
Abstract
The AHNAK nucleoprotein has been determined to exert an anti-obesity effect in adipose tissue and further inhibit adipogenic differentiation. In this study, we examined the role of AHNAK in regulating hepatic lipid metabolism to prevent diet-induced fatty liver. Ahnak KO mice have reportedly exhibited reduced fat accumulation in the liver and decreased serum triglyceride (TG) levels when provided with either a normal chow diet or a high-fat diet (HFD). Gene expression profiling was used to identify novel factors that could be modulated by genetic manipulation of the Ahnak gene. The results revealed that fibroblast growth factor 21 (FGF21) was markedly increased in the livers of Ahnak KO mice compared with WT mice fed a HFD. Ahnak knockdown in hepatocytes reportedly prevented excessive lipid accumulation induced by palmitate treatment and was associated with increased secretion of FGF21 and the expression of genes involved in fatty acid oxidation, which are primarily downstream of PPARα. These results indicate that pronounced obesity and hepatic steatosis are attenuated in HFD-fed Ahnak KO mice. This may be attributed, in part, to the induction of FGF21 and regulation of lipid metabolism, which are considered to be involved in increased fatty acid oxidation and reduced lipogenesis in the liver. These findings suggest that targeting AHNAK may have beneficial implications in preventing or treating hepatic steatosis.
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) has been identified as a common disorder that is characterized by increased hepatic triglyceride (TG) accumulation. NAFLD represents a broad spectrum of liver diseases ranging from nonalcoholic simple fatty liver to nonalcoholic steatohepatitis (NASH), hepatic fibrosis, and cirrhosis1. Hepatic lipid accumulation is known to be regulated by lipid uptake, de novo synthesis, oxidation, and transport of fatty acids to the circulation2. An imbalance in these complex metabolic processes can cause an excessive amount of hepatic TG accumulation.
Fibroblast growth factor 21 (FGF21) is a member of the fibroblast growth factor family consisting of 22 members that have diverse functions in the regulation of physiological homeostasis in metabolic tissues3. FGF21 is primarily synthesized and secreted from the liver4, and the action of circulating FGF21 is mediated through FGF receptors complexed with β-Klotho5. The expression of Fgf21 is then induced by the activation of peroxisome proliferator-activated receptor alpha (PPARα). Nonesterified fatty acids bind to and activate PPARα. Ligand-bound PPARα then forms a heterodimer with retinoid X receptors to induce the expression of Fgf21. In cultured adipocytes, FGF21 signaling is transduced by activating the β-Klotho-FGFR1c complex. However, FGF21 signaling can be transduced in β-Klotho knockout mice, indicating the existence of a β-Klotho-independent FGF21 signaling pathway6. FGF21 has been known to act on adipose tissue to reduce plasma glucose and TGs, which in turn decreases body weight7. FGF21 is a potent regulator of adiponectin secretion in white adipose tissue and exerts body weight-lowering effects in brown adipose tissues and beige cells mediated by the induction of thermogenesis8,9. FGF21 also exerts direct effects on pancreatic islet cells to increase beta-cell function, survival10, and growth plate chondrocytes11. Treatment with FGF21 can reduce hepatic TG accumulation and hepatic steatosis, thus attenuating body weight gain in rodents and primates12,13. Additionally, FGF21 regulates energy homeostasis in adipocytes through an AMP-activated protein kinase (AMPK)/sirtuin 1 (SIRT1)/peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α) signaling cascade, resulting in enhanced mitochondrial oxidative capacity14,15,16. Thus, FGF21 may represent a novel therapeutic molecule based on the findings that it protects animals from diet-induced obesity and reduces hepatic lipid accumulation, enhances glucose metabolism, and thus prevents hepatic steatosis, fibrosis, and NAFLD when administered to diabetic rodents17,18.
AHNAK is a very large protein that acts not only on calcium signaling or ion channels but also on nucleoproteins that are known to regulate a wide variety of biological functions19, such as adipogenesis20, browning21, tumor development22, and adipocyte differentiation23. In a previous study, we found that Ahnak KO mice exhibit strong resistance to high-fat diet (HFD)-induced obesity. Changes in the pattern of urinary metabolites in HFD-fed Ahnak KO mice suggest that the strong resistance to HFD-induced obesity in Ahnak KO mice is related to perturbations in amino acids that are related to fat metabolism24. Although Ahnak KO mice showed strong resistance to diet-induced obesity and hepatic steatosis, the role of AHNAK in the regulation of hepatic steatosis has not been studied.
In the present study, the role of AHNAK in diet-induced fatty liver disease was determined by using Ahnak KO mice. Ahnak KO mice have been shown to not have excessive hepatic lipid accumulation when fed a HFD. Gene expression analysis showed that a HFD upregulated Fgf21 in the liver of Ahnak KO mice but not in WT littermates. Increased hepatic and circulating levels of FGF21 in HFD-fed Ahnak KO mice were associated with increased hepatic expression of genes involved in fatty acid oxidation and decreased expression of genes involved in lipogenesis compared with those in the WT littermates. Primary hepatocytes from Ahnak KO mice consistently exhibited decreased lipid accumulation and were accompanied by increased FGF21 secretion and cellular FGF21 protein levels. In addition, to enhance FGF21 production and secretion, Ahnak-deficient hepatocytes also exhibited increased expression of PPARα protein and its target genes in response to palmitate treatment. Characterization of phenotypic changes in Ahnak KO mice, such as reduced hepatic steatosis, decreased adiposity, and increased energy expenditure, establishes AHNAK as a novel regulator of FGF21 in modulating hepatic fatty acid metabolism. Thus, the present study establishes the role of AHNAK in the regulation of hepatocyte lipid metabolism through PPARα/FGF21 signaling.
Materials and Methods
Experimental animals
Ahnak KO mice were generated by disrupting exon 5 of the Ahnak gene as described16. Ahnak KO mice were obtained by crossing heterozygous breeders. Eight-week-old male KO and wild-type mice were randomly assigned and fed either regular chow or a 60% HFD (D12492; Research Diets Inc., NJ, USA) for 7 weeks. All animals were maintained at 24 ± 2 °C with 12 h of light per day and had free access to water in a specific pathogen-free barrier facility. These procedures were reviewed according to the “Guide for Animal Experiments” (edited by the Korean Academy of Medical Sciences) by the Institutional Animal Care and Use Committee at Seoul National University. The animal protocol was approved by the committee on the Ethics of Animal Experiments at Seoul National University (Permit Number: SNU-131024-6). All of the experiments were conducted to minimize the number of animals used25.
Measurement of metabolic parameters in mice
A comprehensive animal metabolic monitoring system (CLAMS; Columbus Instruments, Columbus, OH) was used to evaluate the activity, food consumption, and energy expenditure of 8-week-old male KO and wild-type mice by switching the feeding method, such as feeding an NCD for 2 days, followed by a HFD for 4 days. Energy expenditure and food intake data were normalized with respect to lean body weight. The energy expenditure and respiratory exchange ratio (RER) were then calculated from gas exchange data [energy expenditure = (3.815 + 1.232 × RER) × VO2]. The RER was determined by the ratio of VCO2 to VO2, which changes depending on the energy source of the animal. When carbohydrates are the only substrate being oxidized, the RQ will be 1.0, whereas it will be 0.7 when only the fatty acids are oxidized. Activity was measured on the x- and z-axes using infrared beams to count the number of beam breaks during a specified measurement period. Feeding was measured by recording the difference in the scale measurement of the center feeder from one time point to another26.
Histochemical study
For histological examination, mouse liver tissues were dissected and fixed in 10% neutral buffered formalin. The samples were subsequently embedded in paraffin or OCT compound. For hematoxylin and eosin (H&E) staining, tissue sections were cut at a thickness of 4 µm and were then stained using a commercial kit (HHS123, Sigma-Aldrich, St. Louis, MO). For Oil Red O staining, frozen samples were sectioned at 5 µm in thickness using a cryostat27. These sections were placed onto slides, washed with 60% isopropanol, and then stained with Oil Red O for 30 min.
Measurement of VLDL secretion and lipid clearance rates
To assess VLDL secretion, serum triacylglycerol (TG) was measured in blood obtained from 4-hour-fasted mice for the indicated times after i.p. injection of Poloxamer 407 (1 mg/g of body weight) solution in PBS28. For measurement of lipid clearance rates, mice were fasted for 12 h and administered olive oil (Sigma-Aldrich, St. Louis, MO) by oral gavage. Blood was collected from the tail vein prior to gavage and 1, 2, 3, and 6 h after treatment. TGs were measured enzymatically with TG reagent (BioVision, Korea).
Primary hepatocyte and HepG2 cell culture
Primary hepatocytes from WT and Ahnak KO mice were isolated using a two-step perfusion technique and cultured as described previously29. After cell attachment, hepatocytes were serum-starved for 4 h and then treated with 250 μM palmitate for 12 h. In some experiments, hepatocytes were treated with AICAR for the indicated time periods. HepG2 cells were purchased from the American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-Life Technologies, Grand Island, NY) containing high glucose, 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin, as described previously30. Control siRNA and siRNA for the Ahnak gene (Bioneer, Korea) were used for gene silencing. A cell steatosis model was established by culturing HepG2 cells in DMEM with BSA-conjugated palmitate (250 μM) for 12 or 24 h.
Quantitative real-time PCR
Total RNA was extracted from the liver using the Total RNA Purification System (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Messenger RNA was reverse-transcribed using AccuPower CycleScript RT PreMix (Bioneer, Daejeon, Korea). Quantitative real-time PCR was performed with SYBR Green dye using the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Cheshire, U.K.). Expression of the respective genes was normalized to the 36B4 signal as an internal control, and relative gene expression was quantitated by the comparative Ct method (ΔΔCt). The primer sequences of target genes are listed in Supplementary Table 1.
Western blot analysis
Total protein was extracted from the liver of each group using protein lysis buffer (Invitrogen, Carlsbad, CA) containing phosphatase inhibitor (GenDEPOT, Barker, TX, USA). Protein extracts were resolved by 10% or 12% SDS-PAGE gels and were later transferred onto PVDF membranes (Millipore, Billerica, MA). The protein bands were detected with antibodies against pAMPK (Thr172), AMPK, pACC, ACC, PPARα, SCD1, CPT1 (Abcam, Cambridge, UK), and GAPDH (Cell Signaling Technologies, Beverly, MA) using the appropriate secondary HRP-conjugated antibodies (Cell Signaling Technologies, Beverly, MA). The blots were developed and imaged using the MicroChemi 4.2 system (DNR Bio-imaging system, Israel).
FGF21 measurement
FGF21 concentrations in plasma and culture medium were measured by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol. To measure FGF21 secretion, the hepatocytes were serum-starved for 4 h, and then the supernatants were collected and preserved in a −80 °C freezer until further analysis.
Statistical analysis
The results are expressed as the means ± SEM. Student’s t tests were used to analyze gene expression differences between WT and Ahnak KO mice as measured by quantitative real-time PCR. Differences were considered significant at p < 0.05.
Results
Ahnak KO mice are resistant to HFD-induced hepatic steatosis
To determine the potential correlation between AHNAK expression and hepatic steatosis, we examined AHNAK expression in the livers of diet-induced and genetically induced obese mice. Ahnak mRNA was found to be significantly increased in the livers of both obese mouse models (Supplementary Fig. 1). To test the function of AHNAK in diet-induced hepatic steatosis, Ahnak KO mice and their wild-type littermates were fed a HFD containing 60% fat or a NCD. When 8-week-old male mice were fed a HFD for 7 weeks, Ahnak KO mice weighed less than WT mice in both the NCD and HFD groups. In addition, HFD-KO mice gained significantly less weight than WT mice (Fig. 1a, b).

a Body weight changes in mice receiving a 60% high-fat diet for 7 weeks. b Macroscopic appearance of the liver and mice. The liver-to-body weight ratio was measured. c H&E staining of WT and KO mice. Oil Red O staining of lipid droplets. Original magnification, x200. d Fasting blood glucose levels, serum cholesterol, and serum and hepatic TG contents were measured. Data were presented as the means ± SEM. *p < 0.05; **p < 0.01.
In a previous study, we reported that the differences in body weight caused by fewer fat pads are due to impaired adipogenesis in KO mice20. Nevertheless, since the liver is a secondary fat storage site and an essential metabolic organ, subsequent studies have been conducted. The ratio of liver-to-body weight was observed to be significantly decreased in KO mice compared with WT mice fed a HFD (Fig. 1b). Histological analysis revealed that WT mice receiving a HFD exhibited severe hepatic steatosis with an accumulation of intracellular lipid droplets, whereas Ahnak KO mice did not display fatty liver symptoms (Fig. 1c). Consistent with the histological results, both hepatic and serum levels of TG decreased in the livers of HFD-fed Ahnak KO mice compared with WT mice. The basal level of blood glucose was also determined to decrease in the HFD-fed Ahnak KO mice compared with HFD-fed WT mice (Fig. 1d). The HFD-fed WT mice had elevated blood glucose levels, whereas HFD-fed Ahnak KO mice did not exhibit hyperglycemia. Total cholesterol levels were also reduced in HFD-fed Ahnak KO mice compared with WT mice (Fig. 1d). Overall, these observations indicate that Ahnak KO mice were significantly protected from HFD-induced obesity and hepatic steatosis.
Ahnak-deficient mice exhibit decreased serum lipid clearance but not VLDL secretion
To test whether lipid transport in the liver was altered in Ahnak KO mice, olive oil was administered to WT and KO mice, and the lipid clearance rate was evaluated. Ahnak KO mice were found to exhibit higher levels of serum TG than WT mice after olive oil treatment in the HFD group (Fig. 2a). However, there were no significant differences in serum TG levels between the WT and KO mice in the NCD group (Supplementary Fig. 2). To determine which molecular mechanism affects lipid transport in Ahnak KO mice, the mRNA expression of genes involved in lipid uptake was measured. The expression of Cd36, a membrane protein that facilitates the uptake of fatty acids and cholesterol, was markedly reduced in the liver of Ahnak KO mice fed either a NCD or a HFD, whereas the expression of fatty acid binding proteins involved in fatty acid uptake, such as Fatp1, Fatp2, and Fabp4, was similar or slightly altered between WT and KO mice (Fig. 2b). The current data suggest that Ahnak KO mice have reduced expression of genes related to fatty acid transport, which could explain the observed impaired lipid transport to the liver and reduced hepatic lipid accumulation (Fig. 1c). Additionally, to examine the effect of Ahnak deficiency on VLDL secretion, the animals were treated with Poloxamer 407, which inhibits lipoprotein lipase activity and VLDL secretion. The VLDL secretion rate was estimated by measuring the serum TG levels at each time point following Poloxamer 407 treatment. The VLDL secretion rate was similar between WT and KO mice fed either a NCD (Supplementary Fig. 2) or a HFD (Fig. 2c). Consistent with the VLDL secretion results, the expression levels of ApoB and Mtp, which promote VLDL assembly and secretion, were also similar between WT and KO mice (Fig. 2b).

a Lipid clearance was estimated by measuring blood TG levels after oral olive oil administration. b Analysis of gene expression involved in VLDL secretion and lipid uptake after a 7-week HFD feeding by quantitative real-time PCR normalized to 36B4 (n = 4–6 mice). c VLDL secretion. Mice were treated with Poloxamer 407 to block VLDL clearance, and VLDL secretion was then estimated by measuring blood TG levels after Poloxamer 407 treatment. Data were presented as the means ± SEM. *p < 0.05; **p < 0.01.
Ahnak deficiency in mice increases the production and secretion of hepatic FGF21
As part of a screening to identify novel secreted proteins that could be modulated by AHNAK genetic manipulation, gene expression profiling revealed that Fgf21 was increased by ~3.2-fold in the livers of Ahnak KO mice compared with those of WT mice under HFD conditions (accession No. GSE70119). Hierarchical clustering revealed that gene expression in the livers of WT mice that were fed a NCD was closely aligned with that of Ahnak KO mice. The livers of HFD-fed Ahnak KO mice had a similar cluster of genes compared with that of the NCD-fed group, whereas the livers of HFD-fed WT mice showed a distinctive differential expression gene (DEG) profile (Supplementary Fig. 3a). A gene ontology analysis of the DEGs revealed that Ahnak deficiency induced several metabolic pathways (Supplementary Fig. 3b). Downregulated genes in the livers of HFD-fed Ahnak KO mice were associated with the immune response. Genes involved in proinflammatory signaling were significantly downregulated in the livers of Ahnak KO mice fed a HFD, including chemokine (C-C motif) ligand 2 (Ccl2), chemokine (C-C motif) ligand 3 (Ccl3), tumor necrosis factor α (Tnfα), and F4/80. However, the expression levels of anti-inflammatory genes were not changed (Supplementary Fig. 3c). As expected, hepatic mRNA expression and protein levels of FGF21 also increased in Ahnak KO mice compared with WT mice fed a HFD (Fig. 3a). However, no difference was determined in Fgf21 expression when the animals were fed a NCD. FGF21 activity is mediated through FGF receptors that interact with β-Klotho. A genetic deficiency of Ahnak was determined to have no effect on the hepatic expression of FGF receptors or β-Klotho when fed either a NCD or a HFD. Interestingly, FGFR1 expression was dramatically increased in the livers of both WT and Ahnak KO mice fed a HFD (Fig. 3a). These results indicate that HFD-induced circulating FGF21 acts primarily through binding to FGFR1. FGF21 is a significant metabolic regulator in several diabetic animal models. FGF21 can protect animals from diet-induced obesity and reduce hepatic lipid accumulation, enhance glucose metabolism, and thus prevent hepatic steatosis, fibrosis, and NAFLD when administered to diabetic rodents8,9,10,12,15,16. To elucidate the role of AHNAK in preventing diet-induced hepatic steatosis, we determined whether Ahnak deficiency regulates HFD-induced production and release of hepatic FGF21. As shown in Fig. 3c, the protein expression of hepatic FGF21 was found to be significantly increased in the livers of HFD-fed Ahnak KO mice. Furthermore, plasma FGF21 and PPARα protein levels were also elevated in Ahnak KO mice receiving a HFD (Fig. 3b, c). AMPK, SIRT1, and PGC1α have been identified as important regulators of mitochondrial biogenesis and function31,32, and LKB1, a serine threonine kinase, directly activates AMPK activity33. In addition, FGF21 regulates energy metabolism by activating the AMPK-SIRT1-PGC1α pathway14,16. Because the expression of FGF21 was increased in the livers of HFD-fed Ahnak KO mice, we determined whether the expression levels of these molecules were altered. Consistent with our hypothesis, we observed increased protein levels of LKB1, SIRT1, and PGC1α and increased amounts of phosphorylated AMPK. In addition, the rate-limiting enzyme of fatty acid oxidation, carnitine palmitoyltransferase 1 (CPT1), was upregulated (Fig. 3c). Despite increased expression of PGC1α, no differences were observed between the HFD-fed WT and Ahnak KO mice in the levels of mitochondrial oxidative phosphorylation complex proteins (Supplementary Fig. 4). These results indicate that pronounced obesity and hepatic steatosis are attenuated in Ahnak KO mice fed a HFD. This may be attributed, in part, to increased expression of genes associated with fatty acid oxidation and induction of FGF21 in the liver.

Measurement of selected inflammatory gene expression in the liver after a 7-week HFD feeding by quantitative real-time PCR and normalization to 36B4. a Analysis of FGF21 expression and its receptors (Fgfr1, Fgfr2, Fgfr4, and coreceptor β-Klotho). b Plasma levels of FGF21 in Ahnak KO and WT mice fed chow and a HFD for 7 weeks. c Immunoblots showing the protein expression levels of FGF21 and PPARα and their downstream targets in HFD-fed Ahnak KO and WT mice. Data were presented as the means ± SEM (n = 4–6 mice). *p < 0.05; **p < 0.01.
Ahnak KO mice exhibit altered hepatic expression of key genes involved in lipid metabolism
FGF21 regulates hepatic fatty acid oxidation and ketogenesis34. To determine the functional consequence of FGF21 induction in Ahnak KO mice receiving a HFD, we measured the expression of key genes involved in lipid metabolism. The mRNA level of PPARα, a key regulator of fatty acid oxidation, was not altered in either NCD- or HFD-fed Ahnak KO mice compared with WT mice. However, PPARα target genes, including Cpt1, carnitine palmitoyltransferase 2 (Cpt2), acyl-coenzyme A oxidase 1 (Acox), acyl-coenzyme A dehydrogenase (Lcad), and Pgc1α, were found to be significantly increased in Ahnak KO mice compared with WT mice (Fig. 4a).

Relative mRNA expression of key genes involved in fatty acid oxidation (a) and lipogenesis (b) as measured by quantitative real-time PCR in the livers of Ahnak KO and WT mice fed chow and a HFD for 7 weeks. Data were presented as the means ± SEM. *p < 0.05; **p < 0.01, versus HFD-fed WT mice; # p < 0.05; ## p < 0.01, versus NCD-fed WT mice.
The lipogenic pathway in the liver has been determined to have been severely compromised during hepatic steatosis. Therefore, we examined the changes in lipogenic pathway-related gene expression. Ahnak KO mice that were fed a HFD showed inactivation of a hepatic lipogenic pathway that included the downregulation of genes required for fatty acid synthesis and TG esterification (Fig. 4b). We observed decreased expression of sterol regulatory element binding transcription factor 1c (Srebf1) mRNA, a key regulator of de novo lipogenesis, consistent with changes observed in the expression of its targets, including fatty acid synthase (Fasn) and stearoyl-coenzyme A desaturase 1 (Scd1). Hepatic mRNA expression of peroxisome proliferator-activated receptor gamma (Pparγ) and its targets, including diacylglycerol O-acyltransferase 1 (Dgat1) and diacylglycerol O-acyltransferase 2 (Dgat2), which catalyze the final step in TG esterification, was also decreased in HFD-fed Ahnak KO mice compared with HFD-fed WT mice. Additionally, the expression of cell death-inducing DNA fragmentation factor alpha subunit-like effector A (Cidea), a lipid droplet-associated protein that has emerged as an important regulator of lipid storage, and the formation of large lipid droplets in adipocytes and hepatocytes35, was also decreased in the livers of HFD-fed Ahnak KO mice compared with WT mice (Fig. 4b). These results indicate that a deficiency in the Ahnak gene in mice affects hepatic gene expression associated with the lipogenic pathway.
HFD-fed Ahnak KO mice exhibit increased whole-body energy expenditure
FGF21 has been identified to exert an effect on energy metabolism and body weight8,9,36. As shown in Fig. 1, compared with their WT littermates fed a HFD, Ahnak KO mice appeared to exhibit decreased fat mass24 accompanied by reduced liver weight. Furthermore, the production and circulation of FGF21 were increased in Ahnak KO mice receiving a HFD (Fig. 3a–c). Therefore, we determined whether Ahnak deficiency in mice affects energy metabolism through increased FGF21 expression. To assess whether the induction of FGF21 is responsible for energy consumption in Ahnak KO mice, calorimetry was assessed in Ahnak KO mice fed a NCD for 2 days, followed by a HFD for 4 days to induce FGF21 expression. As shown in Fig. 5a–c, Ahnak KO mice exhibited an increase in VO2 rates during HFD conditions throughout the light and dark cycles. Daily food intake was similar between WT and Ahnak KO mice for both the NCD and HFD. Comprehensive animal monitoring studies indicated that the rate of oxygen consumption (VO2) was increased in Ahnak KO mice. The calculated daily energy expenditure was also increased in Ahnak KO mice when switched to a HFD. Furthermore, the RER was reduced in Ahnak KO mice during the light and dark phases (Fig. 5c and Supplementary Fig. 5). This suggests that Ahnak KO mice prefer lipids as an energy source rather than carbohydrates compared with WT mice. Consistent with the augmented hepatic and circulating levels of FGF21 in HFD-fed Ahnak KO mice (Fig. 3b, c), Ahnak deficiency was associated with improved energy expenditure (Fig. 5), increased hepatic expression of genes involved in fatty acid oxidation (Fig. 4a), and reduced expression of genes that control lipogenesis (Fig. 4b).

Metabolic parameters were determined using indirect calorimetry for Ahnak KO mice and wild-type littermates placed on a NCD followed by a HFD. a O2 consumption. b RER. c Quantitative values of VO2, RER, food intake, and energy expenditure. Data were presented as the means ± SEM (n = 5). *p < 0.05.
Ahnak deficiency attenuates palmitate-induced lipid accumulation in primary hepatocytes through FGF21 induction
To further analyze the role of Ahnak in lipid accumulation in hepatocytes, we examined palmitate-induced lipid accumulation in primary hepatocytes isolated from Ahnak KO and WT mice. Oil Red O staining showed that lipid droplets in palmitate-treated WT hepatocytes were higher than those in Ahnak-deficient hepatocytes (Fig. 6a).

a Primary hepatocytes were incubated with 0.25 mM palmitate for 24 h. Lipid accumulation was estimated by Oil Red O staining after palmitate incubation. Quantitation of the positive areas of Oil Red O staining is shown. b Relative FGF21 mRNA expression was measured after BSA and palmitate treatment (left). Secreted FGF21 was measured in culture media from WT and Ahnak-deficient hepatocytes (right). c Protein expression levels of FGF21, PPARα, and PGC1α were measured in WT and Ahnak-deficient hepatocytes incubated with 0.25 mM BSA or palmitate for 24 h. d Relative mRNA expression levels of genes involved in lipogenesis and fatty acid oxidation were measured after BSA and palmitate treatment. Data were presented as the means ± SEM. *p < 0.05; **p < 0.01, versus palmitate-treated WT hepatocytes; #p < 0.05; ##p < 0.01, versus BSA-treated WT hepatocytes.
We next examined whether Ahnak deficiency regulates both the production and secretion of FGF21 in primary hepatocytes. As shown in Fig. 6b, the expression of Fgf21 was increased in Ahnak-deficient hepatocytes compared with WT hepatocytes and further increased by palmitate treatment of Ahnak-deficient hepatocytes (Fig. 6b). Consistent with Fgf21 mRNA expression, FGF21 protein levels were also increased in palmitate-treated Ahnak-deficient hepatocytes compared with WT hepatocytes. A concomitant increase in the expression of PPARα protein was also observed in palmitate-treated hepatocytes isolated from Ahnak KO mice (Fig. 6c). To determine the level of FGF21 released from hepatocytes, we measured the concentration of FGF21 in the culture media obtained from Ahnak-deficient and WT hepatocytes. As expected, the level of secreted FGF21 was increased in Ahnak-deficient hepatocytes compared with WT hepatocytes (Fig. 6b).
To identify which molecular mechanism regulates lipid accumulation in Ahnak-deficient hepatocytes, the expression of lipid metabolism genes was measured (Fig. 6d). Srebf1, a key transcription factor regulating de novo lipogenesis, was significantly decreased in BSA- and palmitate-treated Ahnak-deficient hepatocytes compared with WT hepatocytes. Scd1 expression was also significantly decreased in Ahnak KO hepatocytes. Furthermore, genes involved in fatty acid oxidation, such as Acox, Cpt1, Cpt2, and PPARα, were increased in Ahnak KO hepatocytes. These results indicate that palmitate-induced lipid accumulation was attenuated in Ahnak-deficient hepatocytes and may be further attributed to the induction of FGF21 and PPARα expression. Collectively, these data confirm that Ahnak deficiency contributes to the induction of the PPARα-FGF21 hormone axis in hepatocytes. This suggests that Ahnak deficiency improves hepatic steatosis largely through enhanced expression of FGF21 and genes associated with fatty acid oxidation.
Discussion
In the present study, we examined the role of AHNAK in the regulation of hepatic lipid metabolism in diet-induced fatty liver. We found that Ahnak KO mice were resistant to diet-induced obesity and hepatic steatosis when fed a HFD. To identify the molecular mechanism underlying the protection from diet-induced hepatic steatosis, a microarray analysis was performed with liver samples from HFD-fed Ahnak KO and WT mice. The gene expression profiles revealed a similar cluster of genes in the liver of HFD-fed Ahnak KO mice compared with the NCD-fed WT and Ahnak KO mice. A gene ontology analysis of the downregulated DEGs (GSE70119) revealed that the oxidative reduction pathway, including fatty acid oxidation and inflammatory signaling pathways, was highly enriched in Ahnak KO mice. The expression of several genes involved in proinflammatory signaling was decreased in the Ahnak KO mice (Supplementary Fig. 3). The results indicate that excessive lipid accumulation in the liver may induce oxidative stress and the inflammatory response37,38. Thus, attenuated lipid accumulation in the liver of Ahnak KO mice fed a HFD may result in the downregulation of inflammatory signaling.
The most important finding of this study is the identification of FGF21 as a novel regulatory protein modulated by the genetic manipulation of Ahnak. Gene expression profiling revealed that Fgf21 was increased by ~3.2-fold in the livers of Ahnak KO mice compared with WT mice fed a HFD. Ahnak deficiency in mice reportedly results in increased hepatic FGF21 production and release (Fig. 3a–c). It is well documented that FGF21 functions as a metabolic regulator in several diabetic animal models39. Administration of FGF21 can reduce hepatic TG accumulation and thus reverse body weight gain and hepatic steatosis in rodents and primates12,13. Additionally, FGF21 regulates energy homeostasis in adipocytes through an AMPK/SIRT1/PGC1α-dependent mechanism, resulting in enhanced oxidative metabolism and mitochondrial biogenesis14,15. Therefore, we focused on the role of FGF21 in preventing diet-induced hepatic steatosis in Ahnak-deficient mice. As expected, Ahnak KO mice exhibited an increase in hepatic expression and plasma levels of FGF21 compared with WT mice fed a HFD (Fig. 3).
We also found that Ahnak KO mice exhibited reduced expression of Srebf1 (Fig. 4b) and its target genes, leading to the inhibition of hepatic lipid accumulation. Furthermore, Ahnak KO mice showed increased expression of genes involved in fatty acid oxidation through the PPARα/AMPK signaling pathway. Consistent with these results, the excessive lipid accumulation induced by palmitate treatment was prevented in Ahnak-deficient hepatocytes and Ahnak knockdown HepG2 cells, accompanied by increased expression of genes involved in fatty acid oxidation through the increased expression of PPARα protein and AMPK activation. Therefore, our study establishes that Ahnak deficiency in mice attenuates diet-induced hepatic lipid accumulation by activating PPARα/AMPK signaling. These findings also suggest that targeting AHNAK may have beneficial effects on the development of hepatic steatosis.
Hepatic expression of FGF21 is induced directly by PPARα in the liver in response to fasting and PPARα agonists34,40,41. The hepatic PPARα pathway plays a significant role in ketogenesis and fatty acid oxidation42,43. The mechanism is mediated, in part, by PPARα-dependent expression of Fgf21, a gene that is upregulated by Ahnak deficiency in mice. In the present study, we discovered that AHNAK modulates PPARα function, which is involved in fatty acid oxidation. Ahnak deficiency or deletion promotes fatty acid oxidation in the liver, contributing to the improvement in hepatic steatosis. However, PPARα mRNA and protein levels between Ahnak KO and WT mice were not consistent. Although the expression of Pparα mRNA was similar between HFD-fed WT and KO mice (Fig. 4a), hepatic PPARα protein expression was increased in Ahnak KO mice after HFD feeding or palmitate treatment compared with that of WT mice (Figs. 3c and 6c). This inconsistency may be explained by earlier observations showing that PPARα turnover can be regulated by the ubiquitin-proteasome system in a ligand-dependent manner44. In addition, it was recently reported that hepatic PPARα function is controlled by polyubiquitination and proteasome-mediated degradation through the coordinated actions of PAQR3 and HUWE145. Therefore, the discrepancy observed between the expression of PPARα mRNA and protein in Ahnak KO and WT mice could be due to posttranslational modification of PPARα, which is regulated by AHNAK. Furthermore, AHNAK has been identified as a new substrate of the ubiquitin-protein ligase E3C (UBE3C) in the regulation of p53 activity46. However, the direct mechanism linking AHNAK to PPARα stability has yet to be identified.
Circulating FGF21 regulates energy homeostasis in adipocytes through an AMPK/SIRT1/PGC1α-dependent mechanism, resulting in enhanced mitochondrial oxidative capacity14,15. Phosphorylation of AMPK and PGC1α protein expression was increased in the livers of HFD-Ahnak KO mice compared with WT mice. These results indicate that pronounced obesity and hepatic steatosis are attenuated in Ahnak KO mice fed a HFD. This may be attributed, in part, to the induction of FGF21, fatty acid oxidation, and inhibition of lipogenic genes in the liver.
To further understand how Ahnak deficiency inhibits lipid accumulation in the liver, we analyzed the changes in the expression of lipogenic genes. We found that hepatic expression of several genes involved in fatty acid uptake and lipid accumulation was decreased in HFD-fed Ahnak KO mice (Fig. 4a). Ahnak KO mice also exhibited decreased lipid uptake, consistent with decreased Cd36 expression and expression of the lipogenic genes Srebf1, Fasn, Scd1, Dgat1, and Dgat2. SREBF1 and PPARγ are well-known transcriptional regulators of lipogenic gene expression in the liver47. Therefore, Ahnak KO mice may inhibit hepatic lipid accumulation through regulation of lipogenic gene expression. Although HFD feeding increased Srebf1 expression in Ahnak KO mice compared with NCD-fed Ahnak KO mice (Fig. 4b), the expression of Srebf1 in primary hepatocytes of Ahnak KO mice was not changed when comparing the BSA and palmitate treatment groups (Fig. 6d). In addition, the expression patterns of target genes such as Fasn and Scd1 differed in the liver tissue (Fig. 4b) and primary hepatocytes (Fig. 6d), respectively. Because Srebf1 gene expression is regulated by itself, it is difficult to explain this phenomenon based on whether AHNAK affects the transcriptional regulatory roles of SREBF1 or regulates Srebf1 gene expression. To activate SREBF1, cleavage of SREBFs by the site-2 protease (S2P) within a transmembrane segment or near the membrane surface after an initial cleavage by site-1 protease (S1P) should occur due to sterols under HFD conditions. Moreover, it is known that SREBF1-mediated de novo fatty acid synthesis in the liver is negatively regulated by the insulin-induced gene (INSIG) molecule48. It was recently reported that AMPK interacts with and mediates phosphorylation of INSIG, resulting in the inhibition of SREBF1 cleavage and processing and thus attenuating lipogenic gene expression49. In the present study, the phosphorylated active form of AMPK was increased in the livers of HFD-fed Ahnak KO mice (Fig. 3c). Further studies are needed to clarify whether AHNAK has a regulatory role through the AMPK response in activating SREBF1 during HFD conditions. In addition, because whole-body Ahnak KO mice were used in our study, it should be considered that decreased inflammatory signaling in Ahnak KO mice may result in attenuated lipid accumulation. This has been strongly supported by an earlier observation that Ahnak KO mice showed CD4 + T cell inactivation and decreased cytokine secretion50. Thus, attenuated lipid accumulation in the liver of Ahnak KO mice fed a HFD may result in the downregulation of inflammatory signaling.
It is well established that FGF21 has an effect on energy metabolism and body weight8,9,36. To directly assess whether the induction of FGF21 in Ahnak KO mice is responsible for energy fluctuations, indirect calorimetry was used to assess Ahnak KO mice placed on a NCD followed by a HFD. As shown in Fig. 5a–c, Ahnak KO mice exhibited increased VO2 rates under HFD conditions throughout the light and dark cycles but not under NCD conditions. Daily food intake was determined to be similar between WT and Ahnak KO mice for both the NCD and HFD. Comprehensive animal monitoring studies indicated that the rates of oxygen consumption (VO2) were increased in Ahnak KO mice. The calculated daily energy expenditure was also increased in Ahnak KO mice when switched to a HFD. Moreover, the RER was reduced in Ahnak KO mice during the light and dark phases (Fig. 5a–c and Supplementary Fig. 5). This may indicate that Ahnak KO mice use more lipids as an energy source than carbohydrates when compared with WT mice. Overall, the results in Ahnak KO mice were characterized by a decrease in fat mass, an increase in energy expenditure, and no significant alterations in food intake.
To better understand the effects of Ahnak deficiency at the cellular level, we examined lipid accumulation in primary hepatocytes following exposure to palmitate. Ahnak deficiency in hepatocytes resulted in decreased expression of lipogenic genes and increased expression of genes involved in fatty acid oxidation, resulting in significantly attenuated lipid accumulation in palmitate-treated cells (Fig. 6d). We next examined whether Ahnak deficiency regulates the production and secretion of FGF21 in primary hepatocytes. As shown in Fig. 6b, Fgf21 expression was increased in Ahnak-deficient hepatocytes compared with WT hepatocytes, and it was further increased by palmitate treatment of Ahnak-deficient hepatocytes. Consistent with mRNA expression, FGF21 protein levels were also increased in palmitate-treated Ahnak-deficient hepatocytes compared with WT hepatocytes (Fig. 6c). However, no differences were observed between BSA-treated Ahnak-deficient and WT hepatocytes. Consistent with FGF21 expression, PPARα protein was significantly increased in palmitate-treated hepatocytes isolated from Ahnak KO mice (Fig. 6c). To determine the level of secreted FGF21 from hepatocytes, we measured the concentration of FGF21 in culture media obtained from Ahnak-deficient and WT hepatocytes. The concentration of FGF21 was increased in serum-starved Ahnak-deficient hepatocytes compared with WT hepatocytes (Fig. 6b). These results indicate that palmitate-induced lipid accumulation was attenuated in Ahnak-deficient hepatocytes and can be attributed, in part, to the induction of FGF21 and PPARα expression in hepatocytes. These results are consistent with the observed decreased lipid accumulation in Ahnak-deficient hepatocytes treated with palmitate.
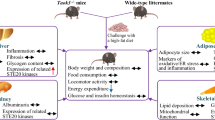
Characterization of the phenotypic changes in Ahnak KO mice, including reduced hepatic steatosis, decreased adiposity, and increased energy expenditure, establishes AHNAK as a novel regulator of FGF21 for the modulation of hepatic fatty acid metabolism. Ahnak deficiency in mice results in increased hepatic FGF21 production and release, reduced lipogenesis, and increased expression of genes involved in the fatty acid oxidation pathway (Fig. 7). The discovery of FGF21 induction in Ahnak KO mice is also supported by the fact that the production and secretion of FGF21 can be increased by Ahnak knockdown in hepatocytes. Therefore, our study establishes the role of AHNAK in regulating hepatocyte lipid metabolism through the PPARα/FGF21 axis. Ahnak deficiency has been reported to suppress lipid accumulation in the liver and may represent a novel therapeutic target to reduce the formation of fatty liver.

High-fat diet-induced obese Ahnak-deficient mice exhibited increased hepatic FGF21 production and release compared with WT HFD-fed mice, thereby inducing the expression of genes involved in the fatty acid oxidation pathway, decreasing lipogenesis, decreasing hepatic steatosis, and increasing energy expenditure.
Data availability
Experimental materials used to generate the results reported in this manuscript are available upon request.
References
Charlton, M. et al. Frequency of nonalcoholic steatohepatitis as a cause of advanced liver disease. Liver Transpl. 7, 608–614 (2001).
Koo, S. H. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 19, 210–215 (2013).
Scott, M., Castleden, C. M., Adam, H. K., Smith, R. P. & Fitzsimons, T. J. The effect of ageing on the disposition of nifedipine and atenolol. Br. J. Clin. Pharm. 25, 289–296 (1988).
Kharitonenkov, A. & Larsen, P. FGF21 reloaded: challenges of a rapidly growing field. Trends Endocrinol. Metab. 22, 81–86 (2011).
Kharitonenkov, A. et al. FGF-21/FGF-21 receptor interaction and activation is determined by betaKlotho. J. Cell Physiol. 215, 1–7 (2008).
Murata, Y., Konishi, M. & Itoh, N. FGF21 as an endocrine regulator in lipid metabolism: from molecular evolution to physiology and pathophysiology. J. Nutr. Metab. 2011, 981315 (2011).
Adams, A. C. et al. The breadth of FGF21’s metabolic actions are governed by FGFR1 in adipose tissue. Mol. Metab. 2, 31–37 (2012).
Fisher, F. M. et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281 (2012).
Holland, W. L. et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 17, 790–797 (2013).
Wente, W. et al. Fibroblast growth factor-21 improves pancreatic beta-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes 55, 2470–2478 (2006).
Wu, S., Levenson, A., Kharitonenkov, A. & De Luca, F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. J. Biol. Chem. 287, 26060–26067 (2012).
Xu, J. et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58, 250–259 (2009).
Kharitonenkov, A. et al. FGF-21 as a novel metabolic regulator. J. Clin. Invest 115, 1627–1635 (2005).
Chau, M. D., Gao, J., Yang, Q., Wu, Z. & Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl Acad. Sci. USA 107, 12553–12558 (2010).
Gimeno, R. E. & Moller, D. E. FGF21-based pharmacotherapy-potential utility for metabolic disorders. Trends Endocrinol. Metab. 25, 303–311 (2014).
Li, Y. et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 146, 539–549 e537 (2014).
Dostalova, I., Haluzikova, D. & Haluzik, M. Fibroblast growth factor 21: a novel metabolic regulator with potential therapeutic properties in obesity/type 2 diabetes mellitus. Physiol. Res. 58, 1–7 (2009).
Kharitonenkov, A. & Shanafelt, A. B. FGF21: a novel prospect for the treatment of metabolic diseases. Curr. Opin. Investig. Drugs 10, 359–364 (2009).
Lee, I. H. et al. AHNAK-mediated activation of phospholipase C-gamma1 through protein kinase C. J. Biol. Chem. 279, 26645–26653 (2004).
Shin, J. H. et al. Obesity resistance and enhanced insulin sensitivity in Ahnak-/- mice fed a high fat diet are related to impaired adipogenesis and increased energy expenditure. PloS ONE 10, e0139720 (2015).
Shin, J. H. et al. AHNAK deficiency promotes browning and lipolysis in mice via increased responsiveness to beta-adrenergic signalling. Sci. Rep. 6, 23426 (2016).
Park, J. W. et al. AHNAK loss in mice promotes type II pneumocyte hyperplasia and lung tumor development. Mol. Cancer Res. 16, 1287–1298 (2018).
Woo, J. K. et al. Essential role of Ahnak in adipocyte differentiation leading to the transcriptional regulation of Bmpr1alpha expression. Cell Death Dis. 9, 864 (2018).
Kim, I. Y. et al. 1H NMR-based metabolomic study on resistance to diet-induced obesity in AHNAK knock-out mice. Biochem Biophys. Res. Commun. 403, 428–434 (2010).
Nam, M. H., Chun, M. S., Seong, J. K. & Kim, H. G. Ensuring reproducibility and ethics in animal experiments reporting in Korea using the ARRIVE guideline. Lab. Anim. Res. 34, 11–19 (2018).
Choi, C. S. et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc. Natl Acad. Sci. USA 104, 16480–16485 (2007).
Yu, J. Y. et al. Effects of DA-5513 on alcohol metabolism and alcoholic fatty liver in rats. Lab. Anim. Res. 34, 49–57 (2018).
Liu, Y. et al. Knockdown of acyl-CoA: diacylglycerol acyltransferase 2 with antisense oligonucleotide reduces VLDL TG and ApoB secretion in mice. Biochim. Biophys. Acta 1781, 97–104 (2008).
Koo, S. H. et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111 (2005).
Shang, J. et al. Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase. Acta Pharm. Sin. 29, 698–706 (2008).
Hardie, D. G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 (2007).
Rodgers, J. T., Lerin, C., Gerhart-Hines, Z. & Puigserver, P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 582, 46–53 (2008).
Shaw, R. J. et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 (2005).
Badman, M. K. et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 (2007).
Jaffe, G. J., Mieler, W. F., Burke, J. M. & Williams, G. A. Photoablation of ocular melanoma with a high-powered argon endolaser. Arch. Ophthalmol. 107, 113–118 (1989).
Veniant, M. M. et al. Pharmacologic effects of FGF21 are independent of the “Browning” of white adipose tissue. Cell Metab. 21, 731–738 (2015).
Desvergne, B. & Wahli, W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 20, 649–688 (1999).
Gawrieh, S., Opara, E. C. & Koch, T. R. Oxidative stress in nonalcoholic fatty liver disease: pathogenesis and antioxidant therapies. J. Investig. Med. 52, 506–514 (2004).
Li, H., Zhang, J. & Jia, W. Fibroblast growth factor 21: a novel metabolic regulator from pharmacology to physiology. Front. Med. 7, 25–30 (2013).
Inagaki, T. et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 (2007).
Liu, J. J., Foo, J. P., Liu, S. & Lim, S. C. The role of fibroblast growth factor 21 in diabetes and its complications: a review from clinical perspective. Diabetes Res. Clin. Pract. 108, 382–389 (2015).
Li, H. H. et al. Modulation of fatty acid and bile acid metabolism by peroxisome proliferator-activated receptor alpha protects against alcoholic liver disease. Alcohol Clin. Exp. Res. 38, 1520–1531 (2014).
Patterson, A. D. et al. Human urinary metabolomic profile of PPARalpha induced fatty acid beta-oxidation. J. Proteome Res. 8, 4293–4300 (2009).
Blanquart, C., Barbier, O., Fruchart, J. C., Staels, B. & Glineur, C. Peroxisome proliferator-activated receptor alpha (PPARalpha) turnover by the ubiquitin-proteasome system controls the ligand-induced expression level of its target genes. J. Biol. Chem. 277, 37254–37259 (2002).
Zhao, Z. et al. Hepatic PPARalpha function is controlled by polyubiquitination and proteasome-mediated degradation through the coordinated actions of PAQR3 and HUWE1. Hepatology 68, 289–303 (2018).
Gu, J. et al. Ubiquitin-protein ligase E3C maintains non-small-cell lung cancer stemness by targeting AHNAK-p53 complex. Cancer Lett. 443, 125–134 (2019).
Pettinelli, P. & Videla, L. A. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab. 96, 1424–1430 (2011).
Rawson, R. B. The SREBP pathway-insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 4, 631–640 (2003).
Han, Y. et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat. Commun. 10, 623 (2019).
Choi, E. W. et al. Ahnak-knockout mice show susceptibility to Bartonella henselae infection because of CD4+ T cell inactivation and decreased cytokine secretion. BMB Rep. 52, 289–294 (2019).
Acknowledgements
This research was supported by the Korea Mouse Phenotyping Project (2013M3A9D5072550) and the Bio and Medical Technology Development Program (2012M3A9B6055344) of the National Research Foundation funded by the Ministry of Science and ICT. This study was partially supported by the Research Institute for Veterinary Science, Seoul National University. We thanks to Yang Jin Joo in KMPC for preparing the summary illustration. This research was supported by the Korea Mouse Phenotyping Project.
Author information
Authors and Affiliations
Contributions
Y.N.K., J.H.S., and J.K.S. were responsible for the experimental design, developed the methodology, and wrote the manuscript. D.S.K. and S.Y.C. were responsible for high-throughput screening and data analysis. M.Y.K., H.J.L., M.R.R.J., and I.Y.K. conceived the experiments and performed the data analysis. M.O.L. and Y.S.B. provided reagents and analyzed the data. M.Y.K. and J.K.S. wrote and revised the manuscript with input from all coauthors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, Y.N., Shin, J.H., Kyeong, D.S. et al. Ahnak deficiency attenuates high-fat diet-induced fatty liver in mice through FGF21 induction. Exp Mol Med 53, 468–482 (2021). https://doi.org/10.1038/s12276-021-00573-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00573-3
- Springer Nature Limited
This article is cited by
-
Weight regain, but not weight loss exacerbates hepatic fibrosis during multiple weight cycling events in male mice
European Journal of Nutrition (2024)
-
SNP discovery and association study for growth, fatness and meat quality traits in Iberian crossbred pigs
Scientific Reports (2022)
-
The critical roles of histone deacetylase 3 in the pathogenesis of solid organ injury
Cell Death & Disease (2021)