
Abstract
Tulips (Tulipa gesneriana) are one of the most widely cultivated bulbous plants with substantial ornamental value. However, the lack of well-documented reference genomes has limited the research progress and molecular breeding of tulips. In the present study, a full-length transcriptome of a commercial tulip cultivar was obtained using single-molecule long-read sequencing (PacBio Iso-Seq). In total, 244,357 full-length transcripts were identified, which had an average length of 2,044 bp and an N50 value of 3,861; 67,350 of these were annotated to databases. An inaugural integrated analysis of the transcriptome and phytohormone profiles during flower opening and petal senescence was performed using Illumina RNA-seq, coupled with Mfuzz (an R pakage, http://mfuzz.sysbiolab.eu) and weighted gene coexpression network analysis (WGCNA). A total of 16 gene coexpression and six transcription factor (TF) modules were constructed. Additionally, 26 hormone analogs were comprehensively profiled. Finally, a prominently novel gene, Tulipa gesneriana Homeobox12-like (TgHB12-like), which encodes an homeodomain–leucine zipper (HD-zip) TF, was identified as a pivotal regulator of petal senescence. Overall, this work facilitates the identification of hormones and TFs in plants related to flower opening and senescence in tulips. It also provides an important and valuable genetic basis for further research in them.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tulip (Tulipa L.) is widely known throughout the world market and has great commercial significance. It has been cultivated for cut flowers as a garden, potted, and landscaping plant. Until now, more than 8,000 tulip cultivars have been bred with a variety of traits regarding flower shapes, colors, lifespan, plant architecture, and resistance to abiotic stress (Van Scheepen 1996; Marasek-Ciolakowska et al. 2012). A majority of the cultivars, which number 1100 and are distributed in the commercial market, belong to Tulipa gesneriana L. and the Darwin hybrid group (Van Tuyl and Van Creil 2007). The latter is a large family of tulips that was bred through interspecific crosses between T. generiana and T. fosteriana and has remarkable resistance.
Flower longevity is an essential horticultural trait in tulips, which is determined based on petal senescence or abscission. A repetitive diurnal petal movement was observed during the flower opening period, which is considered an indicator of the duration of flower bloom in tulips (van Doorn and van Meeteren 2003; Azad et al. 2008). Phytohormones, transcription factors (TFs), and environmental stimuli are critical regulators of flower opening and senescence (Rogers 2013; Ma et al. 2018; Baharudin and Osman 2023). Abscisic acid (ABA) treatment sped up flower senescence in daylilies (Hemerocallis fulva) (Panavas et al. 1998) and daffodils (Narcissus pseudonarcissus) (Hunter et al. 2004). In Lily (Lilium spp.) and gladiolus (Gladiolus palustris), the ABA content increased following flower senescence (Arrom and Munné-Bosch 2012; Kumar et al. 2014). Ethylene plays a dual role in the regulation of flower senescence in tulips (Sexton et al. 2000; Wang et al. 2020). Transcription factors (TFs), in general, are critical regulators of senescence initiation in flowers in hormone-dependent or independent manners. Rosa hybrida MYB transcription factor 108 (RhMYB108) participated in petal senescence in roses induced by ethylene and Jasmonic acid (JA) (Zhang et al. 2019). FOREVER YOUNG FLOWER (FYF) encodes a MADS-box TF that acts as a negative regulator of ethylene responses to repress flower senescence and abscission (Chen et al. 2015). Silencing of Petunia hybrida homeodomain–leucine zipper (PhHD-Zip), which encodes a homeodomain–leucine zipper (HD-Zip) protein, remarkably extended flower longevity in petunias (Chang et al. 2014). Another HD-Zip TF, RhHB1, positively induces petal senescence in roses in an ABA- and ethylene-dependent manner (Lü et al. 2014). PhFBH4,a basic helix–loop–helix (bHLH) TF, regulated ethylene biosynthesis to modulate petal senescence (Yin et al. 2015). Besides, ANTHOCYANIN1 (AN1), another bHLH TF, regulated petal senescence and anthocyanin accumulation in petunias (Prinsi et al. 2016). Phytohormones also play essential roles in flower longevity regulation in tulips. In tulips, ethylene production was enhanced during petal senescence (Wang et al. 2020). An NAM/ATAF/CUC (NAC) TF, TgNAP, elevated salicylic acid (SA) and H2O2 accumulation to promote petal senescence in tulips (Meng et al. 2022). Recently, it was found that tulip TgWRKY75 activates salicylic acid and abscisic acid biosynthesis to synergistically promote petal senescence (Meng et al. 2024). All of these researches indicated that hormones and TFs usually performed their roles synergistically to regulate the lifespan of flowers.
Over the past decade, transcriptomics has been widely used to explore insights into the genetic information of plant species. All Tulipa spp. possess a large-sized genome, and the diploid 2C DNA values varied from 32 to 69 pg in T. clusiana (Clusius tulip) and T. gesneriana, respectively (Zonneveld 2009). In 2012, using 454 pyro-sequencing technology, the first set of transcriptome contigs was reported in tulip spp., which had an average length of 514 bp, and partial transcriptomic data was obtained (Shahin et al. 2012). Molecular markers such as simple sequence repeats (SSRs) and single nucleotide polymorphisms (SNPs) have been developed for genotyping identification in tulips with potential application in molecular breeding (Shahin et al. 2012; Tang et al. 2013). At present, little genomic and transcriptomic information is available in tulips, which limits the progress of molecular breeding research.
Full-length transcriptome sequencing is of great significance as it can overcome the limitations of short-read RNA sequencing without further assembly. This study employed PacBio Isoform-Seq and Illumina RNA-seq technologies to reveal the complexity of the full-length transcriptome of T. gesneriana used as a reference. Here, we report the inaugural release of a full-length reference transcriptome sequence in tulips. Additionally, the transcriptome and phytohormone profiles during flower opening and petal senescence have been comprehensively analyzed. In total, 51 and 49 TFs were identified to be significantly altered during flower opening and petal senescence, respectively. The most enriched pathways in these processes were those related to ABA and auxin, followed by ethylene, cytokinins (CKs), SA, and JA. Furthermore, a HB-Zip TF, TgHB12-like, was functionally characterized as a positive regulator of petal senescence in tulips. This research may contribute to unraveling the characteristics of the T. gesneriana transcriptome and provide a valuable genetic resource for further study in tulips.
Materials and methods
Plant material and growth conditions
A total of 37 tulip cultivars were used in this study, and the detailed information is listed in Table S1. T. gesneriana cultivar ‘Red Power’ was used for PacBio-based Isoform (Iso)-Seq, Illumina RNA-Seq, gene expression, and virus-induced gene silencing (VIGS) assay. The perimeters of all bulbs used in this study were 12–13 cm, which were ordered from the Beijing Sangu Co., Beijing, China. Tulip plants were planted in a greenhouse located on the campus of the Huazhong Agricultural University, Wuhan, China (114°21′E, 30°28′N; altitude 30 m). The growth conditions were maintained at 20°C day/15°C night under a 16 h light/8 h dark photoperiod. The relative humidity was kept at 60%–70%.
Samples collection and RNA preparation
For PacBio Iso-Seq, six tissues, including leaves, flowers, stems, roots, floral buds, and bulb scales of tulip plants, were collected. For Illumina RNA-Seq, flowers at five different developmental stages in triplicates (15 flowers for each stage) were gathered. The flower stages were selected as defined previously (Wang et al. 2020). The total RNA of each tissue was extracted using TRIzol™ reagent (Invitrogen, MA, USA). The RNA integrity number (RIN) of all RNAs is observed to be approximately 7–9. All sequencing experiments were performed at the Biomarker Information Co. Ltd., Beijing, China.
PacBio Iso-Seq, data processing, and correction
For single-molecule real-time (SMRT) sequencing, the RNA samples from all six tissues were pooled in equal amounts for cDNA synthesis using the SMRTer™ PCR cDNA Synthesis Kit (Clontech, CA, USA). Three SMRT libraries of lengths 1–2, 2–3, and 3–6 kb were generated and sequenced using eight SMRT cells on the RS II system (Pacific Biosciences, CA, USA) (Sharon et al. 2013). Full-length (FL) and full-length non-chimeric (FLNC) transcripts were screened with a polyA tail signal and the 5’ and 3’ primers of the cDNA in Regions of Interests (ROIs). The FLNC reads were clustered using the RS_IsoSeq module of SMRT analysis (V2.3.0). The Illumina RNA-Seq data was employed for hybrid error correction implemented with Proovread (v2.13.13) (Hackl et al. 2014). A post-correction accuracy of > 99% of the FL transcripts was obtained and used for further analysis. High-quality Iso-Seq FL transcripts were obtained after removing redundancy (identity > 0.99), filtering the adapter sequences, and low-quality reads (read-length < 50 bp and read-score < 0.75) (Gao et al. 2019). Bowtie2 was used to blast the SMRT transcript sequence reads obtained from each sample (Langmead and Salzberg 2012). Finally, the novel high-quality FL transcript data set for T. gesneriana was acquired. The workflow for SMRT sequencing is shown in Fig. S1.
Identification of SSRs, long non-coding RNAs, and prediction of alternative splice sites
Of the total sequence, 171,242,964 bp long, 76,291 transcripts > 500 bp were employed for SSR identification using MISA (http://pgrc.ipk-gatersleben.de/misa/). Those with more than two perfect SSRs and an interval less than 100 bp were indicated as compound SSRs. The candidate, long no-coding RNA (lncRNA) transcripts, were selected based on length more than 200 nts and exons more than 2, and further screened using the CPC/CNCI/CPAT/Pfam databases (Sun et al. 2013). The Iso-Seq™ data was utilized to run an all-vs-all Basic Local Alignment Search Tool (BLAST). The de novo alternative splice (AS) sites were detected as was previously done in T. gesneriana without the reference genome sequences (Liu et al. 2017).
Coding-domain sequence (CDS) detection and gene functional annotation
TransDecoder (v3.0.0) (https://github.com/TransDecoder/TransDecoder/releases) identified the candidate coding regions within the full-length transcripts (Altschul et al. 1997). Annotation was performed using four databases: Swiss-Prot; Protein family (Pfam); KOG/COG/eggNOG, KEGG, and Gene Ontology (GO); and NCBI nonredundant (NR) database. The TFs were annotated using the Plant TFDB database (http://planttfdb.gao-lab.org) (Jin et al. 2017).
Illumina RNA-Seq, weighted correlation network analysis (WGCNA), and differential expression analysis
Individual tulip cDNA libraries were constructed by the NEBNext® Ultra II™ RNA Library Prep Kit (New England Biolabs, MA, USA) and used for Illumina RNA-Seq. The cDNA libraries were sequenced with the paired-end sequencing method on a HiSeq 4000 sequencing system (Illumina, CA, USA). The raw Illumina-Seq reads were filtered to remove the low-quality reads, adaptor sequences, and ambiguous reads with N > 10%. The clean data were then mapped based on the assembled tulip transcriptome. Gene expression levels in each sample were estimated by RSEM software (http://deweylab.biostat.wisc.edu/rsem/) (Li and Dewey 2011) and quantified as fragments per kilobase of transcript per million (FPKM). WGCNA was used to construct the gene coexpression networks (https://horvath.genetics.ucla.edu/html/CoexpressionNetwork) (Langfelder and Horvath 2008). The differential expression analysis among the flower stages was performed utilizing the DESeq R package (V1.10.1) (Anders and Huber 2010). An false discovery rate (FDR) < 0.05 and log2 |(foldchange)| ≥ 1 was set as the threshold for identifying the significant differential expression. MFUZZ (https://mfuzz.sysbiolab.edu.eu) was used for the clustering of gene expression (Kumar and Futschik 2007).
GO-term and KEGG pathway enrichment analysis
The GO classification was executed using the GOseq R packages (Young et al. 2010). KEGG analysis was performed using differentially expressed genes (DEGs) obtained from each comparison (http://www.genome.jp/kegg/) (Kanehisa et al. 2016). The statistical enrichment of the DEGs identified in the KEGG pathways was accomplished by employing the KOBAS software (http://kobas.cbi.pku.edu.cn/genelist/) (Bu et al. 2021).
Hormone quantification
The phytohormones were comprehensively profiled as previously described (Wang et al. 2017). Briefly, five intact flowers were collected for each flower stage in triplicates totaling 15. Each sample, weighing 200 mg, was ground into a powder, divided into two equal parts, and then transferred into 10 mL centrifuge tubes. One part was prepared for the examination of CKs and acidic phytohormones, while the other one was for that of brassinosteroids (BRs). An LC-30AD HPLC system (Shimadzu, Tokyo, Japan) coupled with an AB SCIEX 4500 Triple Quad™ liquid chromatography-mass spectrometer (MS)/MS (Sciex, Foster City, CA, USA) was used for the quantification of CKs and acidic phytohormones. BRs were estimated according to a previous report (Yu et al. 2016). All quantifications were performed in three biological replicates, and the final concentration of each analog was indicated as the average value ± SE.
VIGS assay
The full-length CDS of F01.PB503 (TgHB12-like) was cloned using the specific primers listed in Table S2. Based on sequence alignment, a fragment with low similarity with other homologs was isolated and sub-cloned into the tobacco rattle virus RNA2 (TRV2) vector. It was then introduced into the Agrobacterium tumefaciens strain GV3101 cells and used for petal disc infiltration. Post-transformation, the expression levels of TgHB12-like and Tulipa gesneriana senescence-related gene6 (TgSAG6) were quantified by RT-qPCR, and electrolyte leakage (EL) was determined. The total anthocyanin content was ascertained as described previously (Yan et al. 2022) calculated by the formula: QAnthocyanins = (A530 – 0.25 × A657)/M; where A530 and A657 were the absorptions at these wavelengths, and M is the weight of the plant material used for extraction.
Phylogenetic analysis and sequence alignment
The proteins encoded by TgHB12-like and its closest orthologs were aligned, and phylogenetic trees were constructed using the MEGA 7.0 software (https://www.megasoftware.net/) with the Maximum Likelihood method based on the bootstrap analysis with 1000 replications.
Transcriptional activation ability assay
The entire length of TgHB12-like was constructed into a pGBKT7 vector. The resulting positive and empty vectors (as negative controls) were employed to co-transform the cells of the yeast strain AH109. The transcriptional activation of these transformed yeast cells was assessed by spotting the transformants on SD/-Trp/-Leu and SD/-Trp/-Leu/-His/-Ade media at 30℃ for three days.
RT-qPCR analysis
Total RNA was extracted using the EASYspin Plus Complex Plant RNA Kit (Vazyme, Nanjing, China). The reverse transcription was performed using 1.0 µg of the total RNA with HiScript® II 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China). The primers for RT-qPCR were designed using the web tool GenScript (https://www.genscript.com/ssl-bin/app/primer). The relative transcription levels were calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001). Tulipa gesneriana beta-actin (TgACTIN) was employed as the reference gene. The primer sequences are listed in Table S2.
Statistical analysis
All experiments were repeated at least thrice, and the means ± standard errors (SEs) of the results of these independent experiments are shown. Bars with asterisks indicate statistically significant differences at P < 0.01 calculated using Student’s t-test. The variations among the stages were assessed using Duncan’s multiple range test (DMRT) and indicated by different letters at *P < 0.05.
Data mining and deposition
The raw PacBio Iso-Seq data were deposited to the NCBI Sequence Read Archive (SRA) database with the BioProject number PRJNA703083. The RNA-Seq data of the five tulip flower stages were uploaded with the accession number GSE136183 (https://www.ncbi.nlm.nih.gov/).
Results
Variations of flower lifespan and physiological changes during the flowering period
As a commercially important cut flower, the flowering lifespan is of significance to tulips. The flowering phases among 37 commercial cultivars were compared to find that they varied between 9.7–21.4 days from bud emergence to petal senescence (Fig. 1a), and the flowering lifespan diverged in tulips with diverse genetic backgrounds (Table S1). The morphological changes were obvious during flower opening and senescence of an intact tulip flower. The first stage (S1) was defined with green floral buds, followed by the second stage (S2) with colored and expanded petals (Fig. 1b). The opening of flowers with rhythmic petal movement was defined as the third stage (S3). The early senescence stage (S4) was designated as the one when a flower lost petal movements and the cup shape. The flowers with obvious wilting and color fading were described to belong to the late senescence stage (S5) (Fig. 1b). The flower diameters increased from 1.03 (S1) to 8.34 cm (S5), while the relative water content (RWC) of petals decreased from 90.75% to 81.5% at the senescent stages (Fig. 1c, d). Besides an enhancement of cell membrane permeability at the last stages, the accumulation of anthocyanin contents was enhanced initially from S1 to S3, but then gradually declined in senescent petals (Fig. 1e, f). All these results indicated that flower opening and senescence in tulips are accompanied by morphological and physiological changes.

Variations in flower lifespan and physiological changes during flower opening and senescence in tulips. a The variations in flower longevity among tulip cultivars (n = 30). b The morphological changes in intact flowers from the stages of floral bud to senescence. c–f Modifications in flower diameter. Relative water content (RWC) (c), electrolyte leakage (d), and anthocyanin contents (e) during flower opening and petal senescence (f). Letters above the columns indicate statistically significant differences at p < 0.05, analyzed by Tukey’s test. Bar = 1.0 cm. S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage
Full-length transcriptome sequencing and assembly
A comprehensive full-length transcriptomic analysis was performed to explore the mechanisms underlying flower opening and senescence, generating a total of 1,202,336 reads (21.58 Gigabit, Gb) across all three cDNA libraries, of which 9,054,775 subreads were obtained. The mean lengths of the three cDNA libraries were 1,781 bp (1–2 kb); 2,868 (2–3 kb); and 3,450 bp (3–6 kb), respectively (Table S3). Finally, 587,619 reads of insert (ROIs) with an average length of 2,861 bp were obtained (Table S4). More than 41.5% (244,357) of the ROIs were full-length non-chimeric (FLNC) reads (Table S5). A total of 91,890 consensus isoforms with a mean length of 2,219 bp were obtained based on the clustering algorithm of iterative clustering for error correction (ICE). Using the Illumina reads, 71,517 high-quality consensus transcripts with an accuracy > 99% were recognized, along with 20,373 low-quality isoforms, which need to be further corrected (Table S6). Ultimately, 76,388 non-redundant transcripts were screened from all the T. gesneriana high-quality transcripts, with 33,505 SSRs identified from 21,904 transcripts and more than one SSR among the 4,480 (20.45%) unique transcripts. The most abundant SSR type was mono-nucleotide repeats (P1, 42.31%) (Fig. S2a). Furthermore, the density of SSRs in each 1.0 Mb of the sequence varied from 0.6 to 77.4 (Fig. S2b). All SSRs and the corresponding primers are listed in Table S7. Due to a lack of a reference genome, only 871 AS events were recognized (Table S8). Long non-coding RNAs (LncRNAs), i.e., those with length > 200 bases, were further predicted to identify 3,114 unique transcripts designated as candidate lncRNAs matching all four databases (Fig. S2c; Table S9).
Functional annotation of transcripts
Annotation was performed using 76,388 non-redundant transcripts, of which 17,513 belonged to KEGG; 29,157 to COG; 33,703 to GO; 44,813 to KOG; 53,217 to Swiss-Prot; 54,680 to Pfam; 65,943 to eggNOG; and 66,864 to the NR databases, respectively. Finally, 67,350 transcripts were annotated to the eight databases. Among all the 76,388 SMRT unique transcripts, 66,635 were identified to possess significant matches to the non-redundant (Nr) protein database. Among the transcripts of all plants, 11 species with matches > 1.0% were sorted, of which Elaeis gunieensis, Phoenix dactylifera, and Asparagus officinalis obtained 27.37, 21.66, and 8.33% of the matched hits, respectively (Fig. 2a). Besides, 7.29% of the hits were from other plant species (Fig. 2a). The number of open reading frames (ORFs) was 70,644 including 39,632 complete ORFs. The distribution of the lengths of the ORF-encoded amino-acid chains is shown in Fig. 2b. The functional annotation of all unique transcripts is illustrated in Fig. 2c and listed in Dataset S1.

National Center for Biotechnology Information (NCBI) non-redundant (Nr)-based distribution of homologous species and annotation of the candidate protein-coding RNAs. a The percentage of the total homologous gene hits distributed among the species. All of the top five species are monocots. b The distribution of the lengths of the open reading frame (ORF)-encoded amino acid sequences. c The gene ontology (GO)-term-based pathway annotation of all transcripts
WGCNA performance during flower opening and senescence in tulip
Illumina RNA-Seq was performed to explore the transcriptome variations among the five developmental stages during flower opening and petal senescence in tulips. A total of 119.46 GB of clean data was obtained from the 15 samples (three replications for each phase) with Q30 > 88.69% (Table S10). The clean data were mapped based on the assembled tulip transcriptome (Table S11). The FPKM values of each sample are shown in Fig. S3a. The correlation between the flower stages and biological replicate samples was evaluated using the Pearson coefficient values (Fig. S3b). WGCNA was performed to investigate the co-expression networks of genes that were associated with the flower developmental stage (Fig. 3a, b). All the genes were partitioned into 16 co-expression modules (Table S12; Fig. 3c), with sizes ranging remarkably from 58 genes in the light-cyan module to 10,003 in the turquoise module (Fig. 3d). All modules were further normalized into five stages to investigate the alterations with respect to the flower developmental stages. Along with flower opening and senescence, the 6,208 unigenes of the brown module were upregulated, whereas 4,907 unigenes of the yellow module were downregulated (Fig. 3d). Other modules included genes enriched in at least one or two specific developmental stages, such as the red (2987) and black modules which represented evidently up- and down-regulated genes at stage S4, respectively (Fig. 3d).

Co-expression network analyses of the differentially expressed genes (DEGs). a Selection of the soft-thresholding powers for scale-free co-expression network (n = 22). b Weighted correlation network analysis (WGCNA) for cluster dendrograms and module assignments. Genes were clustered based on the dissimilarity measure. Colors in the horizontal bar represent the modules, which correspond to the branch modules with highly interconnected gene groups. The gray modules indicate genes that did not fall into any module and were excluded from the subsequent study. Finally, 16 gene modules were identified by WGCNA. c Eigengene adjacency heatmap indicating the 16 modules of co-expressed genes. d The normalized average co-expression patterns of the 16 gene modules in five stages. Module colors are available online in Table S12
Identification of the DEGs related to flower opening and petal senescence
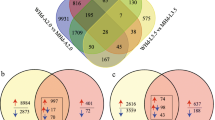
The number of DEGs identified between any two stages is shown in Fig. 4a (Dataset S2). Venn analysis indicated that 1,633 and 1,702 transcripts changed specifically during flower opening (S1 vs. S2 and S1 vs. S3) and in petal senescence (S3 vs. S4 and S3 vs. S5), respectively, while the alteration of 272 transcripts was common during the whole process of flower opening and petal senescence (Fig. 4b; Dataset S2). Heatmaps indicated the expression patterns of the specifically regulated 1,633 and 1,702 DEGs during flower opening (S1 vs. S3) and petal senescence (S3 vs. S5), respectively (Fig. 4c, d). These were subjected to GO-term and KEGG enrichment to understand further the biological and metabolic pathways associated with the above developmental processes. As shown in Fig. S4, the most enriched GO-terms of the 1,633 transcripts were “photosynthesis” and “fatty acid biosynthesis”, but in the 1,702 transcripts, “hydrolysis” and “DNA catabolic pathways” were dominantly enriched (Fig. S4a, b). Moreover, KEGG analysis indicated that most of the 1,633 DEGs annotated into photosynthesis pathways (cluter_frequency = 9.3%, p < 0.001) and amino acid metabolism pathway (cluter_frequency = 7.97%, p < 0.001) (Fig. 4e). Starch and sucrose metabolism (cluter_frequency = 8.3%, p < 0.001), as well as Valine (Val), Leucine (Leu), and Isoleucine (Ile) degradation (cluter_frequency = 8.3%, p < 0.05), were the most enriched pathways among the 1,702 senescence-related DEGs (Fig. 4f).

Identification of the differentially expressed genes (DEGs) among the five developmental stages of tulip flowers. a The numbers of DEGs detected between any two stages. b Venn map indicates the number of co-regulated and/or specifically regulated DEGs during flower opening and petal senescence. c, d Heat map analysis shows the relative expression levels in fragments per kilobase of transcript per million (FPKM) of the 1633 DEGs identified during the flower opening stages (c) and the 1702 co-regulated genes detected during the senescent stages (d). e Kyoto Encyclopedia of Genes and Genome (KEGG) pathway enrichment of the 1633 DEGs recognized among the first three stages (p < 0.05). f KEGG pathway enrichment of the 1702 DEGs identified among the last three stages (p < 0.05)
Phytohormone profiling and identification of hormone-related DEGs involved in flowering
As phytohormones are vital regulators of flower lifespan, the identification of DEGs related to the hormone pathways was then focused on. In the S1 vs. S2 or S1 vs. S3 comparisons, 114 and 112 DEGs were annotated to the hormone pathways, respectively (Fig. 5a). In the S3 vs. S4 or S3 vs. S5 comparisons, 105 and 188 hormone-related DEGs were characterized, respectively (Fig. 5a; Dataset S3). The most enriched pathways were ABA- and auxin-, followed by ethylene-, CKs-, SA-, and JA-related pathways; the lowest enrichment was in the gibberellins (GAs) and BRs associated pathways (Fig. 5b; Table S13). MapMan pathway analysis revealed the comparative expression patterns of unigenes involved in hormones signalling among the various stages (Fig. 5c). The ABA content gradually enhanced from S1 (11.2 ng/g) to S4 (41.4 ng/g) and then declined at S5 (27.7 ng/g) (Fig. 5d). The highest Indole-3-acetic acid (IAA) content was detected at S1 (8.6 ng/g), which reduced markedly at S2 and S3 that showed similar levels, and gradually decreased from S3 to the lowest level at S5 (2.5 ng/g) (Fig. 5e). The content of JA was drastically down-regulated from 27.7 ng/g (S1) to 0.6 ng/g (S2), but then gradually elevated from S2 to S5 (Fig. 5f). The contents of CKs, GAs, and BRs are shown in Table 1. Among all the analogs, the most accumulated types of CKs and GAs were cis-zeatin riboside (cZR) and GA4, respectively. Moreover, typhasterol (TY) was dominant at the first three stages but was hard to detect at the petal senescent stages (Table 1). These results collectively demonstrated that the accumulation of phytohormones and hormone-related transcripts was significantly modified during flower opening and senescence in tulips.

Identification of the hormone-related differentially expressed genes (DEGs) and quantification of the endogenous hormones. a Venn maps show the co-regulated and/or specifically regulated DEGs during flower opening and petal senescence. b The enrichment and distribution of hormones associated with the DEGs detected among comparisons S1 vs. S2, S1 vs. S3, S3 vs. S4, and S3 vs. S5 from (a). c MapMan pathway analysis of the DEGs associated with hormones. d Abscisic acid (ABA) contents. e Indole-3-acetic acid (IAA) contents. f Jasmonic acid (JA) contents. The data represent the mean ± SE of three independent replicates (n = 10). S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage. SA, salicylic acid; BA, benzylaminopurine; GA, gibberellin acid; BR, brassinosteroids
Identification and clustering analysis of TFs involved in flowering
In total, 2113 unigenes were annotated as TFs and assigned to 35 families of TFs (Dataset S4). MapMan pathway analysis indicated that the unigenes encoding TFs and those involved in protein modification and degradation pathways exhibited distinct expression patterns (Fig. S5). Among them, 2038 TFs (FKPM > 0 in at least one stage) were classified into six distinct expression clusters among the five developmental stages (Fig. 6a–f). Clusters 1 and 4 represented TFs, which were markedly elevated at the early and late senescent stages (S4 and S5), respectively (Fig. 6a–d). Those of cluster 2 were mainly diminished at S2 to S4 (Fig. 6b). The enriched genes in clusters 3 and 6 represented those that altered at S1 and S2, respectively (Fig. 6c–f). The TF-encoding unigenes in cluster 5 displayed a continuous up-regulation from S3 to S5 (Fig. 6e). The most identified TF family was MYB, followed by Zinc finger-CCCH, Homeobox, and bHLH (Fig. 6g). The numbers of up- and down-regulated TF-related genes varied from 113 in S4 vs. S3 to 223 in S5 vs. S3 (Fig. 6h). Among the 51 TF-related genes which specifically changed from S1 to S3 during flower opening, the most enriched were of bHLH with ten unigenes, accounting for 19.6%. Additionally, 16 NAC domain- and ten homeobox-domain-containing proteins were dominantly changed, which occupied 32.6% and 20.4% of the 49 TF-related DEGs in the petal senescence process of S3 to S5, respectively (Dataset S4).

Identification of the transcription factor (TF)-related differentially expressed genes (DEGs) during flower opening and senescence. a–f Cluster analysis indicating the relative expression patterns of 2038 TFs. Green or yellow lines correspond to genes with low membership values; purple and blue lines correspond to genes with high membership values. g The numbers of all the identified TFs and families to which they were annotated. h The numbers of the up- and down-regulated differentially expressed TFs in the S1 vs. S2/3 and S3 vs. S4/5 comparisons, respectively. S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage
Identification of the functional role of TgHB12-like in the regulation of petal senescence
Venn map analysis was performed to characterize further the common DEGs identified by hormone-related, TF-related, and WGCNA modules, which were explicitly involved in the regulation of petal senescence (Fig. S6). A unigene, F01.PB503 was specifically identified among the TF- and hormone-related DEGs (Fig. 7a), which was also the only unigene recognized from the brown WGCNA module (Fig. S6b). RT-qPCR revealed that F01.PB503 was up-regulated in S4 and S5 (Fig. 7b). The full-length CDS of F01.PB503 was cloned. The encoded protein contains a conserved homeodomain–leucine zipper (HD-Zipper) domain and had the highest sequence similarities of 54.63% with PdHOX6-like (Phoenix dactylifera) and 55.88% with EgATHB12-like (Elaeis guineensis), respectively (Fig. S7a). On alignment with Arabidopsis homeobox-domain containing proteins, F01.PB503 showed the most similarity with AtHB12 (AT3G61890), which was hence named TgHB12-like (Fig. S7a, b). Moreover, the transcription activation ability of TgHB12-like protein was examined in the yeast strain AH109 (Fig. S7c, d). VIGS was used to characterize the role of TgHB12-like during petal senescence regulation in tulips. These results indicated that the transient suppression of TgHB12-like expression significantly delayed the progress of petal disc senescence (Fig. 7c, d). The values of EL and anthocyanin content, which are physiological indicators of flower senescence, were consistent with the petal disc phenotypes (Fig. 7e, f). Overall, the above results indicated that transcriptomic analysis facilitates the identification of hormones and TFs related to flower opening and senescence in T. gesneriana.

Functional identification of the Tulipa gesneriana homebox (HB) (Homeodomain-leucine zipper) 12-like. a Venn map shows the common and specific differentially expressed genes (DEGs) identified from hormone- and transcription factor (TF)-related transcripts in the senescence stages. b Quantitative real-time PCR (qRT-PCR) confirms the expression of Tulipa gesneriana Homeobox12-like (TgHB12-like). Tulipa gesneriana beta-actin (TgACTIN) was used as an internal control. c Petal disc phenotypes on the 5th day after virus-induced gene silencing (VIGS) assay. The left and right photographs indicate the infiltration with the tobacco rattle virus vector controls (TRV1 and TRV2) and TgHB12-like vector (TRV1 and TRV2-TgHB12-like), respectively. d VIGS assay indicated the relative expression levels of TgHB12-like and Tulipa gesneriana senescence-related gene6 (TgSAG6) genes. e The anthocyanin contents and f ion leakage of petal discs infiltrated with the empty and TgHB12-like vectors on the 5th day of treatment, respectively. The data represent the means ± SEs of three independent replicates (n = 15). S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage
Discussion
The existing Tulipa species built up a vast genome of more than 20 Gb over hundreds of years of cultivation. To date, the complete tulip genome has not been released due to the difficulty in assembling a genome containing a large number of repeats. Nanopore-sequencing technology has been applied to overcome these challenges. A pioneering substantial chromosome-level genome assembly in garlic, ~ 16.24 Gb in total, has been constructed recently (Sun et al. 2020), which provided new clues and possibilities for assembling large genomes. Genome maps offer fundamental information for unraveling the molecular mechanisms involved in trait formation in crop species. Single-molecule long-read sequencing technology facilitates research without the need for native genome information. In the present study, PacBio Iso-Seq of six tissues was performed to obtain a comprehensive profile of the tulip transcriptome (Fig. S1). It consisted of 244,357 full-length transcripts, of which 67,350 were annotated and are of great significance for further research on tulips (Fig. 2; Fig. S2). Molecular marker-assisted selection/breeding is of great importance for improving the breeding efficiency, which depends on the development of molecular markers. In tulips, a genetic map was constructed using SNP and expressed sequence tags-simple sequence repeat (EST-SSR) markers, which identified six putative quantitative trait loci (QTLs) associated with resistance to Fusarium oxysporum (Tang et al. 2015). The genetic diversity and relationships among 280 individuals of 36 wild and cultivated tulip accessions were assessed using 15 highly polymorphic and reproducible genetic microsatellite markers (Pourkhaloee et al. 2018). The prediction of SSRs in this study provides more genetic tools for breeding, identification of germplasm diversity, and the conservation of threatened or rare wild tulip species.
Prolonging the lifespan of flowers is vital for tulip breeding; therefore, it is essential to explore the mechanism underlying flower senescence regulation. This study identified 16 gene co-expression modules that displayed distinct expression patterns during flower opening and senescence (Fig. 3). Of these, turquoise, yellow, and light-cyan modules showed a consistent downward expression pattern, but the blue and brown modules demonstrated an opposite trend from S1 to S5 (Fig. 3d). The 11 other modules were enriched in only one or two specific flower stages, which implied that these candidate genes were involved in the onset of one particular developmental process, i.e., petal expansion or color change. Additionally, 22,551 DEGs were identified between any two stages during flower opening and senescence (Fig. 4). GO analysis indicated that 1,633 DEGs were specifically enriched during flower opening and were mainly annotated into the photosynthesis and fatty acid biosynthesis pathways (Fig. 5; Fig. S4). These results corresponded to the primary morphological changes, including petal color change from green to red and petal size enlargement, which was consistent with previous reports (Müller et al. 2010). In contrast, during the flower senescence stages, the 1,702 specifically enriched DEGs were mainly annotated into hydrolysis and DNA catabolic pathways, which indicated the involvement of a programmed cell death (PCD) process (Fig. 4; Fig. S4). The flowering period typically terminates during petal senescence and PCD; moreover, petal deterioration is accompanied by varying degrees of nutrient remobilization (Verlinden 2003; Rogers 2006; Grosskinsky et al. 2018), which were consistent with the results of this report.
In this study, the phytohormone profile showed dynamic changes in hormone accumulation during flower development. Most analogs of CKs, GAs, and BRs gradually decreased from S1 to S5 (Table 1), which was consistent with various studies indicating that these three hormones may be typical negative regulators of petal senescence (Ma et al. 2018). CKs delayed flower senescence in petunias through rapid repression of ethylene biosynthesis and signal transduction (Trivellini et al. 2015). In the present study, most of the DEGs were enriched in the ABA and auxin pathways (Fig. 4a–c). The ABA contents were the highest in early senescent petals (Fig. 5d), which implied a role of ABA at the point of onset of petal senescence in tulips. However, in lilies, the ABA levels increased in the later stages of flower opening, which implied a species-specific variation in ABA function (Arrom and Munné-Bosch 2012; Cubría-Radío et al. 2017). Interestingly, the trends of change were similar between IAA and JA in the first three stages of tulip flowers, with both showing a remarkably high level at S1 and then declining significantly at S2 and S3 (Fig. 5e, f). The auxin levels substantially increased in wilting petals prior to their abscission in Lilium longiflorum (Lombardi et al. 2015). Nevertheless, the roles of auxin and JA in regulating petal senescence need more defining.
Besides, an integrative analysis was achieved through the identification of WGCNA co-expression modules, TF clusters, and those involved in hormone pathways during flower opening and senescence. Several modules were potentially linked to a specific developmental stage, such as Cluster 1, which is related to flower opening, and Cluster 6, which is related to petal senescence regulation (Fig. 6a–f). Among the six unigenes commonly characterized based on TF- and hormone-related DEGs, two HD-Zipper proteins, F01.PB22197 and F01.PB503 were identified (Fig. 7a; Dataset S4). TgHB12-like was selected not only for its dominantly high expression levels in the senescent stages but also for its common identification by TF-, hormone-, and WGCNA brown-module enrichment. Arabidopsis thaliana Homeobox12 (AtHB12) functioned in ABA sensitivity, root elongation, and leaf expansion (Valdés et al. 2012; Hur et al. 2015). In petunias, silencing of an ABA-induced gene PhHD-Zip reduced the expression of ABA biosynthesis-related genes and delayed flower senescence (Chang et al. 2014). Using the VIGS assay, the positive role of TgHB12-like in petal senescence was identified in tulips (Fig. 7). Hence, the functional identification of TgHB12-like implies the reliability of the network analysis of the transcriptome obtained in this report. Further experimental validation is needed to confirm the functions of other identified candidate genes and gene-network prediction during flower opening and senescence in tulips.
Conclusions
In summary, the first full-length transcriptome specific to T. gesneriana was characterized by 51 TF- and 40 hormone-related unigenes commonly altered during flower opening (S1 to S3). The accumulation of ABA evidently enhanced, unlike those of other hormones, including GAs, JA, IAA, CKs, and BRs. The most enriched KEGG pathways were photosynthesis, fatty acid biosynthesis, and amino acid metabolic pathways in flower opening. However, in reference to petal senescence, most of the common 49 TFs and 36 hormone-related unigenes identified were remarkably up-regulated; the levels of ABA, JA, and CKs were enhanced, unlike those of GAs, BRs, and IAA. The hydrolysis, DNA catabolic, starch, sucrose, and carbon metabolism were the most enriched pathways (Fig. 8). All these results provide an important and valuable genetic basis for further research on the potential mechanism associated with development and adaptation in T. gesneriana. In future studies, the functional analysis of the commonly identified TFs and/or hormone-related DEGs using gene editing needs to be performed to further document the processes underlying flower longevity in tulips.

A summary of the potential regulation of transcription factors, phytohormones, and metabolic pathways during flower opening and senescence in tulips. The dashed lines indicate that the inferences need further investigation. The red and green arrows show an up- or down-regulation. DEGs, differential expressed genes; bHLH, basic helix-loop-helix; MYB, v-Myb avian myeloblastosis viral oncogene homolog; ERF, ethylene responsive factor; GATA, GATA-binding transcription factor; NAC, NAM, ATAF1/2, CUC1/2; BT2, BTB-TAZ DOMAIN PROTEIN; SRM1, Serine/arginine repetitive matrix protein 1; WRKY, W-box binding transcription factor; ABA, abscisic acid; JA, jasmonic acid; Cks, cytokinins; GAs, gibberellins; BRs, brassinosteroids; IAA, indole-3-acetic acid. S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request. The full-length sequences and the Iso-seq data sets that support the findings of this study have been deposited in GenBank with the following accession numbers: PRJNA703083 and GSE136183 (https://www.ncbi.nlm.nih.gov/).
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped blast and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–402. https://doi.org/10.1093/nar/25.17.3389.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. https://doi.org/10.1186/gb-2010-11-10-r106.
Arrom L, Munné-Bosch S. Hormonal changes during flower development in floral tissues of Lilium. Planta. 2012;236:343–54. https://doi.org/10.1007/s00425-012-1615-0.
Azad AK, Ishikawa T, Sawa Y, Shibata H. Intracellular energy depletion triggers programmed cell death during petal senescence in tulip. J Exp Bot. 2008;59:2085–95. https://doi.org/10.1093/jxb/ern066.
Baharudin NF, Osman NI. Plant development, stress responses, and secondary metabolism under ethylene regulation. Plant Stress. 2023;7:100146. https://doi.org/10.1016/j.stress.2023.100146.
Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucl Acids Res. 2021;49:W317–25. https://doi.org/10.1093/nar/gkab447.
Chang X, Donnelly L, Sun D, Rao J, Reid MS, Jiang CZ. A petunia homeodomain-leucine zipper protein, PhHD-Zip, plays an important role in flower senescence. PLoS One. 2014;9:e88320. https://doi.org/10.1371/journal.pone.0088320.
Chen WH, Li PF, Chen MK, Lee YI, Yang CH. Forever young flower negatively regulates ethylene response DNA-binding factors by activating an ethylene-responsive factor to control Arabidopsis floral organ senescence and abscission. Plant Physiol. 2015;168:1666–83. https://doi.org/10.1104/pp.15.00433.
Cubría-Radío M, Arrom L, Puig S, Munné-Bosch S. Hormonal sensitivity decreases during the progression of flower senescence in Lilium longiflorum. J Plant Growth Regul. 2017;36:402–12. https://doi.org/10.1007/s00344-016-9648-4.
Gao Y, Xi F, Zhang H, Liu X, Wang H, Zhao L, et al. Single-molecule real-time (SMRT) isoform sequencing (Iso-Seq) in plants: the status of the bioinformatics tools to unravel the transcriptome complexity. Curr Bioinform. 2019;14:566–73. https://doi.org/10.2174/1574893614666190204151746.
Grosskinsky DK, Syaifullah SJ, Roitsch T. Integration of multi-omics techniques and physiological phenotyping within a holistic phenomics approach to study senescence in model and crop plants. J Exp Bot. 2018;69:825–44. https://doi.org/10.1093/jxb/erx333.
Hackl T, Hedrich R, Schultz J, Förster F. Proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics. 2014;30:3004–11. https://doi.org/10.1093/bioinformatics/btu392.
Hunter DA, Ferrante A, Vernieri P, Reid MS. Role of abscisic acid in perianth senescence of daffodil (Narcissus pseudonarcissus ‘Dutch Master’). Physiol Plant. 2004;121:313–21. https://doi.org/10.1111/j.0031-9317.2004.0311.x.
Hur YS, Um JH, Kim S, Kim K, Park HJ, Lim JS, et al. Arabidopsis thaliana homeobox 12 (ATHB12), a homeodomain-leucine zipper protein, regulates leaf growth by promoting cell expansion and endoreduplication. New Phytol. 2015;205:316–28. https://doi.org/10.1111/nph.12998.
Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucl Acids Res. 2017;45:D1040-5. https://doi.org/10.1093/nar/gkw982.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucl Acids Res. 2016;44:D457–62. https://doi.org/10.1093/nar/gkv1070.
Kumar L, Futschik ME. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. https://doi.org/10.6026/97320630002005.
Kumar M, Singh VP, Arora A, Singh N. The role of abscisic acid (ABA) in ethylene insensitive Gladiolus (Gladiolus grandiflora Hort.) flower senescence. Acta Physiol Plant. 2014;36:151–9. https://doi.org/10.1007/s11738-013-1395-6.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. https://doi.org/10.1186/1471-2105-9-559.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9. https://doi.org/10.1038/NMETH.1923.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. https://doi.org/10.1186/1471-2105-12-323.
Liu X, Mei W, Soltis PS, Soltis DE, Barbazuk WB. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol Ecol Resources. 2017;17:1243–56. https://doi.org/10.1111/1755-0998.12670.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–8. https://doi.org/10.1006/meth.2001.1262.
Lombardi L, Arrom L, Mariotti L, Battelli R, Picciarelli P, Kille P, et al. Auxin involvement in tepal senescence and abscission in Lilium: a tale of two lilies. J Exp Bot. 2015;66:945–56. https://doi.org/10.1093/jxb/eru451.
Lü P, Zhang C, Liu J, Liu X, Jiang G, Jiang X, et al. RhHB1 mediates the antagonism of gibberellins to ABA and ethylene during rose (Rosa hybrida) petal senescence. Plant J. 2014;78:578–90. https://doi.org/10.1111/tpj.12494.
Ma N, Ma C, Liu Y, Shahid MO, Wang C, Gao J. Petal senescence: a hormone view. J Exp Bot. 2018;69:719–32. https://doi.org/10.1093/jxb/ery009.
Marasek-Ciolakowska A, Ramanna MS, Arens P, Van Tuyl JM. Breeding and cytogenetics in the genus tulipa. Floric Ornam Biotechnol. 2012;6:90–7.
Meng L, Yang H, Yang J, Wang Y, Ye T, Xiang L, Chan Z, Wang Y. Tulip transcription factor TgWRKY75 activates salicylic acid and abscisic acid biosynthesis to synergistically promote petal senescece. J Exp Bot. 2024;75:2435–50. https://doi.org/10.1093/jxb/erae021.
Meng L, Yang H, Xiang L, Wang Y, Chan Z. NAC transcriptionfactor TgNAP promotes tulip petal senescence. Plant Physiol. 2022;190:1960–77. https://doi.org/10.1093/plphys/kiac351.
Müller GL, Drincovich MF, Andreo CS, Lara MV. Role of photosynthesis and analysis of key enzymes involved in primary metabolism throughout the lifespan of the tobacco flower. J Exp Bot. 2010;61:3675–88. https://doi.org/10.1093/jxb/erq187.
Panavas T, Walker EL, Rubinstein B. Possible involvement of abscisic acid in senescence of daylily petals. J Exp Bot. 1998;49:1987–97. https://doi.org/10.1093/jxb/49.329.1987.
Pourkhaloee A, Khosh-Khui M, Arens P, Salehi H, Razi H, Niazi A, et al. Molecular analysis of genetic diversity, population structure, and phylogeny of wild and cultivated tulips (Tulipa L.) by genic microsatellites. Horticul Environ Biotech. 2018;59:875–88. https://doi.org/10.1007/s13580-018-0055-6.
Prinsi B, Negri AS, Quattrocchio FM, Koes RE, Espen L. Proteomics of red and white corolla limbs in petunia reveals a novel function of the anthocyanin regulator anthocyanin1 in determining flower longevity. J Proteomics. 2016;131:38–47. https://doi.org/10.1016/j.jprot.2015.10.008.
Rogers HJ. Programmed cell death in floral organs: how and why do flowers die? Annals Bot. 2006;97:309–15. https://doi.org/10.1093/aob/mcj051.
Rogers HJ. From models to ornamentals: how is flower senescence regulated? Plant Mol Biol. 2013;82:563–74. https://doi.org/10.1007/s11103-012-9968-0.
Sexton R, Laird G, van Doorn WG. Lack of ethylene involvement in tulip tepal abscission. Physiol Plantarum. 2000;108:321–9. https://doi.org/10.1034/j.1399-3054.2000.108003321.x.
Shahin A, van Kaauwen M, Esselink D, Visser RGF, van Tuyl JM, Arens P, et al. Generation and analysis of expressed sequence tags in the extreme large genomes Lilium and Tulipa. BMC Genomics. 2012;13:640. https://doi.org/10.1186/1471-2164-13-640.
Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. 2013;31:1009–14. https://doi.org/10.1038/nbt.2705.
Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucl Acids Res. 2013;41:e166. https://doi.org/10.1093/nar/gkt646.
Sun X, Zhu S, Li N, Cheng Y, Liu T, Qiao X, et al. A chromosome-level genome assembly of garlic (Allium sativum) provides insights into genome evolution and allicin biosynthesis. Mol Plant. 2020;13:1328–39. https://doi.org/10.1016/j.molp.2020.07.019.
Tang N, Shahin A, Bijman P, Liu J, van Tuyl J, Arens P. Genetic diversity and structure in a collection of tulip cultivars assessed by SNP markers. Sci Hortic. 2013;161:286–92. https://doi.org/10.1016/j.scienta.2013.07.016.
Tang N, van der Lee T, Shahin A, Holdinga M, Bijman P, Caser M, et al. Genetic mapping of resistance to Fusarium oxysporum f. sp tulipae in tulip. Mol Breed. 2015;35:122. https://doi.org/10.1007/s11032-015-0316-3.
Trivellini A, Cocetta G, Vernieri P, Mensuali-Sodi A, Ferrante A. Effect of cytokinins on delaying petunia flower senescence: a transcriptome study approach. Plant Mol Biol. 2015;87:169–80. https://doi.org/10.1007/s11103-014-0268-8.
Valdés AE, Verns E, Johansson H, Rada-Iglesias A, Engstrom P. The homeodomain-leucine zipper (HD-Zip) class I transcription factors ATHB7 and ATHB12 modulate abscisic acid signalling by regulating protein phosphatase 2C and abscisic acid receptor gene activities. Plant Mol Biol. 2012;80:405–18. https://doi.org/10.1007/s11103-012-9956-4.
van Doorn WG, van Meeteren U. Flower opening and closure: a review. J Exp Bot. 2003;54:1801–12. https://doi.org/10.1093/jxb/erg213.
Van Scheepen J. Classified list and international register of tulip names. Hillegom: Royal General Bulbgrowers’ Association (KAVB); 1996.
Van Tuyl JM, Creij MGM. Tulipa gesneriana and T. hybrids. In: Anderson NO, editor. Flower breeding and genetics. Berlin: Springer-Verlag; 2007. p. 623–41. https://doi.org/10.1007/978-1-4020-4428-1.
Verlinden S. Changes in mineral concentrations in petunia coroll during development and senescence. Hort Sci. 2003;38:71–4. https://doi.org/10.1038/s41438-018-0018-1.
Wang Q, Cai WJ, Yu L, Ding J, Feng YQ. Comprehensive profiling of phytohormones in honey by sequential liquid–liquid extraction coupled with liquid chromatography–mass spectrometry. J Agri Food Chem. 2017;65:575–85. https://doi.org/10.1021/acs.jafc.6b04234.
Wang Y, Zhao H, Liu C, Qu L, Bao M, Wang J, et al. Integrating physiological and metabolites analysis to identify ethylene involvement in petal senescence in Tulipa gesneriana. Plant Physiol Biochem. 2020;149:121–31. https://doi.org/10.1016/j.plaphy.2020.02.001.
Yan S, Li Y, Liu J, Si D, Zhang X. Guideline for extraction, qualitative, quantitative, and stabilityanalysis of anthocyanins. eFood. 2022;4:e59. https://doi.org/10.1002/efd2.59.
Yin J, Chang X, Kasuga T, Bui M, Reid MS, Jiang CZ. A basic helix-loop-helix transcription factor, PhFBH4, regulates flower senescence by modulating ethylene biosynthesis pathway in petunia. Hortic Res. 2015;2:15059. https://doi.org/10.1038/hortres.2015.59.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. https://doi.org/10.1186/gb-2010-11-2-r14.
Yu L, Ding J, Wang YL, Liu P, Feng YQ. Phenylaminomethyl-benzeneboric acid modified tip extraction for determination of brassinosteroids in plant tissues by stable isotope labeling-liquid chromatography mass spectrometry. Anal Chem. 2016;88:1286–93. https://doi.org/10.1021/acs.analchem.5b03720.
Zhang S, Zhao Q, Zeng D, Xu J, Zhou H, Wang F, et al. RhMYB108, an R2R3-MYB transcription factor, is involved in ethylene- and JA-induced petal senescence in rose plants. Hortic Res. 2019;6:131. https://doi.org/10.1038/s41438-019-0221-8.
Zonneveld BJ. The systematic value of nuclear genome size for all species of Tulipa L. (Liliaceae). Plant Syst Evol. 2009;281:217–45. https://doi.org/10.1007/s00606-009-0203-7.
Acknowledgements
We appreciate contributions from all members in Chan’s group who provided valuable suggestions and efforts on discussions to this study, and all reviewers for their comments and suggestions.
Funding
This work was supported by the National Natural Science Foundation of China (32170372) and the Fundamental Research Funds for the Central Universities (Program No. 2662020YLPY010 and 2662016QD026).
Author information
Authors and Affiliations
Contributions
YW (Yanping Wang) designed the research; LM, HY, YL, YW (Yikun Wu), TY and YW (Yaping Wang) performed the experiments; LM and YW (Yanping Wang) analyzed and interpreted the data, wrote the paper; LX contributed valuable discussion; ZC and LQ revised the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
44281_2024_31_MOESM1_ESM.tif
Supplementary Material 1: Figure S1. The workflow of single molecular real-time (SMRT) sequencing and illumine RNA-sequence analysis using various tissues of tulip cultivar ‘Red Power’. Six tissues from 30 individual tulip plants including scale, floral bud, leaf, stem, root and flower were separately collected and pooled together for SMRT sequencing. 15 samples from five stages (stage 1 to stage 5) with three replicates for each stage were used for illumine RNA-sequencing to explore the transcriptome changes during flower opening and petal senescence. FLNC, full-length non-chimeric; AS, alternative splice; SSR, simple sequence repeats; GO, Gene Ontology; KOG,clusters of orthologous groups of proteins ; Pfam, protein families database; COG; eggnog, evolutionary genealogy of genes: non-supervised orthologous groups; NR, non-redundant; CDS, coding-domain sequence; FDR, false discovery rate.
44281_2024_31_MOESM2_ESM.tif
Supplementary Material 2: Fig. S2. Overview of simple sequence repeats (SSRs) and long no-coding RNA (lncRNA) predication of full-length transcripts in Tulipa gesneriana. a The distribution of SSRs with different repeats and motifs. b The number of SSRs per millon bps (Mb) transcripts. P1, mono-nucleotide repeats; P2, di-nucleotide repeats; P3, tri-nucleotide; P4, tetra-nucleotide repeats; P5, penta-nucleotide repeats; C*, compound SSRs; P6, hexa-nucleotide repeats. c Venn diagram of the number of predicted lncRNAs according to coding-non-coding index (cnci), coding potential calculator (cpc), protein families (pfam) and coding potential assessment tool (cpat) analysis.
44281_2024_31_MOESM3_ESM.tif
Supplementary Material 3: Fig. S3. The box diagram of fragments per kilobase of transcript per million (FPKM) values and correlation analysis of expression level in each two samples. a The box plots show FPKM distribution among different samples. b The heat map of expression level correlation of each two samples. T01 to T15 represent 15 samples from five different developmental stages.
44281_2024_31_MOESM4_ESM.tif
Supplementary Material 4: Fig. S4. The Gene Ontology-term (GO-term) pathway analysis. of specifically expressed genes during the flower opening process. a 1633 differential expressed genes (DEGs) identified from the flower opening process (S1 vs. S2 and S1 vs. S3). b 1702 DEGs identified from the petal senescence process (S4 vs. S3 and S5 vs. S3), respectively. S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage.
44281_2024_31_MOESM5_ESM.tif
Supplementary Material 5: Fig. S5. Mapman analysis of differential expressed genes (DEGs). a Heatmaps showed the expression patterns of unigenes involved in transcription factors, protein modification and protein degradation. b Heatmaps showed the expression patterns of unigenes involved in cell division and cycle, as well as plant development pathways. S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage.
44281_2024_31_MOESM6_ESM.tif
Supplementary Material 6: Fig. S6. Venn-map shows the differential expressed genes (DEGs) overly identified from hormone-related, transcription factors (TF)-related and weighted correlation network analysis (WGCNA-related) analysis. The hormone-related and TF-related DEGs used in this analysis were differentially regulated genes in comparison between S4 vs. S3 and S5 vs. S3 as shown in Figs. 3d, 5a and 6a. The clusters identified from WGCNA-network were indicated as different colors. S1, first stage; S2, second stage; S3, third stage; S4, The early senescence stage; S5, the late senescence stage.
44281_2024_31_MOESM7_ESM.tif
Supplementary Material 7: Fig. S7. TgHB12-like protein sequence analysis and transcripional activation detection in yeast strain. a Comparison of the conserved homeodomain-zipper (HD-Zipper) sequences among the most related plant species. b Phylogenetic analysis of the full length sequences of Tulipa gesneriana Homeobox12-like (TgHB12-like) and homologous genes from Arabidopsis. c The schematic diagram of TgHB12-BD (binding domain) plasmid. d Transcriptional activation of TgHB12-like in yeast AH109 strain.
44281_2024_31_MOESM10_ESM.docx
Supplementary Material 10: Table S3. A statistical analysis of the results of the PacBio single-molecule long-read sequencing in Tulipa gesneriana.
44281_2024_31_MOESM12_ESM.docx
Supplementary Material 12: Table S5. A statistical analysis of the full-length non-chimeric (FLNC) reads obtained by the PacBio single-molecule long-read sequencing.
44281_2024_31_MOESM13_ESM.docx
Supplementary Material 13: Table S6. The distribution of the consensus isoforms obtained from the different cDNA libraries.
44281_2024_31_MOESM15_ESM.docx
Supplementary Material 15: Table S11. The alignment of the RNA-Seq data mapped to the full-length transcriptome. The coding-domain sequence and the amino acid sequence encoded by Tulipa gesneriana Homeobox12-like (TgHB12-like).
44281_2024_31_MOESM23_ESM.xlsx
Supplementary Material 23: Table S12. A list of the 16 modules classified based on weighted correlation network analysis (WGCNA) performance.
44281_2024_31_MOESM24_ESM.xls
Supplementary Material 24: Table S13. Quantification of the hormones, cytokinins, gibberellins (GAs), and brassinosteroids (BRs).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meng, L., Yang, H., La, Y. et al. Transcriptional modules and hormonal metabolic pathways reveal the critical role of TgHB12-like in the regulation of flower opening and petal senescence in Tulipa gesneriana. HORTIC. ADV. 2, 18 (2024). https://doi.org/10.1007/s44281-024-00031-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44281-024-00031-w