
Abstract
The liver is a highly regenerative organ, but its regenerative capacity is compromised in severe liver diseases. Hepatocyte-driven liver regeneration that involves the proliferation of preexisting hepatocytes is a primary regeneration mode. On the other hand, liver progenitor cell (LPC)-driven liver regeneration that involves dedifferentiation of biliary epithelial cells or hepatocytes into LPCs, LPC proliferation, and subsequent differentiation of LPCs into hepatocytes is a secondary mode. This secondary mode plays a significant role in liver regeneration when the primary mode does not effectively work, as observed in severe liver injury settings. Thus, promoting LPC-driven liver regeneration may be clinically beneficial to patients with severe liver diseases. In this review, we describe the current understanding of LPC-driven liver regeneration by exploring current knowledge on the activation, origin, and roles of LPCs during regeneration. We also describe animal models used to study LPC-driven liver regeneration, given their potential to further deepen our understanding of the regeneration process. This understanding will eventually contribute to developing strategies to promote LPC-driven liver regeneration in patients with severe liver diseases.
Similar content being viewed by others


Introduction
The liver is an essential and multifunctional organ in vertebrates. It consists of hepatocytes and biliary epithelial cells (BECs) that are differentiated from common progenitor cells called hepatoblasts during development. Hepatocytes, a major cell type in the liver, detoxify various metabolites, regulate glucose and lipid metabolism, synthesize serum proteins, and secrete bile. BECs form the biliary network that transports bile from hepatocytes to the gallbladder1. Upon food ingestion, bile is released from the gallbladder into the duodenum and helps absorb fats in the gut.
The liver is also a highly regenerative organ. It is able to restore its mass and function after injury. Depending on the source of regenerating hepatocytes, there are two modes of liver regeneration: hepatocyte- and LPC-driven liver regeneration2,3. For instance, upon two-third partial hepatectomy, the remaining hepatocytes proliferate to restore the resected liver mass4 (hepatocyte-driven regeneration). On the other hand, when hepatocyte proliferation is compromised, BECs are able to dedifferentiate into liver progenitor cells (LPCs), also known as oval cells. Then, these LPCs later differentiate into hepatocytes2,3 (LPC-driven regeneration). In addition to this BEC-to-LPC dedifferentiation, hepatocytes can dedifferentiate into LPCs and later differentiate back into hepatocytes5.
Given that liver diseases have been a major health concern due to their high prevalence and poor long-term clinical outcome, the regenerative potential of the liver is especially important to note. Globally, approximately two million deaths per year are caused by severe liver diseases, including viral hepatitis, liver cirrhosis, and liver cancer6. Currently, liver transplantation is the only curative option for these life-threatening diseases; however, the shortage of donor livers limits this option. Thus, patients with severe liver diseases often die while waiting for a donor liver6,7. Given that LPC-driven liver regeneration is prevalent in severe liver injury settings, augmenting this regeneration mode should be clinically beneficial to patients with severe liver diseases.
In this review, we focus mainly on LPC-driven liver regeneration by examining recent findings and exploring current knowledge about the activation, origin, and role of LPCs during liver regeneration. We also provide information on the clinical significance and therapeutic potential of LPCs.
Features of LPCs
Hepatocyte-driven liver regeneration is achieved by the proliferation of preexisting hepatocytes4, whereas LPC-driven liver regeneration is achieved by the proliferation and subsequent differentiation of LPCs2,3. Thus, LPC-driven liver regeneration is important in severe liver injury settings accompanied by impaired hepatocyte proliferation. In LPC-driven liver regeneration, hepatocytes or BECs first dedifferentiate into LPCs following LPC proliferation, and the LPCs then differentiate into hepatocytes1,8.
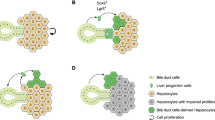
Over time, LPCs have been variously named in rodent and human studies as oval cells, hepatic progenitor cells, liver stem cells, ductular reactions, or atypical ductular cells9. They were first described as oval cells in rats due to their large nuclear-to-cytoplasm ratio and oval-shaped nuclei10. Anatomically, LPCs have been suggested to reside within the canals of Hering, also known as intrahepatic bile ductules, which are positioned between the bile duct and hepatocytes1 (Fig. 1). Although LPCs are not observed in the normal adult liver, they appear and expand in response to severe or chronic liver injury2,3. LPCs express both hepatocyte (KRT8, KRT18, and albumin)11,12,13 and BEC (KRT7, KRT19, EpCAM, and SOX9) markers11,14,15,16. Depending on injury settings, LPCs also express the hepatoblast marker α-fetoprotein (AFP)17, hematopoietic markers, such as CD34, CD90, CD133, c-Kit, CXCR4, and Sca1 (refs. 12,18,19,20,21), or the neuronal marker NCAM22. The various expression of these markers in LPCs suggests their progenitor features and heterogeneous nature. Recently, single-cell RNA sequencing analyses of EpCAM+ hepatic cells isolated from mice fed a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet further revealed the heterogeneity of LPCs23,24.

The liver consists of hepatocytes, BECs, Kupffer cells, HSCs, and endothelial cells. LPCs are thought to arise near bile ductules, also known as the canals of Hering, which are positioned between the bile duct and hepatocytes. In the zebrafish liver, most BECs make up bile ductules.
The molecular mechanisms of LPC activation
Extensive LPC activation and expansion are detected in patients with various liver diseases, including alcoholic or nonalcoholic liver disease25, chronic viral hepatitis26,27, and cholestatic hepatitis28, all of which cause a massive loss of hepatocytes in the liver. LPC activation, also referred to as ductular reactions, involves the proliferation and expansion of LPCs in the periportal regions in the liver, thereby increasing the number of bile ductules. Additionally, LPC activation involves macrophage infiltration, extracellular matrix remodeling, and myofibroblast activation29,30.
The mechanisms of LPC activation have been extensively investigated using animal models of chronic liver injury. Hepatocyte-specific deletion of the inhibitor kappa B kinase with Alb-Cre mice inhibited hepatocyte proliferation and induced apoptosis31. Dying hepatocytes in these mice produced hedgehog ligands that promote the outgrowth of LPCs and myofibroblasts31. Hepatocyte-specific deletion of survivin with Alb-Cre mice also induced hepatocyte apoptosis and inflammation, eliciting LPC activation32. These findings support that the extensive loss of hepatocytes and the impairment of hepatocyte proliferation induce LPC activation. Furthermore, it was recently reported that YAP and mTORC1 signaling are important for LPC activation. YAP and mTORC1 signaling positively regulate the growth of BEC-derived organoids in vitro and the proliferation of BECs and LPCs in mice24. TET1-mediated epigenetic remodeling through YAP signaling was also recently reported to positively control LPC activation33.
Additionally, macrophages are known to associate with LPC activation during regeneration. Dying hepatocytes and their debris are engulfed and removed by Kupffer cells, which are the resident macrophages in the liver. These macrophages secrete tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), a member of the proinflammatory TNF family, and TWEAK binds to its receptor FGF-inducible 14 (Fn14) expressed on LPCs30. Fn14 was increasingly expressed on LPCs in chronic hepatitis C and other human liver diseases30. Moreover, overexpression of TWEAK in hepatocytes stimulated LPC proliferation through the nuclear factor-κB (NF-κB) signaling pathway30,34.
TNFα35, interferon-γ (IFNγ)36, and interleukin-6 (IL-6) (refs. 35,37) signaling are also known to control LPC activation. Moreover, other inflammatory-related proteins, including cyclooxygenase-2 (ref. 38), lymphotoxin beta39,40, and galectin-3 (ref. 41), are reported to regulate LPC activation. Growth factor signaling pathways, such as HGF/c-Met42,43, TGF-β42,44,45, FGF7 (ref. 46), and VEGF47 signaling, are also involved in LPC activation and expansion (Table 1).
Origins of LPCs
Despite the absence of LPC-specific markers that are expressed in LPCs but not in BECs, BECs were hypothesized to be the origin of LPCs due to their phenotypical similarity and locational contiguity48 (Fig. 2). This was later validated by lineage tracing. By genetically labeling nearly all hepatocytes in mice, the Grompe group showed that BECs in mice fed a DDC diet for 2 weeks contributed to LPCs5. The same group also showed, by tracing the lineage of Sox9+ BECs with Sox9-CreERT2 mice, that BECs gave rise to LPCs in DDC diet, choline-deficient, ethionine-supplemented diet (CDE), and CCl4 injury models49. By tracing the lineage of BECs with Krt19-CreERT mice, it was also shown that BECs contribute to LPCs in the DDC and CDE models50,51. Although these lineage-tracing studies have validated BECs as the origin of LPCs, the activated LPCs in the studies did not differentiate into hepatocytes. Later, the fact that LPCs originate from BECs was also confirmed in liver injury models in which LPCs significantly contribute to hepatocytes52. In mice with β1-integrin knocked down in all hepatocytes, thus blocking hepatocyte proliferation, BEC lineage tracing revealed the significant contribution of BECs to hepatocytes in several liver injury models, including DDC52. Hepatocyte-specific overexpression of p21 combined with the liver injury models also exhibited a significant contribution of BECs to hepatocytes52. BEC-to-LPC dedifferentiation was also observed in zebrafish53,54,55. Upon the severe loss of hepatocytes in zebrafish larvae by pharmacogenetic means, BECs dedifferentiated into LPCs and subsequently differentiated into hepatocytes. In this zebrafish model, nearly all hepatocytes in the recovered larvae originated from BECs53.

BECs and hepatocytes can give rise to LPCs in various liver injury settings. HSCs might be an additional source of LPCs in certain liver injury settings.
In addition to BECs, lineage-tracing studies revealed hepatocytes as an additional source of LPCs5,51,56 (Fig. 2). By labeling nearly all hepatocytes in mice, the Grompe group showed that hepatocytes in mice fed a DDC diet for 6, but not 2, weeks contributed to LPCs5. Once the liver injury was gone, these hepatocyte-derived LPCs reverted to hepatocytes5. These data suggest that hepatocytes can contribute to the LPC population by undergoing metaplasia in chronic liver injury settings. Supporting this hepatocyte origin, overexpression of constitutively active YAP1 (ref. 57) or Notch56 in mature hepatocytes converted the hepatocytes to LPCs. Moreover, inhibiting YAP23 or Notch56 signaling in hepatocytes suppressed their conversion to LPCs in the DDC model. Hepatocyte-to-LPC dedifferentiation was also observed in zebrafish58. In Tg(fabp10a:pt-β-catenin) zebrafish larvae, which overexpress a stable form of β-catenin in hepatocytes, a subset of hepatocytes dedifferentiated into LPCs and later differentiated into hepatocytes58, as observed in the mice fed a DDC diet5. Although some lineage-tracing studies suggested hepatic stellate cells (HSCs) as an origin of LPCs and regenerated hepatocytes59,60,61,62, other lineage-tracing studies showed no conversion of HSCs to LPCs or hepatocytes in multiple liver injury settings50,63, raising uncertainty regarding HSCs as an origin of LPCs (Fig. 2).
The beneficial role of LPCs: their differentiation into hepatocytes
Studies with diseased human livers have suggested that BECs dedifferentiate into LPCs and that LPCs can differentiate into hepatocytes11,64,65,66,67 (Fig. 3). Supporting these human studies, lineage-tracing studies in mice52,68,69,70,71 and zebrafish53,54,55 have demonstrated that in severe liver injury settings, BECs first dedifferentiate into LPCs and subsequently differentiate into hepatocytes. For the initial mouse lineage-tracing studies, several BEC-specific, inducible Cre lines, such as Opn-CreERT2, Krt19-CreERT, Hnf1b-CreERT2, and Sox9-CreERT2, were used to trace the fate of BECs in DDC- and CDE-mediated liver injury models. While no contribution of BECs to hepatocytes was observed in the DDC model, a few hepatocytes (<2.5%) originated from BECs in the CDE model5,50,72,73,74. This low percentage of contribution of BECs to hepatocytes raised a question about the significance of LPC-driven regeneration in liver regeneration. However, several groups recently showed a significant contribution of BECs to hepatocytes in severe liver injury settings in which hepatocyte proliferation is greatly compromised. To block hepatocyte proliferation, Mdm2 (ref. 70), β1-integrin52, or β-catenin68 was deleted specifically in hepatocytes, or p21 was overexpressed in hepatocytes52. In addition to these genetic blocks of hepatocyte proliferation, long-term chronic liver injury elicited the natural impairment of hepatocyte proliferation, thereby inducing the differentiation of BEC-derived LPCs into hepatocytes69,71. As previously mentioned in the section on LPC origins, hepatocytes as well as BECs can give rise to LPCs, and hepatocyte-derived LPCs appear to revert to hepatocytes during recovery5. For the zebrafish lineage-tracing studies, the BEC-specific, inducible Cre line, Tg(Tp1:CreERT2), was used in the complete hepatocyte-ablation model. Upon severe hepatocyte loss, BECs dedifferentiate into LPCs, and subsequently, LPCs differentiate into either hepatocytes or BECs53,54,55. Intriguingly, suppressing LPC differentiation in the zebrafish model impaired liver regeneration and recovery75,76, suggesting the beneficial effect of LPC differentiation on liver recovery.

LPCs play a beneficial role in liver regeneration/recovery by differentiating themselves into hepatocytes, whereas they play negative roles by contributing to liver fibrosis and tumor formation, such as HCC and ICC.
Given the beneficial effect of promoting LPC differentiation, elucidation of the molecular mechanisms of LPC differentiation has been attempted. Liver-specific c-Met knockout mice fed a DDC diet exhibited a significant reduction in the number of A6+/EpCAM− hepatocyte-like cells compared to c-Met wild-type mice, suggesting that HGF/c-Met signaling may regulate the differentiation of LPCs into hepatocytes during regeneration43. In addition, treatment with iloprost, a laminin signaling inhibitor, increased a LPC-derived hepatocyte population in the CDE model, suggesting that LPC microenvironment may control its differentiation72. Notch and Wnt/β-catenin signaling are also involved in LPC differentiation29. Wnt3a secreted from macrophages induces the expression of Numb, which inhibits Notch signaling, in LPCs, thereby promoting the differentiation of LPCs into hepatocytes. Jag1 expressed in myofibroblasts increases Notch signaling in LPCs, thereby promoting the differentiation of LPCs into BECs29. In addition to these mouse studies, zebrafish studies revealed additional players that control LPC differentiation. Suppressing BMP signaling inhibited the differentiation of LPCs into hepatocytes, and this LPC differentiation defect was also observed in smad5 and tbx2b zebrafish mutants75,76. Suppressing Hdac1 activity derepressed sox9b expression, thereby inhibiting the differentiation of LPCs into hepatocytes75,76.
The negative roles of LPCs: fibrosis and liver cancer
Hepatic fibrosis, the formation of an abnormally large amount of scar tissue in the liver, occurs in most chronic liver diseases. As a response to liver injury, quiescent HSCs are activated to become profibrogenic myofibroblasts77. This HSC activation is associated with LPC activation78. A correlation between the number of LPCs and the severity of fibrosis in chronic liver diseases25,27,78,79,80 suggests that LPCs may promote hepatic fibrosis (Fig. 3). T helper type 1 (Th1) cells produce IFNγ, which regulates LPC proliferation81. BALB/c mice deficient in Th1 signaling fed a CDE diet exhibited reduced LPC proliferation and fibrosis compared to C57Bl/6 mice, which have normal Th1 signaling. Supplementation of IFNγ increased both LPC number and fibrosis in the CDE model36. In addition, TWEAK, which induces LPC activation, was associated with liver fibrosis82. Administration of TWEAK induced both LPC activation and collagen expression83, whereas Fn14 knockout mice fed a CDE diet exhibited reduced LPC proliferation and collagen expression34. These results together suggest that LPCs could drive liver fibrosis (Fig. 3).
Liver cancer is the sixth most commonly diagnosed cancer and the fourth leading cause of cancer death worldwide in 2018, accounting for approximately 841,000 new cases and 782,000 deaths annually6. Of primary liver cancer cases, 75–85% are hepatocellular carcinoma (HCC), and 10–15% are intrahepatic cholangiocarcinoma (ICC). LPC activation is known to be associated with pathological scarring processes and appears to contribute to liver tumor formation84. LPC markers, such as KRT7, KRT19, OV6, and EpCAM, are detected in HCCs85,86. More importantly, inhibition of LPC proliferation in chronically injured mouse livers reduced tumor development38,87,88. Additionally, functional genomics analysis of human HCCs revealed a Notch-associated signature in one-third of human HCCs89. Persistent Notch signaling activation in mouse livers induced features of human hepatocarcinogenesis, including dysplasia and HCC89. Given that Notch signaling regulates cancer stem cells during hepatocarcinogenesis90, these results together suggest a role for LPCs in HCC formation (Fig. 3).
Given its biliary features, ICC was thought to originate from BECs. Indeed, lineage-tracing studies confirmed the BEC origin91. However, ectopic and persistent activation of Notch and AKT signaling in hepatocytes generated ICC, revealing hepatocytes as an additional origin of ICC92,93. Moreover, neomorphic mutations of IDH1 or IDH2 that acquire abnormal activity to convert α-ketoglutarate to 2-hydroxyglutarate lead to ICC formation by inhibiting the differentiation of LPCs into hepatocytes94. An additional type of primary liver carcinoma that is of interest is combined or mixed hepatocellular cholangiocarcinoma (cHCC-CCA). It has been considered that cHCC-CCA originates from LPCs, which can differentiate into both hepatocytes and BECs95. cHCC-CCA also exhibits stem or progenitor features with downregulation of the hepatocyte differentiation program and a commitment to the biliary lineage96. These findings together suggest that LPCs contribute to ICC formation regardless of their origin.
Animal models for LPC research
Given the high cost and ethical issues of human studies, animal models have been used to study human liver diseases. Among them, rodents are widely used because of their remarkable genetic similarity to humans97. LPCs were first identified in rats98, and their activation was primarily investigated in rat models of chronic liver injury in which hepatocyte proliferation was compromised99. In this model, hepatocyte necrosis was induced by injecting d-galactosamine, and AFP+ LPCs were observed in the periportal area99. LPC activation and proliferation were also observed in the Solt–Farber liver injury model in which 2-acetylaminofluorene was administered to rats followed by two-third partial hepatectomy100,101.
Given the advantage of genetic manipulation in mice, mouse models have been widely used for LPC research97. Mice fed a DDC diet are used to study metabolic liver diseases, sclerosing cholangitis, and biliary fibrosis102. Mice fed a CDE diet exhibit steatosis, inflammation, LPC activation and expansion, portal fibrosis, and HCC103. CCl4 is also widely used as a hepatic toxin that induces chronic liver injury. Repeated injection of CCl4 causes centrilobular necrosis followed by a wound-healing process97. It also induces LPC activation and expansion, fibrosis, and cirrhosis69,97,104. Additionally, administration of thioacetamide induces chronic inflammation, LPC activation and expansion, fibrosis, cirrhosis, and liver cancer71,105,106. Since all these liver toxins induce LPC activation and expansion, these mouse models have been used to investigate the mechanisms of LPC activation and expansion.
As previously mentioned in the section on the beneficial role of LPCs, additional mouse models for BEC/LPC-driven liver regeneration in which BECs significantly contribute to regenerating hepatocytes have recently been established. In these mouse models, liver toxins, such as CDE and DDC, were used to induce liver injury, and hepatocyte proliferation was additionally suppressed52,68. Using the hepatocyte-specific p21-overexpressing model, the positive role of TET1 in LPC-driven liver regeneration was recently reported33. These mouse models will allow one to better understand the molecular mechanisms of LPC-driven liver regeneration, particularly LPC differentiation.
In addition to rodent models, zebrafish have relatively recently been used for LPC research due to their small size, which allows for simple chemical treatment. Various zebrafish liver injury models, including one-third partial hepatectomy107,108, ethanol treatment55,109, oncogene-induced liver cancers110,111,112, and hepatocyte ablation53,54,55, have been established. In particular, the hepatocyte-ablation model has been used to study LPC-driven liver regeneration. Our group developed the Tg(fabp10a:CFP-NTR) zebrafish line that expresses nitroreductase (NTR) under the hepatocyte-specific fabp10a promoter. Since NTR converts the nontoxic prodrug metronidazole (Mtz) into a cytotoxic drug, Mtz treatment specifically ablates hepatocytes in Tg(fabp10a:CFP-NTR) fish. Upon extensive hepatocyte loss, BECs dedifferentiate into LPCs, and LPCs subsequently differentiate into hepatocytes, thereby leading to full liver recovery53. Using this model, we recently reported that BMP signaling regulates LPC-driven liver regeneration through Tbx2a and Id2a75 and that bromodomain and extraterminal proteins (BET), Hdac1, Kdm1a, Sox9b, and Notch3 regulate LPC-driven liver regeneration76,113,114. Using the same zebrafish model, two other groups also reported that mTORC1 signaling regulates LPC-driven liver regeneration115,116.
In the Tg(fabp10a:CFP-NTR) model, LPC-driven liver regeneration occurs robustly and rapidly. Although this rapid regeneration has allowed us to identify small molecules that impair liver regeneration53, it is not suitable for identifying compounds that promote LPC-driven liver regeneration. Our group has recently established a new zebrafish liver injury model for LPC-driven liver regeneration in which LPCs slowly differentiate into hepatocytes58. In this model, Tg(fabp10a:pt-β-catenin) zebrafish larvae, which overexpress a stable form of β-catenin in hepatocytes, exhibited hepatocyte damage by oncogene-induced senescence and apoptosis, LPC activation, fibrosis, and differentiation of LPCs into hepatocytes, leading to the recovery of the liver. The activated LPCs persist for several days and gradually differentiate into hepatocytes. This slow progression of LPC differentiation allows the identification of small molecules that can promote the differentiation of LPCs into hepatocytes. Indeed, using this model, we discovered that treatment with EGFR inhibitors promoted LPC-driven liver regeneration, particularly the differentiation of LPCs into hepatocytes58. These zebrafish models will not only further help to better understand the molecular mechanisms of LPC-driven liver regeneration but also provide significant insights into promoting LPC-driven regeneration in patients with chronic liver diseases.
Conclusions
Although LPC-driven liver regeneration occurs to restore liver parenchyma in chronic liver diseases, it does not appear to occur effectively in patients with advanced liver disease117. A correlation between LPC numbers and disease severity in patients with chronic liver diseases27 implies not only that LPCs are activated in the patients but also that the LPCs ineffectively differentiate into hepatocytes. Persistent LPCs induce inflammation and subsequent fibrosis by secreting proinflammatory cytokines118. Since promoting the differentiation of LPCs into hepatocytes can generate more functional hepatocytes and concomitantly reduce fibrosis, a strategy to promote the differentiation is an attractive therapeutic option for patients with advanced liver disease. To establish such a strategy, it is crucial to identify appropriate target molecules of which manipulation promotes the differentiation of LPCs into hepatocytes. Research using the animal models of chronic and severe liver injury will help to discover such molecules and eventually make the strategy feasible.
References
Stanger, B. Z. Cellular homeostasis and repair in the mammalian liver. Annu. Rev. Physiol. 77, 179–200 (2015).
Duncan, A. W., Dorrell, C. & Grompe, M. Stem cells and liver regeneration. Gastroenterology 137, 466–481 (2009).
Miyajima, A., Tanaka, M. & Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 14, 561–574 (2014).
Michalopoulos, G. K. & DeFrances, M. C. Liver regeneration. Science 276, 60–66 (1997).
Tarlow, B. D. et al. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 15, 605–618 (2014).
Asrani, S. K., Devarbhavi, H., Eaton, J. & Kamath, P. S. Burden of liver diseases in the world. J. Hepatol. 70, 151–171 (2019).
Kim, P. T. & Testa, G. Living donor liver transplantation in the USA. Hepatobiliary Surg. Nutr. 5, 133–140 (2016).
Shin, D. & Monga, S. P. Cellular and molecular basis of liver development. Compr. Physiol. 3, 799–815 (2013).
Sell, S. Comparison of liver progenitor cells in human atypical ductular reactions with those seen in experimental models of liver injury. Hepatology 27, 317–331 (1998).
Tatematsu, M., Ho, R. H., Kaku, T., Ekem, J. K. & Farber, E. Studies on the proliferation and fate of oval cells in the liver of rats treated with 2-acetylaminofluorene and partial hepatectomy. Am. J. Pathol. 114, 418–430 (1984).
Xiao, J. C., Ruck, P., Adam, A., Wang, T. X. & Kaiserling, E. Small epithelial cells in human liver cirrhosis exhibit features of hepatic stem-like cells: immunohistochemical, electron microscopic and immunoelectron microscopic findings. Histopathology 42, 141–149 (2003).
Dan, Y. Y. et al. Isolation of multipotent progenitor cells from human fetal liver capable of differentiating into liver and mesenchymal lineages. Proc. Natl Acad. Sci. USA 103, 9912–9917 (2006).
Li, J. et al. Human hepatic progenitor cells express hematopoietic cell markers CD45 and CD109. Int. J. Med. Sci. 11, 65–79 (2014).
Okabe, M. et al. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development 136, 1951–1960 (2009).
Lee, J. S. et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 12, 410–416 (2006).
Furuyama, K. et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet. 43, 34–41 (2011).
Kuhlmann, W. D. & Peschke, P. Hepatic progenitor cells, stem cells, and AFP expression in models of liver injury. Int. J. Exp. Pathol. 87, 343–359 (2006).
Crosby, H. A., Kelly, D. A. & Strain, A. J. Human hepatic stem-like cells isolated using c-kit or CD34 can differentiate into biliary epithelium. Gastroenterology 120, 534–544 (2001).
Cardinale, V. et al. Multipotent stem/progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes, and pancreatic islets. Hepatology 54, 2159–2172 (2011).
Petersen, B. E. et al. Mouse A6-positive hepatic oval cells also express several hematopoietic stem cell markers. Hepatology 37, 632–640 (2003).
Dorrell, C. et al. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes Dev. 25, 1193–1203 (2011).
Fabris, L. et al. Characterization and isolation of ductular cells coexpressing neural cell adhesion molecule and Bcl-2 from primary cholangiopathies and ductal plate malformations. Am. J. Pathol. 156, 1599–1612 (2000).
Pepe-Mooney, B. J. et al. Single-cell analysis of the liver epithelium reveals dynamic heterogeneity and an essential role for YAP in homeostasis and regeneration. Cell Stem Cell 25, 23–38 e28 (2019).
Planas-Paz, L. et al. YAP, but not RSPO-LGR4/5, signaling in biliary epithelial cells promotes a ductular reaction in response to liver injury. Cell Stem Cell. https://doi.org/10.1016/j.stem.2019.04.005 (2019).
Roskams, T. et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am. J. Pathol. 163, 1301–1311 (2003).
Lowes, K. N., Croager, E. J., Abraham, L. J., Olynyk, J. K. & Yeoh, G. C. Upregulation of lymphotoxin beta expression in liver progenitor (oval) cells in chronic hepatitis C. Gut 52, 1327–1332 (2003).
Lowes, K. N., Brennan, B. A., Yeoh, G. C. & Olynyk, J. K. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am. J. Pathol. 154, 537–541 (1999).
Roskams, T. et al. Hepatic OV-6 expression in human liver disease and rat experiments: evidence for hepatic progenitor cells in man. J. Hepatol. 29, 455–463 (1998).
Boulter, L. et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med. 18, 572–579 (2012).
Jakubowski, A. et al. TWEAK induces liver progenitor cell proliferation. J. Clin. Invest. 115, 2330–2340 (2005).
Jung, Y. et al. Signals from dying hepatocytes trigger growth of liver progenitors. Gut 59, 655–665 (2010).
Li, D. et al. Hepatic loss of survivin impairs postnatal liver development and promotes expansion of hepatic progenitor cells in mice. Hepatology 58, 2109–2121 (2013).
Aloia, L. et al. Epigenetic remodelling licences adult cholangiocytes for organoid formation and liver regeneration. Nat. Cell Biol. 21, 1321–1333 (2019).
Tirnitz-Parker, J. E. et al. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology 52, 291–302 (2010).
Knight, B. et al. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J. Exp. Med. 192, 1809–1818 (2000).
Knight, B., Lim, R., Yeoh, G. C. & Olynyk, J. K. Interferon-gamma exacerbates liver damage, the hepatic progenitor cell response and fibrosis in a mouse model of chronic liver injury. J. Hepatol. 47, 826–833 (2007).
Yeoh, G. C. et al. Opposing roles of gp130-mediated STAT-3 and ERK-1/ 2 signaling in liver progenitor cell migration and proliferation. Hepatology 45, 486–494 (2007).
Davies, R. A., Knight, B., Tian, Y. W., Yeoh, G. C. & Olynyk, J. K. Hepatic oval cell response to the choline-deficient, ethionine supplemented model of murine liver injury is attenuated by the administration of a cyclo-oxygenase 2 inhibitor. Carcinogenesis 27, 1607–1616 (2006).
Ruddell, R. G. et al. Lymphotoxin-beta receptor signaling regulates hepatic stellate cell function and wound healing in a murine model of chronic liver injury. Hepatology 49, 227–239 (2009).
Akhurst, B. et al. Differential lymphotoxin-beta and interferon gamma signaling during mouse liver regeneration induced by chronic and acute injury. Hepatology 41, 327–335 (2005).
Hsieh, W. C. et al. Galectin-3 regulates hepatic progenitor cell expansion during liver injury. Gut 64, 312–321 (2015).
Chen, L. et al. HSCs play a distinct role in different phases of oval cell-mediated liver regeneration. Cell Biochem. Funct. 30, 588–596 (2012).
Ishikawa, T. et al. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology 55, 1215–1226 (2012).
Thenappan, A. et al. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology 51, 1373–1382 (2010).
Dai, Z. et al. Growth differentiation factor 11 attenuates liver fibrosis via expansion of liver progenitor cells. Gut. https://doi.org/10.1136/gutjnl-2019-318812 (2019).
Takase, H. M. et al. FGF7 is a functional niche signal required for stimulation of adult liver progenitor cells that support liver regeneration. Genes Dev. 27, 169–181 (2013).
Franchitto, A. et al. Expression of vascular endothelial growth factors and their receptors by hepatic progenitor cells in human liver diseases. Hepatobiliary Surg. Nutr. 2, 68–77 (2013).
Fausto, N. & Campbell, J. S. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech. Dev. 120, 117–130 (2003).
Tarlow, B. D., Finegold, M. J. & Grompe, M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology 60, 278–289 (2014).
Schaub, J. R., Malato, Y., Gormond, C. & Willenbring, H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 8, 933–939 (2014).
Yanger, K. et al. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell 15, 340–349 (2014).
Raven, A. et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 547, 350–354 (2017).
Choi, T. Y., Ninov, N., Stainier, D. Y. & Shin, D. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology 146, 776–788 (2014).
He, J., Lu, H., Zou, Q. & Luo, L. Regeneration of liver after extreme hepatocyte loss occurs mainly via biliary transdifferentiation in zebrafish. Gastroenterology 146, 789–800.e788 (2014).
Huang, M. et al. Antagonistic interaction between Wnt and Notch activity modulates the regenerative capacity of a zebrafish fibrotic liver model. Hepatology 60, 1753–1766 (2014).
Yanger, K. et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 27, 719–724 (2013).
Yimlamai, D. et al. Hippo pathway activity influences liver cell fate. Cell 157, 1324–1338 (2014).
So, J. et al. Attenuating the Epidermal Growth Factor Receptor–Extracellular Signal-Regulated Kinase–Sex-Determining Region Y-Box 9 Axis Promotes Liver Progenitor Cell-Mediated Liver Regeneration in Zebrafish. Hepatology. https://doi.org/10.1002/hep.31437 (2020). Online ahead of print.
Yang, L. et al. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells 26, 2104–2113 (2008).
Michelotti, G. A. et al. Smoothened is a master regulator of adult liver repair. J. Clin. Invest. 123, 2380–2394 (2013).
Swiderska-Syn, M. et al. Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy. Gut 63, 1333–1344 (2014).
Kordes, C., Sawitza, I., Gotze, S., Herebian, D. & Haussinger, D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J. Clin. Invest. 124, 5503–5515 (2014).
Mederacke, I. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 4, 2823 (2013).
Fellous, T. G. et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology 49, 1655–1663 (2009).
Stueck, A. E. & Wanless, I. R. Hepatocyte buds derived from progenitor cells repopulate regions of parenchymal extinction in human cirrhosis. Hepatology 61, 1696–1707 (2015).
Yoon, S. M. et al. Epithelial cell adhesion molecule (EpCAM) marks hepatocytes newly derived from stem/progenitor cells in humans. Hepatology 53, 964–973 (2011).
Zhou, H., Rogler, L. E., Teperman, L., Morgan, G. & Rogler, C. E. Identification of hepatocytic and bile ductular cell lineages and candidate stem cells in bipolar ductular reactions in cirrhotic human liver. Hepatology 45, 716–724 (2007).
Russell, J. O. et al. Hepatocyte-specific beta-catenin deletion during severe liver injury provokes cholangiocytes to differentiate into hepatocytes. Hepatology 69, 742–759 (2019).
Manco, R. et al. Reactive cholangiocytes differentiate into proliferative hepatocytes with efficient DNA repair in mice with chronic liver injury. J. Hepatol. 70, 1180–1191 (2019).
Lu, W.-Y. et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 17, 971–983 (2015).
Deng, X. et al. Chronic liver injury induces conversion of biliary epithelial cells into hepatocytes. Cell Stem Cell 23, 114–122. e113 (2018).
Espanol-Suner, R. et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 143, 1564–1575.e1567 (2012).
Rodrigo-Torres, D. et al. The biliary epithelium gives rise to liver progenitor cells. Hepatology 60, 1367–1377 (2014).
Jors, S. et al. Lineage fate of ductular reactions in liver injury and carcinogenesis. J. Clin. Invest. 125, 2445–2457 (2015).
Choi, T. Y. et al. Bone morphogenetic protein signaling governs biliary-driven liver regeneration in zebrafish through tbx2b and id2a. Hepatology 66, 1616–1630 (2017).
Ko, S. et al. Hdac1 regulates differentiation of bipotent liver progenitor cells during regeneration via Sox9b and Cdk8. Gastroenterology 156, 187–202.e114 (2019).
Bataller, R. & Brenner, D. A. Liver fibrosis. J. Clin. Invest. 115, 209–218 (2005).
Williams, M. J., Clouston, A. D. & Forbes, S. J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 146, 349–356 (2014).
Clouston, A. D. et al. Fibrosis correlates with a ductular reaction in hepatitis C: roles of impaired replication, progenitor cells and steatosis. Hepatology 41, 809–818 (2005).
Richardson, M. M. et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 133, 80–90 (2007).
Knight, B. et al. Attenuated liver progenitor (oval) cell and fibrogenic responses to the choline deficient, ethionine supplemented diet in the BALB/c inbred strain of mice. J. Hepatol. 46, 134–141 (2007).
Tirnitz-Parker, J. E., Olynyk, J. K. & Ramm, G. A. Role of TWEAK in coregulating liver progenitor cell and fibrogenic responses. Hepatology 59, 1198–1201 (2014).
Kuramitsu, K. et al. Failure of fibrotic liver regeneration in mice is linked to a severe fibrogenic response driven by hepatic progenitor cell activation. Am. J. Pathol. 183, 182–194 (2013).
Libbrecht, L. Hepatic progenitor cells in human liver tumor development. World J. Gastroenterol. 12, 6261–6265 (2006).
Hixson, D. C., Brown, J., McBride, A. C. & Affigne, S. Differentiation status of rat ductal cells and ethionine-induced hepatic carcinomas defined with surface-reactive monoclonal antibodies. Exp. Mol. Pathol. 68, 152–169 (2000).
Libbrecht, L., Desmet, V., Van Damme, B. & Roskams, T. The immunohistochemical phenotype of dysplastic foci in human liver: correlation with putative progenitor cells. J. Hepatol. 33, 76–84 (2000).
Knight, B., Tirnitz-Parker, J. E. & Olynyk, J. K. C-kit inhibition by imatinib mesylate attenuates progenitor cell expansion and inhibits liver tumor formation in mice. Gastroenterology 135, 969–979, 979.e961 (2008).
Lee, K. P. et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl Acad. Sci. USA 107, 8248–8253 (2010).
Villanueva, A. et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 143, 1660–1669.e1667 (2012).
Huang, Q., Li, J., Zheng, J. & Wei, A. The carcinogenic role of the notch signaling pathway in the development of hepatocellular carcinoma. J. Cancer 10, 1570–1579 (2019).
Guest, R. V. et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 74, 1005–1010 (2014).
Sekiya, S. & Suzuki, A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Invest. 122, 3914–3918 (2012).
Fan, B. et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Invest. 122, 2911–2915 (2012).
Saha, S. K. et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114 (2014).
Brunt, E. et al. cHCC-CCA: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 68, 113–126 (2018).
Coulouarn, C. et al. Combined hepatocellular-cholangiocarcinomas exhibit progenitor features and activation of Wnt and TGFbeta signaling pathways. Carcinogenesis 33, 1791–1796 (2012).
Liu, Y. et al. Animal models of chronic liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G449–G468 (2013).
Farber, E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3’-methyl-4-dimethylaminoazobenzene. Cancer Res. 16, 142–148 (1956).
Dabeva, M. D. & Shafritz, D. A. Activation, proliferation, and differentiation of progenitor cells into hepatocytes in the D-galactosamine model of liver regeneration. Am. J. Pathol. 143, 1606–1620 (1993).
Evarts, R. P., Nagy, P., Nakatsukasa, H., Marsden, E. & Thorgeirsson, S. S. In vivo differentiation of rat liver oval cells into hepatocytes. Cancer Res. 49, 1541–1547 (1989).
Shiojiri, N., Lemire, J. M. & Fausto, N. Cell lineages and oval cell progenitors in rat liver development. Cancer Res. 51, 2611–2620 (1991).
Preisegger, K. H. et al. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Invest. 79, 103–109 (1999).
Passman, A. M. et al. A modified choline-deficient, ethionine-supplemented diet reduces morbidity and retains a liver progenitor cell response in mice. Dis. Models Mech. 8, 1635–1641 (2015).
Constandinou, C., Henderson, N. & Iredale, J. P. Modeling liver fibrosis in rodents. Methods Mol. Med. 117, 237–250 (2005).
Boulter, L. et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Invest. 125, 1269–1285 (2015).
Sakurai, T. et al. p38alpha inhibits liver fibrogenesis and consequent hepatocarcinogenesis by curtailing accumulation of reactive oxygen species. Cancer Res. 73, 215–224 (2013).
Goessling, W. et al. APC mutant zebrafish uncover a changing temporal requirement for wnt signaling in liver development. Dev. Biol. 320, 161–174 (2008).
Sadler, K. C., Krahn, K. N., Gaur, N. A. & Ukomadu, C. Liver growth in the embryo and during liver regeneration in zebrafish requires the cell cycle regulator, uhrf1. Proc. Natl Acad. Sci. USA 104, 1570–1575 (2007).
Passeri, M. J., Cinaroglu, A., Gao, C. & Sadler, K. C. Hepatic steatosis in response to acute alcohol exposure in zebrafish requires sterol regulatory element binding protein activation. Hepatology 49, 443–452 (2009).
Mudbhary, R. et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 25, 196–209 (2014).
Wang, J., Leng, X., Wang, G., Wan, X. & Cao, H. The construction of intrahepatic cholangiocarcinoma model in zebrafish. Sci. Rep. 7, 13419 (2017).
Evason, K. J. et al. Identification of chemical inhibitors of beta-catenin-driven liver tumorigenesis in zebrafish. PLoS Genet. 11, e1005305 (2015).
Ko, S. et al. Bromodomain and extraterminal (BET) proteins regulate biliary-driven liver regeneration. J. Hepatol. 64, 316–325 (2016).
Ko, S., Russell, J. O., Molina, L. M. & Monga, S. P. Liver progenitors and adult cell plasticity in hepatic injury and repair: knowns and unknowns. Annu. Rev. Pathol. 15, 23–50 (2019).
Chaturantabut, S. et al. Estrogen activation of G-protein-coupled estrogen receptor 1 regulates phosphoinositide 3-kinase and mTOR signaling to promote liver growth in zebrafish and proliferation of human hepatocytes. Gastroenterology 156, 1788–1804.e1713 (2019).
He, J. et al. Mammalian target of Rapamycin complex 1 signaling is required for the dedifferentiation from biliary cell to bipotential progenitor cell in zebrafish liver regeneration. Hepatology 70, 2092–2106 (2019).
Weng, H. L. et al. Two sides of one coin: massive hepatic necrosis and progenitor cell-mediated regeneration in acute liver failure. Front. Physiol. 6, 178 (2015).
Lukacs-Kornek, V. & Lammert, F. The progenitor cell dilemma: cellular and functional heterogeneity in assistance or escalation of liver injury. J. Hepatol. 66, 619–630 (2017).
Acknowledgements
This work was supported by NIH grants to D.S. (DK101426, DK116993) and by the NIH/NIDDK Digestive Disease Research Core Center grant P30DK120531.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
So, J., Kim, A., Lee, SH. et al. Liver progenitor cell-driven liver regeneration. Exp Mol Med 52, 1230–1238 (2020). https://doi.org/10.1038/s12276-020-0483-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-0483-0
- Springer Nature Limited
This article is cited by
-
A two-step strategy to expand primary human hepatocytes in vitro with efficient metabolic and regenerative capacities
Stem Cell Research & Therapy (2024)
-
The endocannabinoid system promotes hepatocyte progenitor cell proliferation and maturation by modulating cellular energetics
Cell Death Discovery (2023)
-
In Vitro Generated Equine Hepatic-Like Progenitor Cells as a Novel Potent Cell Pool for Equine Metabolic Syndrome (EMS) Treatment
Stem Cell Reviews and Reports (2023)
-
Transplantation of 3D adipose-derived stem cell/hepatocyte spheroids alleviates chronic hepatic damage in a rat model of thioacetamide-induced liver cirrhosis
Scientific Reports (2022)
-
Study protocol of Phase 2 open-label multicenter randomized controlled trial for granulocyte-colony stimulating factor (GCSF) in post-Kasai Type 3 biliary atresia
Pediatric Surgery International (2022)