
Abstract
Multiple system atrophy (MSA) is a neurodegenerative disease with diverse clinical manifestations, including parkinsonism, cerebellar syndrome, and autonomic failure. Pathologically, MSA is characterized by glial cytoplasmic inclusions in oligodendrocytes, which contain fibrillary forms of α-synuclein. MSA is categorized as one of the α-synucleinopathy, and α-synuclein aggregation is thought to be the culprit of the disease pathogenesis. Studies on MSA pathogenesis are scarce relative to studies on the pathogenesis of other synucleinopathies, such as Parkinson’s disease and dementia with Lewy bodies. However, recent developments in cellular and animal models of MSA, especially α-synuclein transgenic models, have driven advancements in research on this disease. Here, we review the currently available models of MSA, which include toxicant-induced animal models, α-synuclein-overexpressing cellular models, and mouse models that express α-synuclein specifically in oligodendrocytes through cell type-specific promoters. We will also discuss the results of studies in recently developed transmission mouse models, into which MSA brain extracts were intracerebrally injected. By reviewing the findings obtained from these model systems, we will discuss what we have learned about the disease and describe the strengths and limitations of the models, thereby ultimately providing direction for the design of better models and future research.
Similar content being viewed by others

Introduction
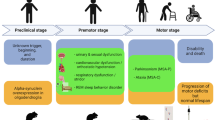
Multiple system atrophy (MSA) is a rapidly progressive sporadic adult-onset neurodegenerative disorder. It was first termed to describe neuronal atrophy found in various diseases, including striatonigral degeneration, olivopontocerebellar atrophy, and Shy-Drager syndrome1. Epidemiologic studies of MSA have shown a prevalence range of 3.4 to 4.9 per 100,000 people, increasing to 7.8 per 100,000 among people older than 40 years of age2. Moreover, it affects men and women equally and has an average age onset of approximately 55–60 years3,4. The mean life expectancy of MSA is 6–10 years following diagnosis5,6.
MSA is characterized by clinical symptoms that are subdivided into extrapyramidal, pyramidal, cerebellar, and autonomic symptoms. Extrapyramidal symptoms include bradykinesia, rigidity, and postural instability, which are similar to the symptoms of Parkinson’s disease (PD). The autonomic symptoms include common autonomic dysfunctions, such as urogenital, gastrointestinal, and cardiovascular failure. Nonmotor symptoms, such as sleep and cognitive disorders, respiratory problems, and emotional/behavioral symptoms, might also occur during disease development7,8. The different symptoms of MSA can be used to categorize the disease into two subtypes: the parkinsonian subtype (MSA-P) and the cerebellar type (MSA-C). MSΑ−P patients exhibit more PD symptoms, whereas MSA-C patients commonly display cerebellar ataxia. More than two-thirds of MSA patients in Western countries are MSA-P patients, while MSA-C is more common in Japan9,10,11 However, MSA-P is more common than MSA-C in Korea, indicating that the subtypes of MSA vary among Asian countries12,13.
MSA is pathologically distinguished by a widespread neuronal loss that is accompanied by gliosis in the basal ganglia, cerebellum, pons, inferior olivary nuclei, and spinal cord. The symptoms of MSA are similar to those of PD, which makes it difficult to distinguish the two diseases. Clinical differential diagnosis is practically possible, although neuropathological confirmation is still required for a definitive diagnosis of MSA (Fig. 1).

The symptoms are subdivided into parkinsonian, cerebellar, and autonomic. Parkinsonian symptoms include motor symptoms, such as bradykinesia, tremor, and postural instability. Patients with more evident parkinsonism are considered to have the parkinsonian subtype of the disease (MSA-P). Patients with cerebellar symptoms, such as ataxia and cerebellar oculomotor dysfunction, are considered to have the cerebellar subtype of the disease (MSA-C). Both subtypes share common autonomic dysfunctions described above.
Multiple system atrophy and glial cytoplasmic inclusions
The important neuropathological hallmark of MSA is the presence of argyrophilic filamentous glial cytoplasmic inclusions (GCIs), predominantly in oligodendrocytes14. GCIs are spherical protein aggregates located near nuclei with a diameter of 5–20 μm and various morphologies. GCIs in oligodendrocytes are usually larger and paler than nonoligodendrocyte-derived GCIs. They are primarily composed of loosely packed filaments of α-synuclein protein that is phosphorylated at residue Ser129 and ubiquitinated15,16. Immunohistochemical studies have identified other proteins that colocalize with α-synuclein. These include p25α/TPPP (tubulin polymerization-promoting protein), α,β-crystallin, tau, LRRK2, cyclin-dependent kinase 5 (cdk5), microtubule-associated protein 5, ubiquitin, and tubulin (reviewed in ref. 17). p25α/TPPP has a vital role in the stabilization of microtubules, the projection of mature oligodendrocytes, and ciliary structures18. It is essential for the differentiation and maturation of oligodendrocytes19. p25α/TPPP is commonly found in myelin sheaths, but during the first stages of MSA, it relocates to the oligodendrocyte soma, resulting in early myelin dysfunction20. The redistribution of p25α in oligodendrocytes causes an increase in the volume of cell bodies, which is a typical characteristic of cells with GCIs. Ultimately, the presence of p25α in the cell body enhances the aggregation of α-synuclein, which may lead to oligodendroglial dysfunction and neuronal degeneration18,21.
Multiple system atrophy and α-synuclein
MSA belongs to a diverse group of neurodegenerative disorders described as α-synucleinopathies, which are similar to PD and dementia with Lewy bodies (DLB). These disorders are characterized by the abnormal accumulation of α-synuclein protein aggregates22,23. α-Synuclein is a predominantly neuronal presynaptic protein present in the brain and is expressed in other tissues at various levels. It is encoded by the SNCA gene, which is linked to PD and has also been associated with an increased risk of PD, DLB, and MSA24.
The presence of GCIs and the excessive accumulation of α-synuclein in the oligodendrocytes are accompanied by neuronal degeneration, brain atrophy, demyelination, and mutation of nerve cells in MSA patients25,26. A study by Peng et al. showed differences between GCI-α-synuclein and LB-α-synuclein27. These inclusions are conformationally and biologically distinct. GCI-α-synuclein is 1000-fold more potent than LB-α-synuclein in seeding the aggregation of monomeric α-synuclein, which may explain the highly aggressive and rapidly progressive nature of MSA symptoms.
Several studies have reported that there is little to no α-synuclein expression in mature oligodendrocytes in the human brain11,28,29. There is no evidence of increased α-synuclein expression in MSA oligodendrocytes. However, a recent study by Asi et al. showed a three-fold increase in SNCA mRNA levels in MSA oligodendrocytes postmortem, although the change did not reach statistical significance; it is still questionable whether the increase in mRNA levels can significantly change the levels of the α-synuclein protein in oligodendrocytes30. In vitro cultures of MSA patient-derived induced pluripotent stem cells (iPSCs) showed that α-synuclein is only expressed in the early stages of oligodendrocyte maturation but not in the premyelination period31.
The mechanisms of the accumulation of α-synuclein in oligodendrocytes are still unknown. Several hypotheses have provided possible explanations as to how GCIs form32. One possibility is that they form through the induced expression and aggregation of α-synuclein in oligodendrocytes and other glial cells under disease conditions, but there is little evidence to support this cell-autonomous mechanism. An alternative explanation is that they form through the uptake of α-synuclein secreted from neurons by oligodendrocytes. Studies have shown the transfer of neuronal α-synuclein, both in co-cultures and through exogenous addition, into oligodendrocytes in vitro and in vivo33,34,35. In MSA, oligodendrocytes might be more prone than neuronal cells to the accumulation of neuron-derived α-synuclein, possibly because the clearance mechanism might not be as efficient as that in neurons. In support of this explanation, Peng et al. suggested that the cellular milieu determines different synucleinopathies, such as LBD and MSA27.
A failure to discard proteins through cellular degradation pathways may lead to the production of toxic aggregates that may incorporate into GCIs and cause oligodendrocyte dysfunction. Oligodendrocytes become enlarged, and nuclei turn pale when myelin degeneration occurs. The propagation of α-synuclein aggregates to other adjacent cells may lead to inflammatory responses by microglia and neurodegeneration (Fig. 2).

Neuron-derived α-synuclein aggregates in the extracellular space are taken up by neighboring glial cells. Microglia and astrocytes, which secrete inflammatory molecules, are activated. Oligodendrocytes undergo demyelination, exposing neuronal axons that may retract or be degenerated by the hostile external environment, causing neurodegeneration.
Astrogliosis and microgliosis in MSA
The activation of astrocytes and microglia has been observed in the brains of MSA patients, as well as in those of transgenic models of MSA11,36,37. Studies have revealed the potential role of the neuron-to-glia transmission of α-synuclein in glial activation in both cell and animal models. Extracellular α-synuclein leads to inflammatory responses in astrocytes and microglia38,39,40. Activated Iba-1-positive microglia and GFAP-positive astrocytes have been shown to localize in the proximity of GCIs41,42. Astrogliosis is an important pathological characteristic of MSA. Treating astrocytes with extracellular α-synuclein induces ERK/MAPKK-dependent astrogliosis42. Activated astrocytes can secrete cytokines, which may trigger microgliosis. Therefore, the proinflammatory function of extracellular α-synuclein in astrocytes may have a crucial role in spreading MSA neuropathology41,43.
Microglia are the primary immunophagocytic cells in the brain. An increased number of activated microglia is found in α-synucleinopathies44. The injection of GCI extract into the mouse brain causes localized microgliosis, as well as astrogliosis42. Toll-like receptors (TLRs), such as TLR2 and TLR4, have been shown to interact with extracellular α-synuclein in microglia39,45. Microglia activated by extracellular α-synuclein then secrete toxic factors that can trigger further neurodegeneration and gliosis46.
Models of multiple system atrophy
Animal toxin models
In vitro and in vivo models have been developed to obtain a better understanding of MSA pathophysiology. The systemic administration and local stereotaxic injection of toxins induced lesions in specific anatomical areas of models to reproduce MSA symptoms, particularly L-DOPA-unresponsive parkinsonism47. The stereotaxic injection of two toxins, namely, 6-hydroxydopamine (6-OHDA) and quinolinic acid (QA), into different regions of the rat brain in sequence had distinct pathological and behavioral outcomes48. 6-OHDA caused striatal dopamine depletion and decreased the number of dopaminergic cells in the substantia nigra pars compacta (SNpc)49. The injection of QA induced the loss of spiny striatal neurons. Behavioral deficits induced by both single and combined lesions also imitated the symptoms of MSA-P. However, there was no significant difference in performance on the rotarod test or drug-induced rotation test50. The inoculation of brain regions with a single toxin also created lesions. The injection of 3-nitropropionic acid (3-NP), a succinate dehydrogenase inhibitor, or 1-methyl-4-phenylpyridinium ion (MPP+), a mitochondrial complex I inhibitor, into the striatum induced combined degeneration of nigral and striatal neurons51,52. The simultaneous or sequential systemic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 3-NP caused motor symptoms and dyskinesia in mice and nonhuman primates that responded poorly to L-DOPA treatment53,54,55,56.
These approaches partially mimicked MSA neuropathology in the nigrostriatal system, which arises in the early to late period of the disease. The lesions only resulted in symptoms within the area of administration and failed to spread outside the basal ganglia47. In addition, toxin-based approaches did not induce GCI pathology, which is one of the essential hallmarks of MSA47.
In vitro genetic models
In vivo toxin models cannot precisely depict disease advancement or the mechanism. MSA pathophysiology has been addressed at the cellular level through modified gene expression. Because one of the main components of GCIs in MSA is α-synuclein, many studies have used in vitro expression of α-synuclein to investigate the disease mechanism32.
U-373 MG cell line and primary mixed rat glial cultures
A study conducted by Stefanova et al. showed that the overexpression of α-synuclein induced cell death in a U-373 MG human glioblastoma astrocytoma cell line and primary oligodendrocytes from mixed rat glial cultures32,57. Glial cells expressing high levels of α-synuclein were highly prone to oxidative stress. Upon treatment with TNFα, a proinflammatory cytokine released by microglia in MSA, significant cytotoxic changes were observed in α-synuclein-expressing cells. This suggested that a toxic environment, along with high levels of α-synuclein in glia, might represent a severe risk for the development of MSA.
OLN-93 cell line
OLN-93 cells, primary oligodendrocytic cells derived from Wistar rat brain cultures, have been used to study MSA pathology32,58. Kragh and colleagues co-expressed α-synuclein and p25α in the OLN-93 cell line. The co-expression resulted in the enhanced expression of IkBα, which sequestered the NF-κB transcription factor p65 in the cytoplasm. The inhibition of NF-κB signaling impeded its cytoprotective effects while causing the retraction of microtubules and triggering the activation of the apoptotic protein caspase-358,59. The phosphorylation of Ser129 in α-synuclein protein was necessary for the process. Human brain tissues from MSA patients also exhibit increased expression of IkBα and NFκB p65 in some oligodendrocytes with GCIs.
CG4 cell line
The central glia 4 (CG4) cell line, a rodent oligodendroglial cell line, has been used to stably express α-synuclein. The overexpression of the protein impaired the maturation of cells into oligodendrocytes. BDNF partially rescued this impairment32,60. α-Synuclein delayed the maturation of oligodendroglia by inhibiting the expression of myelin basic protein (MBP) and myelin gene regulatory factor while increasing the levels of the oligodendroglial differentiation repressor Hes5. The lentiviral transduction of α-synuclein in CG4 cells impaired autophagy and this impairment was reversed by the inhibition of miR-101, a microRNA that regulates autophagy genes such as RAB5A, MTOR, ATG4D, and STMN161.
iPSCs from human MSA patients
The advancement of stem cell technologies has allowed the culturing of patient-derived cells and their differentiation into various cell types, including oligodendrocytes. The expression of α-synuclein was examined by real-time PCR and immunocytochemistry. Oligodendrocyte precursor cells (OPCs) expressed α-synuclein, but the expression levels were decreased as the cells differentiated into mature oligodendrocytes. There was no difference between patient-derived and healthy control iPSCs31.
In vivo genetic models
The transgenic expression of the α-synuclein gene under oligodendrocyte-specific promoters has been used to create mouse models of MSA. Through the use of proteolipid protein (PLP), MBP, and the cyclic nucleotide 3’-phosphodiesterase (CNP) promoters, α-synuclein was exclusively expressed in oligodendrocytes62,63,64. However, transgenic mouse lines overexpressing α-synuclein displayed different pathological features of MSA under different oligodendroglial promoters. In these models, motor and nonmotor symptoms developed to varying degrees. Furthermore, these models exhibited accumulation of α-synuclein and its aggregates in oligodendrocytes similar to GCIs in MSA patients.
In addition, adeno-associated viruses (AAVs) were genetically modified to express α-synuclein, specifically in oligodendrocytes. Using these recombinant viruses, more diverse animal models, such as nonhuman primates, can be established65,66. However, the various symptoms described in each of these models and the pattern and degree of degeneration in the nervous system did not precisely correlate with disease pathology47,67.
PLP-hαSyn transgenic mice
PLP promoter-driven α-synuclein (PLP-hαSyn) transgenic mice bred on a C57/BL6 background exhibited phosphorylation of α-synuclein at Ser129 and aggregation of α-synuclein along with GCI-like inclusions62. Mitochondrial inhibition by 3-NP in these mice induced the degeneration of the striatonigral system and gliosis68. The loss of dopaminergic neurons in the SNpc, Purkinje cells, and neurons in the pons and medulla oblongata, as in the human MSA-C subtype, was also observed. Treatment with 3-NP augmented motor deficits in tg mice compared to non-tg controls at ~12 months. Autonomic symptoms, however, appeared earlier, as early as 2 months of age69. Urinary bladder dysfunction with morphological changes in the bladder wall and increased postvoid residual volumes was detected at 2 months of age70. Heart rate variability, an indication of changed sympathovagal balance, as observed in the human disease, was reduced at 5 months of age71. At the age of 13 months, respiratory failure was observed72. Progressive motor deficits emerged at 6 months and progressed until 18 months of follow-up. The loss of dopaminergic neurons in the SNpc was found during the initial stages. The loss of striatal dopaminergic terminals and DARPP32-positive projection neurons was observed at 12 months73. Gliosis and increased levels of cytokines were also reported.
MBP-hαSyn transgenic mice
Mice with MBP promoter-driven α-synuclein expression in oligodendrocytes exhibited somewhat different pathological features of MSA that those exhibited by PLP-hαsyn and CNP-hαsyn tg mice63. These mice displayed demyelination along with axonal degeneration in the cerebellum, basal ganglia, brain stem, corpus callosum, and neocortex. The high expressor line (line 29) died prematurely at 6 months. The moderate expressor line (line 1) exhibited a mild phenotype with disease onset after 6 months of age. α-Synuclein accumulation was also evident, along with elevated astrogliosis. 3-NP administration increased nitrated and oxidized α-synuclein levels but not the levels of the phosphorylated or total protein74. Neurological deficits were augmented and accompanied by widespread neurodegeneration and behavioral problems. Increases in neuroinflammation were detected in regions of high α-synuclein expression, such as the corpus callosum and the striatum, even before symptoms were evident75. Inflammatory responses were restricted to myeloid cells, and severe astrogliosis was only detected in gray matter regions. RNA sequencing of α-synuclein-expressing primary oligodendrocytes demonstrated upregulation of cytokines and genes important for inflammatory responses, suggesting that neuroinflammation may be critical for disease development. MBP-hαsyn tg mice displayed GDNF deficiency, IκBα and miR-96 upregulation, and a delay in oligodendrocyte maturation59,69,76,77.
CNP-hαSyn transgenic mice
CNP promoter-hαSyn mice exhibited the accumulation of fibrillary human α-synuclein in oligodendrocytes, a loss and demyelination of these cells, and severe gliosis in the brain and spinal cord64,69. The aggregation of endogenous mouse α-synuclein, which is associated with neuronal loss, was also detected in the axons and axon terminals of spinal cord neurons. Motor deficits started at the age of 7 to 9 months, but the phenotypes seem to be different from typical MSA symptoms78,79,80.
In general, the accumulation of α-synuclein in oligodendroglia has been found in all three oligodendrocyte-specific promoter-driven human α-synuclein tg mice. However, each of these mice displayed distinct patterns of pathological changes that do not precisely replicate human MSA pathology47,81.
Viral-mediated oligodendroglial α-synuclein expression models
Bassil and colleagues utilized AAVs to express human α-synuclein under the control of the MBP promoter in adult Sprague-Dawley (SD) rats and nonhuman primates (macaques)65. α-Synuclein expression was observed in ~80% of oligodendrocytes in the injected area of SD rats. The injected rats displayed L-DOPA-unresponsive motor deficits by 6 months of age, while significant dopaminergic cell loss occurred at 3 months of age. Increased amounts of the insoluble and phosphorylated forms of human α-synuclein were detected in the striatum and SNpc of injected rats. The injected macaques showed expression of human α-synuclein in ~60% of oligodendrocytes and ~40% of neurons. It has yet to be determined whether the neuronal α-synuclein came from the direct expression of α-synuclein in neurons or from the oligodendrocyte-to-neuronal transmission of α-synuclein. Longitudinal studies are also required to determine if viral-mediated oligodendroglial α-synuclein expression is useful as an MSA model.
A study by Mandel et al. utilized the oligotrophic AAV vector Olig001, which specifically transduces oligodendrocytes, to express human α-synuclein in nonhuman primates66. The injection of the recombinant vector into the striatum of rhesus macaques induced widespread expression of α-synuclein in more than 90% of oligodendrocytes throughout the striatum by 3 months of age. Phosphorylated α-synuclein was detected, and the GCI-like inclusions in oligodendrocytes were resistant to proteinase K (PK) digestion. Demyelination in the white matter tracts of the corpus callosum and microgliosis in the striatum was also detected.
The viral expression of α-synuclein in animals has advantages over genetic models in that expression can be manipulated temporally. The recombinant virus can be injected at an early stage or later in development. The cell specificity of viral infection has significantly improved over the years. However, more studies of such models in rodents and nonhuman primates are still needed for the development of MSA models.
Transmission models
Numerous studies have reported little to no α-synuclein expression in mature oligodendrocytes in vitro and in vivo11. The mechanisms of the accumulation of α-synuclein aggregates in the oligodendrocytes and astrocytes of MSA patients are still largely unknown. However, recent reports of the transfer of α-synuclein from one cell to another, particularly from neurons to glia, have led to the hypothesis that toxic α-synuclein aggregates secreted from neurons are taken up by oligodendrocytes and disrupts their function35,38. Based on this hypothesis, several groups have developed transmission models.
In vitro culture models of exogenously added α-synuclein aggregates
Pukas et al. utilized primary rat brain oligodendrocytes and an oligodendroglial cell line, OLN-93, and added soluble or preaggregated forms of human recombinant α-synuclein82. Both forms of exogenously added α-synuclein were internalized and formed small intracellular aggregates in oligodendroglial cells. Under oxidative stress, the levels of aggregates increased, and cell viability was reduced. Human α-synuclein preformed fibrils (PFFs) triggered the expression and aggregation of endogenous α-synuclein in rat primary OPCs83. However, when using PFFs, one should consider that the PFFs may differ from neuron-derived α-synuclein aggregates in structure and composition. In addition, there may be essential cofactors secreted from surrounding cells that influence the transmission of the protein.
In vitro co-culture models of α-synuclein-expressing donor cells and naïve recipient cells have been established. Neuron-to-neuron, neuron-to-astrocyte, and neuron-to-microglia transmission of α-synuclein have recently been reported38,39,84. The transfer of α-synuclein to recipient cells caused various cellular changes, including cell toxicity and inflammatory responses. The advantage of using such models is that it is easy to manipulate the expression of specific genes and to detect cellular changes by biochemical methods
The use of co-cultures of neurons and oligodendrocytes was reported recently. Mouse embryonic stem cell (ESC)-derived neural progenitor cells (NPCs) differentiated into OPCs and then into mature oligodendrocytes. These cells were co-cultured with differentiated cortical neurons from mouse ESCs on the same plate. However, unlike in other studies in which neurons overexpressed α-synuclein and were co-cultured with other naïve recipient cells, this study utilized direct lentiviral overexpression of α-synuclein in oligodendrocytes, which induced myelination deficits upon co-culture with naïve neurons85. Neuron-to-oligodendrocyte transmission and its effects remain to be studied.
Stereotaxic injection of recombinant proteins or MSA brain extracts into animal models
Brain extracts from 14 MSA patients were inoculated into the brains of TgM83+/− hemizygous mice expressing the A53T α-synuclein mutant under the prion promoter86. All mice showed neurodegeneration and accumulation of α-synuclein aggregates in neuronal cell bodies and axons at 4 months postinjection, whereas control injections did not induce neurodegeneration at the same age. Interestingly, PD brain extracts did not promote the aggregation of α-synuclein in TgM83 mice, indicating that pathogenic α-synuclein species may differ in MSA and PD. The same group performed comparison experiments with different transgenic animal models expressing wild-type, A30P, and A53T α-synuclein on a mouse α-synuclein knockout background87.
In contrast to the results obtained in TgM83+/− mice, these mice, when injected with MSA brain extracts, did not show any motor deficits, even after 330–400 days. Only A53T-expressing (Tg(SNCA*A53T+/+)) mice, but not others, displayed α-synuclein pathology in neurons and astrocytes in the limbic system. Interestingly, these injected mice retained infectivity after the initial inoculation of human MSA patient samples. Mouse brain extracts from MSA patient extract-injected tg mice were inoculated for the second time into both TgM83+/− and Tg(SNCA*A53T+/+) mice. These second-round synucleinopathy models also developed pathology, and the transmission of α-synuclein was confirmed.
These injection models support the α-synuclein transmission hypothesis, as pathological α-synuclein from diseased patients was transferred to nonsymptomatic mice and induced pathology.
Conclusion
MSA is a neurodegenerative disease with clinical symptoms similar to those of PD and cerebellar ataxia. The complexity and rapid progression of the disease, as well as its unresponsiveness to drugs, such as L-DOPA for parkinsonian symptoms, make MSA a challenging disease to treat. Various models have been generated over the years to study the mechanism of MSA development and progression. In this review, we summarized toxin-induced models, in vitro and in vivo genetic models, and transmission models for studying α-synuclein pathology and behavioral symptoms in MSA (Fig. 3).

In vitro cell models have been used to express α-synuclein in oligodendrocytes. The effects of the addition of exogenous α-synuclein aggregates to oligodendrocytes have been studied in in vitro transmission models. Recent developments in stem cell technologies have allowed the growth of MSA-derived iPSCs and their differentiation into oligodendrocytes. Comparison studies of RNA, protein, and epigenetic changes in normal and patient-derived cells are ongoing. Animal models induced by the injection of toxins exhibit MSA-like symptoms, and oligodendrocyte-specific expression of α-synuclein has been achieved through the generation of tg mice and viral-mediated expression.
The main goal of the genetic models is to generate oligodendrocytes with GCI-like α-synuclein pathology, thereby allowing us to study the roles of α-synuclein in MSA pathogenesis. Two methods have been applied to generate models with oligodendrocytic α-synuclein accumulation. The first method was to ectopically express α-synuclein in oligodendrocytes. α-Synuclein was overexpressed in oligodendroglial cells in culture. Transgenic animal models expressing α-synuclein specifically in oligodendrocytes were created. These direct expression models displayed glial α-synuclein pathology and various CNS phenotypes akin to those of MSA. However, because mature oligodendrocytes have little to no α-synuclein expression, it is not clear whether these ectopic expression models are valid representatives of human MSA.
More recent efforts to develop MSA models have utilized the fact that α-synuclein can be transferred between cells. The exogenous addition of recombinant α-synuclein aggregates to oligodendrocytes resulted in the uptake and induction of endogenous protein aggregate formation. The stereotaxic injection of human MSA brain extracts into the brains of α-synuclein tg mice induced extensive spreading of α-synuclein pathology, neuroinflammation, and neurodegeneration. However, these injected mouse models displayed neuronal α-synuclein pathology instead of oligodendroglial pathology.
Although researchers have been able to recapitulate some aspects of MSA with current models, none of these models present bona fide MSA pathology, which is represented by oligodendroglial GCIs generated from endogenous α-synuclein proteins. Recent studies have suggested that GCIs are generated through the transfer of α-synuclein from neurons to oligodendrocytes. To test this hypothesis, we will have to understand more about the biology of oligodendrocytes in MSA. Critical questions include how oligodendrocytes respond to and process neuron-derived α-synuclein and how normal oligodendrocytes and MSA oligodendrocytes respond differently to α-synuclein. We anticipate that the technical advancement of single-cell analysis and bioinformatics will contribute to answering these questions.
References
Graham, J. G. & Oppenheimer, D. R. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 32, 28–34 (1969).
Schrag, A., Ben-Shlomo, Y. & Quinn, N. P. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 354, 1771–1775 (1999).
Ben-Shlomo, Y., Wenning, G. K., Tison, F. & Quinn, N. P. Survival of patients with pathologically proven multiple system atrophy: a meta-analysis. Neurology 48, 384–393 (1997).
Wullner, U. et al. Features of probable multiple system atrophy patients identified among 4770 patients with parkinsonism enrolled in the multicentre registry of the German Competence Network on Parkinson’s disease. J. Neural Transm. 114, 1161–1165 (2007).
Schrag, A., Wenning, G. K., Quinn, N. & Ben-Shlomo, Y. Survival in multiple system atrophy. Mov. Disord. 23, 294–296 (2008).
Fanciulli, A. & Wenning, G. K. Multiple-system atrophy. N. Engl. J. Med. 372, 249–263 (2015).
Jecmenica-Lukic, M., Poewe, W., Tolosa, E. & Wenning, G. K. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol. 11, 361–368 (2012).
Benrud-Larson, L. M., Sandroni, P., Schrag, A. & Low, P. A. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov. Disord. 20, 951–957 (2005).
Wenning, G. K. et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol. 12, 264–274 (2013).
Yabe, I. et al. MSA-C is the predominant clinical phenotype of MSA in Japan: analysis of 142 patients with probable MSA. J. Neurol. Sci. 249, 115–121 (2006).
Valera, E. & Masliah, E. The neuropathology of multiple system atrophy and its therapeutic implications. Auton. Neurosci. 211, 1–6 (2018).
Kim, H. J. et al. Clinical and imaging characteristics of dementia in multiple system atrophy. Parkinsonism Relat. Disord. 19, 617–621 (2013).
Lee, S. W. & Koh, S. B. Clinical features and disability milestones in multiple system atrophy and progressive supranuclear palsy. J. Mov. Disord. 5, 42–47 (2012).
Papp, M. I., Kahn, J. E. & Lantos, P. L. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J. Neurol. Sci. 94, 79–100 (1989).
Fujiwara, H. et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164 (2002).
Hasegawa, M. et al. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076 (2002).
Jellinger, K. A. & Lantos, P. L. Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update. Acta Neuropathol. 119, 657–667 (2010).
Ovadi, J. & Orosz, F. An unstructured protein with destructive potential: TPPP/p25 in neurodegeneration. Bioessays 31, 676–686 (2009).
Lehotzky, A. et al. Tubulin polymerization-promoting protein (TPPP/p25) is critical for oligodendrocyte differentiation. Glia 58, 157–168 (2010).
Song, Y. J. et al. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am. J. Pathol. 171, 1291–1303 (2007).
Lindersson, E. et al. p25alpha Stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synucleinopathies. J. Biol. Chem. 280, 5703–5715 (2005).
Jellinger, K. A. Lewy body-related alpha-synucleinopathy in the aged human brain. J. Neural Transm. 111, 1219–1235 (2004).
McCann, H., Stevens, C. H., Cartwright, H. & Halliday, G. M. a-synucleinopathy phenotypes. Parkinsonism Relat. Disord. 20, S62–S67 (2014).
Scholz, S. W. et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann. Neurol. 65, 610–614 (2009).
Tu, P. H. et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann. Neurol. 44, 415–422 (1998).
Wakabayashi, K. et al. Accumulation of alpha-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol. 96, 445–452 (1998).
Peng, C. et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557, 558–563 (2018).
Miller, D. W. et al. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J. Neural Transm. 112, 1613–1624 (2005).
Ozawa, T. et al. Analysis of the expression level of alpha-synuclein mRNA using postmortem brain samples from pathologically confirmed cases of multiple system atrophy. Acta Neuropathol. 102, 188–190 (2001).
Asi, Y. T. et al. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia 62, 964–970 (2014).
Djelloul, M. et al. Alpha-synuclein expression in the oligodendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Rep. 5, 174–184 (2015).
Abati, E., Di Fonzo, A. & Corti, S. In vitro models of multiple system atrophy from primary cells to induced pluripotent stem cells. J. Cell Mol. Med. 22, 2536–2546 (2018).
Lee, H.-J. et al. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849 (2008).
Kisos, H., Pukass, K., Ben-Hur, T., Richter-Landsberg, C. & Sharon, R. Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS One 7, e46817 (2012).
Reyes, J. F. et al. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62, 387–398 (2014).
Schwarz, J. et al. Signal changes on MRI and increases in reactive microgliosis, astrogliosis, and iron in the putamen of two patients with multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 60, 98–101 (1996).
Stefanova, N. et al. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov. Disord. 22, 2196–2203 (2007).
Lee, H.-J. et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272 (2010).
Kim, C. et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562 (2013).
Alvarez-Erviti, L., Couch, Y., Richardson, J., Cooper, J. M. & Wood, M. J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 69, 337–342 (2011).
Vieira, B. D., Radford, R. A., Chung, R. S., Guillemin, G. J. & Pountney, D. L. Neuroinflammation in multiple system atrophy: response to and cause of alpha-synuclein aggregation. Front. Cell Neurosci. 9, 437 (2015).
Radford, R. et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to alpha-synuclein inclusions. Mol. Cell Neurosci. 65, 68–81 (2015).
Valdinocci, D., et al. Extracellular interactions of alpha-synuclein in multiple system atrophy. Int. J. Mol. Sci. 19, 4129 (2018).
Croisier, E. & Graeber, M. B. Glial degeneration and reactive gliosis in alpha-synucleinopathies: the emerging concept of primary gliodegeneration. Acta Neuropathol. 112, 517–530 (2006).
Fellner, L. et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 61, 349–360 (2013).
Kim, C., Lee, H. J., Masliah, E. & Lee, S. J. Non-cell-autonomous neurotoxicity of alpha-synuclein through microglial toll-like receptor 2. Exp. Neurobiol. 25, 113–119 (2016).
Fernagut, P. O. & Tison, F. Animal models of multiple system atrophy. Neuroscience 211, 77–82 (2012).
Scherfler, C. et al. Complex motor disturbances in a sequential double lesion rat model of striatonigral degeneration (multiple system atrophy). Neuroscience 99, 43–54 (2000).
Puschban, Z. et al. Evidence for dopaminergic re-innervation by embryonic allografts in an optimized rat model of the Parkinsonian variant of multiple system atrophy. Brain Res. Bull. 68, 54–58 (2005).
Yoon, H. H. et al. Evaluation of a multiple system atrophy model in rats using multitracer microPET. Acta Neurochir. 154, 935–940 (2012).
Waldner, R. et al. No functional effects of embryonic neuronal grafts on motor deficits in a 3-nitropropionic acid rat model of advanced striatonigral degeneration (multiple system atrophy). Neuroscience 102, 581–592 (2001).
Ghorayeb, I. et al. A ‘single toxin-double lesion’ rat model of striatonigral degeneration by intrastriatal 1-methyl-4-phenylpyridinium ion injection: a motor behavioural analysis. Neuroscience 115, 533–546 (2002).
Ghorayeb, I. et al. Toward a primate model of L-dopa-unresponsive parkinsonism mimicking striatonigral degeneration. Mov. Disord. 15, 531–536 (2000).
Ghorayeb, I. et al. Dystonia is predictive of subsequent altered dopaminergic responsiveness in a chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine+3-nitropropionic acid model of striatonigral degeneration in monkeys. Neurosci. Lett. 335, 34–38 (2002).
Fernagut, P. O. et al. Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterisation. Neuroscience 114, 1005–1017 (2002).
Fernagut, P. O., Barraud, Q., Bezard, E., Ghorayeb, I. & Tison, F. Metabolic activity of the subthalamic nucleus in a primate model of L-dopa-unresponsive parkinsonism. Neurol. Res. 32, 1050–1053 (2010).
Stefanova, N., Reindl, M., Poewe, W. & Wenning, G. K. In vitro models of multiple system atrophy. Mov. Disord. 20, S53–S56 (2005).
Kragh, C. L. et al. Alpha-synuclein aggregation and Ser-129 phosphorylation-dependent cell death in oligodendroglial cells. J. Biol. Chem. 284, 10211–10222 (2009).
Kragh, C. L. et al. Prodegenerative IkappaBalpha expression in oligodendroglial alpha-synuclein models of multiple system atrophy. Neurobiol. Dis. 63, 171–183 (2014).
May, V. E. et al. alpha-Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol. Aging 35, 2357–2368 (2014).
Valera, E. et al. MicroRNA-101 modulates autophagy and oligodendroglial alpha-synuclein accumulation in multiple system atrophy. Front. Mol. Neurosci. 10, 329 (2017).
Kahle, P. J. et al. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 3, 583–588 (2002).
Shults, C. W. et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J. Neurosci. 25, 10689–10699 (2005).
Yazawa, I. et al. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45, 847–859 (2005).
Bassil, F. et al. Viral-mediated oligodendroglial alpha-synuclein expression models multiple system atrophy. Mov. Disord. 32, 1230–1239 (2017).
Mandel, R. J. et al. Novel oligodendroglial alpha synuclein viral vector models of multiple system atrophy: studies in rodents and nonhuman primates. Acta Neuropathol. Commun. 5, 47 (2017).
Overk, C. et al. Multiple system atrophy: experimental models and reality. Acta Neuropathol. 135, 33–47 (2018).
Stefanova, N. et al. Oxidative stress in transgenic mice with oligodendroglial alpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am. J. Pathol. 166, 869–876 (2005).
Stefanova, N. & Wenning, G. K. Animal models of multiple system atrophy. Clin. Auton. Res. 25, 9–17 (2015).
Boudes, M. et al. Bladder dysfunction in a transgenic mouse model of multiple system atrophy. Mov. Disord. 28, 347–355 (2013).
Kuzdas, D. et al. Oligodendroglial alpha-synucleinopathy and MSA-like cardiovascular autonomic failure: experimental evidence. Exp. Neurol. 247, 531–536 (2013).
Flabeau, O. et al. Breathing variability and brainstem serotonergic loss in a genetic model of multiple system atrophy. Mov. Disord. 29, 388–395 (2014).
Refolo, V. et al. Progressive striatonigral degeneration in a transgenic mouse model of multiple system atrophy: translational implications for interventional therapies. Acta Neuropathol. Commun. 6, 2 (2018).
Ubhi, K. et al. Mitochondrial inhibitor 3-nitroproprionic acid enhances oxidative modification of alpha-synuclein in a transgenic mouse model of multiple system atrophy. J. Neurosci. Res. 87, 2728–2739 (2009).
Hoffmann, A. et al. Oligodendroglial alpha-synucleinopathy-driven neuroinflammation in multiple system atrophy. Brain Pathol. 29, 380–396 (2019).
Ubhi, K. et al. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J. Neurosci. 30, 6236–6246 (2010).
Ubhi, K. et al. Widespread microRNA dysregulation in multiple system atrophy-disease-related alteration in miR-96. Eur. J. Neurosci. 39, 1026–1041 (2014).
Nakayama, K., Suzuki, Y. & Yazawa, I. Microtubule depolymerization suppresses alpha-synuclein accumulation in a mouse model of multiple system atrophy. Am. J. Pathol. 174, 1471–1480 (2009).
Nakayama, K., Suzuki, Y. & Yazawa, I. Binding of neuronal alpha-synuclein to beta-III tubulin and accumulation in a model of multiple system atrophy. Biochem. Biophys. Res. Commun. 417, 1170–1175 (2012).
Ito, H., Nakayama, K., Jin, C., Suzuki, Y. & Yazawa, I. alpha-Synuclein accumulation reduces GABAergic inhibitory transmission in a model of multiple system atrophy. Biochem. Biophys. Res. Commun. 428, 348–353 (2012).
Ubhi, K., Low, P. & Masliah, E. Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 34, 581–590 (2011).
Pukass, K. & Richter-Landsberg, C. Oxidative stress promotes uptake, accumulation, and oligomerization of extracellular alpha-synuclein in oligodendrocytes. J. Mol. Neurosci. 52, 339–352 (2014).
Kaji, S. et al. Pathological endogenous alpha-synuclein accumulation in oligodendrocyte precursor cells potentially induces inclusions in multiple system atrophy. Stem Cell Rep. 10, 356–365 (2018).
Desplats, P. et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl Acad. Sci. USA 106, 13010–13015 (2009).
Ettle, B. et al. alpha-Synuclein-induced myelination deficit defines a novel interventional target for multiple system atrophy. Acta Neuropathol. 132, 59–75 (2016).
Prusiner, S. B. et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl Acad. Sci. USA 112, E5308–E5317 (2015).
Woerman, A. L. et al. Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol. 137, 437–454 (2019).
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean Government (NRF-2015R1D1A1A01059164, NRF-2017R1A2B4003220, NRF-2016R1A5A2012284, MSIT, NRF-2018R1A2A1A05078261, and NRF-2018R1A5A2025964).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, HJ., Ricarte, D., Ortiz, D. et al. Models of multiple system atrophy. Exp Mol Med 51, 1–10 (2019). https://doi.org/10.1038/s12276-019-0346-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0346-8
- Springer Nature Limited
This article is cited by
-
The ‘α-synucleinopathy syndicate’: multiple system atrophy and Parkinson’s disease
Journal of Neural Transmission (2024)
-
Impaired Sphingolipid Hydrolase Activities in Dementia with Lewy Bodies and Multiple System Atrophy
Molecular Neurobiology (2022)
-
Neuropathology of multiple system atrophy: Kurt Jellinger`s legacy
Journal of Neural Transmission (2021)
-
Insights into the pathogenesis of multiple system atrophy: focus on glial cytoplasmic inclusions
Translational Neurodegeneration (2020)