

Abstract
The common carp, Cyprinus carpio, is one of the most important cyprinid species and globally accounts for 10% of freshwater aquaculture production. Here we present a draft genome of domesticated C. carpio (strain Songpu), whose current assembly contains 52,610 protein-coding genes and approximately 92.3% coverage of its paleotetraploidized genome (2n = 100). The latest round of whole-genome duplication has been estimated to have occurred approximately 8.2 million years ago. Genome resequencing of 33 representative individuals from worldwide populations demonstrates a single origin for C. carpio in 2 subspecies (C. carpio Haematopterus and C. carpio carpio). Integrative genomic and transcriptomic analyses were used to identify loci potentially associated with traits including scaling patterns and skin color. In combination with the high-resolution genetic map, the draft genome paves the way for better molecular studies and improved genome-assisted breeding of C. carpio and other closely related species.
Similar content being viewed by others


Main
Carp (cyprinids) contribute over 20 million metric tons to fish production worldwide and account for approximately 40% of total global aquaculture production and 70% of total freshwater aquaculture production. They have emerged as the most economically important teleost family. In comparison to other major aquaculture species, such as salmon and shrimp, carp are recognized as an ecofriendly fish because most are omnivorous filter-feeders and thus consume much less fish meal and fish oil. As one of the dominant cyprinid species, C. carpio (the common carp) is cultured in over 100 countries worldwide and accounts for up to 10% (over 3 million metric tons) of global annual freshwater aquaculture production1,2. In addition to its value as a food source, C. carpio is also an important ornamental fish species. One of its variants, koi, is the most popular outdoor ornamental fish because of its distinctive color and scale patterns.
Most teleosts have undergone a teleost-specific genome duplication (TSGD) and contain 24 to 25 chromosomes in their haploid genome. The haploid genome of C. carpio has 50 chromosomes3, and molecular evidence suggests that an additional whole-genome duplication (WGD) event tetraploidized the genome4,5,6,7. Although cytogenetic evidence of the allotetraploidization of C. carpio has suggested that 50 bivalents rather than 25 quadrivalents are formed during meiosis6, genome-scale validation is of great importance. Owing to its economic value in aquaculture, C. carpio has been intensively studied in terms of its physiology, development, immunology, disease resistance, selective breeding and transgenic manipulation. In addition, it is also considered an alternative vertebrate fish model to zebrafish (Danio rerio). A variety of C. carpio genome resources have been developed over the past decade, including a large number of genetic markers8,9, genetic maps10,11,12,13, a BAC-based physical map14,15, a large number of ESTs16,17,18 and cDNA microarrays19. Recently, a comparative exomic study of C. carpio and D. rerio has been reported, providing additional genome resourcing data for the research community20.
Using a whole-genome shotgun strategy and combining data from several next-generation sequencing platforms, we have produced a high-quality genome assembly for C. carpio (strain Songpu) and completed the genomic resequencing of 33 C. carpio accessions that represent major domesticated strains and populations. In addition to comparative and evolutionary studies of C. carpio and its closely related species using the genome sequences, we also demonstrate the genetic basis of phenotypic traits on scale patterning and body color determination, on the basis of data from two distinct domesticated strains (Songpu and Hebao). This study on the C. carpio genome provides a valuable resource for the molecular-guided breeding and genetic improvement of the common carp.
Results
Sequencing and assembly
We prepared genomic DNA from a homozygous double-haploid clonal line from the domesticated strain Songpu, which has a documented breeding history. We performed whole-genome shotgun sequencing on three next-generation sequencing platforms, Roche 454, Illumina and SOLiD, using both single-end and paired-end or mate-pair libraries of various insert size ranging from 250 bp to 8 kb (Supplementary Table 1). Our contig assembly was based on single-end pyrosequencing data (CABOG, Celera Assembler with Best Overlap Graph), and the scaffold assembly was based on paired-end and mate-pair sequences from different sequencing platforms and 29,046 paired BAC-end sequences14,15. After gap filling, the contig and scaffold N50 lengths reached 68.4 kb and 1.0 Mb, respectively. The total length of all scaffolds was 1.69 Gb (Table 1). We estimated the genome size to be 1.83 Gb on the basis of K-mer analysis, which is consistent with estimates based on cytogenetic methods3,21 (Supplementary Fig. 1). Thus, the scaffolds covered at least 92.3% of the genome (90.2% if we excluded sequence gaps of 40 Mb in length) and 90% of the assembly containing 2,503 large scaffolds, for a total length of 1.53 Gb.
To validate the genome assembly, we mapped all paired-end and mate-pair reads from different sequencing platforms to the assembly and found that an average of 80.3% of the reads (78.1% of Illumina reads, 74.6% of Life Technologies SOLiD reads, 98% of Roche 454 reads and 98.8% of BAC-end reads) could be mapped (Supplementary Fig. 2 and Supplementary Table 2). To assess the accuracy of the assembly, we aligned our assembly to an assembled BAC and five large scaffolds from previously published genome sequences20; the result demonstrates high consistency between the two data sets (Supplementary Fig. 3). To assess gene coverage, we mapped assembled transcriptome sequences, including publically available ESTs and new mRNA reads from multiple tissues, to the assembly. The effort yielded ∼88.8% coverage of these transcripts by nucleotide sequence similarity (Supplementary Table 3); of all of the mapped genes, 90% were common among the sequenced teleosts (Supplementary Table 4). Owing to the multiple rounds of GWD, C. carpio genes are rich in paralogs, which are thought to interfere with assembly. Therefore, we mapped 19 duplicated genes that were shared among teleosts to the genome and found that 16 of the gene pairs mapped to distinct locations whereas the other 3 collapsed into single genes, given the high similarity among the paralogs (Supplementary Table 5).
Genetic map and markers
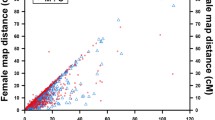
Our attempt to anchor the genome assembly onto a newly updated high-resolution genetic map, constructed using a genetic mapping panel of 107 full siblings produced from the cross of a Songpu pair (Supplementary Note), succeeded in placing a total of 3,470 high-quality SNPs and 773 microsatellite markers (Supplementary Table 6), which clustered into 50 linkage groups (Supplementary Fig. 4) and covered a genetic distance of 3,946.7 cM. The physical coverage of these linkage groups contained 1,456 of the longest scaffolds (∼875 Mb in total length and 16.7 Mb of gaps) (Fig. 1 and Table 1), and the ratio of the median genetic distance to the physical distance was 0.2 Mb/cM (Supplementary Figs. 5 and 6).

(a) The genetic linkage map. (b) Anchors between the genetic markers and the assembled scaffolds. (c) Assembled chromosomes. (d) Gene distribution on each chromosome; red lines indicate genes on the plus strand, and blue lines indicate genes on the minus strand. (e) GC content within a 10-kb sliding window; blue indicates GC content that is higher than average, and black indicates GC content that is lower than average. (f) Repeat content within a 10-kb sliding window.
Genome characterization
The C. carpio genome has a GC content of 37.0%, slightly higher than that of D. rerio but much lower than that of other sequenced teleost genomes (Supplementary Fig. 7 and Supplementary Note). To identify transposable elements (TEs)22, we constructed a C. carpio–specific repeat database. We found that 529 Mb of the assembled contigs (31.3% of the genome assembly) could be attributed to TEs (Table 1 and Supplementary Tables 7 and 8). This proportion of the content is higher than that for most of the sequenced teleost genomes (7.1% in Takifugu rubripes23, 5.7% in Tetraodon nigroviridis24, 30.68% in Oryzias latipes25 and 13.48% in Gasterosteus aculeatus26) but lower than that for the D. rerio genome (59.78%) (Supplementary Table 9). Of the TEs, the fraction of class I TEs (retroelements) was 9.99% of the total genome assembly (4.90% long interspersed nuclear elements (LINEs), 4.35% long terminal repeats (LTRs) and 0.47% short interspersed nuclear elements (SINEs)), whereas that of the class II TEs (DNA transposons) was 17.53%. The most abundant DNA transposon family identified in the C. carpio genome was the hAT superfamily, which had approximately 463,000 copies and accounted for 33% of all identified DNA transposons (consistent with our previous findings from the analysis of BAC-end sequences15). The distribution of the divergence rates for the TEs peaked at 6% in C. carpio and at 8% in D. rerio, suggesting a more recent expansion of these elements in the C. carpio genome (Supplementary Fig. 8).
We used a comprehensive strategy to annotate C. carpio genes by combining ab initio gene prediction (FGENESH and AUGUSTUS), protein-based homology (Supplementary Note) and transcript-based evidence (transcriptomes from multiple tissues and developmental stages) (Supplementary Table 10). All predicted gene structures were integrated with EVidenceModeler (EVM)27 to yield a consensus gene set containing a total of 52,610 protein-coding genes, of which 91.4% were proven to be expressed (Table 1, Supplementary Fig. 9 and Supplementary Tables 11 and 12). This gene number is almost twice that found in D. rerio, confirming the fact that the tetraploid genome retained a large portion of its gene duplicates after the latest WGD. The average gene and coding sequence lengths were 12,145 bp and 1,487 bp, respectively, and C. carpio genes had an average of 7.48 exons per gene (Supplementary Fig. 10 and Supplementary Table 13). In addition, the non-protein-coding genes included 1,012 rRNA, 3,622 tRNA and 914 microRNA (miRNA) genes (Table 1 and Supplementary Table 14).
Genome evolution
C. carpio has 100 chromosomes, approximately twice as many as are found in other cyprinid fish species. Many studies have corroborated the occurrence of either TSGD or the third round of WGD in most ray-finned fishes28,29,30,31,32 and have predicted that TSGD has facilitated the evolutionary radiation and phenotypic diversification of the teleost fishes29,31. The C. carpio genome is believed to have undergone an additional round of genome duplication (4R) and to have thus tetraploidized4,5,6,33. We have identified approximately 50 chromosome bivalents rather than quadrivalents in the meiotic nuclei of C. carpio, suggesting that it is not a true tetraploid species according to karyotyping. The tetraploidy observed in C. carpio seems to result from allotetraploidization (species hybridization) rather than autotetraploidization (genome doubling)6. We aligned 52,610 high-confidence gene models to the 50 C. carpio chromosomes and the D. rerio genome (n = 25 chromosomes) and identified 8,002 orthologous gene pairs with a clear two-to-one orthologous relationship between the two species, respectively (Fig. 2a). The major obscure synteny found on the long arm of D. rerio chromosome 4 is actually in accordance with a recent report that highlighted unique features of this region34: the region shows little orthology with other sequenced teleost genomes and harbors zebrafish-specific gene duplication and a high-density small nuclear RNA (snRNA) cluster that accounts for 53.2% of all snRNAs in the genome. This region most likely emerged in D. rerio after Danio-Cyprinus divergence. In addition, we also observed a number of minor chromosome rearrangements on the carp chromosomes, including on the long arm of chromosome 8 (showing weakened orthology with D. rerio chromosome 4) and the region containing orthologs with D. rerio chromosome 17. We also identified 2,114 best-match reciprocal paralogous gene pairs and built ohnologous blocks on 25 paired chromosomes. A circular representation of ohnolog pairs clearly demonstrates their one-to-one syntenic relationship (Fig. 2b), consistent with previous observations for genome tetraploidization.

(a) Maps of the 50 common carp chromosomes and of the 25 zebrafish chromosomes based on the positions of 8,002 orthologous pairs demonstrate highly conserved synteny for the 2 species and the tetraploidization of the common carp genome. (b) Schematic of major interchromosomal relationships in the common carp genome based on reciprocal best-match relationships. Chromosomes are represented as colored blocks. The positions of duplicated genes on the chromosomes are linked by gray lines. (c) The distribution of the synonymous substitution rates (Ks) of homologous gene groups for intraspecies and interspecies comparisons. The peak (Ks = 0.03) of the paralogous Ks distribution (inset) indicates the recent WGD. (d) The third and fourth genome duplications in the common carp genome were identified by 4dTv analyses. (e) Comparison of the gene repertoire of the Ciona species with those of five teleost and six tetrapod genomes, ranging from the highly conserved chordate genes (the black fraction on the left) to species-specific genes (the rust red fraction on the right). “1:1:1” indicates universal single-copy genes, and “X:X:X” indicates any other orthologous group (missing in one species), where X means one or more orthologs per species. “Patchy” indicates the existence of other orthologs that are present in at least one teleost and one tetrapod genome. The species tree on the left was built on the basis of 941 single-copy orthologs, for which the Ciona species served as the outgroup.
To further provide insight into the tetraploid nature of the genome at the gene level after the 4R WGD event, we investigated the hox gene clusters in C. carpio. This species has almost twice the number of hox clusters as D. rerio35 and the same number of hox gene clusters as the Atlantic salmon (Salmo salar)36, which is an autotetraploid species37 (Supplementary Figs. 11 and 12, and Supplementary Note).
To determine the date of the C. carpio WGD event (4R), we used a total of 5,783 gene families and calculated their synonymous substitution rates (Ks values) (Fig. 2c and Supplementary Note). On the basis of a Ks rate of 3.51 × 10−9 substitutions per synonymous site per year5 and the obtained Ks value of 0.03, we estimated that the latest WGD (4R) happened 8.2 million years ago, a date more recent than the predictions suggested in previous reports5,7. The carp-zebrafish paralogous genes displayed a distinct peak (Ks = 0.45) that corresponded to a divergence time of 128 million years ago. In combination with the duplication time and divergence time predictions, these data suggest that the latest WGD event (4R) occurred long after C. carpio and D. rerio split. Similarly, an analysis of fourfold synonymous third-codon transversion (4dTv) provided additional evidence for an extra round of WGD (Fig. 2d). C. carpio and D. rerio had a peak in common (4dTv = 0.58), which corresponds to the TSGD event (3R). An extra 4dTv peak within the C. carpio paralogous genes (4dTv = 0.1) corresponds to the latest carp-specific WGD (4R).
The annotated gene models of the C. carpio genome are substantially better than those of other completely sequenced fish genomes. To understand the evolutionary relationship of the 52,610 gene models with those of other vertebrates, we performed systematic cross-species comparative analysis and classified the genes according to their similarities. We first used five teleosts (C. carpio, D. rerio, T. rubripes, O. latipes and G. aculeatus), six tetrapods (Homo sapiens, Mus musculus, Sus scrofa, Gallus gallus, Anolis carolinensis and Xenopus tropicalis) and Ciona intestinalis (outgroup) for the comparison (Fig. 2e). We identified 941 single-copy orthologs that were conserved among all investigated species, which only accounted for 1.8% of the predicted gene models of C. carpio. Second, we constructed the species phylogeny using a maximum-likelihood approach with multiple alignments of single-copy orthologs. The remaining gene models (98.2%) were more complex and included many-to-many orthologs (28.0%), non-uniformly occurring, patchy orthologs (11.0%) and undetectable models (6.1%). Third, the predicted C. carpio gene models corresponded to orthologous genes in D. rerio (8,002 orthologous genes), including 2,037 (3.9%) cyprinid-specific gene models. Fourth, we also identified 3,212 species-specific gene models of C. carpio that did not have any homologs in the 10 other vertebrates and the Ciona species examined. This number is higher than the number of species-specific genes in the D. rerio genome, suggesting that a significant number of novel genes were generated in C. carpio after the divergence of C. carpio and D. rerio, likely owing to the latest WGD and independent gene evolution (Supplementary Table 15 and Supplementary Note).
Genetic diversity
C. carpio, as a genetically diverse and successful species, has adapted to various environments across a broad ecological spectrum in Eurasia and has been domesticated for more than 2,000 years. This species has been bred into numerous strains and local populations, producing distinct phenotypic changes in its growth rate, temperature and hypoxia tolerance, body color, scale pattern and body shape, which are partially attributable to genome diversity due to its two WGD (3R and 4R) events31. To investigate its genetic variation, we selected 33 representative C. carpio accessions for genome resequencing, which included 13 accessions of 4 wild populations from the Danube River, the Yellow River, the Heilongjiang (Amur) River and the Chattahoochee River and 20 accessions of 6 domesticated strains from Asia and Europe (including Songpu, Xingguo red, Oujiang color, Hebao, Szarvas 22 and koi) (Supplementary Table 16). With a total of 4,176 million paired-end reads (101-bp read length, 417.6 Gb in total length and 229-fold coverage of the genome; Supplementary Table 17), we identified 18,949,596 candidate SNPs and 1,694,102 small insertion-deletions (indels) (Supplementary Table 18).
To investigate the divergence of the representative C. carpio accessions from diverse geographical habitats and domestic histories, we constructed phylogenetic trees on the basis of the sequence variations (Fig. 3a and Supplementary Fig. 13). It was obvious that the European and Asian accessions formed two distinct clades. One of the strains, Songpu, was also grouped into the European clade as it was bred from mirror carp originally introduced from Europe in the 1950s. Our principal-component analysis (PCA) yielded a similar result (Fig. 3b), showing Asian accessions as a tight cluster that was separate from the European accessions. We further analyzed the population structure using the Bayesian clustering program STRUCTURE38. Because the values of ln likelihood were distinctively high for the models K = 3 and 4 (Supplementary Fig. 14), we show the clusters of K = 3 and 4 in Figure 3c. Almost all the accessions either had common ancestry or showed a single origin of the Eurasian population, and the results agree with the hypothesis that modern C. carpio evolved from the Caspian Sea ancestor and spread into Europe and the eastern mainland of Asia39. There were no uniform patterns covering all the populations, with the exception of two extremely isolated wild populations, Heilongjiang and Oujiang; in other words, extensive genetic admixture has been occurring in both the wild and domesticated C. carpio populations. For instance, Songpu carried admixture from the Asian population (K = 3), a finding supported by the recent history of introgression after its introduction to China. We also observed that the US accessions separated into both the European and Asian clades, and the trend indicates multiple introductions to North America from both Europe and Asia. This observation is also supported by our PCA and population structure analyses.

(a) A maximum-likelihood phylogenetic tree of ten strains of common carp generated on the basis of SNPs. Strain abbreviations: Sp, Songpu; D, Danube; Sz, Szarvas; NA, North American; Y, Yellow River; H, Heilongjiang; O, Oujiang color; Hb, Hebao; X, Xingguo; K, koi. (b) PCA of C. carpio strains. (c) The population structure of common carp strains. Each color represents one ancestral population; each strain is represented by a vertical bar, and the length of each colored segment in each vertical bar represents the proportion contributed by ancestral populations. K = 3 and 4 was used for analysis with the highest ln value.
We performed a further genetic diversity scan comparing the Hebao and Songpu genomes to identify highly different genomic regions. Hebao is one of the typical strains derived from East Asian subspecies (C. carpio haematopterus), whereas Songpu is the strain derived from mirror carp of European subspecies (C. carpio carpio) (Fig. 4a). We predict that these two varieties retain substantial genetic differences, given their distinct body shapes, scale morphogenesis and patterns, and skin color phenotypes. We identified a total of 205 genome regions with the highest (top 1% of πHebao/πSongpu) genetic diversity (12.67 Mb in length) containing 326 candidate genes (Supplementary Tables 19 and 20). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that a significant portion of these candidate genes were associated with epithelial morphogenesis, hair follicle morphogenesis, pigmentation and immune response, including adherens junction signaling, signaling by Rho family GTPases, tight junction signaling, prolactin signaling, fibroblast growth factor (FGF) signaling, interleukin (IL)-6 signaling and other functional pathways (Fig. 4b and Supplementary Table 21). The results are consistent with the phenotype differences in scale pattern and skin color observed for Hebao and Songpu. We investigated the candidate genes and detected 82 genes and 106 genes that harbored nonsynonymous SNPs in the Songpu and Hebao genomes, respectively (Supplementary Tables 22 and 23). We also identified two Songpu genes (fgfr1a1 and lrrc72) and two Hebao genes (zpld1 and nlk) that harbored deletions in coding regions (Supplementary Figs. 15 and 16). All these instances of sequence diversity altering protein-coding sequences provide candidate loci for assessing phenotypic differences between Hebao and Songpu. Notably, the fgfr1a1 gene (encoding FGF receptor 1 a1) on chromosome 34 of the Songpu genome contained a 306-bp specific deletion in intron 10 (228-bp deletion) and exon 11 (78-bp deletion) (Fig. 4c). The deletion had previously been reported as the causative mutation for scale loss and reduction in the mirror carp40. Extensive investigation on large samples from four strains confirmed that the deletion was only found in Songpu (Supplementary Fig. 16).

(a) Typical Songpu and Hebao carp. (b) The distribution of genes involved in KEGG pathways in the regions with the top 1% of πHebao/πSongpu. (c) The diversity (π) distribution on chromosome 34 for Songpu and Hebao showed a 78-bp deletion in exon 11 of the fgfr1a1 gene in Songpu; this deletion results in a reduced-scale phenotype. (d) The pheomelanin synthesis pathway. Transcriptome analysis showed that the slc7a11 gene is significantly more upregulated in the skin of Hebao than that of Songpu. slc7a11 encodes the transmembrane cystine/glutamate exchanger (xCT), which transports cystine into melanocytes to synthesize pheomelanin (yellow to red pigment).
Comparative analysis of the skin transcriptome
To further elucidate the differences in the scale pattern and skin color of Hebao and Songpu, we performed a comparative analysis of the skin transcriptome in both strains using a deep RNA sequencing (RNA-seq) approach. We identified 894 differentially expressed transcripts, including 567 upregulated genes in Hebao and 327 upregulated genes in Songpu. The experiment was validated with quantitative RT-PCR (qRT-PCR) on selected genes (Supplementary Fig. 17). Further analysis showed distinct expression patterns in Hebao and Songpu for many genes associated with the Wnt/β-catenin signaling pathway (Supplementary Table 24), which is an essential pathway in initiating hair follicle formation41. Both mammalian hair and teleost scales are skin appendages, and their formation involves similar developmental pathways. We inferred that the gene expression differences in these two different carp populations were correlated with the reduced-scale phenotype in Songpu and the full-scale phenotype in Hebao.
We also observed a difference in the expression of the slc7a11 gene (encoding solute carrier family 7 member 11), the plasma membrane cystine/glutamate exchanger (xCT) that transports cystine into melanocytes. In the melanogenesis pathway, tyrosine is oxidized to form dopaquinone, which is then intracellularly catalyzed to become eumelanin (brown to black pigment) through polymerization and oxidation reactions. However, cystine and dopaquinone can switch off the eumelanin synthesis pathway and promote the synthesis of pheomelanin (yellow to red pigment)42,43. slc7a11 was expressed at a higher level in Hebao than in Songpu, suggesting that more cystine is transported into the pigment cells in Hebao, resulting in the preponderant synthesis of pheomelanin in the skin of Hebao while eumelanin synthesis is suppressed. Higher pheomelanin accumulation in the pigment cells gives Hebao its red skin appearance (Fig. 4d). However, the genetic basis for differences in slc7a11 expression remains unclear, and further investigation will be necessary to understand the overall role of slc7a11 in color variation.
Discussion
As one of the most representative carp species, C. carpio had a value and global production in 2011 of $5.31 billion and 3.73 million tons, respectively (FAO statistics; see URLs), and the importance of C. carpio has been increasing over the past decade. The species is also widely cultured as an ornamental fish because of its various color and scale patterns. We sequenced and assembled the C. carpio genome from the genome of a gynogenetic individual using multiple next-generation sequencing platforms and a hybrid assembly strategy. The draft genome provides an important genomic resource to study the genetic basis of economically important traits in carp and to facilitate genome-based genetic breeding technologies in common carp aquaculture. The draft genome also provides insight into the latest WGD event of allotetraploidization that occurred approximately 8.2 million years ago, doubling the chromosome number and gene content of C. carpio. The whole-genome resequencing of selected accessions also offers a glimpse into the phylogenetic relationship and population structure of major global accessions of the C. carpio population. Comparison of the genomic diversity of two distinct strains, Songpu and Hebao, coupled with additional transcriptomic studies, has allowed us to identify genetic loci and to determine the molecular basis of scale patterns and skin colors, providing a foundation for further studies using comprehensive approaches to completely define the mechanisms underlying these phenotypes. Thus, the draft genome assembly presented here provides a valuable resource for genetic, genomic and biological studies of C. carpio and for improving the aquaculturally important traits of farmed C. carpio and other key cyprinid species in aquaculture.
Methods
Ethics statement.
This study was approved by the Animal Care and Use Committee of the Centre for Applied Aquatic Genomics at the Chinese Academy of Fishery Sciences.
Genome sequence and assembly.
A gynogenetic Songpu C. carpio was selected as the genomic DNA source for whole-genome sequencing. We constructed 21 shotgun libraries and an 8K mate-pair library according to Roche 454 standard operating procedures. The 22 libraries were sequenced on a Roche 454 genome sequencer using GS FLX Titanium chemistry. We also constructed six paired-end libraries by following the Illumina procedure. Paired-end sequencing of each library was performed on an Illumina HiSeq 2000 instrument to produce the raw data. We then filtered out low-quality and short reads to obtain a set of usable reads. Another library with an 8-kb jumping distance was generated and was sequenced on the SOLiD platform. The published 65,720 clean BAC-end sequences were collected for genome assembly. We assembled the Roche 454 read data set and the Sanger BAC-end sequences into contigs using the Celera assembler44. Reads from the Illumina libraries, the SOLiD libraries and the 8,000 Roche 454 mate-pair libraries were aligned to the genomic sequences, and paired-end relationships between the reads were used to construct scaffolds. BAC-end sequences were mapped to the scaffolds and were used for further scaffolding. Finally, we used the paired-end information from the short paired-end reads to fill the gaps between the scaffolds with Gapcloser45.
Linkage mapping and map integration.
Microsatellite and SNP markers were used for genotyping analysis and linkage map construction. A tailed primer protocol was used to amplify microsatellite alleles46,47. PCR products were analyzed on a 3130xl Genetic Analyzer. Restriction site–associated DNA (RAD) technology48 was used to develop polymorphic SNP markers. JoinMap4.0 software was used to perform the linkage analysis. Linkage between markers was examined by estimating the logarithm of odds (LOD) scores for the recombination rate, and map distances were calculated using the Kosambi mapping function. We then used BLAT (with alignment length coverage of >70%) to align the molecular markers to scaffolds. We linked the scaffolds onto chromosomes with a string of 100 Ns representing the gap between 2 adjacent scaffolds on the basis of a high-resolution genetic map.
Repeat analysis.
Both homology-based and de novo prediction analyses were used to identify the repeat content in the carp genome. For the homology-based analysis, we used Repbase (version 20120418) to perform a TE search with RepeatMasker (3.3.0) and the WuBlast search engine. For the de novo prediction analysis, we used RepeatModeler to construct a TE library. Elements within the library were then classified using a homologous search with Repbase and a Support Vector Machine (SVM) method (TEClass).
Gene prediction and functional annotation.
We used three approaches for gene prediction: ab initio gene prediction, sequence homology–based prediction and expression evidence–based prediction. Briefly, two ab initio prediction software programs, AUGUSTUS49 and FGENESH50, were used to predict genes in the repeat-masked genome sequences. Gene model parameters for the programs were trained from long genes and known teleost genes processed by PASA51. Sequence homology–based gene prediction included both raw and precise alignments. First, protein sequences from the NCBI non-redundant (nr) database and 68 species sequences in Ensembl (version 68)52 were collected to build a database. Assembled genome sequences were aligned to their corresponding protein sequences in the database using BLASTX. Identified homologous proteins were selected and then aligned to the genome with TBLASTN. Adjacent and overlapping matches were merged using Perl scripts, building the longest protein for each genomic sequence region. Each target region in the genome was then extended by 10 kb from both ends of the aligned region to cover potential UTRs. Protein sequences were then aligned to those genome fragments by Genewise53. Transcriptome reads were generated using the Roche Genome Sequencer FLX (previously released data; available from the Sequence Read Archive (SRA) under accessions SRA009366 and SRA050545) and the Illumina HiSeq 2000 (Supplementary Note). Reads were mapped to genomic sequences by TopHat54, and Cufflinks55 was used to produce transcript assemblies. For a gene locus with several alternatively spliced transcripts generated by Cufflinks, the transcript with the longest exon length was chosen. All evidence was merged to form a comprehensive consensus gene set using EVM27. PASA was used to update the EVM consensus predictions by adding UTR annotations. To obtain gene function annotations, BLAST searches were conducted against the NCBI nr, SwissProt and TrEMBL protein databases, and homologs were called with E values of <1 × 10−5. The functional classification of GO categories was performed using the InterProScan56 program. Pathway analysis was performed using the KEGG57 annotation service, the KEGG Automatic Annotation Server (KAAS)58.
Comparative genomic analysis.
Protein-coding genes and coding DNA sequences from 11 species (D. rerio, G. aculeatus, O. latipes, T. rubripes, T. nigroviridis, X. tropicalis, A. carolinensis, H. sapiens, M. musculus, S. scrofa, G. gallus and C. intestinalis) were downloaded from Ensembl (version 68)52. For genes with alternatively spliced variants, only the longest transcript was selected. Any genes encoding proteins of fewer than 30 amino acids were discarded. The OrthoMCL pipeline59 was used to define gene families in the common ancestor of the species. All-against-all similarities were performed using BLASTP, with an E-value cutoff of 1 × 10−5. The well-aligned regions of each gene family, aligned using MUSCLE60, were extracted with Gblocks61. Phylogenetic analysis of the superalignments was performed using a maximum-likelihood method implemented in PhyML62 with the Jones-Taylor-Thornton (JTT) model. C. intestinalis was selected as the outgroup. We used MCScanX63 to identify syntenic blocks for C. carpio and D. rerio, with the gap size set to 15 genes and at least 5 syntenic genes. Circos64 was used for visualization.
To detect the conserved synteny blocks generated by the fourth round of genome duplication, we identified the reciprocal best-match paralogs from the above all-against-all BLASTP comparisons. Two chromosome regions with the gap size set to 15 genes and at least 5 syntenic genes were considered to have been duplicated.
Genome evolutionary analysis.
We used two methods to detect genome duplication signatures. All-against-all BLASTP comparisons (E value < 1 × 10−5) were used to identify pairs of homologous genes. For each homologous gene pair in C. carpio, the synonymous site divergence value (Ks) was calculated using the CodeML program (run mode −2) from the PAML package65. The distributions of Ks values for D. rerio paralogous pairs and pairs between D. rerio and the common carp were analyzed using the same pipeline. We calculated the 4dTv values of paralogous pairs within species and of orthologous pairs between species to give the distribution of the 4dTv value to estimate the speciation and WGD event that occurred during evolutionary history.
Whole-genome resequencing and phylogenetic analysis.
The 10 strains of C. carpio, consisting of 33 individuals, were randomly collected across Europe, North America and China. Danube River carp and Szarvas 22 were collected from the carp live gene bank of the Research Institute for Fisheries, Aquaculture and Irrigation of Hungary (HAKI). North American carp were collected from the Chattahoochee River in the United States. All other strains or wild populations were collected from China, including Songpu carp from the Heilongjiang Fishery Research Institute; Yellow River carp from the Henan Academy of Fishery Sciences; Heilongjiang River carp from Fuyuan county, Heilongjiang province; Hebao carp from Wuyuan county, Jiangxi province; Xingguo red carp from Xingguo county, Jiangxi province; Oujiang color carp from Longquan county, Zhejiang province; and koi from the breeding population of the Beijing Fishery Research Institute. Fin chips and blood samples were collected, and DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen). Genome resequencing was conducted using the Illumina HiSeq 2000 platform. Paired-end reads from each accession were aligned to the reference genome using the Burrows-Wheeler Aligner (BWA)66. After mapping, SNPs were identified on the basis of the mpileup files generated with SAMtools67. The filtering threshold was set to require a read depth of ≥10 and a quality score of ≥20. Genotypes supported by at least two reads and with a minor allele frequency of ≥0.1 were assigned to each genomic position. We performed all-against-all BLASTP for genes in 5 teleosts (C. carpio, D. rerio, T. rubripes, O. latipes and G. aculeatus) to determine the similarity for each gene pair and to identify single-copy genes, obtaining 8,375 homozygous SNPs from 7,709 single-copy genes. A maximum-likelihood tree was constructed with PhyML68 and displayed with MEGA69. PCA was performed with EIGENSOFT70, and homozygous SNPs were used to investigate the population structure using STRUCTURE39 with 2,000 iterations and 2–8 clusters (K). The result of the structure matrix was plotted using DISTRUCT71 software.
Genome diversity analysis and comparison.
We calculated the π distribution for each linkage group using a sliding window method with Tajima's D test in Variscan72 software. The window width was set to 50 kb, and the stepwise distance was 10 kb. π values were compared, and the ratios were sorted. Using the ratio values, we identified the regions with the 1% highest and lowest diversity, and annotated genes were analyzed. Putative SNPs and deletions in the coding regions were identified by mapping the RNA-seq reads to annotated reference genes using BWA and SAMtools. PCR was performed on the deletion regions to verify the identified gene deletions. PCR products were analyzed via electrophoresis on a 2% agarose gel.
RNA sequencing analysis.
RNA was extracted from the skin tissues of 18 Hebao and 18 Songpu individuals and pooled for each strain. RNA-seq reads were generated using the Illumina HiSeq 2000 platform (Supplementary Note). Reads with a low quality score and a read length of less than 10 bp were removed. All cleaned reads were mapped to the assembled reference with Bowtie73. Then, RSEM (RNA-Seq by Expectation Maximization)74 was used to estimate and quantify gene and isoform abundance. Gene expression was measured in fragments per kilobase of exon per million fragments mapped (FPKM)55. Finally, edgeR75 was used to normalize the expression levels in both strains to identify the differentially expressed transcripts by pairwise comparisons. For qRT-PCR validation, total RNA was isolated and purified from all of the samples using the RNeasy kit (Qiagen) and was quantified using a NanoDrop and a Bioanalyzer 2100 (Agilent Technologies). qRT-PCR was performed on the ABI PRISM 7500 Real-Time PCR System with three replicates using the QuantiTect SYBR Green PCR kit (Qiagen). The actb gene was used as the internal reference. Primer information is provided in Supplementary Table 25. Two-sided t tests were used to compare expression levels. GO annotation of the genes was performed on the basis of orthologous relationships with the gene set of D. rerio. Pathway analysis was performed using Ingenuity Pathway Analysis (IPA) tools (Ingenuity Systems).
URLs.
Celera Assembler, http://wgs-assembler.sourceforge.net/; BWA, http://bio-bwa.sourceforge.net/; FGENESH, http://www.softberry.com/; Ensembl, http://www.ensembl.org/; Gene Ontology (GO), http://www.geneontology.org/; KEGG, http://www.genome.jp/kegg/; Repbase, http://www.girinst.org/repbase/index.html; RepeatMasker, http://repeatmasker.org/; Food and Agriculture Organization of the United Nations (FAO) statistics, http://www.fao.org/fishery/culturedspecies/Cyprinus_carpio/.
Accession codes.
The common carp whole-genome shotgun sequencing, genome resequencing and RNA sequencing results have all been deposited in GenBank under project accession PRJNA202478. The genome assembly has been deposited in the European Nucleotide Archive (ENA) under project PRJEB7241.
Accession codes
Primary accessions
BioProject
European Nucleotide Archive
Referenced accessions
Sequence Read Archive
References
FAO Fisheries and Aquaculture Department. The State of World Fisheries and Aquaculture 2006 (Food and Agriculture Organization of the United Nations, Rome, 2007).
Bostock, J. et al. Aquaculture: global status and trends. Phil. Trans. R. Soc. B 365, 2897–2912 (2010).
Hinegardner, R. & Rosen, D.E. Cellular DNA content and the evolution of teleostean fishes. Am. Nat. 106, 621–644 (1972).
Wang, J.T., Li, J.T., Zhang, X.F. & Sun, X.W. Transcriptome analysis reveals the time of the fourth round of genome duplication in common carp (Cyprinus carpio). BMC Genomics 13, 96 (2012).
David, L., Blum, S., Feldman, M.W., Lavi, U. & Hillel, J. Recent duplication of the common carp (Cyprinus carpio L.) genome as revealed by analyses of microsatellite loci. Mol. Biol. Evol. 20, 1425–1434 (2003).
Ohno, S., Muramoto, J., Christian, L. & Atkin, N.B. Diploid-tetraploid relationship among old-world members of the fish family Cyprinidae. Chromosoma 23, 1–9 (1967).
Larhammar, D. & Risinger, C. Molecular genetic aspects of tetraploidy in the common carp Cyprinus carpio. Mol. Phylogenet. Evol. 3, 59–68 (1994).
Ji, P. et al. High throughput mining and characterization of microsatellites from common carp genome. Int. J. Mol. Sci. 13, 9798–9807 (2012).
Xu, J. et al. Genome-wide SNP discovery from transcriptome of four common carp strains. PLoS ONE 7, e48140 (2012).
Zhang, X. et al. A consensus linkage map provides insights on genome character and evolution in common carp (Cyprinus carpio L.). Mar. Biotechnol. (NY) 15, 275–312 (2013).
Zheng, X. et al. A genetic linkage map and comparative genome analysis of common carp (Cyprinus carpio L.) using microsatellites and SNPs. Mol. Genet. Genomics 286, 261–277 (2011).
Zhang, Y. et al. Genetic linkage mapping and analysis of muscle fiber–related QTLs in common carp (Cyprinus carpio L.). Mar. Biotechnol. (NY) 13, 376–392 (2011).
Sun, X.W. & Liang, L.Q. A genetic linkage map of common carp (Cyprinus carpio L.) and mapping of a locus associated with cold tolerance. Aquaculture 238, 165–172 (2004).
Li, Y. et al. Construction and characterization of the BAC library for common carp Cyprinus carpio L. and establishment of microsynteny with zebrafish Danio rerio. Mar. Biotechnol. (NY) 13, 706–712 (2011).
Xu, P. et al. Genomic insight into the common carp (Cyprinus carpio) genome by sequencing analysis of BAC-end sequences. BMC Genomics 12, 188 (2011).
Christoffels, A., Bartfai, R., Srinivasan, H., Komen, H. & Orban, L. Comparative genomics in cyprinids: common carp ESTs help the annotation of the zebrafish genome. BMC Bioinformatics 7 (suppl. 5), S2 (2006).
Zhang, Y. et al. Identification of common carp innate immune genes with whole-genome sequencing and RNA-Seq data. J. Integr. Bioinform. 8, 169 (2011).
Ji, P. et al. Characterization of common carp transcriptome: sequencing, de novo assembly, annotation and comparative genomics. PLoS ONE 7, e35152 (2012).
Williams, D.R. et al. Genomic resources and microarrays for the common carp Cyprinus carpio L. J. Fish Biol. 72, 2095–2117 (2008).
Henkel, C.V. et al. Comparison of the exomes of common carp (Cyprinus carpio) and zebrafish (Danio rerio). Zebrafish 9, 59–67 (2012).
Ojima, Y. & Yamamoto, K. Cellular DNA contents of fishes determined by flow cytometry. La Kromosomo II 57, 1871–1888 (1990).
Kidwell, M.G. & Lisch, D.R. Transposable elements and host genome evolution. Trends Ecol. Evol. 15, 95–99 (2000).
Aparicio, S. et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297, 1301–1310 (2002).
Van de Peer, Y. Tetraodon genome confirms Takifugu findings: most fish are ancient polyploids. Genome Biol. 5, 250 (2004).
Kasahara, M. et al. The medaka draft genome and insights into vertebrate genome evolution. Nature 447, 714–719 (2007).
Jones, F.C. et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61 (2012).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Meyer, A. & Van de Peer, Y. From 2R to 3R: evidence for a fish-specific genome duplication (FSGD). Bioessays 27, 937–945 (2005).
Hoegg, S., Brinkmann, H., Taylor, J.S. & Meyer, A. Phylogenetic timing of the fish-specific genome duplication correlates with the diversification of teleost fish. J. Mol. Evol. 59, 190–203 (2004).
Santini, F., Harmon, L., Carnevale, G. & Alfaro, M. Did genome duplication drive the origin of teleosts? A comparative study of diversification in ray-finned fishes. BMC Evol. Biol. 9, 194 (2009).
Crow, K.D. & Wagner, G.P. Proceedings of the SMBE Tri-National Young Investigators Workshop 2005. What is the role of genome duplication in the evolution of complexity and diversity? Mol. Biol. Evol. 23, 887–892 (2006).
Jaillon, O. et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946–957 (2004).
Zhang, Y. et al. Genome evolution trend of common carp (Cyprinus carpio L.) as revealed by the analysis of microsatellite loci in a gynogentic family. J. Genet. Genomics 35, 97–103 (2008).
Howe, K. et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 (2013).
Amores, A. et al. Zebrafish hox clusters and vertebrate genome evolution. Science 282, 1711–1714 (1998).
Mungpakdee, S. et al. Differential evolution of the 13 Atlantic salmon Hox clusters. Mol. Biol. Evol. 25, 1333–1343 (2008).
Davidson, W.S. et al. Sequencing the genome of the Atlantic salmon (Salmo salar). Genome Biol. 11, 403 (2010).
Pritchard, J.K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Balon, E.K. Origin and domestication of the wild carp, Cyprinus carpio: from Roman gourmets to the swimming flowers. Aquaculture 129, 3–48 (1995).
Rohner, N. et al. Duplication of fgfr1 permits Fgf signaling to serve as a target for selection during domestication. Curr. Biol. 19, 1642–1647 (2009).
Millar, S.E. Molecular mechanisms regulating hair follicle development. J. Invest. Dermatol. 118, 216–225 (2002).
Hoekstra, H.E. Genetics, development and evolution of adaptive pigmentation in vertebrates. Heredity 97, 222–234 (2006).
Ito, S. & Wakamatsu, K. Human hair melanins: what we have learned and have not learned from mouse coat color pigmentation. Pigment Cell Melanoma Res. 24, 63–74 (2011).
Miller, J.R. et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24, 2818–2824 (2008).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Neilan, B.A., Wilton, A.N. & Jacobs, D. A universal procedure for primer labelling of amplicons. Nucleic Acids Res. 25, 2938–2939 (1997).
Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 18, 233–234 (2000).
Sun, X. et al. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 8, e58700 (2013).
Stanke, M., Schoffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Salamov, A.A. & Solovyev, V.V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000).
Campbell, M.A., Haas, B.J., Hamilton, J.P., Mount, S.M. & Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7, 327 (2006).
Flicek, P. et al. Ensembl 2013. Nucleic Acids Res. 41, D48–D55 (2013).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Trapnell, C., Pachter, L. & Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Zdobnov, E.M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A.C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Li, L., Stoeckert, C.J. Jr. & Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Guindon, S., Lethiec, F., Duroux, P. & Gascuel, O. PHYML Online—a web server for fast maximum likelihood–based phylogenetic inference. Nucleic Acids Res. 33, W557–W559 (2005).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Price, A.L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Rosenberg, N.A. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (2004).
Vilella, A.J., Blanco-Garcia, A., Hutter, S. & Rozas, J. VariScan: analysis of evolutionary patterns from large-scale DNA sequence polymorphism data. Bioinformatics 21, 2791–2793 (2005).
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, B. & Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Robinson, M.D., McCarthy, D.J. & Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Acknowledgements
We acknowledge Z. Zhu, N. Li, J. Gui, Z. Bao, G. Zhang, X.L. Zhang, J. Li, Y. Liu, Q. Liu and S. Chen for their support on the common carp genome project. We thank H. Hu, G.Z. Liu, J.H. Yu and C.J. Li for their assistance in sample collection and Y. Wan, T. Sun, W. Liu, L. Jiang, Shu Wang, Y. Zhu, X. Xing, P. Zhou and R. Cui for genotyping and sequencing. We thank J. Postlethwait, G. Hulata, L. David, L. Orban and F. Zhao for their helpful discussions. We acknowledge grant support from the National High-Technology Research and Development Program of China (863 program; 2011AA100401, 2011AA100402 and 2009AA10Z105), the National Department Public Benefit Research Foundation of China (200903045), the National Basic Research Program of China (973 Program; 2010CB126305), the National Natural Science Foundation of China (31302174 and 31101893) and Special Scientific Research Funds for Central Non-Profit Institutes of the Chinese Academy of Fishery Sciences (2009B002 and 2011C016).
Author information
Authors and Affiliations
Contributions
X.S. initiated the common carp genome project. X.S., J.Y., P.X. and X.W. conceived the study. P.X. and X.W. coordinated the project. P.X., J.Y., X. Zhang, J.L., G.L., Y.K. and J. Xu wrote and revised the manuscript and the supplementary information. J.Y., P.X. and X.W. developed the sequencing strategy. X.W., G.S., L.R., X.L., D.Y., L.W. and C.S. performed the library construction and next-generation sequencing. J.L., G.L., G.W., Lipu Song, Lai Song, W.X. and Jintu Wang conducted the genome assembly and assessment. P.X., Z. Zhao, Y.L., Jian Wang and Jintu Wang performed the BAC library construction and BAC-end sequencing. G.L., J.L., Y.K. and G.W. performed the gene prediction, repeat analysis and genome annotation. X. Zhang, Y. Zhang, X. Zheng, Y.K., C. Lu, C. Li, D.C., Y.G. and L.Z. performed the genotyping and genetic linkage mapping. J.L., Y. Zhang, X. Zhang and G.H. performed the genetic mapping and genome integration. Y.K., X. Zheng, D.C., X. Zhang, C. Lu, C. Li and W.L. contributed to gynogenetic common carp preparation and mapping family construction. G.L., J.L., G.W., L.H., S. Wu, K.W., J. Xiao, J. Wu, Z. Zhang and J.S. performed the comparative genomic and genome evolution studies. P.X., J. Xu, Z.J., P.J., Jian Wang, F.S., J.F., C.W., B.W., Q.L. and Y. Zhang collected DNA samples from wild and domesticated common carp populations worldwide. P.X., J. Xu, Z. Zhao, Y.J. and Q.L. performed the whole-genome resequencing, analysis and gene validation. J.L., S. Wang and Y. Zhu performed the noncoding RNA analysis. J. Xu, P.X., P.J. and S. Wang conducted the comparative transcriptome study. G.L., P.X., X.W., Z.F., J.L. and L.X. designed and constructed the genome databases. X.S. supervised the gynogenetic fish preparation, mapping family construction, genotyping and linkage mapping. J.Y. supervised the sequencing, assembly and bioinformatics analysis. Y. Zhou and Z.L. participated in discussions and provided valuable advice.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Figures 1–17 and Supplementary Tables 1–5, 7–18 and 22 (PDF 8873 kb)
Supplementary Tables 6, 19–21 and 23–25
Supplementary Tables 6, 19–21 and 23–25 (XLS 6996 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Xu, P., Zhang, X., Wang, X. et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat Genet 46, 1212–1219 (2014). https://doi.org/10.1038/ng.3098
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3098
- Springer Nature America, Inc.
This article is cited by
-
Evolutionary mechanisms and practical significance of reproductive success and clonal diversity in unisexual vertebrate polyploids
Science China Life Sciences (2024)
-
Transposon signatures of allopolyploid genome evolution
Nature Communications (2023)
-
The vertebrate sialylation machinery: structure-function and molecular evolution of GT-29 sialyltransferases
Glycoconjugate Journal (2023)
-
Negative regulation of interferon regulatory factor 6 (IRF6) in interferon and NF-κB signalling pathways of common carp (Cyprinus carpio L.)
BMC Veterinary Research (2022)
-
A myxozoan genome reveals mosaic evolution in a parasitic cnidarian
BMC Biology (2022)