
Abstract
Glutaric aciduria type II (GAII) is a rare inborn error of metabolism clinically classified into a neonatal-onset form with congenital anomalies, a neonatal-onset form without congenital anomalies and a mild and/or late-onset form (MIM #231680). Here, we report on a GAII patient carrying a homozygous novel c.143_145delAGG (p.Glu48del) mutation in the ETFB gene, who presented with a neonatal-onset form with congenital anomalies and rapidly developed cardiomegaly after birth.
Similar content being viewed by others

Glutaric aciduria type II (GAII), also known as multiple acyl-CoA dehydrogenase deficiency, is an inborn error of metabolism clinically characterized by hypoketotic hypoglycemia and metabolic acidosis, pathologically by fatty infiltration of the liver, heart and kidneys, and biochemically by the accumulation of the metabolites of compounds catalyzed by enzymes that require electron transport flavoprotein (ETF) as an electron acceptor. GAII patients are clinically classified into three types: a neonatal-onset form with congenital anomalies, a neonatal-onset form without congenital anomalies and a mild and/or late-onset form (MIM #231680).1,2 The neonatal-onset form with congenital anomalies is lethal and is characterized by metabolic acidosis, hypoglycemia, hypotonia, ‘sweaty feet’ odor, facial dysmorphisms, polycystic kidneys, hepatomegaly and pulmonary hypoplasia.2 Some cases were noticed to have cardiomegaly after birth.3
ETF is a heterodimer of the ETF alpha subunit (ETFA) and ETF beta subunit (ETFB) that is located in the mitochondrial matrix and serves as the electron acceptor for at least nine flavoprotein dehydrogenases, including acyl-CoA dehydrogenases. It also enables both fatty acid beta-oxidation and amino acid catabolism within the main respiratory chain,4 and is reoxidized by ETF-ubiquinone oxidoreductase in the inner mitochondrial membrane. Most GAII patients have a defect in either ETF or electron transport flavoprotein dehydrogenase (ETFDH).
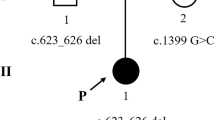
ETFB is located on chromosome 19q13.3 and consists of six exons. GAII patients carrying ETFB mutations can also be classified into the three different clinical forms mentioned earlier.5,6 Here, we present a GAII patient carrying a novel homozygous ETFB mutation, c.143_145delAGG (p.Glu48del), complicated with congenital anomalies and who rapidly developed cardiomegaly after birth.
The female GAII patient, a second child of healthy nonconsanguineous parents, was born at 31 weeks’ gestation by emergency cesarean section because of fetal distress. She had a healthy older brother and no family history of metabolic disease or sudden death in the neonatal period. At 21 weeks’ gestation, ultrasonography revealed that she had enlarged polycystic kidneys. Oligohydramnios gradually developed in the third trimester of pregnancy. Her birth weight was 1,962 g, and her Apgar score was 1 at 1 min and 3 at 5 min. Physical examination revealed low-set ears, micrognathia, redundant skin below the neck, a distended abdomen, a flexed position of both wrists, hypogenitalism and hypotonia.
Ultrasonography of the kidney and brain revealed bilateral enlarged hyperechoic kidneys and large cavum septi pellucidi, respectively. She received high-frequency oscillatory ventilation because of severe pulmonary hypoplasia soon after birth and peritoneal dialysis for profound oliguria 20 h after birth. She also received an intravenous glucose infusion (infusion rate: 10–12 mg/kg/min) after birth; however, severe hypoglycemia (blood glucose levels 16–34 mg/dl at 12–24 h old) was observed. She had persistent moderate metabolic acidosis despite treatment with sodium bicarbonate. At 2 days of age, she was noted to have a ‘sweaty feet’ odor and hyperammonemia (238 μmol/l). Urinary organic acid analysis revealed an increase in the excretion of adipic, suberic, sebacic, ethylmalonic, glutaric and fumaric acids and isovalerylglycine, suggesting a diagnosis of GAII. She was treated with intravenous riboflavin (100 mg/kg/d) and carnitine (100 mg/kg/d) administration. An echocardiogram revealed a slight thickening of the ventricular wall 3 days after birth, which rapidly increased as seen in hypertrophic cardiomyopathy. She developed cardiac dysfunction and died 6 days after birth. An autopsy revealed a thickened ventricle and numerous lipid droplets in the cardiomyocytes by Sudan III staining (Figures 1 and 2).

Heart of the 6-day-old GAII patient with a causative novel ETFB mutation showing a thickened ventricle wall and narrow ventricular lumen.

Fat droplet accumulation in cardiomyocytes by Sudan III staining. Original magnification ×400.
Molecular analysis of genomic DNA extracted from the peripheral blood cells of the patient and her parents was performed after informed consent was obtained. We PCR-amplified all coding regions of ETFA and ETFB using primers designed from genomic data. The patient was revealed to be a homozygote of the novel c.143_145delAGG (p.Glu48del) mutation in ETFB. Her parents were heterozygous for the mutation.
In GAII, lipid accumulation is observed in the tissues including the liver, heart and renal tubular epithelium, which use fatty acids as a primary source of energy. Colevas et al.7 speculated that the malformations might be the consequence of an accumulation of toxic metabolites that is not corrected by placental transfer. Enlarged polycystic kidneys and hepatomegaly are observed in many GAII patients and are often detected in prenatal periods.8 Cardiomegaly is also a complication of many patients, but is not frequently detected during prenatal periods despite common myocardial steatosis.8,9 Most GAII patients, including our case, rapidly develop cardiomegaly after birth. The major energy sources of the fetal heart are lactate and glucose, although this changes to fatty acids soon after birth.10 Such a change in the myocardial energy metabolism could explain the rapid development of cardiomegaly after birth and the absence of rapid cardiac change associated with neonatal-onset GAII.
Clinical phenotypes of GAII patients carrying ETFB mutations range from mild to lethal. Olsen et al.6 studied the genotype–phenotype relationship in GAII by expression experiments and found that null mutations caused the neonatal-onset form with congenital anomalies, whereas mutations resulting in some residual ETF/ETFDH enzyme activity were associated with a milder phenotype. Yotsumoto et al.11 studied 15 Japanese patients with GAII and detected three compound ETFB mutations in four patients: c.[78delG];[490C>T] (p.[Gly28fs];[Arg164Trp]) mutations in a patient with neonatal-onset GAII complicated with congenital anomalies; c.[55A>T];[81delC] (p.[Lys19*];[Gly28fs]) mutations in a patient with neonatal-onset GAII without congenital anomalies; and c.[491G>A];[597+1G>C] (p.[Arg164Gln];[Gly147_Met199del]) mutations in two patients with late-onset GAII. We speculate that the novel ETFB c.143_145delAGG (p.Glu48del) mutation would drastically change the EFTB structure leading to a marked decrease in ETF/ETFDH enzyme activity, as seen for null mutations.
In conclusion, we report the case of a severe form of GAII caused by a novel ETFB mutation. Information about genotype–phenotype relationships is important in the study of genetic disorders such as GAII.
References
References
Gordon N . Glutaric aciduria type I and II. Brain Dev 2006; 28: 136–140.
Frerman FE, Goodman SI . Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: Glutaric acidemia type II. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease, 8th edn. McGraw-Hill: New York, NY, USA, 2001, pp. 2357–2365.
Singla M, Guzman G, Griffin AJ, Bharati S . Cardiomyopathy in multiple Acyl-CoA dehydrogenase deficiency: a clinico-pathological correlation and review of literature. Pediatr Cardiol 2008; 29: 446–451.
Watmough NJ, Frerman FE . The electron transfer flavoprotein: ubiquinone oxidoreductases. Biochim Biophys Acta 2010; 1797: 1910–1916.
Antonacci R, Colombo I, Archidiacono N, Volta M, DiDonato S, Finocchiaro G et al. Assignment of the gene encoding the beta-subunit of the electron-transfer flavoprotein (ETFB) to human chromosome 19q13.3. Genomics 1994; 19: 177–179.
Olsen RKJ, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N . Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat 2003; 22: 12–23.
Colevas AD, Edwards JL, Hruban RH, Mitchell GA, Valle D, Hutchins GM . Glutaric acidemia type II. Comparison of pathologic features in two infants. Arch Pathol Lab Med 1988; 112: 1133–1139.
Kjaergaard S, Graem N, Larsen T, Skovby F . Recurrent fetal polycystic kidneys associated with glutaric aciduria type II. APMIS 1998; 106: 1188–1193.
Hockey A, Knowles S, Davies D, Carey W, Hurst J, Goldblatt J . Glutaric aciduria type II, an unusual cause of prenatal polycystic kidneys: report of prenatal diagnosis and confirmation of autosomal recessive inheritance. Birth Defects Orig Artic Ser 1993; 29: 373–382.
Bartelds B, Knoester H, Smid GB, Takens J, Visser GH, Penninga L et al. Perinatal changes in myocardial metabolism in lambs. Circulation 2000; 102: 926–931.
Yotsumoto Y, Hasegawa Y, Fukuda S, Kobayashi H, Endo M, Fukao T et al. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol Genet Metab 2008; 94: 61–67.
Data Citations
Sudo, Yosuke HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.598 (2015)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sudo, Y., Sasaki, A., Wakabayashi, T. et al. A novel ETFB mutation in a patient with glutaric aciduria type II. Hum Genome Var 2, 15016 (2015). https://doi.org/10.1038/hgv.2015.16
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.16
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.