
Abstract
Genetic alterations have been reported for decades in most human embryonic stem cells (hESCs). Survival advantage, a typical trait acquired during long-term in vitro culture, results from the induction of BCL2L1 upon frequent copy number variation (CNV) at locus 20q11.21 and is one of the strongest candidates associated with genetic alterations that occur via escape from mitotic stress. However, the underlying mechanisms for BCL2L1 induction remain unknown. Furthermore, abnormal mitosis and the survival advantage that frequently occur in late passage are associated with the expression of BCL2L1, which is in locus 20q11.21. In this study, we demonstrated that the expression of TPX2, a gene located in 20q11.21, led to BCL2L1 induction and consequent survival traits under mitotic stress in isogenic pairs of hESCs and human induced pluripotent stem cells (iPSCs) with normal and 20q11.21 CNVs. High Aurora A kinase activity by TPX2 stabilized the YAP1 protein to induce YAP1-dependent BCL2L1 expression. A chemical inhibitor of Aurora A kinase and knockdown of YAP/TAZ significantly abrogated the high tolerance to mitotic stress through BCL2L1 suppression. These results suggest that the collective expression of TPX2 and BCL2L1 from CNV at loci 20q11.21 and a consequent increase in YAP1 signaling promote genome instability during long-term in vitro hESC culture.
Similar content being viewed by others

Introduction
Due to their pluripotency, human embryonic stem cells (hESCs) have great potential in stem cell-based cell therapy; however, their frequent genetic aberrations during in vitro maintenance are considered a major hurdle that compromises the safety of this type of therapy1,2. However, despite these concerns3, few studies have explored the biological consequences, risks and reliable biomarkers of genetic alterations in hESCs4. As demonstrated by several extensive genomic studies, copy number variations (CNVs), including the 20q11.21 locus or trisomy of chromosome 12 or 175,6,7, occur in hPSCs during in vitro culture. One of the most characteristic phenotypic changes of genetically aberrant hESCs is acquired culture adaptation5 for hESC survival under various stress conditions, including culture, genotoxic stress, and single-cell dissociation among others (also referred to as survival advantage)8,9. Such acquired survival traits result from the induction of BCL2L1 (encoding BCL-xL), a typical antiapoptotic gene located at 20q11.218,10 due to amplification of 20q11.2111 and/or dominant mutation of p5312. These alterations impair the major cell death mechanism of hESCs, as the mitochondria are primed to undergo cell death upon genotoxic stress through p53 translocation13,14,15,16. A recent study revealed that escape from mitotic cell death during mitotic errors due to this acquired survival trait (via either induction of BCL2L1 or dominant mutation of NOXA, a proapoptotic factor) leads to aneuploidy9. In addition, a targeting protein for Xklp2 (TPX2), also located in 20q11.21, was suggested to be the putative driver of abnormal mitosis by disrupting spindle dynamics17. However, despite the significance of survival advantage in the phenotypic changes of culture-adapted hESCs and even aneuploidy, the molecular mechanisms underlying this phenomenon, with the exception of BCL2L1 induction, are not fully understood. Moreover, a reduction in serum response factor expression18 or high oxygen concentrations during in vitro culture19 have been associated with hESC genetic alterations.
High susceptibility to genotoxic stress in hESCs is a unique cellular characteristic that has developed as a safeguard for genome integrity20. However, the contemporaneous expression of antiapoptotic factors maintains a fine balance between survival and apoptosis21. It is also worth noting that deletion of Yap1 leads to embryonic lethality22, suggesting that Yap1 is required for the self-renewal and differentiation of mouse ESCs (mESCs)23. A recent study demonstrated that Yap1 in mESCs serves as a safeguard to attenuate mitochondrial apoptosis by upregulating typical antiapoptotic factors, including Bcl2l124. Similarly, Rho-dependent activation of YAP1 promotes the long-term survival of hESCs25. In addition to ESCs, YAP1 activation in cancer cells increases survival through the induction of BIRC5 and BCL2L1, both of which are important for hESC survival26.
In this study, using four types of isogenic hESCs after different periods of in vitro culture8 and with different CNV statuses, we demonstrated that TPX2, located at locus 20q11.21, was highly induced in culture-adapted hESCs and conferred resistance under mitotic stress through BCL2L1 induction. Our findings also revealed that the YAP1 protein stabilization conferred by Aurora A kinase activated by TPX2 induction was responsible for BCL2L1 transcription, resulting in mitotic stress escape and suggesting that additional signaling (e.g., YAP1) would be required for determining culture-adapted phenotypic changes in hESCs other than CNVs.
Materials and methods
Reagents and cell culture
The primary antibodies (Supplementary Table 1) and chemical reagents (Supplementary Table 2) are listed. Human embryonic stem cells (WA09: H9, WiCell Research Institute, CHA3-hESCs) were maintained as previously described8. Medium and reagents for cell culture are listed in Supplementary Table 3. Sequence information of RT-PCR (Supplementary Table 4) and siRNA (Supplementary Table 5) is listed.
Genome-wide gene expression profiling
Assessment of the differences in gene expression between groups was conducted using the R package DESeq2 from the FASTQ files and processed data in the Gene Expression Omnibus (GEO: GSE167495). Differently expressed genes (DEGs) were selected with |log2fold-change | >1 and a false discovery rate (FDR) of <0.01. To identify genomic loci containing the most DEGs, overrepresentation analysis was performed using a hypergeometric test with the positional gene sets from the Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/). After FDR-based multiple testing correction, an enrichment score for each genomic locus was defined as the -log10 Q-value. For GO analysis, GSEA was performed using the GO BP gene sets from MSigDB via the R package fgsea. Significantly upregulated or downregulated GO terms were selected with an adjusted P value of <0.01 and a |normalized enrichment score (NES) | of >2.
Determination of copy number variations
Whole-genome genotyping was performed using the Illumina HumanOmni1-Quad Beadchip (Illumina) containing 1,140,419 genetic markers across the human genome. Samples were processed according to the specifications of the Illumina Infinium HD super assay. Briefly, each sample was whole-genome amplified, fragmented, precipitated, and resuspended in an appropriate hybridization buffer. Denatured samples were hybridized on a prepared BeadChip for a minimum of 16 h at 48 °C. Following hybridization, the bead chips were processed for the single-base extension reaction, stained, and imaged with an Illumina iScan system. Normalized bead intensity data for each sample were loaded into the GenomeStudio software package (Illumina). Signal intensity ratios were calculated using the log R ratio (LRR: log ratio of observed probe intensity to expected intensity; any deviations from zero in this metric are evidence of copy number change), and allelic intensity was determined by the B allele frequency for all samples. Values were exported using Illumina GenomeStudio. Structural variant analysis was performed using the sliding window approach (window size 10).
Live cell imaging
Live cell images were captured by JuLITM Stage (NanoEnTek Inc.). The acquired images were further processed and analyzed with JuLITMSTAT software according to the manufacturer’s instructions.
Statistical analysis
Graphical data are presented as the mean ± S.E.M. Statistical significance for more than three groups was determined using one-way or two-way analysis of variance (ANOVA) followed by a Tukey multiple comparison posttest. Statistical significance between two groups was analyzed using unpaired Student’s t tests. Statistical analysis was performed with GraphPad Prism 8 software (https://www.graphpad.com/scientific-software/prism/). Significance was assumed for p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Supplementary data
Supplementary data are available at EMM online.
Results
The survival advantage of culture-adapted hESCs under mitotic stress
Culture-adapted hESCs become highly resistant to various stressors, such as DNA damage10, single-cell dissociation, and YM1558, ‘a stemtoxic agent’ that selectively induces death in undifferentiated pluripotent stem cells13 through cellular uptake by SLC35F2, a solute carrier protein27,28. In addition to the increased survival of hESCs under genotoxic stress, escape from cell death during mitosis has been suggested to increase aneuploidy during long-term culture9. To examine the effect of the survival trait acquired in long-term in vitro culture, we took advantage of a unique isogenic set of hESCs after different periods of in vitro culture8,17 [e.g., P1 hESCs: fewer than 50 passages (1 year of culture); P2 hESCs: over 100 passages (2 years of culture); P3 hESCs: over 200 passages (4 years of culture); and P4 hESCs: over 300 passages (over 6 years of culture)]. As described previously8,17, a clear survival trait after YM155 treatment was evident in P4 hESCs compared to P1 hESCs (Fig. 1A) and was associated with BCL2L1 induction (Fig. 1B). Next, to examine the escape from mitotic cell death induced by mitotic stresses, which was previously suggested to be a possible cause of aneuploidy in hESCs9, P1 or P4 hESCs were subjected to mitotic stress with typical drugs targeting mitotic spindles such as nocodazole (Noc: a spindle destabilizer) and paclitaxel (Tax: a spindle stabilizer). Similar to YM155 treatment (Supplementary Fig. 1a) and single-cell dissociation (Supplementary Fig. 1b), P4 hESCs, which display acquired survival traits8 as well as abnormal mitosis17, were highly resistant to mitotic stress inducers (Fig. 1C and D).

A After treatment with YM155 (50 nM) for 24 h, the death of P1 and P4 hESCs was determined by Annexin V/7-AAD staining (n = 6 independent experiments; mean ± SEM, two-way ANOVA, ***p < 0.001). B Relative mRNA expression level of BCL2L1 in P1 and P4 hESCs (n = 5 independent biological repeats; independent experiments; mean ± SEM, Mann‒Whitney test, **p < 0.01). C, D Death of P1 and P4 hESCs after treatment with 50 ng/ml nocodazole (Noc: C) or 50 ng/ml paclitaxel (Tax: D) measured by flow cytometry and stained with Annexin V and 7-AAD (n = 5 independent experiments; mean ± SEM, two-way ANOVA, ****p < 0.0001 for C; n = 6 independent experiments, two-way ANOVA, ***p < 0.001 for D).
Except for the role of BCL2L1 induction followed by CNV at locus 20q11.2111 or p53 dominant mutations12, little is known regarding the mechanisms by which the survival trait is acquired during hESC culture adaptation. Furthermore, no previous studies have determined how BCL2L1 transcription becomes upregulated, resulting in the survival advantage trait in culture-adapted hESCs. We observed that the survival trait manifested in P3 hESCs (over 200 passages) and P4 hESCs (over 300 passages) but not P2 hESCs (over 100 passages) under either YM155 treatment (Supplementary Fig. 1a), cell dissociation (Supplementary Fig. 1b) or mitotic stress (Supplementary Fig. 1c), with BCL2L1 expression comparable to that of P1 hESCs (less than 50 passages)8.
BCL2L1 induces survival associated with TPX2
As previously demonstrated, gain of 20q11.21 was the most frequently occurring genome aberration and CNV of ID1, BCL2L1, and HM13 at locus 20q11.21, suggesting that the expression of these genes was a marker of genome aberration5 and may lead to abnormal biological consequences10,11,29. However, intriguingly, not all genes at locus 20q11.21 were transcriptionally active in hESCs with 20q11.21 CNV (e.g., P3 and P4 hESCs) (Fig. 2A). Among the genes in the 20q11.21 locus, TPX2, which was previously shown to induce aberrant mitosis in culture-adapted hESCs17, was concurrently induced with BCL2L1 in P3 and P4 hESCs (Fig. 2B). Another pair of human pluripotent stem cells (hPSCs) at early and late passages (e.g., hCHA3 and BJ-iPSCs), of which the survival trait as well as trisomy at chromosome 12 was manifested in late-passage hPSCs [e.g., passage 181 (P181) for BJ-iPSCs and passage 333 (P333) for CHA3]8, also revealed that hPSCs with gain of 20q11.21 (Supplementary Fig. 1d) at the late passage expressed high levels of TPX2 and BCL2L1 (Supplementary Fig. 1e). Similarly, the BCL-xL protein level was closely associated with the TPX2 protein level in P3 and P4 hESCs, where the survival trait was evident (Fig. 2C). Notably, P4 hESCs with high TPX2 expression also exhibited higher levels of BCL2L1 (Fig. 2D). Considering the roles of TPX2 in cancer malignancy30 and survival/chemoresistance31, it was readily presumed that TPX2 expression may be associated with BCL2L1 induction and the consequent survival traits. As predicted, partial depletion of TPX2 in P4 hESCs (Fig. 2E) and P3 hESCs (Supplementary Fig. 2a) significantly attenuated BCL2L1 expression. Due to the significantly high BCL2L1 and TPX2 coexpression in P3 and P4 hESCs validated by multiple reference genes (Supplementary Fig. 2b and c), alteration of BCL2L1 followed by TPX2 depletion suggests that TPX2 induction somehow regulates BCL2L1 expression and confers the survival trait. Of note, due to the pivotal roles of TPX2 in not only spindle integrity and genome stability but also early embryogenesis32, the establishment of stable TPX2 knockdown in P4 hESCs or TPX2 expression in P1 hESCs was unsuccessful despite multiple trials (data not shown). Thus, alternatively, the doxycycline (Dox)-inducible TPX2 cell line established from P1 hESCs (iTPX2-hESCs) was used to explore the role of TPX2 in BCL2L1 induction (Supplementary Fig. 2d). TPX2 mRNA (Supplementary Fig. 2d) and protein (Supplementary Fig. 2e) induction occurred in a dose-dependent manner. The signal from TPX2 conjugated with green fluorescent protein (GFP) was evident in the mitotic spindle, where TPX2 is located during mitosis33 (Supplementary Fig. 2f). Surprisingly, TPX2 induction was sufficient to increase BCL2L1 mRNA (Fig. 2F and G) and protein expression in a time- (Fig. 2H) and dose-dependent manner (Fig. 2I). It is important to note that iTPX2-hESCs were established from P1 hESCs with normal copy numbers, and therefore, the copy number of iTPX2-hESCs remained normal regardless of Dox treatment, unlike that observed in P4 hESCs (Fig. 2J). The regulatory effect of TPX2 on BCL2L1 was reproduced in iTPX2-hESCs derived from hCHA3 hESCs with consistent results (Supplementary Fig. 2g).

A Graphical representation of chromosome 20 and the 20q11.21 locus with the accompanying genes (left) and relative expression levels of the genes at the 20q11.21 locus in P1, P2, P3, and P4 hESCs (right). B Relative transcript levels of BCL2L1 and TPX2 in P1, P2, P3, and P4 hESCs. C Immunoblotting for TPX2 and BCL-xL from the indicated hESCs; α-tubulin was utilized for equal protein loading. D The relative mRNA expression level of the indicated gene in P1 and P4 hESCs (n = 7 independent biological repeats; mean ± SEM, Mann‒Whitney test, **p < 0.01, *p < 0.05). E Relative mRNA expression levels of TPX2 and BCL2L1 in P4 hESCs 48 h after siRNA administration in the control (NC) and TPX2 groups (siTPX2#3) (n = 5 independent experiments; mean ± SEM, two-way ANOVA, ****p < 0.0001, *p < 0.05). F Relative mRNA expression levels of TPX2 and BCL2L1 in H9-iTPX2-hESCs 24 h after treatment with 0.1 μg/ml Dox (n = 4 independent experiments, mean ± SEM, two-way ANOVA, ****p < 0.0001, *p < 0.05). G Relative mRNA expression levels of TPX2 and BCL2L1 in iTPX2-hESCs at the indicated times after treatment with 0.1 μg/ml Dox. H Immunoblotting of TPX2 and BCL-xL in iTPX2-hESCs at the indicated time after treatment with 0.1 μg/ml Dox (arrow for induced GFP-TPX2, * for endogenous TPX2). Relative band densities of BCL-xL are shown. I Immunoblotting of TPX2 and BCL-xL in iTPX2-hESCs 24 h after treatment with the indicated dose of Dox. Relative band densities of BCL-xL are shown. J Copy number output using the Illumina Asian Screening Array (~700 K) computed by GenomeStudio software provided by Illumina. The two plots shown are for B allele frequency and log R ratio. The copy number gain on chromosome 20 detected in H9-P4 hESCs based on the log R ratio is shown by a red bar.
TPX2 induction rescues mitotic cell death of normal hESCs
We previously demonstrated that TPX2, located at locus 20q11.21 near BCL2L1, is a putative driver of abnormal mitosis17. Using iTPX2-hESCs, cell death under mitotic stress (i.e., Noc) was determined after TPX2 induction by Dox. We noted that only a portion of the cells expressed GFP-TPX2 even after Dox treatment for unknown reasons. Thus, a GFP-negative population was used as an internal control, and cell death among the GFP-positive population (expressing TPX2) of iTPX2-hESCs (Supplementary Fig. 3a) was monitored after mitotic stress (Fig. 3A, left panel). As predicted, GFP-positive cells were more resistant to mitotic stress induced by Noc (Fig. 3A, right panel) than GFP-negative cells. Similar results were obtained at different doses of Noc (Fig. 3B). Resistance to two types of mitotic stress inducers via TPX2 induction was reproduced in other hCHA3-derived iTPX2-hESCs (Supplementary Fig. 3b and c). To rule out the potential prosurvival effect of GFP expression (rather than the effect of TPX2), P1 hESCs expressing enhanced green fluorescent protein (EGFP-P1) were cocultured with P4 hESCs and iTPX2 hESCs (Fig. 3C), as the basal TPX2 levels in P4 hESCs and iTPX2 hESCs [due to leakage of the Tet-O system34] were comparably higher than that in P1 hESCs (Supplementary Fig. 3d). Consistently, hESCs with high TPX2 expression (e.g., P4 and iTPX2) were more resistant to Noc-induced mitotic stress regardless of GFP expression (Fig. 3D and E). To further assess whether high TPX2 expression was responsible for mitotic stress resistance, TPX2 was transiently depleted by multiple siRNAs. P4 hESCs with TPX2 depleted to a level similar to that in P1 hESCs (Supplementary Fig. 3e) sensitized the cells to Noc-induced mitotic stress (Fig. 3F). In addition, TPX2 depletion with independent siRNA (Supplementary Fig. 3f) sensitized P4 hESCs to Noc-induced cell death (Supplementary Fig. 3g).

A Graphical representation of the effect of TPX2 induction on cell death after Noc treatment in hESC-iTPX2 (left) and flow cytometry analysis of PE-Annexin V staining after iTPX2 hESCs were pretreated with 0.1 μg/ml Dox or DMSO (Mock) for 16 h in the presence or absence of nocodazole (50 ng/ml) (Noc Tx). The bar graph shows the death (%) of GFP-negative and GFP-positive cells (right) (n = 4 independent experiments; mean ± SEM, Mann‒Whitney test, *p < 0.05). B Graph of % of cell death determined by 7-AAD staining of the GFP-negative or GFP-positive population after administration of the indicated dose of Noc. C Graphical presentation of the competition assay with coculture of the indicated cell types. D, E Flow cytometry analysis of P1 and P4 hESCs (D, n = 6 independent experiments; mean ± SEM, Two-Way ANOVA, ****p < 0.0001) or P1 and iTPX2 hESCs (E, n = 5, two-way ANOVA, p value < 0.0001) after treatment with 50 ng/ml of Nocodazole for 24 h. Populations were identified by GFP after 2 days of Noc treatment. F Flow cytometry analysis of 7-AAD/Annexin V staining of P4 hESCs after control (siNC) or TPX2 siRNA (siTPX2) treatment (n = 6 independent experiments; mean ± SEM, two-way ANOVA, ****p < 0.0001).
YAP/TEAD4 leads to BCL2L1 expression
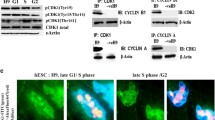
BCL2L1, an antiapoptotic factor highly expressed in P4 hESCs (Fig. 2D), has been shown to be a downstream target of YAP1 in cancer cell models26. Considering the key roles of YAP1 in the survival of ESCs24,25, we hypothesized that the activity of YAP1 may account for the acquisition of survival traits in culture-adapted hESCs. It has been previously demonstrated that Rho and Hippo regulation of YAP/TAZ activation is critical for the survival response of hESCs25. Similarly, two independent gene sets for Hippo signaling were enriched in P1 and P2 hESCs (showing cell death susceptibility) but produced low signaling in P3 and P4 hESCs (showing the survival trait) based on Gene Set Enrichment Analysis (GSEA) (Fig. 4A and Supplementary Fig. 4a). Moreover, YAP1 activity determined by GTIIC reporters was significantly higher in P4 hESCs (Fig. 4B). Considering the pivotal role of YAP1 in the survival of both mouse and human ESCs24,25, the high YAP1 activity acquired during in vitro culture could further enhance the survival signal, thus resulting in a survival advantage. Although the YAP1 mRNA level itself remained unaltered, the BCL2L1 mRNA level was higher in addition to CTGF [encoded by CCN2, a typical YAP1 downstream target gene35,36] in P4 hESCs than in P1 hESCs (Fig. 4C). Despite similarities in YAP1 transcription, the YAP1 protein level was apparently stabilized with lower amounts of phosphorylated YAP1 (at S127) in P4 hESCs with higher TPX2 and TEAD4 expression (Fig. 4D), a finding that was further validated by an anti-active YAP antibody detecting levels of unphosphorylated YAP37(Fig. 4E). Consistently, nuclear YAP1 expression manifested along with TEAD4 protein expression in P4 hESCs (Fig. 4F). In contrast, depletion of YAP1 (Fig. 4G), TAZ (Fig. 4H), or TEAD4 (Fig. 4I) significantly decreased BCL2L1 expression along with CTGF in P4 hESCs. The same result was obtained with different sets of reference genes (Supplementary Fig. 4b). The other sets of hPSCs (i.e., BJ-iPSCs and CHA3) showing survival traits with gain of 20q11.21 (i.e., P181 for BJ-iPSCs and P333 for CHA3) expressed high levels of TPX2 and BCL-xL, corresponding to high levels of the active YAP protein (Supplementary Fig. 4c). On the other hand, the expression of TEAD4 clearly induced the mRNA levels of CTGF and BCL2L1 (Supplementary Fig. 4d) and the protein expression of BCL-xL (Supplementary Fig. 4e). It is also noteworthy that a recent study demonstrated that high YAP activity in culture adapted or genetically variant hPSCs leads to beating the competition against normal hPSCs for further clonal dominance in culture38.

A GSEA of WP_PATHWAYS_REGULATING_HIPPO_SIGANLING (left) and GOBP_REGULATION_OF_HIPPO_SIGNALING (right) of P1/P2 hESCs and P3/P4 hESCs from the RNA-seq data of P1, P2, P3 and P4 hESCs (GSE167495). B 8X GTIIC reporter assay of P1 and P4 hESCs (n = 4 independent experiments; mean ± SEM, Mann–Whitney test, *p < 0.05). C Relative mRNA levels of YAP1, CTGF and BCL2L1 in P1 or P4 hESCs (n = 6 independent experiments; mean ± SEM, two-way ANOVA, **p < 0.01, ***p < 0.001). D Immunoblotting of TPX2, YAP1, TEAD4 and phosphorylation of YAP1 (at serine 127, S127: α-tubulin or β-actin were utilized for equal protein loading. E Immunoblotting for active YAP1 in P1 and P4 hESCs. Vinculin was used as a loading control. F Immunoblotting for YAP1 and TEAD4 in the cytosol or nuclei of P1 or P4 hESCs., α-Tubulin was used for equal protein loading. G–I Relative mRNA expression of the indicated genes in P4 hESCs after control (NC) or siRNA for (G) YAP1 (n = 3 independent experiments; mean ± SEM, two-way ANOVA, ****p < 0.0001) (H) TAZ (n = 4 independent experiments; mean ± SEM, two-way ANOVA, ****p < 0.0001), or (I) TEAD4 (n = 5 independent experiments; mean ± SEM, two-way ANOVA, ***p < 0.001, ****p < 0.0001).
Aurora A stabilizes the YAP1 protein
Previously, Aurora A, a mitotic kinase strongly associated with TPX2 in spindle assembly, was shown to phosphorylate and stabilize YAP1 in cancer9,39. Constitutive activation of Aurora A, which corresponds with TPX2 induction regardless of cell cycle phase, was a distinct cellular phenotype of P3 and P4 hESCs17. P4 hESCs with high Aurora A activity (determined by phospho-Aurora A) and TPX2 protein levels exhibited higher protein levels of BCL-xL and YAP1 (Fig. 5A) than P1 and P2 hESCs (Supplementary Fig. 5a). Of note, P3 and P4 hESCs with higher Aurora A activation at a similar level of mitotic population (determined by the level of phospho-histone H3, pHH3) along with high TPX2 under mitotic stress were evidently resistant to Noc-induced cell death (Supplementary Fig. 5b). Consistently, transient depletion of TPX2 lowered the protein levels of BCL-xL and YAP1 (Fig. 5B). Next, to confirm that the increased YAP1 protein level results from protein stability, the YAP1 protein was monitored after cycloheximide (CHX) treatment to inhibit protein translation in P1 and P4 hESCs (Fig. 5C). Similarly, the increased level of the YAP1 protein was evident in Dox-dependent TPX2 induction (Fig. 5D), whereas the YAP1 mRNA level remained unaffected (Fig. 5E). Aurora A active phosphorylation was clearly induced by TPX2 induction in iTPX2-hESCs, which was concurrent with the increase in YAP1 protein expression (Fig. 5F). Similar to P4 hESCs, the YAP1 protein but not TEAD4 (Supplementary Fig. 5c) was highly stabilized after Dox treatment in iTPX2-hESCs (Fig. 5G). These data suggest that high YAP1 protein levels in culture-adapted hESCs result from high Aurora A activity induced by TPX2.

A Immunoblotting of YAP1, BCL-xL, TPX2, and phospho-Aurora A (pAURKA) in P1 and P4 hESCs. β-Actin was used as a loading control for equal protein concentrations. B Immunoblotting of YAP1, BCL-xL, and TPX2 in P4 hESCs 2 days after the introduction of control (siNC) and TPX2 (siTPX2) siRNA. β-Actin was used as a loading control for equal protein concentrations. C Immunoblotting of YAP1 in P1 or P4 hESCs after 200 μg/ml cycloheximide (CHX) treatment, with β-actin as a loading control for equal protein concentrations. D Immunoblotting for YAP1 or TPX2 after treatment with the indicated dose of Dox for 24 h (arrow for induced GFP-TPX2, * for endogenous TPX2). α-Tubulin was utilized for equal protein loading. E The relative levels of TPX2 or YAP1 mRNA in iTPX2-hESCs after treatment with the indicated dose of Dox for 24 h. F Immunoblotting of YAP1, TPX2, and phospho-Aurora A (pAURKA) after 24 h treatment with 0.1 μg/ml Dox. G Immunoblotting of YAP1 in iTPX2-hESCs with or without 0.1 μg/ml Dox for the indicated time after treatment with 200 μg/ml CHX. β-Actin was utilized as a loading control for equal protein concentrations (top). Graph of the normalized band intensity (n = 4 independent experiments; mean ± SEM, two-way ANOVA, ***p < 0.001, bottom).
Inhibition of Aurora A abrogates the resistance to mitotic stress by YAP1 destabilization
Given that the activity of Aurora A in hESCs with high TPX2 expression appeared to stabilize YAP1, we next tested whether chemical inhibition of Aurora A would destabilize YAP1 and sensitize hESCs with high TPX2 expression to mitotic stress. To this end, we first determined the appropriate concentration of an Aurora A inhibitor (MLN8237: MLN) to inhibit Aurora A in hESCs. Intriguingly, Aurora A activity determined by its active phosphorylation was significantly reduced by 50 nM MLN treatment in P1 hESCs, whereas Aurora A remained active in P4 hESCs with up to 100 nM MLN (Supplementary Fig. 6a). Clear attenuation of active phosphorylation in P4 hESCs was distinct from treatment with 0.5 μM MLN (Fig. 6A). Therefore, all downstream experiments for P4 survival were performed using 0.5 μM MLN. In parallel with Aurora A inhibition by MLN (Fig. 6A), BCL2L1 transcription was reduced along with CTGF and SERPINE1, which are downstream genes of YAP-TEAD435,40 (Fig. 6B). Mitotic resistance in P4 hESCs, as determined by the sub-G1 populations, was also significantly attenuated by additional MLN treatment with Noc (Fig. 6C). The same result was obtained by flow cytometry analysis to determine the live cell population (Fig. 6D). Consistently, marked stabilization of YAP1 by Dox in iTPX2-hESCs was reversed by MLN treatment (Fig. 6E). The reduction in the YAP1 protein by MLN resulted from YAP1 protein destabilization by MLN (Fig. 6F), which further led to downregulation of the BCL-xL protein (Fig. 6G). Accordingly, MLN treatment significantly sensitized the GFP-positive iTPX2-hESC population to mitotic stress (Fig. 6H). In addition, treating P4 hESCs with LY3295668 (LY), a specific inhibitor of Aurora A41, markedly attenuated the level of active YAP1 (Supplementary Fig. 6b), and the BCL-xL protein sensitized P4 hESCs to mitotic stress (Supplementary Fig. 6c, d).

A Immunoblotting for phospho-Aurora A (pAURKA) after treatment with the indicated dose of MLN8237 (MLN) in P1 or P4 hESCs. Vinculin was used as the loading control. B Relative mRNA levels in P4 hESCs after 24 h of treatment with 0.5 μM MLN (n = 4 independent experiments; mean ± SEM, two-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001). C Flow cytometry analysis of DNA contents. The sub-G1 population (red arrow) is presented after 24 h of treatment with nocodazole (Noc: 50 ng/ml) with or without 0.5 μM MLN (left). Graphical presentation of each phase of the cell cycle (right). D Percentages of live cells quantified by flow cytometry with Annexin V/7-AAD staining. After 4 h of pretreatment with MLN, the cells were treated with 50 ng/ml nocodazole with or without 0.5 µM MLN for 24 h (n = 6 independent experiments; mean ± SEM, two-way ANOVA, ****p < 0.0001). E Immunoblotting for YAP1 in iTPX2-hESCs. After 24 h of treatment with 0.1 μg/ml Dox, 0.5 μM MLN was added for 24 h of treatment with or without Dox. β-Actin was used as an equal loading control. F Immunoblotting for YAP1 in iTPX2-hESCs. YAP1 protein levels after 6 h after treatment with CHX (induction with 0.1 μg/ml TPX2 for 24 h prior to the experiment and then pretreatment with 0.5 μM MLN occurred 1 h prior to CHX treatment). The normalized YAP1 protein band intensity is shown (n = 3 independent experiments; mean ± SEM, two-way ANOVA, **p < 0.01). G Immunoblotting for YAP1 or BCL-xL in iTPX2-hESCs 24 h after treatment with 0.5 µM MLN. H The relative of GFP-positive or GFP-negative live iTPX2-hESC populations (n = 4 independent experiments; mean ± SEM, two-way ANOVA. ****p < 0.000).
Discussion
Despite the widespread concerns regarding the frequent genetic aberrations and consequently acquired survival trait of hESCs3, the molecular mechanisms underlying such drastic phenotypic changes have not been fully determined, with the exception of the known role of BCL2L1 induction followed by 20q11.21 CNV8,10,11. Here, we demonstrated that BCL2L1 transcription was induced by YAP1 stabilization due to high Aurora A activity via TPX2 induction at locus 20q11.21. Furthermore, BCL2L1 induction (Fig. 2) followed by YAP1 protein stabilization (Fig. 5) was readily achieved by TPX2 induction without 20q11.21 CNV (Fig. 2I), resulting in survival under mitotic stress (Fig. 3). In addition, human iPSCs with 20q11.21 CNV occurring in a relatively early passage (passage 30) failed to acquire the survival trait (data not shown), whereas late-passage iPSCs (e.g., passage 180) and gain of 20q11.21 (Supplementary Fig. 1d) with high TPX2 and BCL2L1 expression (Supplementary Fig. 1e), showed evident survival traits8. Therefore, it was readily surmised that additional event(s) would occur to trigger BCL2L1 expression in hPSCs with 20q11.21 CNV to gain survival traits.
This result is consistent with a recent study showing that human iPSCs with 20q11.21 CNV only have lesser commitment of the ectodermal lineage in teratoma with minor changes in the expression of genes in 20q11.21 loci42. Therefore, we propose that other cues to activate BCL2L1 promoter activity, such as YAP1 activation, would occur, leading to the acquisition of the survival advantage trait (e.g., YAP1 stabilization by Aurora A activity due to TPX2 induction). A recent study demonstrated that the Hedgehog signal is responsible for TPX2 induction through the FOXM1 transcription factor in cancer cells30. Considering the complexity of the YAP1 activation mechanism, we could not rule out the role of other events [e.g., F-actin stabilization25] in YAP1 activation to induce BCL2L1 expression for culture adaptation of hESCs other than the TPX2-Aurora A axis.
TPX2, with basal expression that is higher in normal hESCs than in differentiated cells17, was critical for the self-renewal of hESCs, as stable depletion of TPX2 failed to be achieved after multiple attempts (data not shown). Thus, only transient depletion of TPX2 with two independent siRNA sequences was performed. These results further support that TPX2 knockout leads to the failure of early embryogenesis prior to blastocyst formation32. As TPX2 induction stabilizes the mitotic spindle and leads to abnormal mitosis17, TPX2 induction occurring in multiple culture-adapted cell lines would serve as a potential driver for not only acquired survival but also further genetic alterations by saving hESCs from cell death after abnormal mitosis.
Unlike other somatic cells, hESCs are highly sensitive to genotoxic stress, which is believed to be a major safeguard system to ensure genome integrity43. This extremely high susceptibility to DNA damage is mostly triggered by high mitochondrial priming to apoptosis16 through p53 mitochondrial translocation13. Thus, p53 dominant-negative mutation12, NOXA mutation9, and BCL2L1 induction10 abrogate the unique genome safeguard mechanism of high mitochondrial priming, which is commonly observed in culture-adapted hESCs. Therefore, the incidence of random mutations, including CNV at 20q11.21 (where TPX2 and BCL2L1 are located), during prolonged culture favors survival via TPX2 induction to activate YAP1-dependent BCL2L1 induction in hESCs with 20q11.21 CNV would be sufficient for dominant selection during multiple bottleneck events44. Thus, continuous loss of high mitochondrial priming and building resistance to mitotic stress may collectively enhance chromosome instability and even induce aneuploidy during long-term hESC culture.
Data availability
Source data are available from the corresponding author upon request. The RNA-seq results have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE 167495. The flow cytometry data were deposited to https://flowrepository.org/id/FR-FCM-Z435.
References
Lund, R. J., Narva, E. & Lahesmaa, R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 13, 732–744 (2012).
Heslop, J. A. et al. Concise review: workshop review: understanding and assessing the risks of stem cell-based therapies. Stem Cells Transl. Med. 4, 389–400 (2015).
Andrews, P. W. et al. Assessing the Safety of Human Pluripotent Stem Cells and Their Derivatives for Clinical Applications. Stem Cell Rep. 9, 1–4 (2017).
Halliwell, J., Barbaric, I. & Andrews, P. W. Acquired genetic changes in human pluripotent stem cells: origins and consequences. Nat. Rev. Mol. Cell Biol. 21, 715–728 (2020).
Baker, D. E. et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 25, 207–215 (2007).
Spits, C. et al. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. 26, 1361–1363 (2008).
Lefort, N. et al. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat. Biotechnol. 26, 1364–1366 (2008).
Cho, S. J. et al. Selective Elimination of Culture-Adapted Human Embryonic Stem Cells with BH3 Mimetics. Stem Cell Rep. 11, 1–13 (2018).
Zhang, J. et al. Anti-apoptotic Mutations Desensitize Human Pluripotent Stem Cells to Mitotic Stress and Enable Aneuploid Cell Survival. Stem Cell Rep. 12, 557–571 (2019).
Avery, S. et al. BCL-XL mediates the strong selective advantage of a 20q11.21 amplification commonly found in human embryonic stem cell cultures. Stem Cell Rep. 1, 379–386 (2013).
Nguyen, H. T. et al. Gain of 20q11.21 in human embryonic stem cells improves cell survival by increased expression of Bcl-xL. Mol. Hum. Reprod. 20, 168–177 (2014).
Merkle, F. T. et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233 (2017).
Lee, M. O. et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl Acad. Sci. USA 110, E3281–E3290 (2013).
TeSlaa, T., Setoguchi, K. & Teitell, M. A. Mitochondria in human pluripotent stem cell apoptosis. Semin. Cell Dev. Biol. 52, 76–83 (2016).
Dumitru, R. et al. Human Embryonic Stem Cells Have Constitutively Active Bax at the Golgi and Are Primed to Undergo Rapid Apoptosis. Mol. Cell. 46, 573–583 (2012).
Liu, J. C. et al. High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 13, 483–491 (2013).
Jeong, H.-C. et al. TPX2 Amplification-Driven Aberrant Mitosis in Long-Term Cultured Human Embryonic Stem Cells. bioRxiv https://doi.org/10.1101/2021.02.22.432205 (2021).
Lamm, N. et al. Genomic Instability in Human Pluripotent Stem Cells Arises from Replicative Stress and Chromosome Condensation Defects. Cell Stem Cell 18, 253–261 (2016).
Thompson, O. et al. Low rates of mutation in clinical grade human pluripotent stem cells under different culture conditions. Nat. Commun. 11, 1528 (2020).
Weissbein, U., Benvenisty, N. & Ben-David, U. Quality control: Genome maintenance in pluripotent stem cells. J. Cell Biol. 204, 153–163 (2014).
Liu, J. C., Lerou, P. H. & Lahav, G. Stem cells: balancing resistance and sensitivity to DNA damage. Trends Cell Biol. 24, 268–274 (2014).
Morin-Kensicki, E. M. et al. Defects in Yolk Sac Vasculogenesis, Chorioallantoic Fusion, and Embryonic Axis Elongation in Mice with Targeted Disruption of Yap65. Mol. Cell Biol. 26, 77–87 (2006).
Lian, I. et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 24, 1106–1118 (2010).
Leblanc, L. et al. Yap1 safeguards mouse embryonic stem cells from excessive apoptosis during differentiation. eLife 7, 1–26 (2018).
Ohgushi, M., Minaguchi, M. & Sasai, Y. Rho-Signaling-Directed YAP/TAZ Activity Underlies the Long-Term Survival and Expansion of Human Embryonic Stem Cells. Cell Stem Cell 17, 448–461 (2015).
Rosenbluh, J. et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473 (2012).
Kim, K. T. et al. Safe scarless cassette-free selection of genome-edited human pluripotent stem cells using temporary drug resistance. Biomaterials 262, 120295 (2020).
Go, Y. H. et al. Structure-Activity Relationship Analysis of YM155 for Inducing Selective Cell Death of Human Pluripotent Stem Cells. Front. Chem. 7, 298 (2019).
Markouli, C. et al. Gain of 20q11.21 in Human Pluripotent Stem Cells Impairs TGF-β-Dependent Neuroectodermal Commitment. Stem Cell Rep. 13, 163–176 (2019).
Wang, Y. et al. The critical role of dysregulated Hh-FOXM1-TPX2 signaling in human hepatocellular carcinoma cell proliferation. Cell Commun. Signal. 18, 116 (2020).
Warner, S. L. et al. Validation of TPX2 as a potential therapeutic target in pancreatic cancer cells. Clin. Cancer Res. 15, 6519–6528 (2009).
Aguirre-Portoles, C. et al. Tpx2 controls spindle integrity, genome stability, and tumor development. Cancer Res. 72, 1518–1528 (2012).
Reid, T. A. et al. Suppression of microtubule assembly kinetics by the mitotic protein TPX2. J. Cell Sci. 129, 1319–1328 (2016).
Meyer-Ficca, M. L. et al. Comparative analysis of inducible expression systems in transient transfection studies. Anal. Biochem. 334, 9–19 (2004).
Zhao, B. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 (2008).
Hansen, C. G., Moroishi, T. & Guan, K. L. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 25, 499–513 (2015).
Guo, X. et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 31, 247–259 (2017).
Price, C. J. et al. Genetically variant human pluripotent stem cells selectively eliminate wild-type counterparts through YAP-mediated cell competition. Dev. Cell 56, 2455–2470.e2410 (2021).
Chang, S. S. et al. Aurora A kinase activates YAP signaling in triple-negative breast cancer. Oncogene 36, 1265–1275 (2017).
Choi, J. Y. et al. TGFbeta promotes YAP-dependent AXL induction in mesenchymal-type lung cancer cells. Mol. Oncol. 15, 679–696 (2021).
Du, J. et al. Aurora A-Selective Inhibitor LY3295668 Leads to Dominant Mitotic Arrest, Apoptosis in Cancer Cells, and Shows Potent Preclinical Antitumor Efficacy. Mol. Cancer Ther. 18, 2207–2219 (2019).
Jo, H. Y. et al. Functional in vivo and in vitro effects of 20q11.21 genetic aberrations on hPSC differentiation. Sci. Rep. 10, 18582 (2020).
Weissbein, U., Benvenisty, N. & Ben-David, U. Genome maintenance in pluripotent stem cells. J. Cell Biol. 204, 153–163 (2014).
Barbaric, I. et al. Time-lapse analysis of human embryonic stem cells reveals multiple bottlenecks restricting colony formation and their relief upon culture adaptation. Stem Cell Rep. 3, 142–155 (2014).
Acknowledgements
We thank Dr. Minh Dang Nguyen at the University of Calgary for kindly providing the TPX2 constructs. This work was supported by a grant from the National Research Foundation of Korea (NRF) (grant number 2020M3a9E4037905), the Korean Fund for Regenerative Medicine funded by the Ministry of Science and ICT, and the Ministry of Health and Welfare (grant number RS-2022-00070316), Republic of Korea and Research Fund from Seoul National University.
Author information
Authors and Affiliations
Contributions
H.J.C. conceived the overall study design, led the experiments and wrote the paper. Y.J.K. conducted the experiments, data analysis and wrote the first draft of the paper. Y.H.G. generated the cell lines and performed the initial characterization. H.C.J. provided the key preliminary data. E.J.K., S.M.K., H.S.C., W.K., H.D.S., and H.L. provided the array data and performed data analysis. All authors contributed to the writing and revision of the paper and endorsed the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, YJ., Go, YH., Jeong, HC. et al. TPX2 prompts mitotic survival via the induction of BCL2L1 through YAP1 protein stabilization in human embryonic stem cells. Exp Mol Med 55, 32–42 (2023). https://doi.org/10.1038/s12276-022-00907-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00907-9
- Springer Nature Limited
This article is cited by
-
Epigenetic repression of CHCHD2 enhances survival from single cell dissociation through attenuated Rho A kinase activity
Cellular and Molecular Life Sciences (2024)
-
Inhibition of the AURKA/YAP1 axis is a promising therapeutic option for overcoming cetuximab resistance in colorectal cancer stem cells
British Journal of Cancer (2024)