed M, Wortmann SB, Korsch E, et al. Perinatal and early infantile symptoms in congenital disorders of glycosylation. Am J Med Genet A. 2013;161A(3):578–84.
Marín-Valencia I, Vilaseca MA, Thió M, García-Cazorla A, Artuch R, Campistol J. Assessment of the perimortem protocol in neonates for the diagnosis of inborn errors of metabolism. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2010;14(2):125–30.
Barone R, Carrozzi M, Parini R, Battini R, Martinelli D, Elia M, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol. 2015;262(1):154–64.
Stancheva-Ivanova MK, Wuyts W, Van Hul E, Radeva BI, Vazharova RV, Sokolov TP, et al. Clinical and molecular studies of EXT1/EXT2 in Bulgaria. J Inherit Metab Dis. 2011;34(4):917–21.
Delgado MA, Martinez-Domenech G, Sarrión P, Urreizti R, Zecchini L, Robledo HH, et al. A broad spectrum of genomic changes in Latin American patients with EXT1/EXT2-CDG. Sci Rep. 2014;4(1):6407.
Geis T, Rödl T, Topaloğlu H, Balci-Hayta B, Hinreiner S, Müller-Felber W, et al. Clinical long-time course, novel mutations and genotype-phenotype correlation in a cohort of 27 families with POMT1-related disorders. Orphanet J Rare Dis. 2019;14(1):179.
Imtiaz F, Worthington V, Champion M, Beesley C, Charlwood J, Clayton P, et al. Genotypes and phenotypes of patients in the UK with carbohydrate-deficient glycoprotein syndrome type 1. J Inherit Metab Dis. 2000;23(2):162–74.
den Hollander B, Rasing A, Post MA, Klein WM, Oud MM, Brands MM, et al. NANS-CDG: delineation of the genetic, biochemical, and clinical spectrum. Front Neurol. 2021;12:668640.
Čechová A, Honzík T, Edmondson AC, Ficicioglu C, Serrano M, Barone R, et al. Should patients with phosphomannomutase 2-CDG (PMM2-CDG) be screened for adrenal insufficiency? Mol Genet Metab. 2021;133(4):397–9.
Stevens E, Carss KJ, Cirak S, Foley AR, Torelli S, Willer T, et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am J Hum Genet. 2013;92(3):354–65.
Witters P, Tahata S, Barone R, Õunap K, Salvarinova R, Grønborg S, et al. Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genet Med. 2020;22(6):1102–7.
Ishigaki K, Ihara C, Nakamura H, Mori-Yoshimura M, Maruo K, Taniguchi-Ikeda M, et al. National registry of patients with Fukuyama congenital muscular dystrophy in Japan. Neuromuscul Disord. 2018;28(10):885–93.
Bowser LE, Young M, Wenger OK, Ammous Z, Brigatti KW, Carson VJ, et al. Recessive GM3 synthase deficiency: natural history, biochemistry, and therapeutic frontier. Mol Genet Metab. 2019;126(4):475–88.
Krooth RS, Macklin MT, Hilbish TF. Diaphysial aclasis (multiple exostoses) on Guam. Am J Hum Genet. 1961;13(3):340–7.
Ciavarella M, Coco M, Baorda F, Stanziale P, Chetta M, Bisceglia L, et al. 20 novel point mutations and one large deletion in EXT1 and EXT2 genes: report of diagnostic screening in a large Italian cohort of patients affected by hereditary multiple exostosis. Gene. 2013;515(2):339–48.
Bayat A, Kløvgaard M, Johannesen KM, Barakat TS, Kievit A, Montomoli M, et al. Deciphering the premature mortality in PIGA-CDG - an untold story. Epilepsy Res. 2021;170:106530.
Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394(6691):388–92.
Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130(Pt 10):2725–35.
Morlino S, Nardella G, Castellana S, Micale L, Copetti M, Fusco C, et al. Review of clinical and molecular variability in autosomal recessive cutis laxa 2A. Am J Med Genet Part A. 2021;185(3):955–65.
Ng BG, Underhill HR, Palm L, Bengtson P, Rozet J-M, Gerber S, et al. DPAGT1 deficiency with encephalopathy (DPAGT1-CDG): clinical and genetic description of 11 new patients. JIMD Rep. 2019;44:85–92.
Goreta SS, Dabelic S, Dumic J. Employment of single-strand conformation polymorphism analysis in screening for α-1,3 glucosyltransferase gene mutation A333V in Croatian population. J Clin Lab Anal. 2011;25(2):65–70.
Quelhas D, Martins E, Azevedo L, Bandeira A, Diogo L, Garcia P, et al. Congenital disorders of glycosylation in Portugal—two decades of experience. J Pediatr. 2021;231:148–56. https://doi.org/10.1016/j.jpeds.2020.12.026.
de Magalhães APPS, Burin MG, de Souza CFM, de Bitencourt FH, Sebastião FM, Silva TO, et al. Transferrin isoelectric focusing for the investigation of congenital disorders of glycosylation: analysis of a ten-year experience in a Brazilian center. J Pediatr (Rio J). 2020;96(6):710–6.
Westphal V, Xiao M, Kwok P-Y, Freeze HH. Identification of a frequent variant in ALG6, the cause of congenital disorder of glycosylation-Ic. Hum Mutat. 2003;22(5):420–1.
Lipiński P, Bogdańska A, Tylki-Szymańska A. Congenital disorders of glycosylation: prevalence, incidence and mutational spectrum in the Polish population. Mol Genet Metab Rep. 2021;27:100726.
Alsubhi S, Alhashem A, Faqeih E, Alfadhel M, Alfaifi A, Altuwaijri W, et al. Congenital disorders of glycosylation: the Saudi experience. Am J Med Genet Part A. 2017;173(10):2614–21.
Hennekam RC. Hereditary multiple exostoses. J Med Genet. 1991;28(4):262–6.
Schollen E, Kjaergaard S, Legius E, Schwartz M, Matthijs G. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). Eur J Hum Genet. 2000;8(5):367–71.
Bjursell C, Wahlström J, Berg K, Stibler H, Kristiansson B, Matthijs G, et al. Detailed mapping of the phosphomannomutase 2 (PMM2) gene and mutation detection enable improved analysis for Scandinavian CDG type I families. Eur J Hum Genet. 1998;6(6):603–11.
Citro V, Cimmaruta C, Monticelli M, Riccio G, Hay Mele B, Cubellis MV, et al. The analysis of variants in the general population reveals that PMM2 is extremely tolerant to missense mutations and that diagnosis of PMM2-CDG can benefit from the identification of modifiers. Int J Mol Sci. 2018;19(8):2218.
Quelhas D, Quental R, Vilarinho L, Amorim A, Azevedo L. Congenital disorder of glycosylation type Ia: searching for the origin of common mutations in PMM2. Ann Hum Genet. 2007;71(Pt 3):348–53.
Hansen L, Lind-Thomsen A, Joshi HJ, Pedersen NB, Have CT, Kong Y, et al. A glycogene mutation map for discovery of diseases of glycosylation. Glycobiology. 2015;25(2):211–24.
Bjursell C, Erlandson A, Nordling M, Nilsson S, Wahlström J, Stibler H, et al. PMM2 mutation spectrum, including 10 novel mutations, in a large CDG type 1A family material with a focus on Scandinavian families. Hum Mutat. 2000;16(5):395–400.
Schollen E, Kjaergaard S, Martinsson T, Vuillaumier-Barrot S, Dunoe M, Keldermans L, et al. Increased recurrence risk in congenital disorders of glycosylation type Ia (CDG-Ia) due to a transmission ratio distortion. J Med Genet. 2004;41(11):877–80.
Briones P, Vilaseca MA, Schollen E, Ferrer I, Maties M, Busquets C, et al. Biochemical and molecular studies in 26 Spanish patients with congenital disorder of glycosylation type Ia. J Inherit Metab Dis. 2002;25(8):635–46.
Molinari F, Foulquier F, Tarpey PS, Morelle W, Boissel S, Teague J, et al. Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am J Hum Genet. 2008;82(5):1150–7.
Ben Ayed I, Ouarda W, Frikha F, Kammoun F, Souissi A, Ben Said M, et al. SRD5A3-CDG: 3D structure modeling, clinical spectrum, and computer-based dysmorphic facial recognition. Am J Med Genet A. 2021;185(4):1081–90.
Lebredonchel E, Houdou M, Potelle S, de Bettignies G, Schulz C, Krzewinski Recchi M-A, et al. Dissection of TMEM165 function in Golgi glycosylation and its Mn(2+) sensitivity. Biochimie. 2019;165:123–30.
Jaeken J, Lefeber DJ, Matthijs G. Clinical Utility Gene Card for: PGM3 defective congenital disorder of glycosylation. Eur J Hum Genet. 2019;27(11):1757–60.
Jaeken J, Lefeber DJ, Matthijs G. Clinical Utility Gene Card for: GALNT3 defective congenital disorder of glycosylation. Eur J Hum Genet. 2018;26(8):1230–3.
Jaeken J, Lefeber DJ, Matthijs G. Clinical utility gene card for: Peters plus syndrome. Eur J Hum Genet. 2016;24(8). https://doi.org/10.1038/ejhg.2016.32.
D’Souza Z, Taher FS, Lupashin VV. Golgi inCOGnito: from vesicle tethering to human disease. Biochim Biophys acta Gen Subj. 2020;1864(11):129694.
Feichtinger RG, Hüllen A, Koller A, Kotzot D, Grote V, Rapp E, et al. A spoonful of L-fucose—an efficient therapy for GFUS-CDG, a new glycosylation disorder. EMBO Mol Med. 2021;13(9):e14332.
Péanne R, de Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. 2018;61:643–63 Elsevier Masson SAS.
Giurgea I, Michel A, Le Merrer M, Seta N, de Lonlay P. Underdiagnosis of mild congenital disorders of glycosylation type Ia. Pediatr Neurol. 2005;32(2):121–3.
Prevalence and incidence of rare diseases: bibliographic data. Available from: www.orpha.net. Cited 2021 May 9.
Sarkhail P. SSIEM 2019: Annual Symposium of the Society for the Study of Inborn Errors of Metabolism, Rotterdam, The Netherlands, 3-6 September 2019. J Inherit Metab Dis. 2019;42(Suppl 1):1–479.
Haanpää MK, Ng BG, Gallant NM, Singh KE, Brown C, Kimonis V, et al. ALG11-CDG syndrome: expanding the phenotype. Am J Med Genet A. 2019;179(3):498–502.
Ziburová J, Nemčovič M, Šesták S, Bellová J, Pakanová Z, Siváková B, et al. A novel homozygous mutation in the human ALG12 gene results in an aberrant profile of oligomannose N-glycans in patient’s serum. Am J Med Genet A. 2021;185(11):3494–501.
van de Loo KFE, van Dongen L, Mohamed M, Gardeitchik T, Kouwenberg TW, Wortmann SB, et al. Socio-emotional problems in children with CDG. In: Zschocke J, Gibson K, Brown G, Morava E, Peters V, editors. JIMD reports, vol. 11. Berlin: Springer; 2013. p. 139–48. https://doi.org/10.1007/8904_2013_233.
Paprocka J, Jezela-Stanek A, Boguszewicz Ł, Sokół M, Lipiński P, Jamroz E, et al. The first metabolome analysis in children with epilepsy and ALG13-CDG resulting from c.320A>G variant. Children. 2021;8(3):251.
Schorling DC, Rost S, Lefeber DJ, Brady L, Müller CR, Korinthenberg R, et al. Early and lethal neurodegeneration with myasthenic and myopathic features: a new ALG14-CDG. Neurology. 2017;89(7):657–64.
González-Domínguez CA, Fiesco-Roa MO, Gómez-Carmona S, Kleinert-Altamirano API, He M, Daniel EJP, et al. ALG1-CDG caused by non-functional alternative splicing involving a novel pathogenic complex allele. Front Genet. 2021;12:1579.
Asteggiano CG, Papazoglu M, Bistué Millón MB, Peralta MF, Azar NB, Spécola NS, et al. Ten years of screening for congenital disorders of glycosylation in Argentina: case studies and pitfalls. Pediatr Res. 2018;84(6):837–41.
Papazoglu GM, Cubilla M, Pereyra M, de Kremer RD, Pérez B, Sturiale L, et al. Mass spectrometry glycophenotype characterization of ALG2-CDG in Argentinean patients with a new genetic variant in homozygosis. Glycoconj J. 2021;38(2):191–200.
Bian Y, Qiao C, Zheng SG, Qiu H, Li H, Zhang ZT, et al. ALG3-CDG: lethal phenotype and novel variants in Chinese siblings. J Hum Genet. 2020;65(12):1129–34.
Farolfi M, Cechova A, Ondruskova N, Zidkova J, Kousal B, Hansikova H, et al. ALG3-CDG: a patient with novel variants and review of the genetic and ophthalmic findings. BMC Ophthalmol. 2021;21(1):249.
Paketci C, Edem P, Hiz S, Sonmezler E, Soydemir D, Sarikaya Uzan G, et al. Successful treatment of intractable epilepsy with ketogenic diet therapy in twins with ALG3-CDG. Brain Dev. 2020;42:539–45.
Davis K, Webster D, Smith C, Jackson S, Sinasac D, Seargeant L, et al. ALG9-CDG: new clinical case and review of the literature. Mol Genet Metab Rep. 2017;13:55–63.
Lipiński P, Rokicki D, Bogdańska A, Lesiak J, Lefeber DJ, Tylki-Szymańska A. ATP6AP1-CDG: follow-up and female phenotype. JIMD Rep. 2020;53(1):80–2.
Vogt G, El Choubassi N, Herczegfalvi Á, Kölbel H, Lekaj A, Schara U, et al. Expanding the clinical and molecular spectrum of ATP6V1A related metabolic cutis laxa. J Inherit Metab Dis. 2021;44(4):972–86.
Vaes L, Tiller GE, Pérez B, Boyer SW, Berry SA, Sarafoglou K, et al. PMM2-CDG caused by uniparental disomy: case report and literature review. JIMD Rep. 2020;54:16–21.
Woods AG, Woods CW, Snow TM. Congenital disorders of glycosylation. Adv Neonatal Care Off J Natl Assoc Neonatal Nurses. 2012;12(2):90–5.
Lorenz D, Kress W, Zaum A-K, Speer CP, Hebestreit H. Report of two siblings with spondylodysplastic Ehlers-Danlos syndrome and B4GALT7 deficiency. BMC Pediatr. 2021;21(1):293.
Girard M, Poujois A, Fabre M, Lacaille F, Debray D, Rio M, et al. CCDC115-CDG: a new rare and misleading inherited cause of liver disease. Mol Genet Metab. 2018;124(3):228–35.
Salazar M, Miyake N, Silva S, Solar B, Papazoglu GM, Asteggiano CG, et al. COG1-congenital disorders of glycosylation: milder presentation and review. Clin Genet. 2021;100(3):318–23.
Wang X, Han L, Wang X-Y, Wang J-H, Li X-M, Jin C-H, et al. Identification of two novel mutations in COG5 causing congenital disorder of glycosylation. Front Genet. 2020;11:168.
Komlosi K, Gläser S, Kopp J, Hotz A, Alter S, Zimmer A, et al. Neonatal presentation of COG6-CDG with prominent skin phenotype. JIMD Rep. 2020;55(1):51–8.
Pi S, Gong J, Xiao W, Xiao B, Mao X, Long H. The second DDOST-CDG patient with lactose intolerance, developmental delay, and situs inversus totalis. J Hum Genet. 2022;67:103–6. https://doi.org/10.1038/s10038-021-00974-2.
Sabry S, Vuillaumier-Barrot S, Mintet E, Fasseu M, Valayannopoulos V, Héron D, et al. A case of fatal type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J Rare Dis. 2016;11(1):84.
Schmale GA, Conrad EU 3rd, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Am. 1994;76(7):986–92.
Paprocka J, Jezela-Stanek A, Tylki-Szymańska A, Grunewald S. Congenital disorders of glycosylation from a neurological perspective. Brain Sci. 2021;11(1):88.
Bursle C, Brown D, Cardinal J, Connor F, Calvert S, Coman D. DMP1-CDG (CDG1e) with significant gastrointestinal manifestations; phenotype and genotype expansion. JIMD Rep. 2017;34:27–32.
van Tol W, Michelakakis H, Georgiadou E, van den Bergh P, Moraitou M, Papadimas GK, et al. Toward understanding tissue-specific symptoms in dolichol-phosphate-mannose synthesis disorders; insight from DPM3-CDG. J Inherit Metab Dis. 2019;42(5):984–92.
Fu J, Ma M, Song J, Pang M, Yang L, Li G, et al. Novel mutations in DPM3 cause dystroglycanopathy with central nervous system involvement. Clin Genet. 2019;96(6):590–1.
Hüllen A, Falkenstein K, Weigel C, Huidekoper H, Naumann-Bartsch N, Spenger J, et al. Congenital disorders of glycosylation with defective fucosylation. J Inherit Metab Dis. 2021;44(6):1441–52.
Chiara Manzini M, Gleason D, Chang BS, Sean Hill R, Barry BJ, Partlow JN, et al. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum Mutat. 2008;29(11):E231–41.
Puckett RL, Moore SA, Winder TL, Willer T, Romansky SG, Covault KK, et al. Further evidence of Fukutin mutations as a cause of childhood onset limb-girdle muscular dystrophy without mental retardation. Neuromuscul Disord. 2009;19(5):352–6.
Chung W, Winder TL, LeDuc CA, Simpson LL, Millar WS, Dungan J, et al. Founder Fukutin mutation causes Walker–Warburg syndrome in four Ashkenazi Jewish families. Prenat Diagn. 2009;29(6):560–9.
Murakami T, Hayashi YK, Noguchi S, Ogawa M, Nonaka I, Tanabe Y, et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol. 2006;60(5):597–602.
Zilmer M, Edmondson AC, Khetarpal SA, Alesi V, Zaki MS, Rostasy K, et al. Novel congenital disorder of O-linked glycosylation caused by GALNT2 loss of function. Brain. 2020;143(4):1114–26.
Sun L, Zhao L, Du L, Zhang P, Zhang M, Li M, et al. Identification of two novel mutations in the GALNT3 gene in a Chinese family with hyperphosphatemic familial tumoral calcinosis. Bone Res. 2016;4(1):1–6.
Kışla Ekinci RM, Gürbüz F, Balcı S, Bişgin A, Taştan M, Yüksel B, et al. Hyperphosphatemic familial tumoral calcinosis in two siblings with a novel mutation in GALNT3 gene: experience from Southern Turkey. J Clin Res Pediatr Endocrinol. 2019;11(1):94–9.
Huh S-Y, Kim H-S, Jang H-J, Park Y-E, Kim D-S. Limb-girdle myasthenia with tubular aggregates associated with novel GFPT1 mutations. Muscle Nerve. 2012;46(4):600–4.
Maselli RA, Arredondo J, Nguyen J, Lara M, Ng F, Ngo M, et al. Exome sequencing detection of two untranslated GFPT1 mutations in a family with limb-girdle myasthenia. Clin Genet. 2014;85(2):166–71.
Finsterer J. Congenital myasthenic syndromes. Orphanet J Rare Dis. 2019;14(1):57.
Raphael AR, Couthouis J, Sakamuri S, Siskind C, Vogel H, Day JW, et al. Congenital muscular dystrophy and generalized epilepsy caused by GMPPB mutations. Brain Res. 2014;1575:66–71.
Bharucha-Goebel DX, Neil E, Donkervoort S, Dastgir J, Wiggs E, Winder TL, et al. Intrafamilial variability in GMPPB-associated dystroglycanopathy: broadening of the phenotype. Neurology. 2015;84(14):1495–7.
Diaz J, Kane TD, Leon E. Evidence of GMPPA founder mutation in indigenous Guatemalan population associated with alacrima, achalasia, and mental retardation syndrome. Am J Med Genet A. 2020;182(3):425–30.
Sparrow DB, Chapman G, Wouters MA, Whittock NV, Ellard S, Fatkin D, et al. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet. 2006;78(1):28–37.
Blommaert E, Péanne R, Cherepanova NA, Rymen D, Staels F, Jaeken J, et al. Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype. Proc Natl Acad Sci. 2019;116(20):9865 LP–9870.
Okamoto N, Ohto T, Enokizono T, Wada Y, Kohmoto T, Imoto I, et al. Siblings with MAN1B1-CDG showing novel biochemical profiles. Cells. 2021;10(11):3117. https://doi.org/10.3390/cells10113117.
Kemme L, Grüneberg M, Reunert J, Rust S, Park J, Westermann C, et al. Translational balancing questioned: unaltered glycosylation during disulfiram treatment in mannosyl-oligosaccharide alpha-1,2-mannnosidase-congenital disorders of glycosylation (MAN1B1-CDG). JIMD Rep. 2021;60(1):42–55.
Sakhi S, Cholet S, Wehbi S, Isidor B, Cogne B, Vuillaumier-Barrot S, et al. MAN1B1-CDG: three new individuals and associated biochemical profiles. Mol Genet Metab Rep. 2021;28:100775.
Balasubramanian M, Johnson DS. MAN1B-CDG: novel variants with a distinct phenotype and review of literature. Eur J Med Genet. 2019;62(2):109–14.
Li M, Xu Y, Wang Y, Yang X-A, Jin D. Compound heterozygous variants in MOGS inducing congenital disorders of glycosylation (CDG) IIb. J Hum Genet. 2019;64(3):265–8.
Abdel Ghaffar TY, Ng BG, Elsayed SM, El Naghi S, Helmy S, Mohammed N, et al. MPI-CDG from a hepatic perspective: report of two Egyptian cases and review of literature. JIMD Rep. 2020;56:20–6.
Mühlhausen C, Henneke L, Schlotawa L, Behme D, Grüneberg M, Gärtner J, et al. Mannose phosphate isomerase deficiency-congenital disorder of glycosylation (MPI-CDG) with cerebral venous sinus thrombosis as first and only presenting symptom: a rare but treatable cause of thrombophilia. JIMD Rep. 2020;55(1):38–43.
Dabaj I, Sudrié-Arnaud B, Lecoquierre F, Raymond K, Ducatez F, Guerrot A-M, et al. NGLY1 deficiency: a rare newly described condition with a typical presentation. Life (Basel, Switzerland). 2021;11(3):187.
Donoghue SE, White SM, Tan TY, Kowalski R, Morava E, Yaplito-Lee J. Galactose treatment of a PGM1 patient presenting with restrictive cardiomyopathy. JIMD Rep. 2021;57(1):29–37.
Edvardson S, Murakami Y, Nguyen TTM, Shahrour M, St-Denis A, Shaag A, et al. Mutations in the phosphatidylinositol glycan C (PIGC) gene are associated with epilepsy and intellectual disability. J Med Genet. 2017;54(3):196–201.
Makrythanasis P, Kato M, Zaki MS, Saitsu H, Nakamura K, Santoni FA, et al. Pathogenic variants in PIGG cause intellectual disability with seizures and hypotonia. Am J Hum Genet. 2016;98(4):615–26.
Vetro A, Pisano T, Chiaro S, Procopio E, Guerra A, Parrini E, et al. Early infantile epileptic-dyskinetic encephalopathy due to biallelic PIGP mutations. Neurol Genet. 2020;6(1):e387.
Nguyen TTM, Murakami Y, Wigby KM, Baratang NV, Rousseau J, St-Denis A, et al. Mutations in PIGS, encoding a GPI transamidase, cause a neurological syndrome ranging from fetal akinesia to epileptic encephalopathy. Am J Hum Genet. 2018;103(4):602–11.
Bayat A, Knaus A, Juul AW, Dukic D, Gardella E, Charzewska A, et al. PIGT-CDG, a disorder of the glycosylphosphatidylinositol anchor: description of 13 novel patients and expansion of the clinical characteristics. Genet Med. 2019;21(10):2216–23.
Horn D, Krawitz P, Mannhardt A, Korenke GC, Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: expanded clinical spectrum. Am J Med Genet Part A. 2011;155(8):1917–22.
Hogrebe M, Murakami Y, Wild M, Ahlmann M, Biskup S, Hörtnagel K, et al. A novel mutation in PIGW causes glycosylphosphatidylinositol deficiency without hyperphosphatasia. Am J Med Genet Part A. 2016;170(12):3319–22.
Yıldırım M, Koçak Eker H, Doğan MT. A homozygous mutation in the POMT2 gene in four siblings with limb-girdle muscular dystrophy 2N. Turk Arch Pediatr. 2021;56:68–71.
Feng Y, Shi P, Liu N, Kong X. Variant analysis of SEC23B gene in 4 families with congenital dyserythropoietic anemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021;38(8):727–30.
Quelhas D, Correia J, Jaeken J, Azevedo L, Lopes-Marques M, Bandeira A, et al. SLC35A2-CDG: novel variant and review. Mol Genet Metab Rep. 2021;26:100717.
Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change! Biochim Biophys Acta. 2009;1792(9):825–6.
Wilson MP, Quelhas D, Leão-Teles E, Sturiale L, Rymen D, Keldermans L, et al. SLC37A4-CDG: second patient. JIMD Rep. 2021;58(1):122–8.
Riley LG, Cowley MJ, Gayevskiy V, Roscioli T, Thorburn DR, Prelog K, et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. 2017;40(2):261–9.
Castiglioni C, Feillet F, Barnerias C, Wiedemann A, Muchart J, Cortes F, et al. Expanding the phenotype of X-linked SSR4-CDG: connective tissue implications. Hum Mutat. 2021;42(2):142–9.
Gordon-Lipkin E, Cohen JS, Srivastava S, Soares BP, Levey E, Fatemi A. ST3GAL5-related disorders: a deficiency in ganglioside metabolism and a genetic cause of intellectual disability and choreoathetosis. J Child Neurol. 2018;33(13):825–31.
Manoochehri J, Dastgheib SA, Khamirani HJ, Mollaie M, Sharifi Z, Zoghi S, et al. A novel frameshift pathogenic variant in ST3GAL5 causing salt and pepper developmental regression syndrome (SPDRS): a case report. Hum Genome Var. 2021;8(1):33.
Shrimal S, Ng BG, Losfeld M-E, Gilmore R, Freeze HH. Mutations in STT3A and STT3B cause two congenital disorders of glycosylation. Hum Mol Genet. 2013;22(22):4638–45.
Chang IJ, Byers HM, Ng BG, Merritt JL 2nd, Gilmore R, Shrimal S, et al. Factor VIII and vWF deficiency in STT3A-CDG. J Inherit Metab Dis. 2019;42(2):325–32.
Vajro P, Zielinska K, Ng BG, Maccarana M, Bengtson P, Poeta M, et al. Three unreported cases of TMEM199-CDG, a rare genetic liver disease with abnormal glycosylation. Orphanet J Rare Dis. 2018;13(1):4.
Munot P, McCrea N, Torelli S, Manzur A, Sewry C, Chambers D, et al. TRAPPC11-related muscular dystrophy with hypoglycosylation of alpha-dystroglycan in skeletal muscle and brain. Neuropathol Appl Neurobiol. 2021;48(2):e12771. https://doi.org/10.1111/nan.12771.
Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis. 2017;40(4):569–86.
Garshasbi M, Kahrizi K, Hosseini M, Nouri Vahid L, Falah M, Hemmati S, et al. A novel nonsense mutation in TUSC3 is responsible for non-syndromic autosomal recessive mental retardation in a consanguineous Iranian family. Am J Med Genet A. 2011;155A(8):1976–80.
Schreml J, Durmaz B, Cogulu O, Keupp K, Beleggia F, Pohl E, et al. The missing “link”: an autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum Genet. 2014;133(1):29–39.
Taylan F, Yavaş Abalı Z, Jäntti N, Güneş N, Darendeliler F, Baş F, et al. Two novel mutations in XYLT2 cause spondyloocular syndrome. Am J Med Genet Part A. 2017;173(12):3195–200.
Matthijs G, Schollen E, Bjursell C, Erlandson A, Freeze H, Imtiaz F, et al. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia). Hum Mutat. 2000;16(5):386–94.
Fernlund E, Andersson O, Ellegård R, Årstrand HK, Green H, Olsson H, et al. The congenital disorder of glycosylation in PGM1 (PGM1-CDG) can cause severe cardiomyopathy and unexpected sudden cardiac death in childhood. Forensic Sci Int Genet. 2019;43:102111.
Amish Population 2021. Available from: https://worldpopulationreview.com/state-rankings/amish-population. Cited 2021 Dec 27.
NSI. Population and demographic processes in 2020. Sofia: Republic of Bulgaria National Statistical Institute; 2021. https://nsi.bg/en.
Hixson L, Hepler BB, Kim MO. The native Hawaiian and other Pacific Islander population: 2010 2010 Census Briefs; 2012.
Croatian Bureau of Statistics - Republic of Croatia. Available from: https://www.dzs.hr/default_e.htm. Cited 2021 Dec 27.
Population - Statistics Denmark. 2021. Available from: https://www.dst.dk/en/Statistik/emner/borgere/befolkning. Cited 2021 Dec 27.
Population, total - Estonia | Data. Available from: https://data.worldbank.org/indicator/SP.POP.TOTL?end=2020&locations=EE&start=2019&view=map. Cited 2021 Dec 27.
Population: size and growth - Statistiek Vlaanderen. Available from: https://www.statistiekvlaanderen.be/en/population-size-and-growth-0. Cited 2021 Dec 27.
Series 001641607 Demography - population at the beginning of the month - France (including Mayotte since 2014) | Insee. Available from: https://www.insee.fr/en/statistiques/serie/001641607?idbank=001641607#Tableau. Cited 2021 Dec 27.
Population census / 2020 population census / Basic complete tabulation on population and households. 2021. Available from: https://www.e-stat.go.jp/en/stat-search/files?page=1&layout=datalist&toukei=00200521&tstat=000001136464&cycle=0&tclass1=000001136466&stat_infid=000032142402&tclass2val=0. Cited 2021 Dec 27.
StatLine - population dynamics; month and year. Available from: https://opendata.cbs.nl/statline/#/CBS/en/dataset/83474ENG/table?ts=1616407710127. Cited 2021 Dec 27.
Black B, Dooley J, Pyper A, Reed M. Multiple hereditary exostoses. An epidemiologic study of an isolated community in Manitoba. Clin Orthop Relat Res. 1993;287:212–7.
First Nation Profiles. 2021. Available from: https://fnp-ppn.aadnc-aandc.gc.ca/fnp/Main/Search/FNRegPopulation.aspx?BAND_NUMBER=327&lang=eng. Cited 2021 Dec 27.
Cierniak-Piotrowska M, Dąbrowska A, Stelmach K. Population. Size and structure and vital statistics in Poland by territorial division in 2021. As of 30th June. Warsaw; 2021.
Population, total - Saudi Arabia. 2021. Available from: https://data.worldbank.org/indicator/SP.POP.TOTL?locations=SA. Cited 2021 Dec 27. Accessed 27 Dec 2021.
Population growth in Sweden remains low in the first six months of 2021. Available from: https://www.scb.se/en/finding-statistics/statistics-by-subject-area/population/population-composition/population-statistics/pong/statistical-news/population-statistics-januaryjune-2021/. Cited 2021 Dec 27.
Adrese Dayalı Nüfus Kayıt Sistemi Sonuçları, 2020. 2021. Available from: https://data.tuik.gov.tr/Bulten/Index?p=Adrese-Dayalı-Nüfus-Kayıt-Sistemi-Sonuçları-2020-37210&dil=1. Cited 2021 Dec 27.
Population estimates for the UK, England and Wales, Scotland and Northern Ireland - Office for National Statistics. Available from: https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates/bulletins/annualmidyearpopulationestimates/mid2020. Cited 2021 Dec 27.
Census - table results. Available from: https://data.census.gov/cedsci/table?y=2021&tid=PEPPOP2021.NST_EST2021_POP. Cited 2021 Dec 27.
Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet. 2014;94(2):161–75.
Zhang Z, Huang T-L, Ma J, He W-J, Gu H. Clinical and whole-exome sequencing findings in two siblings from Hani ethnic minority with congenital glycosylation disorders. BMC Med Genet. 2019;20(1):181.
Enns GM, Steiner RD, Buist N, Cowan C, Leppig KA, McCracken MF, et al. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr. 2002;141(5):695–700.
Mehlman CT, Nematbakhsh AR, Crawford AH, Berlin RE. Spinal deformity associated with carbohydrate-deficient glycoprotein syndrome (Jaeken’s syndrome): a report of three cases. Spine (Phila Pa 1976). 2003;28(7):E132–5.
Thong MK, Fietz M, Nicholls C, Lee MH, Asma O. Congenital disorder of glycosylation type Ia in a Malaysian family: clinical outcome and description of a novel PMM2 mutation. J Inherit Metab Dis. 2009;32(Suppl 1):S41–4.
Iijima K, Murakami F, Nakamura K, Ikawa S, Yuasa I, Motosumi H, et al. Hemostatic studies in patients with carbohydrate-deficient glycoprotein syndrome. Thromb Res. 1994;76(2):193–8.
Garel C, Baumann C, Besnard M, Ogier H, Jaeken J, Hassan M. Carbohydrate-deficient glycoprotein syndrome type I: a new cause of dysostosis multiplex. Skeletal Radiol. 1998;27(1):43–5.
Kjaergaard S, Skovby F, Schwartz M. Absence of homozygosity for predominant mutations in PMM2 in Danish patients with carbohydrate-deficient glycoprotein syndrome type 1. Eur J Hum Genet. 1998;6(4):331–6.
Godfrey C, Escolar D, Brockington M, Clement EM, Mein R, Jimenez-Mallebrera C, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol. 2006;60(5):603–10.
Newell JW, Seo N-S, Enns GM, McCraken M, Mantovani JF, Freeze HH. Congenital disorder of glycosylation Ic in patients of Indian origin. Mol Genet Metab. 2003;79(3):221–8.
Dercksen M, Crutchley AC, Honey EM, Lippert MM, Matthijs G, Mienie LJ, et al. ALG6-CDG in South Africa: genotype-phenotype description of five novel patients. JIMD Rep. 2013;8:17–23.
McKusick VA, Gross MB. OMIM entry - * 311770 - phosphatidylinositol glycan anchor biosynthesis class A protein; PIGA. 2018. Available from: https://www.omim.org/entry/311770. Cited 2021 Jul 18.
Frontiers in Congenital Disorders of Glycosylation (FCDGC) | Rare Diseases Clinical Research Network. Available from: https://rdcrn.org/fcdgc. Cited 2022 Jan 24.
Makhseed N, Dhaunsi G, Jaeken J. Distinct features of congenital disorder of glycosylation type IIx in Kuwait: a case report. J Child Neurol. 2012;27(2):222–4.
Westphal V, Enns GM, McCracken MF, Freeze HH. Functional analysis of novel mutations in a congenital disorder of glycosylation Ia patient with mixed Asian ancestry. Mol Genet Metab. 2001;73(1):71–6.
Lieu MT, Ng BG, Rush JS, Wood T, Basehore MJ, Hegde M, et al. Severe, fatal multisystem manifestations in a patient with dolichol kinase-congenital disorder of glycosylation. Mol Genet Metab. 2013;110(4):484–9.
Sorte H, Mørkrid L, Rødningen O, Kulseth MA, Stray-Pedersen A, Matthijs G, et al. Severe ALG8-CDG (CDG-Ih) associated with homozygosity for two novel missense mutations detected by exome sequencing of candidate genes. Eur J Med Genet. 2012;55(3):196–202.
Rymen D, Peanne R, Millón MB, Race V, Sturiale L, Garozzo D, et al. MAN1B1 deficiency: an unexpected CDG-II. PLoS Genet. 2013;9(12):e1003989.
Pajusalu S, Vals M-A, Mihkla L, Šamarina U, Kahre T, Õunap K. The estimated prevalence of N-linked congenital disorders of glycosylation across various populations based on allele frequencies in general population databases. Front Genet. 2021;12:719437.
Jaeken J, van Eijk HG, van der Heul C, Corbeel L, Eeckels R, Eggermont E. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta. 1984;144(2–3):245–7.
Cano A, Alcalde C, Belanger-Quintana A, Cañedo-Villarroya E, Ceberio L, Chumillas-Calzada S, et al. Transferrin isoforms, old but new biomarkers in hereditary fructose intolerance. J Clin Med. 2021;10(13):2932.
Gianniki M, Nikaina I, Avgerinou G, Kanaka-Gantenbein C, Siahanidou T. Transient cytopenias as a rare presentation of classic galactosemia. Cureus. 2022;14:e23101.
Kim M-J, Yum M-S, Seo GH, Lee Y, Jang HN, Ko T-S, et al. Clinical application of whole exome sequencing to identify rare but remediable neurologic disorders. J Clin Med. 2020;9(11):3724.
Gilissen C, Hoischen A, Brunner HG, Veltman JA. Disease gene identification strategies for exome sequencing. Eur J Hum Genet. 2012;20(5):490–7.
Acknowledgements
We thank the NOVA Sci & Tech Volunteer program— CDG & Allies—PPAIN for their input.
Funding
This work is financed by national funds from FCT-Fundação para a Ciência e a Tecnologia, I.P., in the scope of the project UIDP/04378/2020 and UIDB/04378/2020 of the Research Unit on Applied Molecular Biosciences- UCIBIO and the project LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy-i4HB. Rita Francisco (SFRH/BD/124326/2016) and Carlota Pascoal (SFRH/BD/138647/2018) acknowledge the funding from the Fundação para a Ciência e Tecnologia (FCT), Portugal. Sandra Brasil was supported by CDG & Allies—PPAIN funding.
Author information
Authors and Affiliations
Contributions
VRF, RF, TR, and PAV were responsible for the conception and design of this study. TR developed a script in order to perform the literature search. AP, ABL, MG, MA, and AG contributed to the screening of the publications and extraction of data. AP compiled and analysed the data. AP wrote the original draft of the manuscript. AG contributed to the writing of the manuscript. TR and AP generated the figures used in the main text. RF, CP, CRF, JJ, SB, PS, VRF, and PAV revised, commented, and edited on the draft manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table 1S.
Number of findings per keyword combinations.
Additional file 2: Figure 1S.
Number of reported CDG patients per CDG.
Additional file 3: Table 2S.
Summary of collected patients reports.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piedade, A., Francisco, R., Jaeken, J. et al. Epidemiology of congenital disorders of glycosylation (CDG)—overview and perspectives. J Rare Dis 1, 3 (2022). https://doi.org/10.1007/s44162-022-00003-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44162-022-00003-6