
Abstract
Triple-negative breast cancer (TNBC) remains difficult to treat and urgently needs new therapeutic options. Nintedanib, a multikinase inhibitor, has exhibited efficacy in early clinical trials for HER2-negative breast cancer. In this study, we examined a new molecular mechanism of nintedanib in TNBC. The results demonstrated that nintedanib enhanced TNBC cell apoptosis, which was accompanied by a reduction of p-STAT3 and its downstream proteins. STAT3 overexpression suppressed nintedanib-mediated apoptosis and further increased the activity of purified SHP-1 protein. Moreover, treatment with either a specific inhibitor of SHP-1 or SHP-1-targeted siRNA reduced the apoptotic effects of nintedanib, which validates the role of SHP-1 in nintedanib-mediated apoptosis. Furthermore, nintedanib-induced apoptosis was attenuated in TNBC cells expressing SHP-1 mutants with constantly open conformations, suggesting that the autoinhibitory mechanism of SHP-1 attenuated the effects of nintedanib. Importantly, nintedanib significantly inhibited tumor growth via the SHP-1/p-STAT3 pathway. Clinically, SHP-1 levels were downregulated, whereas p-STAT3 was upregulated in tumor tissues, and SHP-1 transcripts were associated with improved disease-free survival in TNBC patients. Our findings revealed that nintedanib induces TNBC apoptosis by acting as a SHP-1 agonist, suggesting that targeting STAT3 by enhancing SHP-1 expression could be a viable therapeutic strategy against TNBC.
Similar content being viewed by others


Introduction
Triple-negative breast cancer (TNBC) is distinguished by the paradox of a favorable chemotherapeutic response but higher rates of early recurrence and worse outcomes compared to non-TNBC as well as its heterogeneous molecular profiling.1, 2 Currently, there is no approved targeted therapy for TNBC despite the suggestion of various strategies targeting the heterogeneous molecular pathways or subtypes, such as PARP inhibitors for BRCA-deficient subtypes and anti-androgen molecules for androgen receptor-expressing subtypes.1, 2 The identification of novel therapeutic targets and the development of new agents for TNBC remains a challenging task and represents an unmet need.
Src homology region 2 domain containing phosphatase 1 (SHP-1) is a non-receptor protein tyrosine phosphatase (PTP) and a tumor suppressor gene in different cancer types, including breast cancer.3 SHP-1 has an N-terminal Src homology-2 (N-SH2) domain, a C-terminal catalytic PTP domain, and a C-terminal SH2 domain (C-SH2).3, 4 SHP-1 has an autoinhibitory conformation through the interaction between its N-SH2 domain and its C terminus.5 SHP-1 participates in cell growth and survival signaling by dephosphorylating several crucial kinases such as BCR-ABL,6 insulin receptor,7 lymphocyte-specific protein tyrosine kinase,8 phosphoinositide 3-kinase8 as well as affecting the JAK/STAT pathway.3 Signal transducer and activator of transcription 3 (STAT3) regulates the proliferation, metastasis, angiogenesis and drug resistance of cancer cells.9, 10 Constitutive STAT3 activity has been reported in almost 70% of breast cancers,11 particularly in TNBC.12 In addition, abnormally activated STAT3 increases invasion and metastasis in TNBC.13 As a result, targeting STAT3 might be a potential therapeutic approach for treating TNBC.
Nintedanib is a multitargeted angiokinase inhibitor against many growth factor receptors, including PDGFR, FGFR, VEGFR,14 as well as the proto-oncogenes RET, FTL3 and Src,15, 16 with anti-angiogenic activity. This novel angiokinase inhibitor is currently in phase II–III clinical trials for the treatment of advanced ovarian cancer (NCT01610869), non-small-cell lung cancer (NCT00806819) and metastatic colorectal cancer (NCT02393755). In breast cancer research, the combination of nintedanib with paclitaxel promoted tumor remission in 50% of patients with early-stage HER-2-negative breast cancer.17 Despite its promising multitargeted angiokinase inhibitor activity, the molecular mechanism of the anti-tumor effect of nintedanib is poorly understood.
Therefore, we investigated the molecular mechanisms by which nintedanib enhances apoptosis to clarify the role of angiokinase inhibition in its anti-proliferative effects against TNBC. Nintedanib significantly inhibited tumor growth in a TNBC-bearing animal model via the SHP-1/p-STAT3 pathway.
Materials and methods
Cell culture and western blotting
MDA-MB-231, MDA-MB-468 and HCC-1395 cell lines were obtained from the ATCC (American Type Culture Collection, Rockville, MD, USA) and maintained in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, HyClone, Thermo Scientific, Waltham, MA, USA). Cell lysates treated with nintedanib were prepared and analyzed by western blot as previously reported.18 Antibodies targeting p-JAK2 (Tyr1007/1008), JAK2, p-SRC (Tyr416), SRC, p-STAT3 (Tyr705), STAT3, survivin, poly (ADP-ribose) polymerase (PARP) and cleaved caspase 3 were purchased from Cell Signaling (Danvers, MA, USA). SHP-1, cyclin D1 and Mcl-1 antibodies were purchased from Abcam (Cambridge, MA, USA).
Cell cycle distribution and apoptosis assay
Cells were arrested at G0/G1 phase by serum starvation for 48 h in medium containing 0.5% FBS. Then, the synchronized cells were given fresh medium containing 10% FBS for 16 h to allow reentry into the cell cycle, and the cells were then incubated in the presence or absence of nintedanib. Cellular DNA content that represented the cell cycle phase (sub-G1, G0/G1, S or G2/M) was detected by flow cytometry via propidium iodide staining. For the apoptosis assay, cells were incubated in the presence or absence of nintedanib for 48 h. Then, the cells were washed with cold PBS, resuspended in annexin V binding buffer (BD Pharmingen, San Diego, CA, USA), and treated with APC-conjugated annexin V (BD Pharmingen) and propidium iodide (Sigma, St Louis, MO, USA). The apoptotic cells were detected by flow cytometry.
Gene knockdown and SHP-1 phosphatase activity
siRNAs targeting SHP-1 and scrambled control siRNAs were purchased from Dharmacon (Thermo Scientific, Chicago, IL, USA). Cells were transfected with siRNAs using Lipofectamine 2000 (Invitrogen Life Technologies, Rockville, MD, USA) to knock down gene expression. For SHP-1 activity assay, a RediPlate 96 EnzChek Tyrosine Phosphatase Assay Kit (Molecular Probes, Carlsbad, CA, USA) was used following the manufacturer’s instructions.
Xenograft tumor growth
Female BALB/c nu/nu mice (5–7 weeks of age) were obtained from the National Laboratory Animal Center (Taipei, Taiwan, Republic of China). All animal experiments performed in this study were conducted in accordance using protocols approved by the Institutional Animal Care and Use Committee at Taipei Veterans General Hospital. Each mouse was subcutaneously injected in the dorsal flank with 5 × 106 cells as previously described.19 The volume of xenografted tumors was calculated using the standard formula width2 × length × 0.52. Based on the previous studies, effective and well-tolerated doses of nintedanib in mice were calculated in the range of 30–100 mg kg−1.14, 20 A dose of 30 mg kg−1 per day was considered close enough to the maximum tolerated dose in mice without eliciting an obvious effect on the phenotype;21 therefore, we chose this dose for the animal studies. When tumors reached 100–200 mm3 in size, mice were orally administered nintedanib (30 mg kg−1) three times a week. Control mice received vehicle. All the mice were killed on day 33, and the xenografted tumors were harvested and assayed for subsequent experiments.
Immunohistochemical staining
In brief, 4-μm-thick paraffin-embedded tissue sections from patients with TNBC were placed on poly-L-lysine-coated slides and prepared and stained as previously described.18 The expression levels of p-STAT3 and SHP-1 were semiquantitatively assessed based on the staining intensity by a board-certified pathologist.
Surgical samples
A total of 74 patients with TNBC who underwent surgical treatment at the Department of Surgery at Taipei Veterans General Hospital were enrolled in this study. Informed consent was obtained from all the patients, and this study was approved by the Institutional Review Board in Taipei Veterans General Hospital prior to initiation.
In silico survival analysis using a publicly available database
A Kaplan–Meier survival analysis of patients with breast cancer based on SHP-1 (gene name PTPN6, Affymetrix probe ID 206687, Budapest, Hungary) mRNA expression was obtained from the Kaplan–Meier plotter online database (http://kmplot.com/analysis/index).22, 23, 24 Relapse-free survival curves were plotted for patients with TNBC (n=249) and all breast cancer subtypes (n=3554). An autoselected optimal cutoff was chosen in the analysis.
Statistical analysis
Data analysis was based on nonparametric tests, and the differences were considered statistically significant if P<0.05. The results were presented as the mean±s.d. or s.e. For survival analysis, disease-free survival curves of patients were generated by the Kaplan–Meier method and compared using a log-rank test. All statistical analyses were conducted using SPSS for Windows software, version 12.0 (SPSS, Chicago, IL, USA).
Results
Nintedanib exerts an anti-proliferative effect on TNBC cell lines
To examine the anti-tumor ability of nintedanib on human TNBC cells, we first tested the effects of nintedanib on cell growth in three TNBC cell lines: MDA-MB-231, MDA-MB-468 and HCC-1395. As shown in Figure 1a, nintedanib treatment inhibited cell proliferation in a dose-dependent manner. Inhibition of cell proliferation could be the result of apoptosis induction, cell cycle arrest and/or growth inhibition. We therefore investigated whether nintedanib could induce apoptosis or affect the cell cycle distribution in TNBC cells. The results indicated that nintedanib significantly increased the differential apoptotic effects (Figure 1b) in these TNBC cells in a dose-dependent manner. Nintedanib treatment had a mild effect on the population size in the G0/G1, S and G2/M phases but significantly increased the subG1 cell population (Supplementary Figure 1). To further test the cytotoxic effect of nintedanib on normal MCF-10A human breast epithelial cells, the MTT assay and flow cytometry analysis were performed. Nintedanib exerted milder anti-proliferative activity and a more minimal apoptosis-inducing effect on MCF-10A cells compared to those of the TNBC cell lines (Figure 1b and Supplementary Figure 2, lower panel). Our results showed that nintedanib exerted more cytotoxic effects on TNBC cells than on normal breast MCF-10A cells.

Nintedanib exerts anti-proliferative activity in TNBC cell lines. (a) Dose-dependent effects of nintedanib on cell viability in MDA-MB-231 (left), MDA-MB-468 (middle) and HCC-1395 (right) cells. Cells treated with nintedanib at the indicated doses for 48 h were measured by an MTT assay. (b) Dose-dependent effects of nintedanib on apoptosis in MDA-MB-231 (left), MDA-MB-468 (middle) and HCC-1395 (right) cells. Cells were treated with nintedanib at the indicated doses and times. Apoptotic cells were analyzed by flow cytometry, and the means of at least three independent experiments performed in triplicate are shown. The data are shown as the mean±s.d.
Nintedanib enhances cell apoptosis by decreasing p-STAT3 levels in TNBC cells
Given the potential anti-tumor activity of nintedanib, we next focused on nintedanib in TNBC treatment and examined the underlying molecular mechanisms of its function. The results showed that nintedanib inhibited the expression levels of activated STAT3 (p-STAT3) and its downstream pro-proliferative molecules Mcl-1, cyclin D1 and survivin at the indicated doses (Figure 2a). Importantly, PARP is an important marker of caspase 3-mediated apoptosis and was activated in cells treated with nintedanib (Figure 2a). Moreover, nintedanib induced considerable time-dependent apoptosis and reduction of p-STAT3 levels (Figure 2b). We also examined the effects of nintedanib on STAT3 phosphorylation in the cytoplasm and nucleus. As shown in Supplementary Figure 3, nintedanib treatment had no obvious effect on total STAT3 levels but significantly decreased the phosphorylation of STAT3 in both the cytoplasm and nucleus. These results suggested that STAT3 inhibition may be the major mechanism of nintedanib-induced apoptosis in TNBC. To further validate the role of STAT3, we transiently transfected MDA-MB-231 cells with a STAT3 overexpression vector to determine the molecular changes induced by nintedanib. Ectopic expression of STAT3 increased p-STAT3 protein levels and decreased nintedanib-mediated apoptosis, suggesting that activated STAT3 signaling counteracts the anti-tumor activity of nintedanib (Figure 3a).

Nintedanib induces cell apoptosis by inhibiting STAT3 signaling. (a) Dose-dependent and (b) time-dependent effects of nintedanib on STAT3-related protein levels. Cells were treated with nintedanib at the indicated doses and times. Whole-cell extracts were analyzed by western blot analysis using anti-p-STAT3, anti-STAT3, anti-cyclin D1, anti-survivin, anti-Mcl-1, anti-PARP and anti-actin antibodies. Quantification of the protein levels was performed by the ImageJ software. The means of at least three independent experiments performed in triplicate are shown. *P<0.05; **P<0.01; ***P<0.001. The data are shown as the mean±s.d.

SHP-1 is a target of nintedanib in TNBC cells. (a) Ectopic STAT3 expression increased p-STAT3 levels and inhibited nintedanib-induced apoptosis. (b) The protective effects of an SHP-1 inhibitor (PTP III) on nintedanib-induced apoptosis. TNBC cells were pretreated with 25 μM PTP III for 30 min and co-incubated with 15 μM nintedanib for another 48 h. (c) Silencing SHP-1 reversed the apoptotic effects (upper panel) and p-STAT3 inhibition (lower panel) induced by nintedanib. (d) Cells were treated with 5 μM nintedanib for 48 h, and cell lysates were assayed for phosphatase activity (upper panel). Cells were treated with 15 μM nintedanib for 16 h, after which whole-cell extracts were analyzed by western blot analysis using anti-SHP-1, anti-PTPRD and anti-actin antibodies (lower panel). (e) MDA-MB-231 cells were transfected with vectors expressing either wild-type or mutant SHP-1; ΔN1 (deletion of the autoinhibitory N-SH2 domain) and D61A mutants (point mutation of D61A on SHP-1). Then, the cells were treated with either nintedanib or DMSO for another 36 h. Apoptotic cells were analyzed by flow cytometry (upper panel). Whole-cell extracts were prepared and analyzed by western blot analysis using anti-p-STAT3, anti-Myc and anti-actin antibodies (lower panel). Quantification of the protein levels was performed by the ImageJ software. The means of at least three independent experiments performed in triplicate are shown. *P<0.05; **P<0.01; ***P<0.001. The data are shown as the mean±s.d.
Nintedanib targets SHP-1 in TNBC cells
According to previous studies, SHP-1 serves as a crucial negative regulator in STAT3 signaling;25 thus, we determined whether SHP-1 is involved in nintedanib-induced STAT3 suppression. We used a specific SHP-1 inhibitor (PTP III) as well as knocked down SHP-1 via transient transfection of SHP-1-targeted siRNAs to analyze the effect of nintedanib on TNBC cells. Blocking SHP-1 via either PTP III or siRNA abolished nintedanib-mediated cell death (Figure 3b and c). In addition, nintedanib treatment significantly increased SHP-1 activity in MDA-MB-468 cells (Figure 3d, upper panel). As STAT3 is a direct substrate of PTPRD, we also analyzed the expression of PTPRD in TNBC cells with nintedanib treatment. As expected, PTPRD was decreased in nintedanib-treated TNBC cells (Figure 3d, lower panel).
Previous studies have indicated that the autoinhibitory structure of the N-SH2 and PTP domains is the major regulator of SHP-1 activity.3, 5, 26 Therefore, we used two mutant SHP-1 constructs to mimic the open-form structure of SHP-1 and examine the effect of nintedanib on SHP-1 autoinhibition. TNBC cells transduced with ΔN1 and D61A mutant SHP-1-expressing vectors were insensitive to nintedanib-mediated STAT3 inhibition (Figure 3e). We also found that cells ectopically expressing mutant SHP-1 had much lower endogenous p-STAT3 levels compared to control cells (Figure 3e, lower). Therefore, nintedanib potentiates SHP-1 activity by relieving its autoinhibitory conformation to suppress p-STAT3 levels.
Nintedanib suppresses xenograft tumor growth of TNBC cells via SHP-1/STAT3 signaling
To validate the effect of nintedanib on the tumor growth of TNBC cells, nude mice bearing MDA-MB-231 tumors were orally administered either nintedanib or vehicle to assess the growth of the xenografted tumors. The average tumor size (Figure 4a) and tumor weight (Figure 4b) were reduced in mice administered nintedanib. However, there were no differences in body weight between the drug-treated mice and the control mice (Figure 4c). Moreover, we investigated whether nintedanib-mediated inhibition of tumor growth in MDA-MB-231 cells was dependent on the SHP-1/p-STAT3 pathway. The data showed that p-STAT3 protein levels were reduced (Figure 4d), whereas cleaved PARP levels (Figure 4d) and SHP-1 activity were increased (Figure 4e) in the tumor samples from mice treated with nintedanib. Notably, the downregulation of p-STAT3 was also accompanied by elevated TUNEL staining in vivo (Figure 4f). Taken together, these data indicate that nintedanib inhibits TNBC tumor growth via SHP-1/p-STAT3 signaling.

Nintedanib diminishes xenograft tumor growth of TNBC cells. Mice were orally administered nintedanib (30 mg kg−1 body weight) three times a week as described in the Methods section. Control mice received vehicle. (a) Growth curves, (b) tumor weight and (c) body weight of xenograft mice bearing MDA-MB-231 tumors (n=6). The data are shown as the mean±s.e. (d) The protein levels of p-STAT3, STAT3 and PARP as assessed by western blot analysis in MDA-MB-231 tumors. (e) SHP-1 activity in MDA-MB-231 tumors (n=6). The data are shown as the mean±s.d. (f) H&E and IHC staining for p-STAT3 and the apoptosis index based on TUNEL staining in mice with MDA-MB-231 xenograft tumors and administered either vehicle or nintedanib (× 200).
SHP-1 levels are downregulated whereas p-STAT3 protein levels are upregulated in tumor samples, and SHP-1 transcripts are associated with better disease-free survival in patients with TNBC
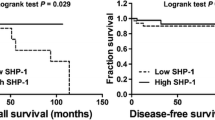
To assess whether any significant differences in SHP-1 and p-STAT3 expression exist in TNBC carcinoma specimens compared with normal tissues, IHC data were analyzed. We evaluated the SHP-1 and p-STAT3 protein levels in patients with TNBC. The levels of SHP-1 were downregulated in the tumor samples (Figure 5a, left panel), whereas the levels of p-STAT3 were upregulated (Figure 5a, right panel). Furthermore, the relationship between SHP-1 and p-STAT3 in tumor tissues from patients with TNBC were analyzed using the Pearson correlation analysis. The relative expression of SHP-1 was inversely correlated with the protein levels of p-STAT3 in TNBC specimens (Figure 5b). The survival analysis from the Kaplan–Meier plotter online data set of TNBC cases demonstrated that higher levels of SHP-1 transcripts were associated with better disease-free survival in patients with breast cancer (Figure 5c, right panel), especially among TNBC patients (Figure 5c, left panel).

Clinical relevance of SHP-1 and p-STAT3. (a) The levels of SHP-1 (left) and p-STAT3 (right) in tumor samples from patients with TNBC (normal tissues, n=11; tumor tissues, n=74). *P<0.05. The data are shown as the mean±s.e. (b) The correlation between SHP-1 and p-STAT3 expression in tumors from TNBC patients was analyzed using Pearson correlation analysis (n=57, P=0.047). (c) A Kaplan–Meier survival analysis of SHP-1 mRNA expression in breast cancer patients was obtained from the Kaplan–Meier plotter online database. Relapse-free survival curves were plotted for patients with TNBC (n=249, P<0.001) (left panel) and all breast cancer subtypes (n=3554, P<0.001) (right panel). An autoselected best cutoff value was chosen in this analysis.
Discussion
TNBC tends to behave more aggressively and have a higher risk of distant recurrence than other subtypes of breast cancer.27, 28 Due to a lack of established therapeutic targets, it is difficult to develop new targeted drugs for TNBC. STAT3 activation has been linked to multiple malignant tumors, including breast, ovarian, head and neck, prostate, pancreas and brain cancers.29 STAT3 is highly expressed and activated in most breast cancers,11 especially TNBC.12 High levels of p-STAT3 are correlated with worse outcomes in patients with invasive breast cancers.30 In this study, we found that the SHP-1 levels were downregulated in most tumors and correlated with higher levels of p-STAT3 expression. In addition, we searched the Kaplan–Meier plotter online database and found that the SHP-1 transcript levels correlate with better relapse-free survival among TNBC patients (Figure 5). Apparently, the SHP-1/p-STAT3 signaling axis might be a potential therapeutic target as well as a clinical prognostic indicator in patients with TNBC.
Our findings that nintedanib regulates the STAT3 pathway provide a rationale for its use to treat TNBC. We found that nintedanib was effective in blocking STAT3 activation, thus leading to the suppression of numerous proteins involved in cancer survival and proliferation (Figure 2). Here we found evidence that STAT3 inhibition is linked to nintedanib-induced SHP-1 activation in both in vitro experiments and a preclinical TNBC xenograft tumor model (Figures 3d and 4e). Blocking SHP-1 diminished the nintedanib-inhibited STAT3 effect, which provided further evidence that SHP-1 is the key modulator in nintedanib-mediated STAT3 inhibition (Figure 3). We also found that nintedanib did not further increase SHP-1 activity in cells expressing SHP-1 mutant clones and that these cells were insensitive to nintedanib-mediated p-STAT3 inhibition (Figure 3), suggesting that nintedanib restrains STAT3 activation by regulating SHP-1 autoinhibition. Therefore, the SHP-1/p-STAT3 pathway could be a crucial target for predicting the response to nintedanib.
Various PTPs have been reported to regulate the STAT3 pathway, including SHP-1,31, 32 SHP-233 and PTEN.34 However, as shown in Supplementary Figure 4, nintedanib treatment had no obvious effect on either PTEN or SHP-2 but suppressed the phosphorylation of JAK2 (Tyr1007/1008) and Src (Tyr 416). SHP-1 is known to directly bind to JAK2 kinase to induce its dephosphorylation and inhibition.35 It is possible that nintedanib decreased the p-JAK2 levels by increasing the SHP-1 activity. Alternatively, nintedanib is a multitargeted angiokinase inhibitor against PDGFR, FGFR, VEGFR and non-receptor tyrosine kinases such as Src.15 Since PDGFβ has been reported to induce the JAK2–STAT3 pathway by activating Src,36 nintedanib might inhibit JAK2 by directly inhibiting PDGFβ and Src.
Several lines of evidence have revealed that nintedanib exerts effective antitumor activities in many human tumor xenograft models, including NSCLC, HCC, renal cell carcinoma, colon cancer, ovarian cancer, pancreatic ductal adenocarcinoma and prostate cancer.15, 37, 38, 39 Nintedanib was also observed to strengthen the antitumor response of standard chemotherapeutic drugs. Our previous study demonstrated that nintedanib-induced apoptosis of hepatocellular carcinoma cells is independent of its anti-angiokinase activity via the SHP-1/p-STAT3 pathway.38
There have been various strategies targeting oncogenic STAT3 activity that use many designated agents or natural compounds (for example, JAK inhibitors) or small molecules that directly block functional STAT3 dimerization (via SH2 domains).40, 41 The approach of enhancing SHP-1, a negative regulator of STAT3 phosphorylation, provides an alternative method independent of JAK inhibition. We previously identified that the multi-angiokinase inhibitor sorafenib can act as a direct enhancer of SHP-1, which proves the concept of the SHP-1/p-STAT3 strategy.42, 43 This was further supported by our finding that regorafenib, whose chemical structure is highly similar to that of sorafenib and differs only by a fluoro substitution on the sorafenib moiety,44 can also directly enhance SHP-1 activity.43 Accordingly, we generated a series of sorafenib derivatives that are devoid of the angiokinase (VEGFR/PDGFR) inhibitory activity and classified them as SHP-1 agonists.45 We have shown that the direct SHP-1 agonist SC-43 is effective in TNBC cells.18 In addition to nintedanib, SHP-1 enhancers that have been preclinically examined include sorafenib analogues (SC agents such as SC-1, SC-49, SC-60, SC-78 and SC-43) as well as dovitinib and obatoclax analogues such as SC-2001.19, 46, 47, 48, 49, 50 Interestingly, the chemical structure of nintedanib is different from both sorafenib and regorafenib as well as the sorafenib derivatives, and future structural studies on the crystallization of SHP-1/agonist complexes may help delineate the detailed interactions between these agents and SHP-1.
In conclusion, we demonstrated that nintedanib activated SHP-1 by directly relieving the autoinhibition of SHP-1 and significantly inducing TNBC cell apoptosis via SHP-1-dependent p-STAT3 inhibition (Figure 6). Our findings provide a greater understanding of the molecular mechanism of nintedanib-driven regulation and identify the SHP-1/p-STAT3 pathway as a predictor of the nintedanib response, which can maximize the clinical benefits for TNBC treatment. However, future studies to elucidate the mechanism by which nintedanib inhibits p-STAT3 signaling via the disruption of SHP-1 autoinhibition may lead to further progress in a new targeted therapy for TNBC.

Schematic of the molecular mechanisms of nintedanib activity with regard to the SHP-1/p-STAT3 pathway. In TNBC cells, SHP-1 is autoinhibited, which results in the activation of p-STAT3 signaling and subsequent cell proliferation. Nintedanib induced significant anti-tumor activity by relieving SHP-1 autoinhibition to inhibit p-STAT3 signaling.
References
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121: 2750–2767.
Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA . New strategies for triple-negative breast cancer—deciphering the heterogeneity. Clin Cancer Res 2014; 20: 782–790.
Wu C, Guan Q, Wang Y, Zhao ZJ, Zhou GW . SHP-1 suppresses cancer cell growth by promoting degradation of JAK kinases. J Cell Biochem 2003; 90: 1026–1037.
Lopez-Ruiz P, Rodriguez-Ubreva J, Cariaga AE, Cortes MA, Colas B . SHP-1 in cell-cycle regulation. Anticancer Agents Med Chem 2011; 11: 89–98.
Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ et al. Crystal structure of human protein-tyrosine phosphatase SHP-1. J Biol Chem 2003; 278: 6516–6520.
Tauchi T, Ohyashiki K, Yamashita Y, Sugimoto S, Toyama K . SH2-containing phosphotyrosine phosphatase SHP-1 is involved in BCR-ABL signal transduction pathways. Int J Oncol 1997; 11: 471–475.
Bousquet C, Delesque N, Lopez F, Saint-Laurent N, Esteve JP, Bedecs K et al. sst2 somatostatin receptor mediates negative regulation of insulin receptor signaling through the tyrosine phosphatase SHP-1. J Biol Chem 1998; 273: 7099–7106.
Cuevas B, Lu Y, Watt S, Kumar R, Zhang J, Siminovitch KA et al. SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J Biol Chem 1999; 274: 27583–27589.
Vitale G, Zappavigna S, Marra M, Dicitore A, Meschini S, Condello M et al. The PPAR-gamma agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-beta treated pancreatic cancer cells. Biotechnol Adv 2012; 30: 169–184.
Abubaker K, Luwor RB, Escalona R, McNally O, Quinn MA, Thompson EW et al. Targeted disruption of the JAK2/STAT3 pathway in combination with systemic administration of paclitaxel inhibits the priming of ovarian cancer stem cells leading to a reduced tumor burden. Front Oncol 2014; 4: 75.
Kim SR, Seo HS, Choi HS, Cho SG, Kim YK, Hong EH et al. Trichosanthes kirilowii ethanol extract and cucurbitacin D inhibit cell growth and induce apoptosis through inhibition of STAT3 activity in breast cancer cells. Evid Based Complement Alternat Med 2013; 2013: 975350.
Walker SR, Xiang M, Frank DA . Distinct roles of STAT3 and STAT5 in the pathogenesis and targeted therapy of breast cancer. Mol Cell Endocrinol 2014; 382: 616–621.
Lee HJ, Seo NJ, Jeong SJ, Park Y, Jung DB, Koh W et al. Oral administration of penta-O-galloyl-beta-D-glucose suppresses triple-negative breast cancer xenograft growth and metastasis in strong association with JAK1-STAT3 inhibition. Carcinogenesis 2011; 32: 804–811.
Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 2015; 45: 1434–1445.
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 2008; 68: 4774–4782.
Reck M . Nintedanib: examining the development and mechanism of action of a novel triple angiokinase inhibitor. Expert Rev Anticancer Ther 2015; 15: 579–594.
Quintela-Fandino M, Urruticoechea A, Guerra J, Gil M, Gonzalez-Martin A, Marquez R et al. Phase I clinical trial of nintedanib plus paclitaxel in early HER-2-negative breast cancer (CNIO-BR-01-2010/GEICAM-2010-10 study). Br J Cancer 2014; 111: 1060–1064.
Liu CY, Tseng LM, Su JC, Chang KC, Chu PY, Tai WT et al. Novel sorafenib analogues induce apoptosis through SHP-1 dependent STAT3 inactivation in human breast cancer cells. Breast Cancer Res 2013; 15: R63.
Su JC, Tseng PH, Wu SH, Hsu CY, Tai WT, Li YS et al. SC-2001 overcomes STAT3-mediated sorafenib resistance through RFX-1/SHP-1 activation in hepatocellular carcinoma. Neoplasia 2014; 16: 595–605.
Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B . Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 2014; 349: 209–220.
European Medicines Agency Vargatef: EPAR—Public Assessment Report, Procedure No. EMEA/H/C/002569/0000 (European Medicines Agency, UK, 2015) http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002569/WC500179972.pdf.
Gyorffy B, Schafer R . Meta-analysis of gene expression profiles related to relapse-free survival in 1,079 breast cancer patients. Breast Cancer Res Treat 2009; 118: 433–441.
Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010; 123: 725–731.
Gyorffy B, Surowiak P, Budczies J, Lanczky A . Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013; 8: e82241.
Zhang J, Somani AK, Siminovitch KA . Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol 2000; 12: 361–378.
Yang J, Liang X, Niu T, Meng W, Zhao Z, Zhou GW . Crystal structure of the catalytic domain of protein-tyrosine phosphatase SHP-1. J Biol Chem 1998; 273: 28199–281207.
Chacon RD, Costanzo MV . Triple-negative breast cancer. Breast Cancer Res 2010; 12 (Suppl 2): S3.
Yadav BS, Chanana P, Jhamb S . Biomarkers in triple negative breast cancer: a review. World J Clin Oncol 2015; 6: 252–263.
Santoni M, Massari F, Del Re M, Ciccarese C, Piva F, Principato G et al. Investigational therapies targeting signal transducer and activator of transcription 3 for the treatment of cancer. Expert Opin Investig Drugs 2015; 24: 809–824.
Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001; 20: 2499–2513.
Poole AW, Jones ML . A SHPing tale: perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal 2005; 17: 1323–1332.
Kim YS, Seo DW, Kong SK, Lee JH, Lee ES, Stetler-Stevenson M et al. TIMP1 induces CD44 expression and the activation and nuclear translocation of SHP1 during the late centrocyte/post-germinal center B cell differentiation. Cancer Lett 2008; 269: 37–45.
Kim H, Hawley TS, Hawley RG, Baumann H . Protein tyrosine phosphatase 2 (SHP-2) moderates signaling by gp130 but is not required for the induction of acute-phase plasma protein genes in hepatic cells. Mol Cell Biol 1998; 18: 1525–1533.
Sun S, Steinberg BM . PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J Gen Virol 2002; 83: 1651–1658.
Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC, Yi T . Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol 1996; 16: 6985–6992.
Masamune A, Satoh M, Kikuta K, Suzuki N, Shimosegawa T . Activation of JAK-STAT pathway is required for platelet-derived growth factor-induced proliferation of pancreatic stellate cells. World J Gastroenterol 2005; 11: 3385–3391.
Kutluk Cenik B, Ostapoff KT, Gerber DE, Brekken RA . BIBF 1120 (nintedanib), a triple angiokinase inhibitor, induces hypoxia but not EMT and blocks progression of preclinical models of lung and pancreatic cancer. Mol Cancer Ther 2013; 12: 992–1001.
Tai WT, Shiau CW, Li YS, Chang CW, Huang JW, Hsueh TT et al. Nintedanib (BIBF-1120) inhibits hepatocellular carcinoma growth independent of angiokinase activity. J Hepatol 2014; 61: 89–97.
Awasthi N, Hinz S, Brekken RA, Schwarz MA, Schwarz RE . Nintedanib, a triple angiokinase inhibitor, enhances cytotoxic therapy response in pancreatic cancer. Cancer Lett 2015; 358: 59–66.
Chai EZ, Shanmugam MK, Arfuso F, Dharmarajan A, Wang C, Kumar AP et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther 2016; 162: 86–97.
Furtek SL, Backos DS, Matheson CJ, Reigan P . Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol 2016; 11: 308–318.
Tai WT, Cheng AL, Shiau CW, Huang HP, Huang JW, Chen PJ et al. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J Hepatol 2011; 55: 1041–1048.
Fan LC, Teng HW, Shiau CW, Lin H, Hung MH, Chen YL et al. SHP-1 is a target of regorafenib in colorectal cancer. Oncotarget 2014; 5: 6243–6251.
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011; 129: 245–255.
Su JC, Chen KF, Chen WL, Liu CY, Huang JW, Tai WT et al. Synthesis and biological activity of obatoclax derivatives as novel and potent SHP-1 agonists. Eur J Med Chem 2012; 56: 127–133.
Chen KF, Chen HL, Liu CY, Tai WT, Ichikawa K, Chen PJ et al. Dovitinib sensitizes hepatocellular carcinoma cells to TRAIL and tigatuzumab, a novel anti-DR5 antibody, through SHP-1-dependent inhibition of STAT3. Biochem Pharmacol 2012; 83: 769–777.
Chen KF, Su JC, Liu CY, Huang JW, Chen KC, Chen WL et al. A novel obatoclax derivative, SC-2001, induces apoptosis in hepatocellular carcinoma cells through SHP-1-dependent STAT3 inactivation. Cancer Lett 2012; 321: 27–35.
Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH, Lin MW et al. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol Cancer Ther 2012; 11: 452–463.
Su JC, Tseng PH, Hsu CY, Tai WT, Huang JW, Ko CH et al. RFX1-dependent activation of SHP-1 induces autophagy by a novel obatoclax derivative in hepatocellular carcinoma cells. Oncotarget 2014; 5: 4909–4919.
Su JC, Mar AC, Wu SH, Tai WT, Chu PY, Wu CY et al. Disrupting VEGF-A paracrine and autocrine loops by targeting SHP-1 suppresses triple negative breast cancer metastasis. Sci Rep 2016; 6: 28888.
Acknowledgements
This work was supported by grants from the Taiwan Clinical Oncology Research Foundation; the Yen Tjing Ling Medical Foundation (CI-104-07); the Ministry of Science and Technology, Taiwan (MOST 103-2325-B-075-002, MOST 104-2628-B-075-001-MY3, 105-2314-B-002-190-MY2); the National Health Research Institutes, Taiwan (NHRI-EX106-10608BI); Yang-Ming Branch of Taipei City Hospital (10601-62-020); Taipei Veterans General Hospital (V104C-151, V105C-067, V106C-101); the TVGH-NTUH Joint Research Program (VN105-09 and VN106-07); the Taipei Veterans General Hospital and National Taiwan-University Hospital, and the Ministry of Health and Welfare, Executive Yuan, Taiwan (MOHW105-TDU-B-211-134003 and MOHW106-TDU-B-211-144-003 for the Center of Excellence for Cancer Research at Taipei Veterans General Hospital). This study was also partially supported by the Chong Hin Loon Memorial Cancer and the Biotherapy Research Center of National Yang-Ming University, Taipei, Taiwan.
Author contributions
LMT and KFC are the corresponding authors and were responsible for coordinating and editing the manuscript. CYL and TTH drafted the manuscript. CTH, CYL, CHL, KYL, WCT and TIC conducted the in vitro experiments. PYC, WLW, MHC and CHL conducted the animal experiments. CYL, TTH, CWS, LMT and KFC helped with data interpretation and statistical analysis. CYL, TTH, PYC, CTH, CHL, WLW, KYL, WCT, JCS and MHC prepared the figures. All authors made substantial contributions to either the conception or design of the work. All authors have read, critically revised for intellectual content, and approved the final manuscript. All authors agreed on the accuracy and integrity of all parts of the work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Liu, CY., Huang, TT., Chu, PY. et al. The tyrosine kinase inhibitor nintedanib activates SHP-1 and induces apoptosis in triple-negative breast cancer cells. Exp Mol Med 49, e366 (2017). https://doi.org/10.1038/emm.2017.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.114
- Springer Nature Limited
This article is cited by
-
ETHE1 dampens colorectal cancer angiogenesis by promoting TC45 Dephosphorylation of STAT3 to inhibit VEGF-A expression
Cell Death & Disease (2024)
-
Interfering B cell receptor signaling via SHP-1/p-Lyn axis shows therapeutic potential in diffuse large B-cell lymphoma
Molecular Medicine (2022)
-
Nintedanib induces senolytic effect via STAT3 inhibition
Cell Death & Disease (2022)
-
Long noncoding RNA PENG upregulates PDZK1 expression by sponging miR-15b to suppress clear cell renal cell carcinoma cell proliferation
Oncogene (2020)
-
STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review
Journal of Experimental & Clinical Cancer Research (2019)