
Abstract
Rose (Rosa) is a prominent ornamental plant that holds substantial economic and social significance. Roses originating from different regions exhibit intricate phenotypic and genetic characteristics, but the majority of rose genetic resources are poorly characterized. In this study, 192 genotypes of the genus Rosa were examined using 33 phenotypic traits and 10 pairs of SSR markers. Compared to wild species, both old garden and modern roses exhibited a significant level of diversity, with flower color having the highest degree of diversity and style morphology having the lowest degree of diversity. This phenomenon may be attributed to the limited utilization of wild roses due to their simpler ornamental traits and the frequent phenotypic and molecular infiltration between old garden roses and modern roses. Following a inaugural comprehensive evaluation employing principal component analysis, R. chinensis ‘Zihongxiang’, R. hybrida ‘Burgundy Iceberg’, R. hybrida ‘Conrad F. Meyer’, R. rugosa ‘Gaohong’ and R. floribunda ‘Sheherazad’ were selected as core germplasm resources for future breeding. Moreover, three tetraploid roses, namely R. hybrida ‘Midnight Blue’, R. floribunda ‘Sheherazad’, and R. hybrida ‘Couture Rose Tilia’, with significant differences in both phenotypic and molecular profiles were selected and reciprocally intercrossed. Ultimately, two populations were obtained exhibiting significant variation in flower size, annual stem color, stem pickle density, and leaf number. Furthermore, our results indicated that the traits of flower diameter, flower height, petal width, and petal number may potentially be controlled by two major-effect loci. In conclusion, this study provides novel insights into the evolutionary patterns of Rosa germplasm resources. It paves the way for identifying core genotypes that carry distinct ornamental characteristics and possess immense value for breeding novel varieties in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Roses, belonging to the genus Rosa of the Rosaceae family, are favored for their vibrant flower colors, captivating fragrances, and versatile applications as ornamental plants. Approximately 30,000 to 35,000 cultivated rose varieties have been bred (Qi et al. 2018), with extensive utilization in landscape architecture, floral arrangements, culinary endeavors, pharmaceutical manufacturing, perfumery, and various other industries. As a result, rose plants and their flowers possess a multitudinous cultural and economic value (Raymond et al. 2018). Rose plants are deciduous or evergreen, possessing erect and climbing stems, often accompanied by prickles. Their predominantly odd-pinnate compound leaves are arranged alternately. The rose plant flowers are perfect, possessing both male and female reproductive organs (Basu et al. 2015). Its fruits are aggregated achenes, commonly referred to as the ‘rose fruit’ (Zieliński et al. 2010). Based on the botanical taxonomy of wild roses, the genus Rosa has been subdivided into the subgenera Hulthemia, Rosa, Platyrhodon, and Hesperhodos. Furthermore, the most populous subgenus, Rosa, has been further subdivided into 10 sections and 6 subsections (Wissemann 2017). China is widely recognized as the primary source and breeding ground for most rose germplasm resources, distributed across various provinces and cities throughout the country. Notably, the southwest region, particularly the Sichuan and Yunnan provinces, serves as a significant genetic diversity hub of roses in China (Li 1994; Tang et al. 2008; Zhao and Zhang 2003; Jian et al. 2013). Various breeding methods are utilized to improve roses, encompassing traditional cross-breeding, molecular breeding, and others. Notably, the cross-breeding technique has emerged as the most prevalent approach (Cheng 2000; Smulders et al. 2019). Rose breeding and the generation of rose varieties can be divided into three distinct periods. From prehistoric times to 1875, this initial period was characterized by the natural process of interspecific hybridization and the emergence of new varieties solely through natural pollination. The second period, from 1875 to 1967, encompassed the development of a yellow-colored petal, continuously flowering variety by Pemet in 1900. The third period, from 1967 and continuing to the present day, involves extensive cross-breeding of Chinese, European, and Middle Eastern rose varieties, which served as the genetic foundation for the creation of the ‘modern rose variety’ (De Vries and Dubois 1996; Bendahmane et al. 2013; Lidia and Irina 2009).
The research on genetic diversity provides insights into the stability of several traits as well as the changes that occur during evolution, leading to distinct groups of genotypes. This exploration holds significance in cultivating and safeguarding germplasm resources (Yanchuk 2001; Glaszmann et al. 2010). Genetic diversity analysis has also unveiled the evolution of traits within the Rosa genus (Meng et al. 2011). Phenotypic traits serve as the most perceptible manifestation of genetic diversity (Gulsen et al. 2007). Statistical analysis of phenotypic traits is a prevalent approach for assessing plant genetic diversity, as these traits serve as the foundation for identification, classification, and scientific studies (Silberstein et al. 2003). Observing phenotypes allows us to investigate the molecular mechanisms underlying diverse phenotypic traits in distinct regions and assess the advantages of various hybrid combinations (Singh et al. 2013). However, it is important to note that phenotypic markers have certain limitations, as they are prone to frequently irreversible variations induced by environmental variables as a result of adaptation to adverse conditions. This drawback is both an opportunity and a barrier to scientific research. While it can hamper the precision of research on plant botanical classification research, certain variations can be harnessed for breeding purposes (Sensoy et al. 2007). The instability of phenotypic markers can be surmounted by employing molecular markers, widely employed for discerning genetic relationships among species (Ben-Meir and Vainstein 1994). Furthermore, molecular markers can aid in distinguishing between various sections within the Rosa genus and unveiling genetic similarities (Liorzou et al. 2016; Tan 2017; Yang 2020). The utilization of molecular markers has become increasingly prevalent in genetic map construction (Cao et al. 2000) in both human and plant species (Zietkiewicz et al. 1994).
The size of the flower corolla is the primary ornamental component in roses, significantly influencing the quality and economic value of cut roses, and is determined by various factors, encompassing flower diameter, flower height, petal length, petal width, and petal number. Therefore, it is crucial to research the quantitative traits affecting rose flower characters. These traits have been shown to be influenced by major genes, minor polygenes, or a combination of both, known as trait inheritance affected by major genes and polygenes (Gai et al. 2003). The rose, a perennial woody garden plant, to a great extent, has a wide range of phenotypes in the F1 generation. In outcrossing plants, the F1 generation can be considered equivalent to the F2 generation of inbred crops, thus serving as a suitable generation (F2) for segregation analysis (Yang et al. 2020). Consequently, this approach has facilitated the effective genetic examination of traits in polyploid outcrossing plants within the Rosaceae family (Calle et al. 2020; Shi et al. 2020; Lau et al. 2022).
During long-term evolution and artificial selection, a wide range of traits in germplasm resources of the genus Rosa have emerged because of the interplay between their genetic makeup and external factors. To facilitate the accelerated growth of the rose industry, it is imperative to prioritize the generation of novel genetic resources and genetic enhancement. In contrast to previous studies, this study has collected and evaluated a more substantial assemblage of Rosa germplasm resources, encompassing a comprehensive representation of most sections, totaling 192 genotypes. Additionally, a more comprehensive analysis of phenotypic traits pertaining to the stem, leaf, and flower was undertaken, surpassing previous studies. This study provides essential information on germplasm resources that can facilitate hybrid breeding, laying the foundation for genetic improvement and germplasm innovation in roses. Moreover, it accelerates the advancement of molecular-assisted breeding approaches, with the identification of novel market-trait associations enhancing the effectiveness of rose breeding.
Materials and methods
Plant materials
In this study, a total of 192 genus Rosa genotypes were collected, comprising 38 wild rose genotypes (W), 83 old garden rose genotypes (C), and 71 modern rose genotypes (M). All plants were cultivated in open-air fields at the ornamental plant agricultural station of Huazhong Agricultural University, located in Wuhan City, Hubei province, China. The plants were more than two years old and received regular water and fertilizer management throughout the period of phenotypic assessment. The Latin names and origin of the collected germplasm resources are presented in Table S1. In this study, two hybrid populations were created in the spring of 2020 by crossing the tetraploid rose cultivar R. hybrida ‘Midnight Blue’ (M10) with R. hybrida ‘Sheherazad’ (M19) and R. hybrida ‘Couture Rose Tilia’ (M29). Following winter sowing, two F1 populations were obtained in 2021, consisting of 105 F1 generation plants from the M10 × M19 cross and 89 F1 generation plants from the M10 × M29cross. All materials were cultivated in the ornamental plant agricultural station of Huazhong Agricultural University under standardized maintenance and management practices.
Assessment of plant phenotypic traits
In this study, we evaluated 33 phenotypic traits in 192 genotypes of the genus Rosa. The abbreviations used for these traits are as follows: flowering frequency (FF), overall flower amount (OFA), inflorescence (Inflo), flower color (FC), flower scent (FS), petal number (PN), flower type (FT), calyx tube surface (CS), sepal morphology (SM), petal shape (PeS), state of the front edge of the petal (SFEP), petal velvet (PV), stamen number (SN), pistil status (PiS), anther color (AC), stigma color (SC), style length (SL), style morphology (StM), flower diameter (FD), flower height (FH), petal length (PL), petal width (PW), annual stem color (ASC), stem prickle density (SPD), stem prickle morphology (SPM), number of leaflets (LN), leaf texture (LT), leaf color (LC), leaf edge serration (LES), apical leaflet shape (ALS), leaf tip shape (LTS), stipule shape (SS) and leaf area (LA).
The phenotypic trait classification was based on guidelines for evaluating the distinctness, uniformity, and stability in rose plants (Rosa sp.) and the botanical classification of Floral of China and adapted in accordance with the specific circumstances. The measurement parameters and assigned values for each trait are listed in Table S2. The fundamental principle is to assign a value of 0 when a trait is not present and then assign values on a progressive basis. FC, AC, SC, ASC, and LC were measured using the Royal Horticultural Society Color Chart (RHSCC). The quantitative traits FD, FH, PL, PW, LN, and LA were measured in three times for each rose genotype, and the average values were calculated for the statistical analyses. The quantitative trait PN was transformed into a categorical variable. The phenotypic distribution of FC, PN, and FT is presented in Figure 1. Figures S4 display the flower front and side views of certain germplasms.

Classification of three major floral phenotypic traits in the studied Rosa sp. genotypes. (a) Petal color classification of the Rosa sp. genotypes: a) White; b) Green; c) Yellow; d) Orange; e) Pink; f) Rose red; g) Red; h) Blue-violet; i) Purple red; j) Variable color; k) Multicolor. (b) Petal number: a) Single; b) Semidouble; c) Double; d) Fully double. (c) Flower type of the Rosa sp. genotypes: a) Flat; b) Cup-shaped; c) Spherical; d) Protruding; e) Altar-shaped; f) Rosette; g) Quartered rosette; h) Pompon; i) Anemone; j) Button eye. The scale bar =1cm
Statistical analysis was conducted on PN, FD, FH, PL, PW, ASC, SPD, and LN for a total of 105 M10 × M19 F1 generation plants and 89 M10 × M29 F1 generation plants. The values and measurement standards for each trait are presented in Table S3. The stem and leaf traits were measured in January 2022, while the floral traits were observed and recorded during three distinct flowering periods: from March to May 2022, September to November 2022, and March to May 2023. The data pertaining to flower traits were analyzed from F1 generation offsprings and exhibited a minimum of three phenotypically stable and similar flowers. Based on these criteria, the final numbers of individuals in the two populations for statistical analysis of flower traits were 51 in the M11 × M19 F1 and 60 in the M11 × M29 F1 population.
Data analysis
Genetic diversity analysis was performed according to the method proposed in the past research (Rui et al. 2018). The maximum (Max), minimum (Min), mean (μ), and standard deviation (δ) of the measured values of each quantitative trait were calculated. The coefficient of variation (CV) was calculated as [formula (1)].
All data calculations were performed using Excel 2021 software (Microsoft, China). Shannon-Weaver diversity index [formula (2)] was used to analyze the rose germplasm diversity (Liu et al. 2009; Tena Gashaw et al. 2016; Yirgu et al. 2022).
In the formula, Pi is the frequency at which the ith level of a trait occurs.
Since traits followed a normal distribution, the four classes (M − 1.2818S), (M − 0.5246S), (M + 0.5246S), and (M + 1.2818S) were divided into 5 grades so that each probability of the grades 1-5 was approximately 10%, 20%, 40%, 20%, and 10%, respectively (Liu 1996), the M indicates mean, and the S indicates standard deviation.
Cluster analysis
Cluster analysis on the results of phenotypic traits and molecular markers was performed using the Ntsys 2.10 software. Firstly, the statistical data of 192 Rosa genotypes were converted into a format compatible with the software, and the experimental data were normalized. Then, the genetic distance between the corresponding data was calculated, and groups were assigned according to the genetic distance. The Origin 2021 software was used to plot frequency histograms to analyze the variation of each numerical trait in the two hybrid populations.
The data points from the qualitative traits were marked as 1 and 0 according to their presence or absence in the assessed genotypes (Liu et al. 2016), and the grading criteria were the same as those used in calculating the genetic diversity coefficient. The binary 1/0 matrix was imported into NTSYSpc 2.10 software to calculate the genetic similarity coefficient (GS) matrix by routine DICE of SIMQUAL (Dice 1945). The GS matrix was converted to a genetic distance (GD) matrix in Excel 2021 software (GD=1-GS), and the resulting distance matrix was equivalent to Nei and Li's genetic distance (GDNL) matrix (Nei and Li 1979; Soleimani et al. 2002). MEGA11 was used to draw the clustering analysis diagram of the GDNL matrix by using an unweighted pair-group method with arithmetic mean (UPGMA).
DNA extraction
A modified CTAB method was used to extract genomic DNA from the 192 Rosa genotypes. Specific operations are as follows: (1) 35 new leaves were collected from the test material, placed into tubes with steel balls from grinding, and then were quickly placed into liquid nitrogen. The grinding equipment was used to fully grind samples before DNA isolation. (2) CTAB was mixed with mercaptoethanol in a ratio of 50:1, and the mixture was preheated in a water bath at 65℃ for 10 min. (3) 500 μl of the preheated solution was added into each tube with the ground samples. After mixing evenly, the tubes were incubated in a water bath at 65℃ for 30 min, and then the samples were removed and cooled to the room temperature. (4) An equal volume of chloroform: isoamyl alcohol 24:1 solution was added, and the samples were mixed thoroughly and left to stand for 10 min. (5) The samples were then centrifuged at 10000 r/min for 10 min. (6) Approximately 500 μL of supernatant was transferred into a new centrifugal tube. (7) The steps 4 to 6 were repeated two or three times. (8) Anhydrous ethanol was added to the centrifuge tubes. (9) Then, the samples were centrifuged at 10000 r/min for 10 min. (10) The supernatant was removed, leaving a white precipitate. 1 ml 75% ethanol was added to each sample tube, and then centrifuged for 10 min at 10000 r/min. (11) The ethanol in the sample tube was evaporated, leaving a white precipitate. (12) The precipitated DNA was dissolved in ddH2O preheated at 37℃.
After DNA extraction, 1% agarose gel electrophoresis and NanoDrop One spectrophotometer were used (Thermo Fisher Scientific, USA) to assess DNA quality and concentration. Each sample was then diluted to a concentration of 20–50 ng/μL for PCR amplification.
SSR analysis
We selected 10 SSR molecular markers located on different rose chromosomes to assess DNA polymorphisms between the genotypes (Table 1) (Zhang et al. 2006; Hibrand Saint-Oyant et al. 2008). Genotyping with all 10 pairs of SSR molecular markers was carried out by a Biotechnology Company. The 192 Rosa genotypes were genotyped by the SSR markers, and the resulting products were examined using the QIAxcel Advanced automatic capillary electrophoresis apparatus.
SSR genotyping and clustering
The SSR molecular marker amplification results were recorded using a 1/0 matrix. The band presence or absence at the same migration location was represented by 1 and 0, respectively (Soleimani et al. 2002). The methods of generating the GS matrix and GDNL matrix and constructing the cluster map were consistent with those of phenotypic trait clustering.
Correlation and principal component analysis
Correlation analysis and principal component analysis (PCA) were performed using the IBM SPSS Statistics 27 software, and Origin 2021 was used to plot the correlation heatmap and the 3D PCA loading plot. The analysis was performed according to previously published methods (Jin et al. 2021; Yaghini et al. 2013). Specifically, according to the eigenvalue of the principal component and the principal component loading values of 29 traits, the trait coefficients were calculated and implemented into the equation as F1~F6. The trait coefficient was calculated by dividing the principal component loading values by the arithmetic square root of the principal component characteristic values. By considering the contribution rates of PC1~PC6, a comprehensive score formula, denoted as F, was constructed. \({\text{F}}=0.177\times {\text{F}}1+0.126\times {\text{F}}2+0.096\times {\text{F}}3+0.068\times {\text{F}}4+0.065\times {\text{F}}5+0.061\times {\text{F}}6\) The comprehensive score of the 192 Rosa genotypes was obtained after conversion.
Segregation analysis
Segregation Analysis (SEA v2.0.1) was performed for the quantitative traits based on a mixed major gene-polygene inheritance model (Wang et al. 2022). The mixed major gene-polygene inheritance model was evaluated based on the single-generation segregation analysis method of the plant quantitative traits. The maximum likelihood value (MLV) and Akaike's information criterion (AIC) of various genetic models were obtained by combining the distribution of the phenotype frequency with 11 genetic models based on the phenotypic data of the F1 population (pseudo F2). Simultaneously, these models were used to test the goodness-of-fit. Equal distribution (U12, U22, and U32), Smirnov (nW2), and Kolmogorov (Dn) tests were conducted to identify the optimal model. When selecting the optimal model, one or several modeles were selected as candidates of the optimal model according to the AIC minimum principle. Subsequently, considering the results of the goodness-of-fit test, the candidate model with the least number of goodness-of-fit test results reaching the significance level (P<0.05) was selected as the optimal genetic model for this trait (Wang et al. 2023).
Results
Genetic diversity and variation analysis based on phenotypic traits
The frequency distribution of six quantitative traits closely adhered to the normal distribution. Through following the methodology proposed by Liu et al. (1996), the six quantitative traits, which were deemed to follow a normal distribution, were subsequently classified into five classes by using four reference points. Consequently, the quantitative traits distribution were transformed to a qualitative trait distribution, enabling the calculation of the genetic diversity coefficient. The coefficient of variation (CV) for the six quantitative traits ranged from 28.42% to 80.21%, whereas the H' index ranged between 1.05 and 1.46 (Fig. 2a, Table S4). These findings suggest the occurrence of a substantial amount of variation in the genus Rosa. Notably, four floral traits (FD, FH, PL, and PW) demonstrate high genetic diversity similarity, indicating a potential genetic relationship. Additionally, the coefficient of variation for LA was the highest (80.21%), with a range of 131.89~6.28 cm2, indicating a substantial variation in leaf area within the rose genotypes assessed.

Genetic diversity and variation analysis of the Rosa sp. genotypes. (a) Bar chart of the diversity index for quantitative traits. (b) Bar chart of the diversity index for qualitative traits. (c) The inflorescence types of wild roses, old garden roses, and modern roses. (d) The flower-color types of wild roses, old garden roses, and modern roses. (d) The petal-number types of wild roses, old garden roses, and modern roses. (f) The flower types of wild roses, old garden roses, and modern roses. FF, flowering frequency; OFA, overall flower amount; Inflo, inflorescence; FC, flower color; FS, flower scent; PN, petal number; FT, flower type; CS, calyx tube surface; SM, sepal morphology; PeS, petal shape; SFEP, state of the front edge of petal; PV, petal velvet; SN, stamen number; PiS, pistil status; AC, anther color; SC, stigma color; SL, style length; StM, style morphology; FD, flower diameter; FH, flower height; PL, petal length; PW, petal width; ASC, annual stem color; SPD, stem prickle density; SPM, stem prickle morphology; LN, number of leaflets; LT, leaf texture; LC, leaf color; LES, leaf edge serration; ALS, apical leaflet shape; LTS, leaf tip shape; SS, stipule shape; LA, leaf area
The H' values of 27 qualitative traits in the Rosa germplasms ranged from 1.91 to 0.14 (Fig. 2b, Table S5), suggesting a high genetic diversity. Among these traits, FC presented the highest H' value, indicating a broad variation in flower color. This can be attributed to the global distribution of the genus Rosa germplasm resources and the influence of diverse geographical environments and artificial selection breeding, contributing to significant differentiation in flower color. Conversely, StM exhibited the lowest H' value, suggesting a relatively low variability in style morphology among the different rose varieties.
The flower is widely recognized as the most significant organ in ornamental plants (Wang et al. 2010), and its ornamental value is primarily determined by its floral traits (Nybom 2009, Datta 2018, Hibrand Saint-Oyant et al. 2018). In this study, we analyzed the variation of Inflo, FC, PN, and FT within the 192 Rosa sp. genotypes. Our findings revealed that Inflo could be categorized into five distinct types. Among the single-flower plants, modern roses constituted the majority, followed by old garden roses, with wild roses being the least (Fig. 2C, Table S6).
The classification of FC encompassed eleven distinct types (Fig. 1a), effectively including the majority of the variability in flower color. Modern roses presented the highest diversity, comprising ten color types. In contrast, wild roses presented a relatively limited range, primarily pink and white hues. On the other hand, old garden roses presented greater variability in FC, with a predominant presence of pink and purplish red shades and a lower representation of yellow (Fig. 2d and Table S7). Notably, the degree of flower color variation progressively increased from wild roses to old garden roses and ultimately to modern roses (Fig. 2c–f).
PN was classified into four distinct types, as depicted in Fig. 1b. Among these types, 116 genotypes presented full double petals, with old garden and modern roses accounting for nearly half of the studied population. A gradual increase in PN from wild to modern roses was evident, resulting in a greater abundance of double-flower genotypes (Fig. 2e, Table S8). Similarly, FT could be categorized into ten types, as illustrated in Fig. 1c. The old garden roses encompassed six distinct flower types. In comparison, modern roses presented eight flower types, while only four were presented in wild roses (Fig. 2f and Table S9). These observed changes in PN and FT variability align with the evolutionary trajectory and breeding the history of roses.
Correlation analysis among traits
The degree and the significance of correlations between phenotypic traits were assessed using the Pearson correlation coefficient and the respective p-values. Among the 19 phenotypic traits analyzed, the Pearson correlation coefficients ranged from - 0.35 to 0.89, as depicted in Fig. 3. Notably, PW exhibited a positive correlation with PL at 0.89, the highest correlation coefficient among the traits. Conversely, the most significant negative correlation coefficient (-0.35) was observed between LC and LT. This study primarily investigated the relationship between floral traits, specifically PL, PW, FD, and FH. These traits, which exert a direct influence on the determination of flower size, exhibited highly significant positive correlations (Fig. 3). The highly positive correlation observed can be attributed to the co-inheritance of closely linked genes, as suggested by previous research conducted by Debener and Linde (2009) and Shupert et al. (2007). Notably, a negative correlation between PN and SN was observed.

Correlation heat maps between 19 phenotypic traits among the 192 Rosa sp. genotypes. Red represents positive correlations, and blue represents negative correlations. The darker color represents a greater absolute value of the correlation coefficient. * P<0.05, and ** P<0.01. FF, flowering frequency; OFA, overall flower amount; Inflo, inflorescence; FC, flower color; FS, flower scent; PN, petal number; FT, flower type; CS, calyx tube surface; SM, sepal morphology; PeS, petal shape; SFEP, state of the front edge of petal; PV, petal velvet; SN, stamen number; PiS, pistil status; AC, anther color; SC, stigma color; SL, style length; StM, style morphology; FD, flower diameter; FH, flower height; PL, petal length; PW, petal width; ASC, annual stem color; SPD, stem prickle density; SPM, stem prickle morphology; LN, number of leaflets; LT, leaf texture; LC, leaf color; LES, leaf edge serration; ALS, apical leaflet shape; LTS, leaf tip shape; SS, stipule shape; LA, leaf area
Principal component analysis of phenotypic traits
The genus Rosa exhibits a high degree of diversity across numerous plant traits, including flower color, flower types, plant types, leaf types, and more. In this study, we collected data on 33 traits from a substantial sample of 192 Rosa sp. genotypes. This dataset is considered extensive. To simplify the datasets obtained and facilitate subsequent genotype clustering or classification for pattern identification, a principal component analysis (PCA) was conducted. PCA serves as a valuable initial analytical and clustering method by reducing the dimensionality of the complete dataset (Ringer 2008; Yaghini et al. 2013). Consequently, a three-dimensional scatter plot was generated using the data from 19 phenotypic traits in the 192 Rosa sp. genotypes, allowing for the visualization of relationships between different germplasm types (Fig. 4).

PCA plot of 192 Rosa sp. genotypes based on variability on 19 phenotypic traits. PCs were calculated with one mean value per variety. Percentages in brackets represent the variance explained by each principal component. The dots of different colors represent the different germplasm groups. C, old garden roses; M, modern roses; W, wild roses. The circles correspond to the 95% confidence ellipse for each germplasm group. The arrows represent the Eigenvectors corresponding to the 19 traits
The eigenvalues of the top six components had values greater than 1, with substantial decline in the slope evident in the scree plot (Fig. S1). The first three principal components accounted for 40.0% of the observed phenotypic variation. Principal component one (PC1) (17.7%) predominantly differentiated the genotypes due to variability in flower size attributions, including FD, FH, PL, PW, and LA. PC2 (12.6%) primarily differentiated the genotypes due to variability in stem and leaf attributions, such as LC, LT, LN, SPD, ASC, and FF. PC3 (9.6%) primarily differentiated the genotypes due to variability in flower organ color and number attributions, such as SN, AC, and PN (Table S12).
A significant overlap between the old garden and modern roses was observed, with an almost complete overlap (Fig. 4). Modern roses have predominantly derived from old garden roses, resulting in highly similar phenotypes. Conversely, most wild roses presented no overlap with either of the two germplasm groups and their phenotypes differed significantly. The loading scores (Fig. S2) were extracted, revealing that the five traits FD, FH, PL, PW, and LA were predominantly responsible for the variation across PC1. Their loading scores were consistent with the distribution direction observed in old and modern gardens. Similarly, the three traits SPD, ASC, and FF, predominantly responsible for the variation across PC2, presented values reflecting the distribution pattern of old garden roses and modern roses. Among the traits responsible for the variation across PC3, SN presented loading scores reflecting the distribution of wild roses across PC3, while PN aligned with the distribution observed in old garden roses and modern roses. This demonstrated that wild roses exhibited notable variation in flower size, number of flower organs, flowering frequency, number of leaflets, annual stem color, and stem prickles density, thereby enabling their differentiation based on these traits. It is noteworthy to mention that modern roses have predominantly originated from old garden roses, leading to highly similar phenotypes.
Comprehensive evaluation based on phenotypic traits
The correlation between the F score and the desirable trait values was positive. The composite scores displayed significant variability, ranging from -1.175 to 1.464 (Table S13). Among these scores, the top five materials (R. chinensis ‘Zihongxiang’, R. hybrida ‘Burgundy Iceberg’, R. hybrida ‘Conrad F. Meyer’, R. rugosa ‘Gaohong’, R. floribunda ‘Sheherazad’) demonstrated exceptional flower morphological traits and desirable flower types, making them ideal candidates for breeding purposes.
Furthermore, a total of 15 traits presented a significant positive correlation with the overall F score, with correlation coefficients ranging from -0.172 to 0.755 (Fig. S3). Specifically, a positive correlation was observed between twelve attributes (PL, PW, FD, LA, FS, SPD, ASC, FH, LT, PV, SN, SC, and AC) and the overall F score. Notably, the correlation coefficient between PL and PW exceeded 0.7, indicating a tight interrelationship. Consequently, based on our findings, the floral traits, including flower size, annual stem color, leaf area, and leaf texture, significantly influenced the overall phenotypic characteristics of roses.
Cluster analysis using phenotypic trait markers
The breeding of the first hybrid tea rose, R. hybrida ‘La France’, in 1867 marked the initiation of a prosperous era in modern garden rose breeding. Through the combined assessment of phenotypic and molecular markers, we found that wild roses, old garden roses, and modern roses are closely related regarding both phenotypic and molecular aspects as they were integrally exploited and utilized during the modern rose breeding history.
Utilizing 33 phenotypic traits, a cluster analysis was conducted encompassing all genotypes, resulting in a genetic distance ranging from 0.15 to 0.91. The 192 genotypes were categorized into seven groups, denoted as Group I to Group VII, based on a genetic distance of 0.29 (Fig. 5). Group I comprises only 3 genotypes, characterized by predominantly simple flowers, a strong flower scent, full double flowers, and a limited number of stamens. Group II consists of 2 genotypes, primarily presenting a single flowering period per year, a low overall flower count, cup-shaped flowers, wavy or pleated petal tips, and fewer prickles on the stem. Group III encompasses 13 genotypes, predominantly presenting full double flowers with few stamens and long petals. Group IV consists of 32 genotypes, predominantly comprising wild roses and old garden roses, with only one modern rose genotype. These materials primarily present a single flowering period per year with heart-shaped petals. Group V comprises 15 genotypes, most characterized by a large overall flower number, a smooth surface of the calyx tube, yellow anthers, and a low flower height. Group VI encompasses 23 genotypes, including only old and modern garden roses. These genotypes have primarily single flowers with a strong flower scent, full double petals, and biserrate leaves. Group VII comprises 104 genotypes, which are predominantly old garden and modern roses, with only four wild rose genotypes. The rose genotypes in Groups I and II present distinct phenotypic characteristics, such as multiple flowering periods per year, full double flowers, cup-shaped flowers, and light-yellow stigma. Notably, these groups consist of only 5 rose genotypes, suggesting their exceptional uniqueness and potentially constrained exploitation potential in breeding programs. Notably, the C05 (R. rugosa ‘Gaohong’) cultivar in Group II demonstrates remarkable phenotypic traits, positioning it as the fourth-highest scorer in terms of the overall evaluation. Consequently, this cultivar holds significant potential for future utilization in breeding programs.

Cluster dendrogram of the 192 Rosa sp. genotypes based on the phenotypic trait markers. The old garden roses are indicated by green circles, the modern roses are indicated by red triangles, and the wild roses are indicated by blue stars. The three parents of the F1 segregating populations are framed in red
Cluster analysis using SSR markers
In this study, a total of ten pairs of SSR primers were employed to amplify genomic DNA extracted from the 192 Rosa sp. genotypes. The amplified products were subsequently detected. Based on the results, the number of alleles at the ten SSR loci varied between 8 and 20. A total of 131 alleles were identified by amplifying the ten pairs of SSR primers, resulting in an average of 13.1 alleles per locus. Notably, the RW5D11 locus exhibited the highest number of alleles, with 20 alleles identified. Conversely, the CTG21 displayed the lowest allele number, with only eight alleles detected. The mean effective number of alleles across all loci was calculated to be 6.14, ranging from 3.85 to 10.00. The mean polymorphism information index (PIC) was 0.82, ranging from 0.74 to 0.90. Based on the established criterion of PIC≥0.5 (Botstein et al. 1980), it can be concluded that all of the aforementioned loci exhibited a significant level of polymorphism.
A cluster analysis was conducted using 10 SSR markers. The genetic distance varied from 0.03 to 1.00, classifying the 192 germplasms into seven primary groups based on a genetic distance of 0.34. These groups were denoted as Group I to Group VII (Fig. 6). Group I consisted of 20 genotypes, predominantly comprising wild roses and a few old garden rose varieties. Group II included 15 genotypes, consisting of 13 wild roses, 1 old garden rose, and 1 modern rose variety. Group III encompassed 14 genotypes, consisting of 7 old garden roses, 4 modern roses, and 3 wild rose genotypes. Group IV consisted of only 4 genotypes, consisting of 2 wild roses and 2 old garden rose varieties. Group V consisted of only 4 genotypes, comprising 1 modern rose and 3 old garden roses. Group VI encompassed 57 genotypes, with most being modern rose varieties. Group VII comprised 78 genotypes, with most being old garden roses. Overall, the wild rose accessions were predominantly intermingled with a few old and modern rose varieties. The majority of the former are grouped together, as are the latter. In contrast to the phenotypic marker based cluster analysis, the cluster analysis based on molecular markers revealed a more distinct single cluster encompassing the three rose germplasm groups.

Cluster dendrogram of the 192 Rosa sp. genotypes based on the SSR molecular marker genotyping. The old garden roses are indicated by green circles, the modern roses are indicated by red triangles, and the wild roses are indicated by blue stars. The three parents of the F1 segregating populations are framed in red
Two F1 populations revealed abundant variation and extensive segregation
Based on the cluster analysis results, three tetraploid modern rose genotypes, specifically M10, M19, and M29, were selected as parents due to their substantial genetic dissimilarity in phenotypic traits and molecular characteristics. Consequently, two F1 hybrid populations were obtained. The three parents presented significant variation in flower, stem, and leaf phenotypes (Fig. 7a–c). Based on the findings on the germplasm panel, the traits responsible for the variation across the first three principal components in the PCA, which also demonstrated significant differences in the F1 hybrid populations, were selected for phenotypic analysis, namely PN, FD, FH, PL, PW, ASC, SPD, and LN. The objective of creating a hybrid population is to generate novel varieties with exceptional traits while establishing a framework to facilitate the breeding process through the analysis of the inheritance of the traits of interest in the population.

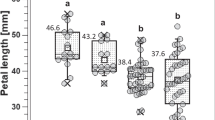
Diagram of crosses between the parental genotypes, frequency distribution histograms, and correlation analysis in the two F1 hybrid populations. (a) The floral traits of the three parental lines M10, M19, M29. (b) The leaf traits of the three parental lines M10, M19, M29. (c) The stem traits of the three parental lines M10, M19, and M29. (d) Frequency distribution of variation types and diversity index of qualitative traits in the M10 × M19 F1 hybrid population. (e) Frequency distribution of variation types and diversity index of qualitative traits in the M10 × M29 F1 hybrid population. (f) Histogram of flower diameter frequency distribution in the M10 × M19 F1 hybrid population. (g) Histogram of flower height frequency distribution in the M10 × M19 F1 hybrid population. (h) Histogram of petal length frequency distribution in the M10 × M19 F1 hybrid population. (i) Histogram of petal width frequency distribution in the M10 × M19 F1 hybrid population. (j) Histogram of flower diameter frequency distribution in the M10 × M29 hybrid population. (k) Histogram of flower height frequency distribution in the M10 × M29 F1 hybrid population. (l) Histogram of petal length frequency distribution in the M10 × M29 F1 hybrid population. (m) Histogram of petal width frequency distribution in the M10 × M29 F1 hybrid population. The gray column in (f)–(m) represents the frequency distribution, and the curve corresponds to the normal distribution curve. The values of the yellow and red triangles in contact with the horizontal coordinate represent the trait values in the maternal and paternal lines, respectively. (n)–(o) Trait correlation heat map in the M10 × M19 and M10 × M29 F1 hybrid populations, respectively. The lower part of the triangle indicates the Pearson correlation value; the higher part of the triangle indicates the significance, * P<0.05, ** P<0.01. The scale bar=1 cm. M10, R. hybrida ‘Midnight Blue’; M19, R. hybrida ‘Sheherazad’'; M29, R. hybrida ‘Couture Rose Tilia’. PN, petal number; ASC, annual stem color; SPD, stem prickle density; LN, number of leaflets
By examining the frequency distribution and genetic diversity index (H') of four qualitative traits in two F1 hybrid populations, we identified 14 variant types across these traits (Table S14). The observed values for H', ranging from 0.53 to 1.16 and 0.28 to 1.29 in the two hybrid populations, respectively, exceeded 1, indicating substantial variation in qualitative traits (Fig. 7d–e). The two F1 hybrid populations presented the greatest variation in SPD and the highest genetic diversity index, suggesting that the genetics underlying SPD are intricate and encompass a wide array of variants. Moreover, the variation regarding the FD, FL, PL, PW, and PN traits in the two F1 hybrid populations was assessed (Table S15). The coefficient of variation in the M10 × M19 hybrid population ranged from 15.82% to 65.49% and from 15.02% to 40.67% in the M10 × M29 hybrid population. These results indicate a medium or higher level of variation in the quantitative traits within the two F1 hybrid populations.
The frequency histogram (Fig. 7f–m) and the kurtosis and skewness values (Table S15) of the four quantitative traits, namely FD, FH, PL, and PW, indicated a continuous multi-peak skewed distribution. The inheritance pattern of these quantitative traits aligned with a mixed major-effect genes-polygene model. Consequently, we applied a mixed inheritance model incorporating major-effect genes and polygenes for the analysis. The AIC values of 11 genetic models were computed for each trait, and a candidate model was chosen for each trait in the two F1 populations based on the minimum AIC values observed (Table S16). The goodness-of-fit test results of the selected candidate models did not achieve statistical significance (P≥0.05) (Table 2), suggesting that the frequency distribution of the traits within the populations closely matched the theoretical distribution based on the candidate genetic models. Consequently, the candidate genetic models were deemed optimal. Based on the chosen optimal genetic models, our findings revealed that PN in the M10 × M19 hybrid population might potentially be controlled by one major-effect locus, while the remaining traits in the two hybrid populations might be controlled by two major-effect loci.
Correlation analysis among traits
Correlation analysis was performed on eight traits within the two F1 hybrid populations (Fig. 7 n–o). In the M10 × M19 hybrid population, out of the correlations assessed on 28 trait pairs, ten trait pairs presented an extremely significant positive correlation, two pairs a highly significant negative correlation, four pairs a significant positive correlation, and one pair a significant negative correlation. Among the correlations assessed on 28 trait pairs within the M10 × M29 hybrid population, eight trait pairs presented a highly significant positive correlation, one pair a highly significant negative correlation, one pair a significant positive correlation, and two pairs a significant negative correlation. Notably, the traits PN, FH, FD, PL, and PW were positively correlated, highly and significantly in both F1 hybrid populations, mirroring the correlations observed in the 192 Rosa sp.genotypes collection. The strong positive correlation between them might be caused by a shared genetic determinant of these traits.
Discussion
Determination of rose core germplasms
The utilization of statistical analysis on phenotypic trait data allows for the more comprehensive evaluation of germplasm resources, thereby facilitating the identification and utilization of genotypes with desirable traits that can be used in the breeding process or cultivated. In this study, the materials presentedd comprehensive scores ranging from -1.175 to 1.464. Notably, the top five genotypes with the highest scores were R. chinensis ‘Zihongxiang’ (C31), R. hybrida ‘Burgundy Iceberg’ (M32), R. hybrida ‘Conrad F. Meyer’ (C55), R. rugosa ‘Gaohong’ (C05), and R. floribunda ‘Sheherazad’ (M19). These genotypes can be considered as the core germplasm collection for future rose breeding practices (Nadeem et al. 2014). In the results of cluster analysis utilizing phenotypic traits, it was observed that all four core genotypes, with the exception of M19, were classified under Group VI. This group primarily consists of old and modern garden roses, suggesting that the materials within Group VI present exceptional phenotypic characteristics. Conversely, in the cluster analysis results based on molecular marker genotyping, two modern rose genotypes, M32 and M19, were classified in Group VI, while the remaining three old garden rose genotypes were classified under Group VII. Furthermore, these three old garden rose genotypes were genetically distinct from the wild roses. This finding suggests that a significant number of breeding lines with high-quality, desirable traits have been generated through long-term rose breeding practices.
The comprehensive phenotypic and molecular marker data from the 192 Rosa sp. genotypes, their genetic group classification, and the populations resulting from high-quality parental lines crossing offer a foundation for future investigations into the molecular mechanisms underlying ornamental traits in roses. In recent years, research on roses had progressively elucidated the molecular mechanisms governing the development and inheritance of their exceptional traits and the regulatory interactions between traits and the environment (Yan et al. 2023), and elucidated an intricate light-mediated regulatory network governing the biosynthesis of anthocyanin in rose petals, using the cultivar R. hybrida ‘Burgundy Iceberg’ as the experimental material. The findings of this study indicated that R. hybrida ‘Burgundy Iceberg’ exhibited the highest score regarding the desirable traits among modern roses, both in terms of phenotypic and molecular marker-based associations, when compared to other contemporary rose varieties. In 2021, a significant milestone was achieved with the completion of the first high-quality genome assembly of R. rugosa at the chromosome level. Comparative analysis with R. chinensis revealed the specific expansion and retention of stress-related genes in R. rugosa, potentially contributing to its adaptation capacity in stressful environments (Chen et al. 2021). Additionally, based on our phenotypic observations, R. rugosa presented superior disease resistance compared to R. chinensis. Therefore, future research efforts can be directed toward investigating the disparity in disease resistance between R. rugosa and R. chinensis to enhance the disease resistance of cultivated varieties. The duration of ornamental plants' attractiveness and ornamental value is determined by the timing and duration of flowering, which indirectly impacts their economic value. Flowering time can be categorized into three types: once-and-only flowering (OF), occasional or re-flowering (OR), and repeated or continuous flowering (CF). Recent studies have suggested that the regulation of rose flowering time may involve homologues of TFL1, RCSPL1-RCTAf15B, and KSN allelic heterozygosity (Iwata et al. 2012; Kurokura et al. 2013; Yu et al. 2023; Bai et al. 2021). Most old garden roses present repeated or continuous flowering during the growing season, whereas most wild roses present once-and-only flowering in a season. Most of the 38 wild roses examined in this study presented once-and-only flowering. However, certain species, such as R. cymosa, R. bracteata, R. igantea, R. gallica, and R. rugosa ‘Purple Branch’, presented repeated or continuous flowering during the growing season. These particular species can serve as valuable germplasm resources for future investigations into flowering time and duration.
Similarities and differences between two cluster analysis approaches
Cluster analysis entails the division of individuals into numerous groups, thereby revealing the degree of relatedness and genetic similarity (Diday and Simon 1976). The methods employed in cluster analysis present inherent consistency, wherein a specific algorithm is employed to aggregate similar individuals into a single class while simultaneously distinguishing those with significant dissimilarities. The progression of cluster analysis, spanning from examining phenotypic traits to exploring genetic diversity through molecular markers, generally gravitates towards simplicity and precision. These methods presents complementary features to improve the identification and appropriate classification of genetic diversity within plant populations.
Our study delved into the interconnections among Rosa genotypes, considering phenotypic traits and molecular markers. Based on a combined analysis of phenotypic traits and molecular markers, the 192 rose genotypes were classified into seven distinct groups. The outcomes of two clustering analyses demonstrated the grouping of wild roses into a single class, signifying their preservation of unique phenotypic traits and notable differentiation from old and modern garden roses. The findings of this study suggest that the molecular markers examined have the potential to accurately assess kinship and similarity differences at the molecular level. The clustering analysis results of 65 Rosa spp. genotypes assessed by six STMS markers aligned with the horticultural classification results, which were consistent with the findings of our study (Scariot et al. 2006). However, some uncertainties persist despite the general classification of old garden rose, modern rose, and wild rose genotypes into three distinct groups. These observations highlight the challenges associated with the horticultural classification of cultivars (Liorzou et al. 2016). The wild roses analyzed in this study exhibit several distinctive phenotypic traits, including a once-per-season flowering, single-petal flowers, a high leaflet number, and trichome-like features on the calyx tube surface. These traits serve to differentiate them from both old garden and modern garden rose genotypes. The classification of old and modern garden roses dates back to 1867, and the clustering analysis conducted in this study confirms the longstanding close relationship between them, considering their breeding history. Furthermore, they share similarities in both phenotypic and molecular marker profiles. At the same time, old garden rose genotypes hold great importance in the evolutionary trajectory and breeding strategies employed to improve modern garden roses.
Significantly, discernible disparities were observed in the clustering outcomes of the two methodologies. Specifically, the genetic distance range derived from phenotypic trait clustering ranged from 0.15 to 0.91, a notably narrower span than the range of 0.03 to 1.00 observed by molecular marker clustering. This discrepancy indicates the accuracy and reliability of molecular markers, as they remain unaffected by external environmental factors (Yang et al. 2020). Muitiple simple sequence repeat (SSR) markers have been developed and extensively employed in the examination of genetic diversity in ornamental plants and cash crops (Meng et al. 2009; Panwar et al. 2015; Amar et al. 2011), thereby facilitating the genetic classification of both wild and cultivated roses (Scariot et al. 2006; Meng et al. 2009; Panwar et al. 2015).
In the current study, notable differences in phenotypic characteristics were observed among the wild, old garden, and modern rose genotypes. Additionally, many of these rose genotypes were grouped in a single cluster at the molecular level, indicating that the conservation of numerous repetitive elements within the Rosa genus plants is preserved over the long-term evolution and selective breeding, despite the considerable variation at a genome-wide level. The clustering analysis revealed that many wild roses did not contribute to the generation of contemporary garden roses. Subsequently, it is recommended that further studies be conducted into the exceptional traits of both old garden roses and wild roses to integrate them into modern rose breeding practices (Zlesak 2006).
The heredity of rose traits is complex
In this study, we have discovered a significant overlap and correlation between traits of old garden roses and modern roses. Initially, during the early stages of hybrid tea rose breeding, only ten rose species, namely R. chinensis, R. foetida, R. gallica, R. gigantea, R. moschata, R. multiflora, R. phoenicea, R. rugosa, R. rubra and R. wichurana (Crespel and Mouchotte 2003), were utilized as parental plants. However, the intercrossing among different genotypes progressively intensified as breeding techniques advanced. This phenomenon was further intensified by environmental shifts and deliberate selection (De Vries and Dubois 1996), gradually increased phenotypic trait variability in the rose germplasm resources (Fig. 2c–f).
Flower color, flower type, and floral scent of ornamental plants have received increased attention from researchers (Bendahmane et al. 2013). In the natural environment, bright flower colors are crucial in attracting pollinating insects (Davies et al. 2012). In contemporary times, flower color has also become a significant factor in consumer selection (Behe et al. 1999; Wijayani et al. 2017). The pigments present in rose petals primarily consist of anthocyanins, flavonols, and carotenoids (Wan et al. 2019). The presence and quantity of these three pigments determine the wide range of colors observed in roses, thereby contributing to the intricate mechanisms controlling rose color formation. Among the 192 Rosa sp. genotypes assessed in this study, the petal color variation and its genetic diversity index were the highest. This suggests that the genetic mechanisms underlying flower color are intricate, and multiple genes likely influence the differentiation in color between various species.
The flower, the primary ornamental component of the Rosa genus, has consistently garnered attention in studies on the genetic and regulatory mechanisms governing petal number. This study reveals a significant negative correlation between petal number (PN) and sepal number (SN) across a substantial collection of 192 Rosa sp. genotypes. The negative correlation between the floral organ number in different whorls may be attributed to the differential expression of B and C genes in the ABC model, resulting in the transformation of petals in whorl 2 into stamens in whorl 3 (Coen and Meyerowitz 1991; Weigel and Meyerowitz 1994; Kitahara et al. 2004; Francois et al. 2018). Previous studies have considered the number of petals as a quantitative trait regulated by multiple quantitative trait loci (QTLs) (Hibrand Saint-Oyant et al. 2008, Debener et al. 2001, Roman et al. 2015). In recent years, it has been suggested that two closely linked loci on LG3 (~27.80–33.83 Mbp) control petal number in double flowers (Hibrand Saint-Oyant et al. 2018; Schulz et al. 2021; Rawandoozi et al. 2023).
In cultivated rose species, the practice of negative selection against the presence of rose stem pickles is commonly observed (Chaanin 2003). In this study, a markedly pronounced negative correlation was identified between SPD and ASC. Previous studies have suggested that prickles on stems may be governed by the inheritance of multiple major-effect dominant genes (Debener and Linde 2009; Linde et al. 2006). Moreover, the association between stem prickles and stem color remains ambiguous, and their apparent negative correlation is worth further investigation in future research.
Conclusions
In summary, an integrated analysis was undertaken to evaluate the genetic diversity of 192 Rosa sp. genotypes through phenotypic and molecular markers. This analysis has erected a fundamental basis for understanding the genetic phenotypic trait variation within the genus Rosa, as well as for the identification and utilization of core germplasm resources, the selection of appropriate parental lines for hybrid breeding, and the development of novel varieties with desirable traits. In future research, cluster analysis results can be utilized to identify appropriate parental lines from diverse classification roses, facilitating the development of hybrid populations. Overall, this study not only advances the exploration of the genetics and inheritance of valuable ornamental traits but also establishes a foundation for the investigation and exploitation of genetic diversity in other ornamental plant germplasm resources.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. Data will be shared upon request by the readers.
References
Amar MH, Biswas MK, Zhang ZW, Guo WW. Exploitation of SSR, SRAP and CAPS-SNP markers for genetic diversity of Citrus germplasm collection. Sci Hortic. 2011;128:220–7. https://doi.org/10.1016/j.scienta.2011.01.021.
Bai M, Liu J, Fan C, Chen Y, Chen H, Lu J, et al. KSN heterozygosity is associated with continuous flowering of Rosa rugosa Purple branch. Hort Res. 2021;8:26. https://doi.org/10.1038/s41438-021-00464-8.
Basu SK, Zandi P, Cetzal-Ix W, Sengupta R. The genus Rosa: An aristocrat from the plant family with class, color and fragrance. Iran Soc Environ. 2015.
Behe B, Nelson R, Barton S, Hall C, Safley CD, Turner S. Consumer preferences for geranium flower color, leaf variegation, and price. HortScience. 1999;34:740–2. https://doi.org/10.21273/HORTSCI.34.4.740.
Bendahmane M, Dubois A, Raymond O, Bris ML. Genetics and genomics of flower initiation and development in roses. J Exp Bot. 2013;64:847–57. https://doi.org/10.1093/jxb/ers387.
Ben-Meir H, Vainstein A. Assessment of genetic relatedness in roses by DNA fingerprint analysis. Sci Hortic. 1994;58:115–21. https://doi.org/10.1016/0304-4238(94)90132-5.
Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980;32:314–31.
Calle A, Balas F, Cai L, Iezzoni A, López-Corrales M, Serradilla MJ, et al. Fruit size and firmness QTL alleles of breeding interest identified in a sweet cherry ‘Ambrunés’ × ‘Sweetheart’ population. Mol Breed. 2020;4086. https://doi.org/10.1007/s11032-020-01165-1.
Cao Y, Wang GY, Wang SC, Wei YL, Lu J, Xie YJ, et al. Construction of a genetic map and location of quantitative trait loci for dwarf trait in maize by RFLP markers. Chi Sci Bull. 2000;45:247–50. https://doi.org/10.1007/BF02884683.
Chaanin A. Breeding| Selection strategies for cut roses. In: Roberts AV, Debener T, Gudin S, editors. Encyclopedia of rose science. Oxford, UK: Elsevier Ltd; 2003. p. 33–41. https://doi.org/10.1016/b0-12-227620-5/00099-9.
Chen F, Su LY, Hu SY, Xue JY, Liu H, Liu GH, et al. A chromosome-level genome assembly of rugged rose (Rosa rugosa) provides insights into its evolution, ecology, and floral characteristics. Hort Res. 2021;8:141. https://doi.org/10.1038/s41438-021-00594-z.
Cheng JS. Genetic breeding of garden plants. 1rded. Beijing: China Forestry Publishing House; 2000.
Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353:31–7. https://doi.org/10.1038/353031a0.
Crespel L, Mouchotte J. Methods of cross-breeding. In: Roberts AV, Debener T, Gudin S, editors. Encyclopedia of rose science. Oxford, UK: Elsevier Ltd; 2003. p. 30–3. https://doi.org/10.1016/B978-0-12-809633-8.05005-6.
Datta SK. Breeding of new ornamental varieties:rose. Curr Sci. 2018;114:1194–206. https://doi.org/10.18520/cs/v114/i06/1194-1206.
Davies KM, Albert NW, Schwinn KE. From landing lights to mimicry: the molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct Plant Biol. 2012;39:619–38. https://doi.org/10.1071/FP12195.
De Vries DP, Dubois LAM. Rose breeding: past, present, prospects. Acta Hortic. 1996;424:241–8. https://doi.org/10.17660/ActaHortic.1996.424.43.
Debener T, Linde M. Exploring complex ornamental genomes: the rose as a model plant. Crit Rev Plant Sci. 2009;28:267–80. https://doi.org/10.1080/07352680903035481.
Debener T, Von Malek B, Mattiesch L, Kaufmann H. Genetic and molecular analysis of important characyers in rose. Acta Hortic. 2001;547:45–9. https://doi.org/10.17660/ActaHortic.2001.547.5.
Dice LR. Measures of the amount of ecologic association between species. Ecology. 1945;26:297–302. https://doi.org/10.2307/1932409.
Diday E, Simon JC. Clustering Analysis. In: Fu KS, editor. Digital pattern recognition. Berlin, Heidelberg: Springer; 1976. p. 47–94. https://doi.org/10.1007/978-3-642-96303-2_3.
Francois L, Verdenaud M, Fu XP, Ruleman D, Dubois A, Vandenbussche M, et al. A miR172 target-deficient AP2-like gene correlates with the double flower phenotype in roses. Sci Rep. 2018;8:12912. https://doi.org/10.1038/s41598-018-30918-4.
Gai J, Zhang Y, Wang J. Genetic system of quantitative traits in plant. Beijing: Science Press; 2003.
Glaszmann JC, Kilian B, Upadhyaya HD, Varshney RK. Accessing genetic diversity for crop improvement. Curr Opin Plant Biol. 2010;13:167–73. https://doi.org/10.1016/j.pbi.2010.01.004.
Gulsen O, Karagul S, Abak K. Diversity and relationships among Turkish okra germplasm by SRAP and phenotypic marker polymorphism. Biologia. 2007;62:41–5. https://doi.org/10.2478/s11756-007-0010-y.
Hibrand Saint-Oyant L, Ruttink T, Hamama L, Kirov I, Lakhwani D, Zhou NN, et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat Plants. 2018;4:473–84. https://doi.org/10.1038/s41477-018-0166-1.
Hibrand-Saint Oyant L, Crespel L, Rajapakse S, Zhang L, Foucher F. Genetic linkage maps of rose constructed with new microsatellite markers and locating QTL controlling flowering traits. Tree Genet Genomes. 2008;4:11–23. https://doi.org/10.1007/s11295-007-0084-2.
Iwata H, Gaston A, Remay A, Thouroude T, Jeauffre J, Kawamura K, et al. The TFL1 homologue KSN is a regulator of continuous flowering in rose and strawberry. Plant J. 2012;69:116–25. https://doi.org/10.1111/j.1365-313X.2011.04776.x.
Jian HY, Zhang T, Wang QG, Li SB, Zhang H, Tang KX. Karyological diversity of wild Rosa in Yunnan, southwestern China. Genet Resour Crop Ev. 2013;60:115–27. https://doi.org/10.1007/s10722-012-9820-z.
Jin N, Jin L, Luo S, Tang Z, Liu Z, Wei S, et al. Comprehensive evaluation of amino acids and polyphenols in 69 varieties of green cabbage (Brassica oleracea L. var. capitata L.) based on multivariate statistical analysis. Molecules. 2021;26:5355. https://doi.org/10.3390/molecules26175355.
Kitahara K, Hibino Y, Aida R, Matsumoto S. Ectopic expression of the rose AGAMOUS-like MADS-box genes ‘MASAKO C1 and D1’ causes similar homeotic transformation of sepal and petal in Arabidopsis and sepal in Torenia. Plant Sci. 2004;166:1245–52. https://doi.org/10.1016/j.plantsci.2003.12.040.
Kurokura T, Mimida N, Battey NH, Hytönen T. The regulation of seasonal flowering in the Rosaceae. J Exp Bot. 2013;64:4131–41. https://doi.org/10.1093/jxb/ert233.
Lau J, Young EL, Collins S, Windham MT, Klein PE, Byrne DH, et al. Rose rosette disease resistance loci detected in two interconnected tetraploid garden rose populations. Front Plant Sci. 2022;13:916231. https://doi.org/10.3389/fpls.2022.916231.
Li XW. Two big biodiversity centers of Chinese endemic genera of seed plants and their characteristics in Yunnan province. Acta Bot Yunnanica. 1994;16:221–7.
Lidia A, Irina SN. Theoretical considerations upon the origin and the origin and nomenclature of the present rose cultivars. J Plant Dev. 2009;16:103–8.
Linde M, Hattendorf A, Kaufmann H, Debener T. Powdery mildew resistance in roses: QTL mapping in different environments using selective genotyping. Thero Appl Genet. 2006;113:1081–92. https://doi.org/10.1007/s00122-006-0367-2.
Liorzou M, Pernet A, Li S, Chastellier A, Thouroude T, Michel G, et al. Nineteenth century French rose (Rosa sp.) germplasm shows a shift over time from a European to an Asian genetic background. J Exp Bot. 2016;67:4711–25. https://doi.org/10.1093/jxb/erw269.
Liu ZZ, Guo RH, Zhao JR, Cai YL, Wang FG, Cao MJ, et al. Genetic diversity of two important groups of maize landraces with same name in China revealed by M13 tailed-primer SSRs. Agr Sci China. 2009;8:15–23. https://doi.org/10.1016/s1671-2927(09)60004-3.
Liu Y, Chen T, Zhang J, Wang J, Wang H, Tang HR, et al. Genetic diversity analysis of Chinese cherry landraces (Prunus pseudocerasus) based on phenotypic traits. Acta Hortic Sin. 2016;43:2119–32. https://doi.org/10.16420/j.issn.0513-353x.2016-0092.
Liu MJ. Studies on the variations and probability gradings of major quantitative characters of Chinese jujube. Acta Hortic Sin. 1996:105-9.
Meng J, Li DZ, Yi TS, Yang JB, Zhao XF. Development and characterization of microsatellite loci for Rosa odorata var. gigantea Rehder & E. H. Wilson (Rosaceae). Conserv Genet. 2009;10:1973–6. https://doi.org/10.1007/s10592-009-9871-7.
Meng J, Fougere-Danezan M, Zhang LB, Li DZ, Yi TS. Untangling the hybrid origin of the Chinese tea roses: evidence from DNA sequences of single-copy nuclear and chloroplast genes. Plant Syst Evol. 2011;297:157–70. https://doi.org/10.1007/s00606-011-0504-5.
Nadeem M, Wang X, Akond M, Awan FS, Riaz A, Younis A. Hybrid identification, morphological evaluation and genetic diversity analysis of Rosa × hybrida by SSR markers. Aust J Crop Sci. 2014;8:183–90. https://doi.org/10.3316/informit.197817439482917.
Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA. 1979;76:2569–5273. https://doi.org/10.1073/pnas.76.10.5269.
Nybom H. Introduction to Rosa. In: Folta KM, Gardiner SE, editors. Genetics and genomics of Rosaceae. New York: Springer; 2009. p. 339–51. https://doi.org/10.1007/978-0-387-77491-6_16.
Panwar S, Singh KP, Namita N, Sonah H, Deshmukh R, Sharma T. Identification and characterization of microsatellites in ESTs of Rosa species: insight in development of SSR markers. Indian J Agric Sci. 2015;85:429–33. https://doi.org/10.56093/ijas.v85i3.47189.
Qi WC, Chen X, Fang PH, Shi SC, Li JJ, Liu XT, et al. Genomic and transcriptomic sequencing of Rosa hybrida provides microsatellite markers for breeding, flower trait improvement and taxonomy studies. BMC Plant Biol. 2018;18:119. https://doi.org/10.1186/s12870-018-1322-5.
Rawandoozi Z, Young EL, Liang SY, Wu X, Fu QY, Hochhaus T, et al. Pedigree-based QTL analysis of flower size traits in two multi-parental diploid rose populations. Front Plant Sci. 2023;14:1226713. https://doi.org/10.3389/fpls.2023.1226713.
Raymond O, Gouzy J, Just J, Badouin H, Verdenaud M, Lemainque A, et al. The Rosa genome provides new insights into the domestication of modern roses. Nat Genet. 2018;50:772–7. https://doi.org/10.1038/s41588-018-0110-3.
Ringnér M. What is principal component analysis. Nat Biotechnol. 2008;26:303–4. https://doi.org/10.1038/nbt0308-303.
Roman H, Rapicault M, Miclot AS, Larenaudie M, Kawamura K, Thouroude T, et al. Genetic analysis of the flowering date and number of petals in rose. Tree Genet Genomes. 2015;11:85. https://doi.org/10.1007/s11295-015-0906-6.
Rui WJ, Wang XM, Zhang QN, Hu XY, Hu XH, Fu JJ, et al. Genetic diversity analysis of 353 tomato germplasm resources by phenotypic traits. Acta Hortic Sin. 2018;45:561–70. https://doi.org/10.16420/j.issn.0513-353x.2017-0274.
Scariot V, Akkak A, Botta R. Characterization and genetic relationships of wild species and old garden roses based on microsatellite analysis. J Am Sor Hortic. 2006;131:66–73. https://doi.org/10.21273/JASHS.131.1.66.
Schulz D, Linde M, Debener T. Detection of reproducible major effect QTL for petal traits in garden roses. Plants. 2021;10:897. https://doi.org/10.3390/plants10050897.
Sensoy S, Büyükalaca S, Abak K. Evaluation of genetic diversity in Turkish melons (Cucumis melo L.) based on phenotypic characters and RAPD markers. Genet Resour Crop Ev. 2007;54:1351–65. https://doi.org/10.1007/s10722-006-9120-6.
Shi P, Xu Z, Zhang SY, Wang XJ, Ma XF, Zheng JC, et al. Construction of a high-density SNP-based genetic map and identification of fruit-related QTLs and candidate genes in peach [Prunus persica (L.) Batsch]. BMC Plant Biol. 2020;20:438. https://doi.org/10.1186/s12870-020-02557-3.
Shupert DA, Byrne DH, Brent Pemberton H. Inheritance of flower traits, leaflet mumber and prickles in roses. Acta Hortic. 2007;751:331–5. https://doi.org/10.17660/ActaHortic.2007.751.42.
Silberstein L, Kovalski I, Brotman Y, Perin C, Dogimont C, Pitrat M, et al. Linkage map of Cucumis melo including phenotypic traits and sequence-characterized genes. Genome. 2003;46:761–73. https://doi.org/10.1139/g03-060.
Singh N, Vasudev S, Kumar Yadava D, Kumar S, Naresh S, Ramachandra Bhat S, et al. Assessment of genetic diversity in Brassica juncea (Brassicaceae) genotypes using phenotypic differences and SSR markers. Rev Biol Trop. 2013;61:1919–34. https://doi.org/10.56093/ijas.v91i6.114299.
Smulders MJM, Arens P, Bourke P, Debener T, Linde M, Riek J, et al. In the name of the rose: a roadmap for rose research in the genome era. Hort Res. 2019;6:65. https://doi.org/10.1038/s41438-019-0156-0.
Soleimani VD, Baum BR, Johnson DA. AFLP and pedigree-based genetic diversity estimates in modern cultivars of durum wheat [Triticum turgidum L. subsp durum (Desf.) Husn.]. Thero Appl Genet. 2002;104:350–7. https://doi.org/10.1007/s001220100714.
Tan JR, Wang J, Luo L, Yu C, Xu TL, Wu YY, et al. Genetic relationships and evolution of old Chinese garden roses based on SSRs and chromosome diversity. Sci Rep. 2017;7:15437. https://doi.org/10.1038/s41598-017-15815-6.
Tang KX, Qiu XQ, Zhang H, Li SF, Wang QG, Jian HY, et al. Study on genetic diversity of some Rosa germplasm in Yunnan based on SSR markers. Acta Hortic Sin. 2008;35:1227–32.
Tena Gashaw E, Mekbib F, Ayana A. Genetic diversity among sugarcane genotypes based on qualitative traits. Adv Agri. 2016;148:8909506. https://doi.org/10.1155/2016/8909506.
Wan HH, Yu C, Han Y, Guo XL, Luo L, Pan HT, et al. Determination of flavonoids and carotenoids and their contributions to various colors of rose cultivars (Rosa spp.). Front Plant Sci. 2019;10:123. https://doi.org/10.3389/fpls.2019.00123.
Wang Y, Ma Y, Dai S. The molecular mechanism in regulation of flowering in ornamental plants. J Integr Plant Biol. 2010;45:641–53. https://doi.org/10.3969/j.issn.1674-3466.2010.06.001.
Wang JT, Zhang YW, Du YW, Ren WL, Li HF, Sun WX, et al. SEA v2.0: an R software package for mixed major genes plus polygenes inheritance analysis of quantitative traits. Acta Agron Sin. 2022;48:1416–24. https://doi.org/10.3724/SP.J.1006.2022.14088.
Wang Y, Liu ZS, Yang XQ, Wang ZY, Ma L, Tu HX, et al. Inheritance analysis of fruit-related traits in Chinese cherry [Cerasus pseudocerasus (Lindl.) G.Don] breeding progenies. Sci Hortic. 2023;307:111519.
Weigel D, Meyerowitz EM. The ABCs of floral homeotic genes. Cell. 1994;78:203–9. https://doi.org/10.1016/0092-8674(94)90291-7.
Wijayani A, Muafi M, Sukwadi R. Market actor’s response towards flower colours in determining the economic value of Chrysanthemum flowers. J Bus Retal Manag Res. 2017;12:69–75. https://doi.org/10.24052/jbrmr/v12is01/martfcidtevocf.
Wissemann V. Conventional taxonomy (wild roses). In: Roberts AV, Debener T, Gudin S, editors. Reference module in life sciences. Elsevier; 2017. p. 1–6. https://doi.org/10.1016/B978-0-12-809633-8.05017-2.
Yaghini H, Shirani M, Archangi A, Sorkheh K, Chaleshtori SB, Sangi SE, et al. Phenotypic diversity and relationships of fruit quality traits in inter-specific almond × peach backcrosses breeding progenies. Euphytica. 2013;194:305–24. https://doi.org/10.1007/s10681-013-0893-3.
Yan YH, Zhao JX, Lin SN, Li ML, Liu JY, Raymond O, et al. Light-mediated anthocyanin biosynthesis in rose petals involves a balanced regulatory module comprising transcription factors RhHY5, RhMYB114a, and RhMYB3b. J Exp Bot. 2023;74:5783–804. https://doi.org/10.1093/jxb/erad253.
Yanchuk AD. A quantitative framework for breeding and conservation of forest tree genetic resources in British Columbia. Can J For Res. 2001;31:566–76. https://doi.org/10.1139/x00-133.
Yang CY, Ma YJ, Cheng BX, Zhou LJ, Yu C, Luo L, et al. Molecular evidence for hybrid origin and phenotypic variation of Rosa section Chinenses. Genes. 2020;11:996. https://doi.org/10.3390/genes11090996.
Yirgu M, Kebede M, Feyissa T, Lakew B, Woldeyohannes AB. Morphological variations of qualitative traits of barley (Hordeum vulgare L.) accessions in Ethiopia. Heliyon. 2022;8:e10949.
Yu R, Xiong ZY, Zhu XH, Feng PP, Hu ZY, Fang RX, et al. RcSPL1–RcTAF15b regulates the flowering time of rose (Rosa chinensis). Hort Res. 2023;10:uhad083. https://doi.org/10.1093/hr/uhad083.
Zhang LH, Byrne DH, Ballard RE, Rajapakse S. Microsatellite marker development in rose and its application in tetraploid mapping. J Am Soc Hortic Sci. 2006;131:380–7. https://doi.org/10.21273/JASHS.131.3.380.
Zhao L, Zhang D. Ornamental plant resources from China. Acta Hortic. 2003;620:365–75. https://doi.org/10.17660/ActaHortic.2003.620.46.
Zieliński J, Tomaszewski D, Guzicka M, Maciejewska-Rutkowska I. Stomata on the pericarp of species of the genus Rosa L. (Rosaceae). Plant Syst Evol. 2010;284:49–55. https://doi.org/10.1007/s00606-009-0234-0.
Zietkiewicz E, Rafalski A, Labuda D. Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics. 1994;20:176–83. https://doi.org/10.1006/geno.1994.1151.
Zlesak DC. Rose. In: Anderson NO, editor. Flower Breeding and Genetics. Dordrecht: Springer; 2006. p. 695–740. https://doi.org/10.1007/978-1-4020-4428-1.
Acknowledgements
Not applicable.
Funding
This work was supported by funding from National Key R&D Program of China (2021YFD1200205 and 2019YFD1000400).
Author information
Authors and Affiliations
Contributions
XF conceived and coordinated this project; XY, BH and HG performed the experiments with contributions from JZ, SY, QW, MB1 and MB2; HG and BH wrote the manuscript, under the supervision of and with contributions from XF and XY. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. Prof. Mohammed Bendahmane and Prof. Manzhu Bao are both editorial board members of Horticulture Advances, and were not involved in the journal’s review or decisions related to this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary table 1. Information of 192 Rosa germplasm resources. Supplementary table 2. 33 phenotypic traits statistical assignment and measurement standards. Supplementary table 3. 9 phenotypic traits statistical assignment and measurement standards of two hybrid populations. Supplementary table 4. Genetic diversity analysis of 6 quantitative characters. Supplementary table 5. Genetic diversity analysis of 27 quality traits. Supplementary table 6. Variation analysis of inflorescence types. Supplementary table 7. Variation analysis of flower color types. Supplementary table 8. Variation analysis of petal number types. Supplementary table 9. Variation analysis of flower types. Supplementary table 10. Polymorphism information of 10 SSR primers. Supplementary table 11. The total variance of principal components.Supplementary table 12. Principal component load value information of 33 traits. Supplementary table 13. Comprehensive scores of 192 species of Rosa germplasm. Supplementary table 14. Frequency distribution and diversity index of variation types for 4 quality traits. Supplementary table 15. Descriptive analysis parameters of five traits. Supplementary table 16. The AIC values of 5 quantitative traits from two hybrid populations. Supplementary fig. 1. The scree plot of principal component analysis. Supplementary fig. 2. Loding plot of 19 phenotypic traits for the first three PCs in 192 genus Rosa germplasm resources. Supplementary fig. 3. Correlation analysis of comprehensive score F and phenotypic traits. Supplementary fig. 4. Front view and side view of some materials flower.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guan, H., Huang, B., Yan, X. et al. Identification of distinct roses suitable for future breeding by phenotypic and genotypic evaluations of 192 rose germplasms. HORTIC. ADV. 2, 5 (2024). https://doi.org/10.1007/s44281-023-00024-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44281-023-00024-1