
Abstract
The inadequate transport of drugs into the tumor tissue caused by its abnormal vasculature is a major obstacle to the treatment of cancer. Anti-vascular endothelial growth factor (anti-VEGF) drugs can cause phenotypic alteration and maturation of the tumor’s vasculature. However, whether this consistently improves delivery and subsequent response to therapy is still controversial. Clinical results indicate that not all patients benefit from antiangiogenic treatment, necessitating the development of criteria to predict the effect of these agents in individual tumors. We demonstrate that, in anti-VEGF-refractory murine tumors, vascular changes after VEGF ablation result in reduced delivery leading to therapeutic failure. In these tumors, the impaired response after anti-VEGF treatment is directly linked to strong deposition of fibrillar extracellular matrix (ECM) components and high expression of lysyl oxidases. The resulting condensed, highly crosslinked ECM impeded drug permeation, protecting tumor cells from exposure to small-molecule drugs. The reduced vascular density after anti-VEGF treatment further decreased delivery in these tumors, an effect not compensated by the improved vessel quality. Pharmacological inhibition of lysyl oxidases improved drug delivery in various tumor models and reversed the negative effect of VEGF ablation on drug delivery and therapeutic response in anti-VEGF-resistant tumors. In conclusion, the vascular changes after anti-VEGF therapy can have a context-dependent negative impact on overall therapeutic efficacy. A determining factor is the tumor ECM, which strongly influences the effect of anti-VEGF therapy. Our results reveal the prospect to revert a possible negative effect and to potentiate responsiveness to antiangiogenic therapy by concomitantly targeting ECM-modifying enzymes.
Similar content being viewed by others


Introduction
Treating solid tumors is complicated by the inadequate transport of systemically administered drugs into the tumor tissue.1 A wide range of antitumor agents, both cytotoxic drugs and newer targeted therapies, has been shown to accumulate at much higher concentrations in non-target organs than in the tumor.2, 3, 4, 5, 6 Furthermore, the distribution of drugs within the tumor is heterogeneous, leaving many tumor cells protected from therapeutically effective drug concentrations.7, 8, 9, 10, 11
The abnormal vasculature observed in many tumors has been discussed as a major factor in reducing tumor drug delivery.12, 13 Tumor blood vessels are often tortuous, leaky, strongly ramified, poorly supported by pericytes and lack a hierarchical structure, which causes an often insufficient and uneven blood flow. This aberrant vessel phenotype results from an imbalanced, strongly pro-angiogenic microenvironment. Agents that dampen the angiogenic signaling, such as inhibitors of the vascular endothelial growth factor (VEGF) pathway, improve blood vessel functionality. In preclinical models, increased pericyte coverage, reduced interstitial pressure and diminished vessel leakiness are observed after antiangiogenic therapy.14, 15, 16 The improved vessel quality should indeed increase drug delivery into the tumor, and this 'vessel normalization' hypothesis may explain why antiangiogenic drugs that show little efficacy as monotherapy in patients improve response to concomitant chemotherapy.17 However, antiangiogenic drugs were developed to inhibit the formation of the tumor vasculature, and reduction in vessel density is also a well-documented result of antiangiogenic treatment.16, 18, 19 Reduced vessel density decreases transport into the tumor and increased tumor hypoxia is commonly reported after VEGF ablation.19 Few studies have carefully examined the consequences of antiangiogenic therapy not on surrogate markers but on tumor drug delivery, with the contrarian results that the effects could be either positive or negative.20, 21, 22, 23, 24, 25 It can be assumed that whether the 'improved vessel quality' or the 'reduced vessel density' effect predominates after antiangiogenic therapy may be affected by many factors. Among those are: the dosage of the antiangiogenic drug, the duration of the treatment, and characteristics of the particular tumor. However, to date, we still lack stratifying criteria to predict how antiangiogenic therapy affects drug delivery in individual tumors.
To be effective, an antineoplastic drug must not only be transported into the tumor but it also has to reach the individual tumor cell at a critical concentration. Although the effectiveness of drug transport into the tissue is determined by vascular characteristics, transport within the tissue occurs by diffusion. The rate of diffusion within the tissue depends on the characteristics of the extracellular matrix (ECM). Tumors are often characterized by increased deposition of ECM proteins. Intriguingly, the ECM can compromise up to 60% of the tumor volume. Furthermore, subsequent enzymatic modification can turn the tumor ECM more rigid. The negative impact of the dense and rigid tumor ECM on the diffusion of macromolecular agents has been established.26, 27 Less is known on its effect on small-molecule drugs. Although composition, quantity and modification of the ECM differs widely between different tumors, experimental data on how this influences drug efficacy and outcome of therapy are limited. Moreover, the likely interconnection between vascular parameters, which can be affected by antiangiogenic treatment, and diffusion controlling ECM parameters has not been studied in detail.
Here we describe a model of syngeneic, implantable fibrosarcoma that is resistant to monoclonal antibody mG6-31-driven VEGF-ablation therapy.28 Blockade of VEGF in this model reduces microvessel density, increases tumor hypoxia and impairs drug delivery leading to reduced responsiveness to the chemotherapeutic drugs. We demonstrate that these resistant tumors express high levels of lysyl oxidases, resulting in a high degree of ECM crosslinking and impeded drug permeation by increasing the ECMs' barrier function. These results highlight a strong interconnection between vascular and ECM parameters in their influence of drug transport in tumors. We show for the first time that inhibition of lysyl oxidases substantially improves drug delivery and potentiates the therapeutic response to drugs used to treat malignant diseases. Furthermore, alteration of the ECM via lysyl oxidase inhibition reverts the potential negative effect of anti-VEGF therapy on drug delivery, and combination of lysyl oxidase inhibition with anti-VEGF therapy synergistically improves response to cytotoxic treatment.
Results
MT6 sarcomas are resistant to VEGF ablation
In contrast to the situation in patients, most murine tumors are responsive to VEGF ablation and react with significant growth reduction. Only few examples for murine tumors that react poorly to anti-VEGF therapy are known.29, 30 Interpretation of results from combination treatments in responsive models is difficult as both anti-VEGF and the chemotherapeutic are individually effective in reducing tumor growth. Thus an increased effectiveness by combining both is impossible to pinpoint to a specific mechanism. Consequently, the effects of anti-VEGF therapy on drug delivery and efficacy have to be studied in models that are unresponsive to VEGF-ablation monotherapy.
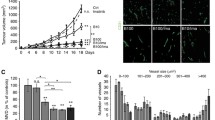
Murine fibrosarcomas established by implanting MT6 cells into C57Bl/6 J mice31 failed to respond to anti-VEGF treatment with mG6-31—an antibody that binds both the human and the murine form of VEGF-A.28 When given at 5 mg/kg body weight (BW) every 6 days, mG6-31 did not significantly reduce tumor growth (Figures 1a and b). An effective doubling of the dosage (7.5 mg/kg BW, q4d) did not improve the outcome (Supplementary Figure S1). Although tumor growth was not significantly affected, mG6-31 treatment strongly reduced microvessel density (Figure 1c), improved vessel maturation (indicated by increased pericyte coverage, Figure 1d) and increased staining for the hypoxia marker carbonic anhydrase IX (CA IX; Figure 1e). Taken together, these results demonstrate that the mG6-31 antibody is effective in the MT6 tumors at causing the expected vascular changes, though the tumors fail to react with a reduction in growth. This mimics the situation observed in patients and makes the MT6 tumors one a rare good model to follow the effect of anti-VEGF treatment on drug delivery.

MT6 sarcomas are resistant to VEGF ablation but react with vascular changes. (a, b) Treatment of established MT6 tumors with mG6-31 (5 mg/kg BW i.p., on days 12 and 18 indicated by black arrowheads) did not affect tumor growth, measured using a caliper or by weighting excised tumors. (c) Sections of tumors were stained for pan endothelial cell antigen (PanEC). Treated tumors display drastically reduced vessel density. Amount of PanEC-positive vessels was counted in whole tumor sections (n=4). Scale bar: 1.00 mm. (d) After mG6-31 treatment, remaining vessels showed increased coverage with NG2-positive pericytes, which was again quantified after imaging whole double immunofluorescent (PanEC and NG2) stained tumor sections (n=5). (e) Treated tumors showed stronger staining for CA IX, indicating increased hypoxia (scale bar: 50 μm, n=5).
VEGF ablation impedes efficacy of chemotherapy in MT6 tumors
Because growth of the MT6 sarcomas is not affected by mG6-31 treatment, it is a suitable model to study the effect of anti-VEGF treatment on drug delivery and efficacy. The different cancers of mesenchymal origin, typically grouped together as sarcomas, respond poorly to systemic treatment with chemotherapies (reviewed in Constantinidou et al.32). Addition of VEGF ablation to the chemotherapeutic regimen could potentially improve the situation of sarcoma patients. We devised a combination treatment of well-established MT6 tumors with mG6-31 and doxorubicin, the most commonly used therapeutic in the management of sarcomas.33 In contrast to the expectations, combination of mG6-31 (5 mg/kg BW, q6d) with doxorubicin (5 mg/kg BW, on days 3 and 5 of a 6-day cycle) was less effective in reducing tumor growth than doxorubicin alone (Figures 2a and c). The same was observed for liposomal doxorubicin (doxil, Figures 2b and c). Doxorubicin induces DNA double-strand breaks and thereby p53-mediated apoptosis.34 Immunohistochemistry showed significantly less staining for p53 and the apoptosis marker cleaved caspase-3 (Supplementary Figure S2) after combination treatment compared with tumors treated with chemotherapy alone, indicating reduced cytotoxic efficacy following anti-VEGF therapy.

VEGF ablation leads to therapeutic failure in MT6 fibrosarcoma. (a–c) Established MT6 sarcomas were treated with mG6-31 (5 mg/kg BW, i.p.) or a control antibody (IgG) and consecutively with two rounds of either doxorubicin or doxil (note: data from a six-armed study was divided into two graphs for clarity). Tumors did not react significantly to mG6-31 treatment alone, and animals pretreated with mG6-31 did respond less to cytotoxic treatment. Treatment days are indicated with black (mG6-31) and red (doxorubicin/doxil) arrowheads. (d–f) MT6 tumors treated with mG6-31 and injected with doxorubicin, doxil or 3H-paclitaxel. mG6-31 pretreatment reduced accumulation of all three drugs in the tumor. (g) 3D angiography using Alexa647-labeled Isolectin (IL-GS-IB4) shows reduction in perfused vessel density in mG6-31-treated tumors. (h) Distribution of H33342 in whole tumor sections was also reduced after mG6-31 treatment (n=4). * indicates statistical significance of doxorubicin/doxil treatment groups versus double treatment groups, *P<0.05, **P<0.01. All error bars: ±s.e.m.
We next tested whether the reduced cytotoxic efficacy is caused by reduced drug delivery into the tumor after mG6-31 treatment. MT6-tumor bearing mice were treated with mG6-31 and, 2 days later, injected with chemotherapeutics or Hoechst 33342 (H33342) for microscopic drug distribution studies. Fluorescent quantification of tumor-extracted doxorubicin showed strongly reduced deposition of the drug after mG6-31 treatment, whether it was used as free base or as liposomal formulation (Figures 2d and e). When the experiment was repeated on day 22 postimplantation after two rounds of mG6-31, doxorubicin accumulation was still significantly lower in mG6-31-treated tumors than in controls, indicating a persistent reduction in delivery (Figure 2e). Paclitaxel, injected as 3H-labeled drug and quantified by β-scintillation, also showed reduced accumulation in treated tumors (Figure 2f). As in the previous experiments, tumor growth was not affected by mG6-31 pretreatment, ruling out differences in drug accumulation based on tumor size. H33342 distribution analysis revealed reduced dye dispersal in tumors after mG6-31 treatment (Figures 2g and h). Thus the vascular changes caused by VEGF ablation reduces the total amount of drugs delivered to the tumors in the MT6 model, thus protecting a substantial part of the tumors from the effect of systemically applied cytotoxic agents. The reduced drug delivery after VEGF ablation in combination with the refractoriness toward anti-VEGF monotherapy reflects the situation seen in at least a subset of patients21 and is a distinctive feature of the MT6 model.
To examine why drug delivery was reduced in the MT6 model, we decided to compare it to a tumor model that responds to VEGF sequestration with increased sensitivity to subsequent chemotherapy. To use a model with characteristics strongly different from the MT6 tumors, we decided on murine 4T1 mammary carcinomas. Orthotopically implanted 4T1 tumors reacted to mG6-31 treatment with reduced tumor growth and a combination of mG6-31 and doxorubicin was more effective than either mG6-31 or the chemotherapeutic alone (Figures 3a and b). We tested accumulation of doxorubicin and 3H-paclitaxel in treated 4T1 tumors 2 days after the first dose of mG6-31 (day 14 after tumor implantation, Figures 3c and d). Neither of the drugs was deposited at higher concentrations after mG6-31 treatment in 4T1 tumors or other organs. However, when whole tumor sections were assessed for distribution of H33342, a larger proportion of the tumor stained positive (Figure 3e). This indicates that vascular changes after mG6-31 treatment caused a more uniform distribution of small-molecule drugs, although the total amount of drug deposited per tumor mass was not changed.

Effect of VEGF ablation on drug distribution and drug tissue penetration is context dependent. (a) 4T1 tumors were treated with mG6-31 or a control IgG and consecutively treated with two rounds of doxorubicin. Treatment days are indicated with black (mG6-31) and red (doxorubicin/doxil) arrawheads. (b) Weight of 4T1 tumors excised 26 days after implantation and 14 days of treatment. (c, d) Pretreatment with mG6-31 did not change amounts of doxorubicin or 3H-paclitaxel delivered to 4T1 tumors and normal organs. Drug accumulation assays were performed on day 14 after tumor implantation, 2 days after the initial mG6-31 treatment. (e) Distribution of H33342 in whole tumor sections was improved after mG6-31 treatment. (f) Immunostaining for CD31 in 4T1 and MT6 tumors treated with mG6-31or an IgG control antibody (scale bar: 100 μm). (g, h) Quantification of relative area stained positive for CD31 and quantification of microvessel density in MT6 and 4T1 tumors treated with mG6-31 or control IgG. (i, j) Isolectin GS-4B staining of perfused vessels (Z-projections: 45 slides each with 0.98-μm spacing, scale bar: 100 μm) showed a more tortuous and convoluted vessel network in the MT6 tumors. mG6-31 reduced the degree of ramification in MT6 tumors. (k–m) Quantification of H33342 penetration depth from vessel surface and tissue volume supplied with detectable amounts of H33342 in correlation to vessel surface area in MT6 and 4T1 tumors treated with mG6-31 or control IgG. * indicates statistical significance versus control, '#' of double treatment groups versus both single treatment groups. *P<0.05; **,##P<0.01; ***,###P<0.001. All error bars: ±s.e.m., n=4 if not otherwise indicated. NS, not significant.
To further elucidate the reasons for the differential effect of mG6-31 pretreatment in the two tumor models, we first verified that there were no intrinsic differences in sensitivity to the chemotherapeutic between 4T1 and MT6 tumor cells (Supplementary Figure S3). The observed differential effect on drug delivery can also be caused by differences in vascular density or vessel quality in the tumor models. Thus mG6-31 treatment of 4T1 and MT6 tumors was repeated in parallel, and the tumors were excised 4 days after a single dose of mG6-31. Staining for the vessel marker CD31 revealed more positive staining in MT6 tumors (Figures 3f and g). However, microvessel density was not different (Figure 3h) as vessels were larger in the MT6 tumors than in 4T1 tumors. A detailed 3D analysis of perfused blood vessels stained with isolectin GS-IB4 showed that blood vessels in MT6 tumors are larger, more tortuous and more ramified (Figures 3j and k). Treatment with mG6-31 led in both models to a pruned down, less chaotic vasculature. Perfused vessel density was similarly decreased in both tumor models after VEGF ablation (Supplementary Figure S4). Thus neither the vascular parameters before treatment nor the changes affected on these parameters by VEGF ablation indicated a cause for the differential effect on drug delivery in the two tumor models. However, a striking difference between 4T1 and MT6 tumors was the extent of H33342 penetration into the tumor parenchyma (Figures 3l and m). In untreated 4T1 tumors, the dye penetrated much deeper from the vessels into the surrounding tissue (21.30±2.005 versus 8.914±0.5670 μm in MT6). Treatment with mG6-31 improved dye penetration in 4T1 tumors significantly (35.20±3.871 μm), whereas the increase was minor in MT6 tumors (10.99±0.7584 μm). Consequently, the tumor volume that was supplied with H33342 in relationship to the vessel surface area was much higher in 4T1 tumors (Figure 3n). mG6-31 treatment improved this ratio in 4T1 tumors even further, while the changes in the MT6 tumors were insignificant. In summary, mG6-31 strongly reduces the perfused vessel density in MT6 sarcomas without significantly improving drug penetration into the tumor parenchyma. This results in less tumor cells being reached and affected by the delivered cytotoxic drug, reducing treatment efficacy. In the 4T1 model, better tissue penetrance is further enhanced after mG6-31 treatment, explaining why after mG6-31 treatment a larger proportion of the tumor is supplied with drugs despite reduced vessel density. These results indicate that tissue permeability is one of the factors that determine whether VEGF ablation improves delivery and response to concomitantly applied drugs.
The ECM of non-responsive MT6 tumors is less permeable for small-molecule drugs
Possible causes for the different tissue permeability are differences in the ECM amount or its composition between MT6 and 4T1 tumors. Trichrome and picrosirius red staining of MT6 tumor sections showed homogeneous deposition of collagenous matrix forming a fine interconnected network surrounding individual cells (Figure 4a). Conversely, in 4T1 tumors collagen is often associated with blood vessels, the network forming large irregular clusters of cells. We next isolated ECM from both tumors. Cellular components were removed by high salt extraction (Figure 4b) and the remaining mainly insoluble material was re-suspended in buffer, yielding a suspension of the complete, native ECM components (cECM). Alternatively, we extracted the ECM precipitate with urea buffer, to obtain the urea soluble fraction of the tumors’ ECM. MT6 tumors yielded significantly higher amounts of cECM proteins (Figure 4c); however, the extracts from 4T1 tumors proved to be more soluble in the urea buffer. We coated membranes of transwell inserts with these extracts and tested the permeability for doxorubicin (Figure 4d). cECM from MT6 tumors was significantly less permeable than cECM from 4T1 tumors (Figures 4e and f), while the urea soluble fractions from both tumors did not differ in their permeability (Figure 4g). Thus the ECM of MT6 sarcomas is indeed less permeable and the components responsible for the low permeability are difficult to solubilize.

ECM quantity and characteristics differ significantly between responsive and non-responsive tumors. (a) Difference in the deposition of collagenous matrix proteins in 4T1 and MT6 tumors visualized by Masson-Goldner trichrome (MGTR) and picrosirius red staining (PSR) (scale bar: 50 μm). (b) Schematic view of ECM isolation process. (c) Protein quantification of matrix extracts from 4T1 and MT6 tumors, using either resolubilization in urea buffer (usECM) or resuspension of complete ECM extracts (cECM). (d) Schematic view of matrix permeability assay. (e, f). Permeability of cECM from 4T1 and MT6 tumors for doxorubicin in the transwell assay was recorded by continuous fluorescence measurement. (g) Relative permeability of usECM extracts calculated from the slope of the signal. (h) Relative mRNA expression of ECM proteins and lysyl oxidases (LOX, LOXL1–LOXL4) in 4T1 versus MT6 cells. (i) Overall lysyl oxidase activity measured in the supernatant of cultured MT6 and 4T1 cells. (j) Relative mRNA expression of lysyl oxidases in MT6 tumors versus the expression levels in 4T1 tumors. (k) Measurement of doxorubicin permeability of matrigel (MG) and collagen I-coated transwell membranes pretreated with rhmLOX or rhLOXL2. All error bars: ±s.e.m. NS, not significant.
To start analyzing the difference between the ECM composition, mRNA expression analysis for a range of common protein components of the tumor ECM and for proteins that modify and stabilize the ECM in cultured MT6 and 4T1 cells was performed (Figure 4h). The expression of most ECM proteins in MT6 and 4T1 cells differs substantially. However, most striking was the significantly higher expression of the lysyl oxidases LOX, LOXL1, LOXL2 and LOXL4 in MT6 cells. The only family member that shows slightly stronger expression in 4T1 cells was LOXL3. Lysyl oxidases crosslink ECM proteins such as collagens and elastins leading to increased tensile strength. We hypothesized that this stiffening of the ECM impedes drug permeability and distribution. Together with the observed high content of ECM proteins this could contribute to the negative effect of vessel depletion by anti-VEGF therapy on drug delivery in the MT6 tumors. Importantly, in contrast to ECM proteins, the ECM-modifying enzymes of the lysyl oxidase family are directly pharmacologically targetable with small-molecule inhibitors. From the limited amount of experimental data available, it seems that the different LOX family members have an only moderately distinct substrate pattern. Consequently, the overall oxidase activity would be the determining factor controlling the permeability of the ECM for small molecules. We measured lysyl oxidase activity in the supernatants of MT6 and 4T1 cells using 2,5-diaminopentane, a substrate recognized by all LOX-family members (Figure 4j).35, 36, 37 MT6 cells show a much stronger overall lysyl oxidase activity, corroborating the mRNA expression results. Lysyl oxidases are strongly expressed by fibroblasts and actual lysyl oxidase activity in tumors may be largely derived from fibrosis and tumor-associated fibroblasts. We therefore analyzed lysyl oxidase mRNA expression in MT6 and 4T1 tumors (Figure 4k). Although differences were smaller than in cell culture, MT6 tumors still expressed significantly higher LOX, LOXL1 and LOXL4 levels.
Lysyl oxidase-catalyzed crosslinking increases the barrier function of the ECM
We next tried to clarify whether the high lysyl oxidase activity in MT6 sarcomas can also be observed in other cancers, rendering LOX inhibition a promising option to improve drug delivery in a broad range of tumors. We first quantified mRNA levels of the different lysyl oxidases in different murine breast carcinomas and sarcoma cells (Supplementary Figure S4). MT6 cells did not display exceptional overall expression levels. As the overall lysyl oxidase activity is most likely determining the degree of ECM crosslinking and drug retention, we measured activity in the supernatant of the different cell lines. Again activity secreted by MT6 cells into the medium was not significantly higher than with other lines of murine or human origin (Supplementary Figure S4B). Patient samples of fibrosarcomas and breast carcinomas confirmed that mRNA expression levels of the lysyl oxidases vary widely between different tumors (Supplementary Figure S5). Although fibrosarcomas expressed higher average levels of LOX and LOXL1–LOXL2, in several of the carcinomas mRNA levels were comparable to the strongest expressing sarcomas. LOXL4 showed higher expression in breast carcinomas, whereas LOXL3 was not detectable in most tumors (data not shown). Also, the expression levels of the individual lysyl oxidases were not strictly correlated. Thus high expression of one family member might compensate for the relative absence of another one.
To further substantiate the ability of lysyl oxidases to increase drug retention of the ECM, we constructed vectors for recombinant expression of human lysyl oxidases. Both mature human LOX (rhmLOX) and human LOXL2 (rhLOXL2) were successfully expressed and purified in an active form via immobilized metal-ion affinity chromatography (Supplementary Figure S6). Diluted matrigel was combined with either rhmLOX or rhLOXL2 and used for coating of transwell membranes (Figure 4l). Treatment with rhmLOX reduced doxorubicin penetration through the matrigel layer significantly to 69.5±1.5%. The effect was not detectable after treatment with 3-aminopropionitrile fumarate (βAPN), an inhibitor of all five lysyl oxidases. The same was observed using rhLOXL2 to catalyze crosslinking (71.1±3.9%) and by substituting collagen I for matrigel. Chemical protein crosslinking also reduced permeability through coated membranes (Supplementary Figure S7).
Lysyl oxidase inhibition improves drug delivery into tumors
The reported redundancy of the lysyl oxidase family members in their biological function and substrate spectra, as well as our results that expression of individual family members are highly diversely expressed in tumors, suggested that a successful treatment strategy by reducing ECM crosslinking should be based on an inhibitor that is effective against all five lysyl oxidases. Thus, to test whether inhibition of lysyl oxidase activity could improve drug delivery in vivo, we treated established tumors with the pan-lysyl oxidase inhibitor βAPN. The treatment was performed by intraperitoneal (i.p.) injections with 100 mg/kg BW βAPN in saline38 and did not affect the growth of MT6 tumors, whereas the growth of 4T1 tumors was reduced (Figure 5a). However, doxorubicin concentrations after a single bolus injection were found to be significantly improved in both tumor models after treatment, having nearly doubled in the treated MT6 tumors (1.596±0.3361 versus 0.8257±0.1391%ID/g in control animals, Figure 5b). In other organs, doxorubicin accumulation was not significantly changed after βAPN treatment. Injected Hoechst 33342 was more evenly distributed throughout the tumor after βAPN treatment (Figures 5c and d). Improved oxygenation resulted in reduced expression of the hypoxia-inducible factor 1α-regulated genes CA IX and VEGF-A (Figure 5e). Recently, it was reported that LOX positively regulates VEGF-A expression and thereby stimulates angiogenesis.39 However, in contrast to this reported direct effect of LOX activity on VEGFA expression, in vitro neither 4T1 nor MT6 cells reacted to βAPN with changed VEGF-A mRNA expression levels (Supplementary Figure S8). These results demonstrated that the reduced levels VEGF-A in the treated tumors are indeed an effect of improved oxygenation. Finally, improved supply of the tumors was indicated by a markedly reduction in central necrosis after βAPN treatment (Figures 5f and g). To examine whether the improvement in drug and oxygen supply to the tumors was indeed caused by a better permeability of the ECM after LOX(L) inhibition, we isolated cECM from both tumors after treatment. Total interference reflection microscopy40 demonstrated the expected strong reduction of crosslinked collagen fibrils in the cECM of treated tumors (Supplementary Figure S7). In transwell-coating experiments, cECM preparations showed significantly better permeability for doxorubicin after βAPN treatment, indicating that lysyl oxidases have a critical role in forming a chemo-protective barrier (Figure 5h).

Lysyl oxidase inhibition improves drug accumulation and distribution within tumors. (a) Treatment of established MT6 and 4T1 tumors with βAPN (100 mg/kg BW i.p., treatment days are indicated by arrowheads). (b) Doxorubicin accumulation in βAPN-treated MT6 and 4T1 tumors versus control tumors and in normal organs of βAPN-treated MT6-bearing C57Bl/6 J mice versus organs from control animals. (c) 3D confocal micrographs of MT6 and 4T1 tumors injected with H33342 and IL-IB4-A647 after βAPN treatment (Z-projections: 30 slides each with 0.90-μm spacing, SB: 100 μm). (d) Quantification of the H33342-positive area in MT6 tumors after βAPN treatment (n=4). (e) mRNA levels for the hypoxia marker CA IX and for VEGF-A in MT6 and 4T1 tumors after βAPN-treatment (n=5). (f) Whole mount sections of MT6 and 4T1 tumors display a strong reduction of central necrosis after APN treatment (NA=necrotic area, SB=1.00 mm). (g) Quantification of necrotic areas in MT6 and 4T1 tumor sections (n=8). (h) Relative permeability of usECM extracts obtained from control and APN treated MT6 and 4T1 tumors. Transwell inserts were coated with 3 μg/mm2 of respective usECM extracts and tested for doxorubicin permeability (n=3). * indicates statistical significance versus control. All error bars: ±s.e.m.
Lysyl oxidase inhibition reverts the negative impact of VEGF ablation on drug delivery in MT6 sarcomas
After having established that inhibition of lysyl oxidases improved drug delivery and distribution, we tested whether this affects therapeutic success in a combination therapy setup and might even reverse the negative effect of VEGF ablation on drug delivery in MT6 fibrosarcomas. Animals with established MT6 tumors were pretreated with βAPN before starting doxorubicin-based therapy (Figure 6a). Treatment with βAPN significantly improved tumor response toward chemotherapy (average tumor size 270.9±57.8 versus 518.6±75.1 mm3). Addition of mG6-31 to the βAPN/doxorubicin regimen further improved therapeutic efficacy (92.8±37.2 mm3). In an additional experiment, we verified that combination of mG6-31 and βAPN did not affect tumor growth (Supplementary Figure S7). In addition to excluding a simple additive effect in the triple treatment group, the results of the study also demonstrated that the refractoriness of the MT6 tumors towards VEGF ablation is not caused by their dense, highly crosslinked ECM.

Lysyl oxidase inhibition reverses the negative effect of VEGF ablation on drug transport in MT6 fibrosarcoma. (a) Treatment of established MT6 tumors with doxorubicin (5 mg/kg BW i.p.), βAPN (100 mg/kg BW i.p.) and mG6-31 (5 mg/kg BW i.p) at the indicated days (black, blue and red arrowheads, respectively). Combining doxorubicin with βAPN improved efficacy; addition of mG6-31 reduced growth of the tumors further. (b) Schedule for short-term βAPN treatment of MT6 tumors. (c) H33342 penetration (n=9) was measured by 3D confocal imaging in MT6 tumors after short-term treatment. (d) Doxorubicin delivery into MT6 tumors of the different treatment groups was quantified by extraction (n=6–8). (e) Schematic model of the combined effect of vessel maturation and ECM permeability on drug delivery: The effect of antiangiogenic treatment is characterized by improvement of vessel maturation resulting in improved perfusion and transport through the individual blood vessel. In tumor tissue with high lysyl oxidase activity, drug diffusivity is strongly impaired and tissue drug penetration is widely independent from drug levels supplied by the individual vessels. Inhibition of lysyl oxidases can improve drug diffusion and restore increased drug delivery after antiangiogenic treatment. All error bars: ±s.e.m. # indicates statistical significance of double treatment group versus control and single treatment group, § of triple treatment group versus double treatment group. #,§P<0.05; ##P<0.01.
To test whether the improved response to chemotherapy after combining lysyl oxidase inhibition with antiangiogenic therapy is caused by improved drug delivery, we treated established MT6 sarcomas for the short period of 4 days with βAPN in combination with a single dose of mG6-31 (Figure 6b) and examined distribution of Hoechst 33342 within the tumor. In tumors treated with the combination of βAPN and mG6-31, the Hoechst dye indeed permeated significantly deeper into the tumor tissue than in control tumors and in tumors from mice that received βAPN alone (Figure 6c). Doxorubicin given as a bolus dose also accumulated significantly stronger in the tumors treated with the βAPN/mG6-31 combination (Figure 6d).
Discussion
Combining drugs that inhibit the VEGF pathway, such as Avastin, with chemotherapeutic treatment regimens is meanwhile an integral part in the management of various malignant diseases. Nevertheless, the therapeutic benefit of adding anti-VEGF agents observed in large patient cohorts is only moderate, indicating that not all patients benefit equally from these agents. We need a better understanding of how these agents influence transport and efficacy of cytotoxic drugs and in which context they are most effective. This will enable us to predict which patients will benefit from adding vascular-targeted therapies to their treatment and in which patients antiangiogenic therapy might be detrimental.
The refractoriness of MT6 tumors to anti-VEGF therapy does not only reflect the situation in patients but also permits unperturbed observation of secondary effects, such as drug transport and effectiveness. Recently, Van der Veldt et al.21 have demonstrated that bevacizumab (Avastin) leads to decreased perfusion and delivery of docetaxel in non-small-cell lung cancer patients. However, the experimental setup in this study did not allow for assessment of whether therapeutic efficacy was also reduced or whether non-small-cell lung cancer patients would still benefit from the combination by a possible additive effect. The MT6 model displays the same response to anti-VEGF that is observed in these patients, and our results indicate for the first time that such a reduction in drug delivery after anti-VEGF therapy might indeed compromise therapeutic activity. If VEGF ablation leads to impaired delivery and distribution of cytotoxic drugs but has no measurable antitumor effect by itself, combination of both might indeed be less effective than the cytotoxic drug alone. However, if the antiangiogenic drug itself inhibits tumor growth, a combination with a cytotoxic drug may still prove beneficial, as Cesca et al.23 reported for vandatenib that also reduced paclitaxel transport into xenografts, but a combination of both was still better than each therapeutic alone.
In our experiments, the vascular changes after VEGF ablation improved response to chemotherapy in 4T1 breast carcinomas. The varying results using anti-VEGF/cytotoxic combination therapy indicate that the effect of antiangiogenic therapy is indeed strongly context dependent.
Our results experimentally underline the inter-relationship between vessel characteristics that control tumor perfusion and matrix properties that regulate drug diffusion within the tissue. In the MT6 model, the key factor leading to reduced accumulation and effectiveness of cytotoxic drugs after mG6-31 treatment was clearly the dense, highly crosslinked ECM. The MT6 matrix was altered nearly impermeable to small-molecule drugs by high lysyl oxidase levels, an effect further corroborated in several in vitro experiments. Consequently, only in combination with lysyl oxidases inhibition, VEGF ablation was able to improve drug delivery into MT6 tumors. In addition to lysyl oxidases, it is likely that also other ECM-modifying enzymes and the composition and quantity of ECM components themselves have a strong impact on drug permeability and thereby on the effect of antiangiogenic therapeutics. Besides the drastically different lysyl oxidase levels, the differences in ECM content and composition probably contributed to the differential drug permeability observed in 4T1 and MT6 tumors. Hyaluronic acid has been shown to impede drug diffusion by a very different mechanism: as it binds large amounts of water, a high hyaluronic acid content results in high interstitial pressure and collapsed blood vessels. Enzymatic removal using exogenous hyaluronidase can reduce the tumor’s interstitial pressure, subsequently improving vessel perfusion and drug efficacy.41, 42 As an intuitive next step to improve drug delivery, the concept of combining matrix modification with antiangiogenic therapy has also been put forward.43, 44
The enzymatically active lysyl oxidases are linked to more invasive and metastatic behavior: in several preclinical models, inhibition of lysyl oxidase with βAPN or antibodies resulted in a therapeutic effect, most markedly suppression of metastasis.38, 45, 46, 47, 48 Our results demonstrate that, beside this effect on tumor cell behavior, lysyl oxidase inhibitors might have benefits as auxiliary drugs with the potency to increase the efficacy of systemic treatment. In addition to improving drug distribution, treatment with βAPN also improved tumor oxygenation. This is in accordance with previous reports that apparent diffusion constant and pO2 in tumors is correlated.11, 49 Improved oxygenation by itself reduces aggressiveness of treated tumors.50, 51 Recently, it has been shown that reduced hypoxia result in even further ECM changes by decreased PLOD2 expression, which subsequently reduces metastasis.52
It is important to note that uptake of chemotherapeutics in other organs was not significantly altered by prolonged βAPN treatment, although some of these organs (for example, heart, muscle) express high levels of lysyl oxidases. In the fast growing murine tumors, the ECM is continuously newly formed and remodeled. When lysyl oxidases are inhibited and cannot crosslink the freshly formed ECM, it will alter its characteristics, including its drug-retention abilities. In the resting, normal tissue, this aspect of lysyl oxidase activity might have a lesser role and might influence the ECM's quality only after more chronic inhibition. It is known that lysyl oxidase activity is necessary to maintain functional and mature connective tissue. Its inhibition by substances such as βAPN can cause diseases, such as lathyrism.53 Targeting tumor-specific upstream regulators of lysyl oxidase expression might be an alternative way to improve ECM characteristics. Recently, Liu et al.8 reported that inhibition of transforming growth factor-β in experimental mammary carcinomas improved delivery of doxorubicin and doxil. Transforming growth factor-β is a known positive regulator of all lysyl oxidase family members.54, 55
Similar to the defective vasculature, the dense tumor ECM impairs transport and distribution. Both characteristics thereby simultaneously protect the tumor from treatment and contribute to a more malignant phenotype. Auxiliary drugs that change these characteristics could improve efficacy of the whole array of antitumor drugs currently at our disposal. Moreover, a better understanding of the intricate mechanisms that protect the tumor from the negative impact of our treatment efforts could lead to a strategic approach toward cancer therapy that aims at defusing those protective traits and making tumors more vulnerable.
Material and methods
If not otherwise indicated, chemicals were purchased from Sigma Aldrich (Munich, Germany) or Carl Roth (Karlsruhe, Germany). βAPN was purchased from Sigma Aldrich. Protein concentrations were determined with the Pierce BCA Kit (Thermo Fisher, Rockford, IL, USA) using a 30-min incubation time at 60 °C. The anti-VEGF antibody mG6-31 and a control IgG (anti-Ragweed) were provided by Genentech (San Francisco, CA, USA).
Cell culture
MT6 (CRL-2805), 4T1 (CRL-2539), MDA-MB-231 (HTB-26), MDA-MB-435S (HTB-129), MCF7 (HTB-22), ZR-75-1 (CRL-1500) and LLC (CRL-1642) cells were obtained from ATCC (Manassas, VA, USA). AC755, EMT6 and 38290 TTT cells have been purchased from NCI Tumor Repository (http://ncifrederick.cancer.gov/Services/NcifRepositories.aspx). E0771 cells have been purchased from Tebu-Bio (Offenbach, Germany). All tumor cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco/Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum and Penicillin/Streptomycin at 37 °C, 5% CO2. Cell lines were tested for mycoplasma contamination.
Tumor models and treatment
All experiments involving animals were reviewed and approved by the institutional animal care and use committee at MSKCC or by the Regional Administration of Unterfranken, Würzburg, Germany. All animal experiments were performed according to ethical standards and guidelines.
Tumor engraftment
MT6 (106 cells in phosphate-buffered saline) fibrosarcomas were generated by subcutaneous injection in the dorsal region of female C57Bl/6 J mice. 4T1 (105 cells in phosphate-buffered saline) breast adenocarcinomas were generated by injection of cells into the inguinal mammary fat pad of female Balb/c mice. Balb/c mice were purchased from Charles River (Sulzfeld, Germany) and C57Bl/6J from Jackson Laboratories (Bar Harbor, ME, USA).
All animals in the individual experiments were of the same age and sex. For each experiment, tumor-bearing mice were randomly assigned to the different treatment groups just prior to the start of treatment. In treatment studies where tumor growth was a critical outcome, assessment of tumor size was performed blinded by a second researcher.
Exclusion of data
Animals that never developed tumors owing to take rate <100% were excluded from the studies. All data from animals that died or had to be killed prior to the scheduled termination of the experiment were excluded.
Tumor treatment
Treatment of fully established tumors started on day 12 after implantation. mG6-31 or an anti-Ragweed control antibody were given at 5 mg/kg every 6 days on days 12, 18 and 24 by i.p. injection. Doxorubicin/doxil (both at 5 mg/kg BW free doxorubicin) was administered by i.p. injection on days 14, 16, 20, 22, 26, 28, 32 and 34. Control substance for doxorubicin was 0.9% NaCl.
3-APN was administered at 100 mg/kg or 30 mg/kg BW in 0.9% NaCl by daily i.p. injection. Control substance was 0.9% NaCl.
Tumor growth was followed by measuring perpendicular diameters of the tumors with a vernier calliper. Tumor volume was calculated using the equation V=π/6 × l × w2. In addition, tumors were excised postmortem and weighted. Only tumors that could be excised completely without additional invaded tissue were used for weight measurements.
References
Kratz F. Drug delivery in oncology − challenges and perspectives. In: Kratz F, Steinhagen H, Senter P (eds). Drug Delivery in Oncology − Challenges and Perspectives in Drug Delivery in Oncology – from Research Concepts to Cancer Therapy, vol. 1. VCM: Weinheim, Germany; 2011, pp LIX-LXXXV.
Gangloff A, Hsueh WA, Kesner AL, Kiesewetter DO, Pio BS, Pegram MD et al. Estimation of paclitaxel biodistribution and uptake in human-derived xenografts in vivo with (18)F-fluoropaclitaxel. J Nucl Med 2005; 46: 1866–1871.
Kesner AL, Hsueh WA, Htet NL, Pio BS, Czernin J, Pegram MD et al. Biodistribution and predictive value of 18 F-fluorocyclophosphamide in mice bearing human breast cancer xenografts. J Nucl Med 2007; 48: 2021–2027.
Staffhorst RW, van der Born K, Erkelens CA, Hamelers IH, Peters GJ, Boven E et al. Antitumor activity and biodistribution of cisplatin nanocapsules in nude mice bearing human ovarian carcinoma xenografts. Anticancer Drugs 2008; 19: 721–727.
Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E . Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res 2009; 69: 873–878.
Phillips PG, Yalcin M, Cui H, Abdel-Nabi H, Sajjad M, Bernacki R et al. Increased tumor uptake of chemotherapeutics and improved chemoresponse by novel non-anticoagulant low molecular weight heparin. Anticancer Res 2011; 31: 411–419.
Kato Y, Okollie B, Artemov D . Noninvasive 1H/13C magnetic resonance spectroscopic imaging of the intratumoral distribution of temozolomide. Magn Reson Med 2006; 55: 755–761.
Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K et al. TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci USA 2012; 109: 16618–16623.
Minko T, Kopeckova P, Pozharov V, Jensen KD, Kopecek J . The influence of cytotoxicity of macromolecules and of VEGF gene modulated vascular permeability on the enhanced permeability and retention effect in resistant solid tumors. Pharm Res 2000; 17: 505–514.
Teicher BA, Herman TS, Holden SA, Wang YY, Pfeffer MR, Crawford JW et al. Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science 1990; 247 (Pt 1): 1457–1461.
Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF . The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res 2005; 11 (Pt 1): 8782–8788.
Lin KY, Maricevich M, Bardeesy N, Weissleder R, Mahmood U . In vivo quantitative microvasculature phenotype imaging of healthy and malignant tissues using a fiber-optic confocal laser microprobe. Transl Oncol 2008; 1: 84–94.
Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA 1998; 95: 4607–4612.
Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK . Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res 2004; 64: 3731–3736.
Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 2004; 6: 553–563.
Willett CG, Boucher Y, Di Tomaso E, Duda DG, Munn LL, Tong RT et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 2004; 10: 145–147.
Jain RK . Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307: 58–62.
Hansen-Algenstaedt N, Stoll BR, Padera TP, Dolmans DE, Hicklin DJ, Fukumura D et al. Tumor oxygenation in hormone-dependent tumors during vascular endothelial growth factor receptor-2 blockade, hormone ablation, and chemotherapy. Cancer Res 2000; 60: 4556–4560.
Rapisarda A, Hollingshead M, Uranchimeg B, Bonomi CA, Borgel SD, Carter JP et al. Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol Cancer Ther 2009; 8: 1867–1877.
Escorcia FE, Henke E, McDevitt MR, Villa CH, Smith-Jones P, Blasberg RG et al. Selective killing of tumor neovasculature paradoxically improves chemotherapy delivery to tumors. Cancer Res 2010; 70: 9277–9286.
Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell 2012; 21: 82–91.
Arjaans M, Oude Munnink TH, Oosting SF, Terwisscha van Scheltinga AG, Gietema JA, Garbacik ET et al. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res 2013; 73: 3347–3355.
Cesca M, Frapolli R, Berndt A, Scarlato V, Richter P, Kosmehl H et al. The effects of vandetanib on paclitaxel tumor distribution and antitumor activity in a xenograft model of human ovarian carcinoma. Neoplasia 2009; 11: 1155–1164.
Turley RS, Fontanella AN, Padussis JC, Toshimitsu H, Tokuhisa Y, Cho EH et al. Bevacizumab-induced alterations in vascular permeability and drug delivery: a novel approach to augment regional chemotherapy for in-transit melanoma. Clin Cancer Res 2012; 18: 3328–3339.
Heskamp S, Boerman OC, Molkenboer-Kuenen JD, Oyen WJ, van der Graaf WT, van Laarhoven HW . Bevacizumab reduces tumor targeting of antiepidermal growth factor and anti-insulin-like growth factor 1 receptor antibodies. Int J Cancer 2013; 133: 307–314.
Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK . Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA 2011; 108: 2909–2914.
McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res 2006; 66: 2509–2513.
Liang WC, Wu X, Peale FV, Lee CV, Meng YG, Gutierrez J et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J Biol Chem 2006; 281: 951–961.
Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol 2010; 28: 585–593.
Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med 2013; 19: 1114–1123.
Sandhu JK, Haqqani AS, Birnboim HC . Effect of dietary vitamin E on spontaneous or nitric oxide donor-induced mutations in a mouse tumor model. J Natl Cancer Inst 2000; 92: 1429–1433.
Constantinidou A, Pollack S, Loggers E, Rodler E, Jones RL . The evolution of systemic therapy in sarcoma. Expert Rev Anticancer Ther 2013; 13: 211–223.
Mendenhall WM, Indelicato DJ, Scarborough MT, Zlotecki RA, Gibbs CP, Mendenhall NP et al. The management of adult soft tissue sarcomas. Am J Clin Oncol 2009; 32: 436–442.
Lorenzo E, Ruiz-Ruiz C, Quesada AJ, Hernandez G, Rodriguez A, Lopez-Rivas A et al. Doxorubicin induces apoptosis and CD95 gene expression in human primary endothelial cells through a p53-dependent mechanism. J Biol Chem 2002; 277: 10883–10892.
Rodriguez HM, Vaysberg M, Mikels A, McCauley S, Velayo AC, Garcia C et al. Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J Biol Chem 2010; 285: 20964–20974.
Kim S, Kim Y . Variations in LOXL1 associated with exfoliation glaucoma do not affect amine oxidase activity. Mol Vis 2012; 18: 265–270.
Fogelgren B, Polgar N, Szauter KM, Ujfaludi Z, Laczko R, Fong KS et al. Cellular fibronectin binds to lysyl oxidase with high affinity and is critical for its proteolytic activation. J Biol Chem 2005; 280: 24690–24697.
Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006; 440: 1222–1226.
Baker AM, Bird D, Welti JC, Gourlaouen M, Lang G, Murray GI et al. Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Cancer Res 2013; 73: 583–594.
Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA 2011; 108: 16369–16374.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013; 62: 112–120.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR . Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21: 418–429.
Jain RK . Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 2014; 26: 605–622.
Stylianopoulos T, Jain RK . Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci USA 2013; 110: 18632–18637.
Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med 2010; 16: 1009–1017.
Bondareva A, Downey CM, Ayres F, Liu W, Boyd SK, Hallgrimsson B et al. The lysyl oxidase inhibitor, beta-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLoS One 2009; 4: e5620.
Erler JT, Giaccia AJ . Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res 2006; 66: 10238–10241.
Wilgus ML, Borczuk AC, Stoopler M, Ginsburg M, Gorenstein L, Sonett JR et al. Lysyl oxidase: a lung adenocarcinoma biomarker of invasion and survival. Cancer 2011; 117: 2186–2191.
Dunn JF, Ding S, O'Hara JA, Liu KJ, Rhodes E, Weaver JB et al. The apparent diffusion constant measured by MRI correlates with pO2 in a RIF-1 tumor. Magn Reson Med 1995; 34: 515–519.
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008; 13: 206–220.
Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res 2003; 63: 1138–1143.
Eisinger-Mathason TS, Zhang M, Qiu Q, Skuli N, Nakazawa MS, Karakasheva T et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov 2013; 3: 1190–1205.
Wilmarth KR, Froines JR . In vitro and in vivo inhibition of lysyl oxidase by aminopropionitriles. J Toxicol Environ Health 1992; 37: 411–423.
Xie J, Jiang J, Zhang Y, Xu C, Yin L, Wang C et al. Up-regulation expressions of lysyl oxidase family in anterior cruciate ligament and medial collateral ligament fibroblasts induced by transforming growth factor-beta 1. Int Orthop 2012; 36: 207–213.
Feres-Filho EJ, Choi YJ, Han X, Takala TE, Trackman PC . Pre- and post-translational regulation of lysyl oxidase by transforming growth factor-beta 1 in osteoblastic MC3T3-E1 cells. J Biol Chem 1995; 270: 30797–30803.
Acknowledgements
We thank Robert Benezra (Cancer Biology and Genetics Program, Memorial Sloan-Kettering Cancer Center) for helpful discussion; Thomas Jarchau (Institute for clinical Biochemistry and Pathobiochemistry, Univeritätsklinikum Würzburg) for a thorough correction of the manuscript; and Afshar Barlas, Mesruh Turkekul (Molecular Cytology Core facility, Memorial Sloan-Kettering Cancer Center) and Oliver Reinhardt (Biology Master Course, Universität Würzburg) for help with experiments. Funding was provided by the Deutsche Forschungsgemeinschaft (DFG Grant Nos. HE3565/1-1, HE3565/2-1 and HE3565/3-1 to EH).
Author contributions
FR, HH, MW, SV, FEE, SK, IW, MT, SG, ZG and EH conducted the experiments. OP, KM, DAS, AR, ZG and EH planned the experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Röhrig, F., Vorlová, S., Hoffmann, H. et al. VEGF-ablation therapy reduces drug delivery and therapeutic response in ECM-dense tumors. Oncogene 36, 1–12 (2017). https://doi.org/10.1038/onc.2016.182
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.182
- Springer Nature Limited
This article is cited by
-
Normalization of Snai1-mediated vessel dysfunction increases drug response in cancer
Oncogene (2024)
-
Image-based modeling of vascular organization to evaluate anti-angiogenic therapy
Biology Direct (2023)
-
Nanoparticle-mediated TRPV1 channel blockade amplifies cancer thermo-immunotherapy via heat shock factor 1 modulation
Nature Communications (2023)
-
The roles and limitations of bevacizumab in the treatment of ovarian cancer
International Journal of Clinical Oncology (2022)
-
Hyper-activation of Aurora kinase a-polo-like kinase 1-FOXM1 axis promotes chronic myeloid leukemia resistance to tyrosine kinase inhibitors
Journal of Experimental & Clinical Cancer Research (2019)