
Abstract
Death is the inevitable fate of all living organisms, whether at the individual or cellular level. For a long time, cell death was believed to be an undesirable but unavoidable final outcome of nonfunctioning cells, as inflammation was inevitably triggered in response to damage. However, experimental evidence accumulated over the past few decades has revealed different types of cell death that are genetically programmed to eliminate unnecessary or severely damaged cells that may damage surrounding tissues. Several types of cell death, including apoptosis, necrosis, autophagic cell death, and lysosomal cell death, which are classified as programmed cell death, and pyroptosis, necroptosis, and NETosis, which are classified as inflammatory cell death, have been described over the years. Recently, several novel forms of cell death, namely, mitoptosis, paraptosis, immunogenic cell death, entosis, methuosis, parthanatos, ferroptosis, autosis, alkaliptosis, oxeiptosis, cuproptosis, and erebosis, have been discovered and advanced our understanding of cell death and its complexity. In this review, we provide a historical overview of the discovery and characterization of different forms of cell death and highlight their diversity and complexity. We also briefly discuss the regulatory mechanisms underlying each type of cell death and the implications of cell death in various physiological and pathological contexts. This review provides a comprehensive understanding of different mechanisms of cell death that can be leveraged to develop novel therapeutic strategies for various diseases.
Similar content being viewed by others
Introduction
Cell death is a biological process that results in the cessation of cell function and, eventually, cell death1. Its main function is to maintain tissue homeostasis by removing nonfunctional, damaged, and harmful cells2. Although this is a natural process involved in tissue formation, maintenance, and repair, it can also be triggered in response to injury, disease, or damage, leading to pathological cell death3,4,5.
Until the 19th century, death was understood only at the individual organism level, and the concept of cell death was not readily accepted by physicians and biologists. After the development of light microscopy, tissue sectioning practices, and staining techniques, death at the cellular level was recognized6,7. Despite cellular theory advocates such as botanist Mathias Jakob Schleiden and zoologist Theodore Schwann in the 1830s–1860s, the use of microscopes in medicine was limited. The application of technical advances in light microscopy and the concept that organisms are composed of cells to medicine was led by the German pathologist Rudolf Virchow6. In his papers published in 18558 and a book, “Cellular Pathology,” based on a compilation of lectures he gave in 18589, Virchow formalized cellular pathology as the fundamental basis of pathology. This new perspective radically changed the way pathology was viewed1,6. Virchow first recognized necrosis as death at the cellular level during his study of cellular changes accompanying tissue damage caused by inflammation6,10.
In 1842, German anatomist Carl Vogt first proposed that spontaneous cell death was a physiological phenomenon. He reported that cell death during metamorphosis in the midwife toad eliminated the notochord and allowed vertebrae to develop7. In 1882, the Russian biologist Elie Metchnikoff, considered a pioneer in modern immunology, observed that phagocytic cells engulfed dying cells in several organisms11. Metchnikoff’s discovery meaningfully contributed to our understanding of the role of the immune system in eliminating dying cells and maintaining tissue homeostasis. Later, in 1972, pathologist John F. Kerr and his colleagues discovered a kind of cell death that differed from necrosis, which they named “apoptosis”12,13. The discovery of apoptosis was a fundamental hallmark in the study of cell death and expanded our understanding of various types of cell death.
Traditional classifications of cell death include necrosis and programmed cell death (PCD). Necrosis, a nonprogrammed form of cell death, is often caused by traumatic injury; PCD, a controlled form of cell death, results from a series of molecular events in response to various physiological or developmental signals14. Apoptosis is a well-characterized PCD mechanism15. Other types of PCD, including autophagic cell death16, lysosomal cell death17, mitoptosis18, paraptosis19, pyroptosis20, NETosis21, necroptosis22, immunogenic cell death23, entosis24, methuosis25, parthanatos26, ferroptosis27, autosis28, alkaliptosis29, oxeiptosis30, cuproptosis31, and erebosis32, have also been identified (Fig. 1; Table 1). To date, the study of cell death is a major field of research in biology2. Morphological features are the primary basis for the traditional classification of cell death. In 2018, the Nomenclature Committee on Cell Death (NCCD) published comprehensive information, which expanded our knowledge of cell death pathways, and the assays commonly used in cell death study2. It emphasized the importance of accurately characterizing and differentiating different types of cell death and highlighted the importance of molecular pathways, genetic factors, biochemical markers, and functional criteria. With increasing understanding of the complexity of cell death, this classification system became more complicated and now includes additional categories. Understanding the various types of cell death and their regulatory mechanisms is essential for evaluating the pathogenesis of various illnesses, such as cancer, neurodegenerative diseases, and autoimmune disorders4,5.

This timeline depicts the important discoveries and advancements in cell death research, including the recognition of multiple forms of cell death.
In this historical review, we provide an overview of different types of cell death and the morphological and biochemical characteristics of cells undergoing these types of cell death. We also discuss the current understanding of the molecular mechanisms underlying the different types of cell death and the significance of these mechanisms in normal physiology and disease. We hope that this review serves as an excellent resource for researchers investigating cell death.
Discovery of diverse types of cell death
Necrosis
Necrosis is an uncontrolled form of cell death that is triggered in response to injury, trauma, or infection33. The term “necrosis” comes from the Greek words “nekros,” meaning dead or corpse, and “osis,” meaning process, referring specifically to destruction and degeneration34. It is unclear who initially coined the term necrosis, but the earliest reference to necrosis recently found on PubMed is “Observation on Necrosis” written by Bouffelin, a surgeon in the Polish army, in 178635. In the literature, necrosis refers to tissue necrosis caused by infection and inflammation, not cell death. As mentioned in the Introduction, Rudolf Virchow was the first scientist to use the term necrosis at the cellular level. He defined necrosis at the cellular level as “the mortified cell is left in its external form” and “necrobiosis or shrinkage necrosis being where the cell vanishes and can no longer be seen in its present form”6,10. In 1877, Carl Weigert and Julius Konheim described certain lesions as exhibiting coagulative necrosis, a basic form of necrosis, and necrobiosis1,6.
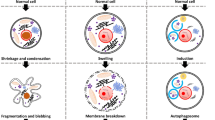
The cellular mechanisms that lead to necrosis are complex and not yet fully understood; however, they generally involve a series of events that result in the breakdown of cellular components and the release of cell contents into the extracellular space (Fig. 2A)36. In contrast to apoptosis, necrosis is frequently associated with inflammation and damage to surrounding tissues because the intracellular contents released by dying cells can activate the immune system and harm neighboring cells37.

A Hallmarks of necrosis and apoptosis are illustrated. Necrosis is an uncontrolled and pathological form of cell death, marked by cell swelling, membrane rupture, and intracellular content release, leading to inflammation and tissue damage. In contrast, apoptosis is a tightly controlled form of cell death that involves characteristic morphological features, such as cell shrinkage, chromatin condensation, membrane blebbing, nuclear fragmentation, and apoptotic body formation. B The two signaling pathways that lead to apoptosis are described. The extrinsic pathway is initiated by the binding of death ligands, such as tumor necrosis factor (TNF)-α or Fas ligand (FasL), to death receptors, which activates caspase 8. The intrinsic pathway, regulated by the Bcl-2 family, is triggered by intracellular stressors, such as DNA damage and oxidative stress, resulting in the release of cytochrome c from mitochondria and activation of caspase 9. The two pathways ultimately converge on caspase 3, which mediates the execution of apoptosis.
Apoptosis
Apoptosis is characterized by the organized breakdown of cells in response to particular signals38. The term “apoptosis” comes from the Greek words “apo,” meaning leaf, and “ptosis,” meaning “falling off,” which describe the process of cells undergoing controlled self-destruction and detachment from the surrounding tissue12. The apoptotic process was first described by Carl Vogt in 1842, and it was rediscovered and named “apoptosis” by Kerr in 197239. As mentioned in the Introduction, Carl Vogt was the first to present the concept of spontaneous cell death as a physiological phenomenon7. In 1885, Walther Flemming described and illustrated a cellular process called chromatolysis, which he discovered while studying the degeneration of ovarian follicles; Franz Nissen, who observed pigment degradation in lactating mammary glands, published similar results1,6. In 1914, Ludwig Gräffer published a paper proposing the premise that some mechanism must balance mitosis and that this process likely involves the process Fleming described as chromatolysis; this paper was subsequently rediscovered by Alfred Glücksmann in 19511. Subsequently, experimental pathologists began to investigate more intensively how cells in different organs die during development and in response to stimuli or injuries.
We now know that spontaneous cell death, such as chromatolysis, is caused by apoptosis. The description of apoptosis as a distinct form of cell death that differs from necrosis was formalized in the early 1970s by Australian pathologist John Kerr and his colleagues6,39. As a graduate student in London in 1962, Kerr found that when the portal venous blood supply to the liver is cut off, a different type of death occurs in cells around distal hepatic veins. This form of cell death was characterized by cytoplasmic shrinkage and condensed nuclear chromatin fragments, in addition to certain manifestations of classical necrosis. Upon further investigation using histochemical and electron microscopy, the newly discovered mode of cell death appeared to be nondegenerative in nature and was called shrinkage necrosis40. In 1972, Kerr was on sabbatical at the University of Aberdeen in Scotland working with Professor Currie and graduate student Andrew Wiley, who realized that a newly characterized type of cell death was regulated by hormones and played an essential role in normal development. Recognizing the inappropriateness of using the term necrosis for cell death under physiological conditions, they proposed that this process be called apoptosis41,42.
Apoptosis is characterized by cell shrinkage, chromatin condensation, and fragmentation into small membrane-bound apoptotic bodies, which are phagocytosed by adjacent parenchymal cells, neoplastic cells, or macrophages15 (Fig. 2A). Apoptosis is a genetically regulated and controlled cell death process, whereas necrosis is an uncontrolled cell death process caused by external stimuli. Apoptosis leads to cell fragmentation and removal by phagocytic cells, whereas necrosis resulted in cell membrane rupture and inflammation43 (Fig. 2A).
Two main pathways lead to the activation of apoptosis: the extrinsic and intrinsic pathways44. The extrinsic pathway is activated by extracellular ligands (tumor necrosis factor [TNF]-α and Fas ligand [FasL]) binding to death receptors, namely, TNF and Fas receptors, respectively45,46. After extracellular ligands bind to death receptors, death-inducing signaling complexes (DISCs) are formed and recruit and activate initiator caspases, such as caspase-8 and caspase-1047. These initiator caspases then cleave and activate effector caspases, such as caspase-3, -6, and -7, leading to the degradation of intracellular components and the induction of apoptosis48. The intrinsic pathway is activated by intracellular stressors, such as DNA damage, oxidative stress, and loss of survival signaling, which lead to permeabilization of the outer mitochondrial membrane49. This pathway is regulated by the Bcl-2 family of antiapoptotic proteins (Bcl-2 and Bcl-xL), proapoptotic proteins (Bax and Bak), and BH3-only proteins (Bim and Bid)50. In response to intracellular stress, the activation of proapoptotic BH3-only proteins inhibits antiapoptotic proteins, allowing Bax and Bak to form mitochondrial pores and release cytochrome c into the cytosol51. The released cytochrome c forms an apoptosome with apoptotic protease activating factor-1 (Apaf-1) and activates caspase-9 (Fig. 2B)52.
Autophagic cell death
Autophagic cell death, also known as type 2 cell death, occurs as a result of the activation of the autophagy pathway53. Autophagy is a cellular process in which cytoplasmic components, including organelles and macromolecules, are sequestered in double-membrane autophagosomes and targeted for lysosomal degradation54. The term “autophagy” comes from the Greek words “auto,” meaning self and “phagy,” meaning eating or devouring, and describes the process by which cells degrade and recycle their components55. The term “autophagy” has been used since the mid-19th century, but Christian de Duve defined the word as it is currently used in 1963 based on his research on lysosomal functions56. The mechanisms underlying autophagy were deduced in the 1990s with the identification of autophagy-related genes by Yoshinori Ohsumi57. Autophagic cell death was first described by Timo J. Nevalainen in 197558.
Under normal conditions, autophagy helps maintain cell homeostasis and recycle nutrients while removing toxic cellular components59. However, under certain conditions, such as nutrient deprivation, oxidative stress, or exposure to cytotoxic agents, autophagy can become dysregulated and result in cell death60.
The first step in the activation of autophagy is the formation of an isolation membrane, or phagophore, which is a double-membrane structure that sequesters cytoplasmic components to be degraded61. Phagophore formation requires the action of the unc-51-like kinase 1 (ULK1) complex, which is composed of several proteins, namely, ULK1, ATG13, FIP200/RB1CC1, and ATG10162. The ULK1 complex is regulated by several signaling pathways, including the mTOR pathway. Under normal conditions, the activation of mTOR suppresses autophagy by inhibiting the formation of the ULK1 complex; however, under stress or nutrient-deprivation conditions, mTOR inhibition leads to the formation of the ULK1 complex and the initiation of autophagy63. The phagophore expands and sequesters the cytoplasmic components to be degraded and then fuses with lysosomes to form autolysosomes, where the sequestered contents are degraded by lysosomal enzymes64. Autophagy is regulated by a complex network of proteins, including the ATG family of proteins, Beclin-1, and microtubule-associated protein light chain 3 (LC3)65. ATG proteins are involved in various steps in the autophagy pathway, including phagophore formation, elongation, and closure66. The Beclin-1 protein is required for the formation of the phagophore, whereas the LC3 protein is involved in the elongation and closure of the phagophore and the maturation of the autophagosome (Fig. 3A)61. Autophagy activation initially leads to the development of large cytoplasmic vacuoles, which in turn cause autophagic cell death16. Autophagy promotes both cell survival and cell death67. Notably, in some cases, autophagy contributes to cancer cell resistance to chemotherapy68. However, in other cases, autophagy can induce cell death and inhibit tumor growth69.

A The figure shows three types of autophagy, namely, macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy involves the formation of double-membrane vesicles that engulf cytoplasmic components and organelles and then fuse with lysosomes to form autolysosomes. Microautophagy involves engulfing cytoplasmic components and organelles directly into lysosomes. Chaperone-mediated autophagy degrades specific proteins via chaperone proteins that transport them to lysosomes. B The figure highlights the contrast between autophagy and autosis, two processes involving autophagy. While autophagic cell death is a result of excessive autophagy, autosis is characterized by three distinct phases characterized by cells with unique morphological features and is triggered by various signals, such as Na+/K+-ATPase, Tat-Beclin 1, and hypoxia–ischemia.
According to the NCCD, autophagic cell death is a regulated form of cell death that relies on autophagy machinery and can be prevented only by blocking autophagy2. Autophagic cell death has been implicated in various physiological and pathological processes, including cancer, neurodegeneration, ischemic injury, and heart disease70,71,72,73,74. However, the precise role of autophagy in cell death is still unclear16. Therefore, further research is necessary to clarify the role of autophagy in cell death. This research may have important implications for the understanding and treatment of various diseases.
Autosis
Autosis is an autophagy-dependent type of cell death that was discovered in 2013, and its name is derived from the Greek words “autos,” meaning self, and “osis,” meaning a process or condition75. Autosis is characterized by unique cell morphology and depends on cellular Na+/K+-ATPase activity75. Autosis is triggered by various signals, such as cerebral hypoxia–ischemia, nutrient deprivation, and autophagy-inducing peptides (Tat-Beclin)75.
The morphological features of autosis are acquired in three distinct phases: phase 1a, marked by a dilated and fragmented endoplasmic reticulum (ER) and an increase in the number of autophagosomes, autolysosomes, and empty vacuoles; phase 1b, involving swelling of the perinuclear space (PNS) in the presence of cytoplasmic materials and electron-dense mitochondria; and phase 2, marked by a reduced number of cytoplasmic organelles, focal nuclear concavity, and PNS ballooning (Fig. 3B)76. Autosis is triggered in vitro in Tat-Beclin 1-treated cells and in vivo in the brains of neonatal rats undergoing challenged with hypoxia–ischemia75. Rubicon levels significantly increase during autosis, which prevents the fusion of autophagosomes with lysosomes and inhibits autophagosome maturation and degradation77,78. Autosis is likely caused by the excessive accumulation of autophagosomes, which can deplete intracellular organelle membranes, such as ER and mitochondrial membranes, leading to reduced organelle function and, in the case of mitochondria, depolarization and loss of mitochondrial membrane potential79,80. Cardiac glycosides that inhibit the Na+/K+-ATPase pump can prevent autosis and contribute to the treatment of heart injuries75. However, the molecular mechanisms underlying the regulation of autosis by Na+/K+-ATPases remain unclear.
A recent study revealed that myxoma virus can infect and proliferate in human tumor cells but not in normal cells and that infected chimeric antigen receptor (CAR)-T and T-cell receptor (TCR)-T cells can efficiently trigger autotic cell death both in vitro and in vivo81. Autosis has also been observed in patients with severe liver diseases, including acute liver insufficiency associated with severe anorexia nervosa82. Although much remains unknown about autosis, its discovery has opened new avenues of research into the complex and diverse mechanisms underlying PCD.
Lysosomal cell death
Lysosomal cell death results from lysosomal membrane permeabilization, which causes the release of lysosomal enzymes into the cytoplasm and activation of cell death pathways83. The concept of lysosomal cell death was first proposed by Christian de Duve in the late 1990s84. Lysosomes are organelles that contain hydrolases critical for degrading intracellular and extracellular material. Under normal physiological conditions, lysosomes play roles in maintaining cellular homeostasis85,86. However, after the lysosomal membrane is damaged, lysosomal hydrolases are released into the cytoplasm, triggering various cell death pathways87.
Lysosomal cell death can be induced by various stimuli, including changes in lysosomal pH, oxidative stress, and lysosomotropic agents88. Lysosomal proteases, such as cathepsins, have been identified as potential causes of lysosomal cell death because they can be released into the cytoplasm and activate the lysosomal apoptotic pathway by cleaving Bid and degrading antiapoptotic Bcl-2 homologs following lysosomal injury and targeted destabilization of the lysosomal membrane (Fig. 4)89,90.

This figure illustrates lysosomal cell death caused by lysosomal membrane permeabilization and the release of lysosomal enzymes into the cytoplasm, leading to the activation of apoptotic cell death pathways. Lysosomal cell death can be induced by stimuli, such as changes in lysosomal pH, oxidative stress, and lysosomotropic agents. The release of lysosomal proteases, such as cathepsins, activates the lysosomal apoptotic pathway by cleaving Bid and degrading antiapoptotic Bcl-2 homologs.
Lysosomal cell death is thought to be involved in various pathological conditions, such as neurodegenerative diseases, cancer, and age-related disorders, and the inhibition of lysosomal cell death may hold therapeutic potential for these diseases91. Further research is necessary to fully understand the mechanisms underlying lysosomal cell death and its role in pathological conditions.
Mitoptosis
Mitoptosis, also known as mitochondrial suicide, is a PCD that involves dysfunctional mitochondria and was first proposed by Vladimir P. Skulachev in 199992. Dysfunctional mitochondria are associated with numerous diseases, including cancer, neurodegenerative disorders, and metabolic diseases93. Mitoptosis and mitophagy (autophagic degradation of mitochondria) are crucial for preventing the accumulation of dysfunctional mitochondria, which can lead to various cellular pathologies94.
Mitoptosis is triggered by mitochondrial dysfunction and reactive oxygen species (ROS) production95. Mitoptosis is involved in several biological processes, such as cell differentiation, hematopoietic stem cell self-renewal, metabolic remodeling, and elimination of paternal mitochondria in organisms for which mitochondrial DNA is maternally inherited96. The steps involved in mitoptosis include the fission of mitochondrial filaments to form spherical mitochondria, clustering of these spherical mitochondria in the perinuclear area, occlusion of these mitochondrial clusters via a membrane that forms a "mitoptotic body," decomposition of mitochondria inside this body into small membrane vesicles, protrusion of the body from the cell, and finally, disruption of the boundary membrane (Fig. 5A)95. Notably, autophagy is not involved in the mitoptotic process. Different forms of mitoptosis have been observed, including inner- and outer-membrane mitoptosis18. In inner membrane mitoptosis, only the internal matrix and cristae of mitochondria are degraded, and the external mitochondrial envelope remains unaltered18. During outer membrane mitoptosis, the internal cristae of mitochondria swell and undergo fragmentation, and the outer mitochondrial membrane bursts, releasing the remnants of cristae into the cytoplasm18.

This figure illustrates the crucial processes of mitoptosis and mitophagy that maintain mitochondrial quality and prevent cell pathology. A Mitoptosis is characterized by several events, including mitochondrial fission, the clustering of spherical mitochondria in the perinuclear area, enwrapping of these clusters by a membrane to form a “mitoptotic body,” decomposition of mitochondria into small vesicles, protrusion of the body from the cell, and disruption of the boundary membrane. This process is driven by mitochondrial dysfunction and reactive oxygen species (ROS) production. B Mitophagy is a selective autophagic mechanism for the degradation of damaged or unnecessary mitochondria. This procedure requires activation of general autophagy and priming of injured mitochondria via the Pink1/Parkin signaling pathway. Autophagosomes engulf targeted mitochondria, which are then digested and degraded in lysosomes.
In contrast to mitoptosis, mitophagy selectively degrades damaged mitochondria through autophagy, which requires the induction of general autophagy and priming of damaged mitochondria mediated by the Pink1/Parkin signaling pathway (Fig. 5B)97. The main difference between mitoptosis and mitophagy is that mitoptosis targets dysfunctional mitochondria, which are subsequently degraded inside the “mitoptotic body,” leading to membrane disruption. In contrast, mitophagy selectively degrades damaged or otherwise undesirable mitochondria. Although both processes are important for maintaining cellular homeostasis by eliminating dysfunctional or excessive mitochondria, their mechanisms of action differ.
Mitoptosis is important for the elimination of damaged or dysfunctional mitochondria and the maintenance of cellular homeostasis18. Recent research on mitoptosis has been focused on understanding the mechanisms that regulate this process and developing strategies for targeting dysfunctional mitochondria in disease contexts.
Immunogenic cell death
Immunogenic cell death (ICD) is a type of PCD in which an immune response is triggered by the release of damage-associated molecular patterns (DAMPs) from dying cells, which attract immune cells to the site of cell death98. The ICD concept was first proposed by the group led by Guido Kroemer and Laurence Zitvogel in 200599.
During ICD, dying tumor cells express calreticulin on their surface, which functions as an “eat me” signal to dendritic cells (DCs) and other phagocytic cells100. This signaling promotes phagocytosis of the dying cells by the DCs, leading to the activation of an immune response. ICD also involves the release of DAMPs, such as ATP, high-mobility group box 1, and heat shock proteins (HSPs), from dying cells101. These DAMPs activate DCs and other immune cells, thereby promoting antigen presentation and immune activation102. Moreover, IFNγ and TNFα released by effector T cells attract and activate other immune cells, including natural killer cells and macrophages, which detect and eradicate cancer cells (Fig. 6)103.

This figure illustrates the mechanism of ICD and its potential as a cancer therapeutic strategy. During ICD, dying cells release damage-associated molecular patterns (DAMPs), such as ATP, high-mobility group box 1 (HMGB1), and heat shock proteins (HSPs), which activate dendritic cells (DCs) and other immune cells, promoting antigen presentation and immune activation. Effector T cells release interferon (IFN)-γ and TNFα, which activate other immune cells, such as natural killer cells and macrophages that detect and eliminate cancer cells.
ICD has emerged as a promising strategy for cancer therapy. It potentially enhances the effectiveness of cancer treatments, such as chemotherapy and radiotherapy, which in turn induce ICD in cancer cells104. ICD-based therapies provide long-lasting protection against cancer recurrence and metastasis by promoting immune responses against cancer cells.
Pyroptosis
Pyroptosis is a type of PCD that involves inflammation and is mediated by caspase-1105. It was first discovered by Brad Cookson and Molly Brennan in 2001; it is a novel form of caspase-1-dependent PCD in immune cells, such as macrophages and dendritic cells, that defends the body against intracellular pathogens106. However, in contrast to other forms of PCD, pyroptosis contributes to tissue damage in inflammatory disorders107.
Pyroptosis is initiated by the activation of pattern recognition receptors (PRRs) in response to pathogen-associated molecular patterns (PAMPs) or DAMPs, which trigger inflammasome assembly108. The inflammasome is a protein complex consisting of PRRs, adaptor proteins, and caspase-1, with caspase-1 cleaving gasdermin D (GSDMD) to produce an N-terminal GSDMD fragment that forms membrane pores109. Caspase-1 also activates the proinflammatory interleukins, interleukin-1 beta (IL-1β) and interleukin-18 (IL-18) activity, also via proteolysis110. The actions of caspases result in the release of proinflammatory cytokines and the recruitment of immune cells to the site of infection or injury (Fig. 7A)111.

A Pyroptosis is characterized by cell swelling, plasma membrane rupture, and the release of proinflammatory cytokines, such as interleukin (IL)-1β and IL-18. Pyroptosis is triggered by the activation of inflammasomes, cytoplasmic complexes that sense danger signals, and initiate a caspase-1-dependent cascade that ultimately leads to cell death. B NETosis is a process in which neutrophils release DNA fibers coated with antimicrobial peptides to trap and kill pathogens. During NETosis, neutrophils undergo marked morphological changes, including chromatin decondensation, nuclear envelope rupture, and granule mixing, leading to the formation of neutrophil extracellular traps (NETs). The release of NETs is triggered by various stimuli, such as pathogens, cytokines, and immune complexes. C Necroptosis is mediated by death receptors. Upon activation of death receptors, such as TNFR1, receptor-interacting protein kinase 1 (RIPK1) binds to RIPK3 to form a necrosome. The necrosome complex promotes the oligomerization and phosphorylation of the mixed lineage kinase domain-like protein (MLKL). The oligomeric form of MLKL is translocated from the cytosol to the plasma membrane, leading to the formation of membrane pores and subsequent plasma membrane rupture. This results in the release of damage-associated molecular patterns (DAMPs), which trigger inflammation.
Pyroptosis has been implicated in several pathological conditions, including infectious diseases, autoimmune disorders, cancer, and neurodegenerative diseases112,113. Studies have shown that inhibition of pyroptosis can alleviate inflammation and tissue damage in the contexts of these conditions114,115. Therefore, targeting pyroptosis may be a potential therapeutic strategy for the treatment of inflammatory diseases.
NETosis
NETosis is a type of PCD characterized by the release of neutrophil extracellular traps (NETs) into the extracellular space116. NETs are web-like structures composed of chromatin, histones, and granular proteins that are released by neutrophils, a type of white blood cell, to capture and kill invading pathogens, including bacteria, viruses, and fungi117. NETosis was first described by Volker Brinkmann et al. in 2004118.
The mechanism of NETosis activation involves a series of complex molecular events, including ROS production, nuclear envelope disassembly, chromatin decondensation, and NET release (Fig. 7b)116. One of the key events in NETosis is the activation of the NADPH oxidase complex, which depends on an increase in the cytoplasmic concentration of Ca2+ and subsequent ROS production116. When ROS are activated, protein complexes known as “azurosomes” dissociates from azurophil granules dissociates and causes NE, cathepsin G, azurocidin, and MPO to be released into the cytosol, where they contribute to chromatin decondensation and nuclear envelope disintegration119. Another important factor in NETosis is peptidyl-arginine deaminase 4 (PAD4), which is transferred from the cytoplasm to the nucleus to catalyze the citrullination of histones, leading to chromatin decondensation120. Histones can also undergo acetylation during NETosis; however, the role of this process is not clearly understood121. In the final stage of NETosis, pores are formed in the plasma membrane, and chromatin is released into the extracellular environment, and NETs are formed. GSDMD plays a critical role in the formation of these membrane pores. In contrast to pyroptosis, in which GSDMD is activated through caspase-induced cleavage, NETosis is activated mainly by NE122,123. In addition, both the ER and mitochondria play important roles in NETosis. NETosis is initiated by calcium release from the neutrophil ER, which triggers the assembly of the NADPH oxidase complex and the generation of ROS124,125,126. Mitochondrial ROS production also promotes NETosis, potentially by regulating NADPH oxidase activity127. Overall, the coordinated activation of multiple pathways and organelles is required for successful NETosis.
NETosis plays an important role in the innate immune response because it allows neutrophils to directly combat pathogens128. However, excessive or inappropriate NETosis can contribute to the development of inflammatory and autoimmune diseases, such as sepsis, rheumatoid arthritis, lupus, and cancer129,130. Therefore, regulation of NETosis is a topic of ongoing research in the field of immunology.
Necroptosis
Necroptosis is a form of PCD that differs from necrosis and apoptosis in terms of morphology and biochemistry131. Necroptosis was first identified and described by Dr. Francis Chan et al. in 2005132. It is mediated by a signaling cascade involving the activation of receptor-interacting protein kinase 1 (RIPK1) and RIPK3 and the formation of a complex called the necrosome133.
TNFα and TNFR1 ligation triggers a well-characterized necroptosis-inducing pathway134. Under normal conditions, stress signals activate caspase-8, leading to the initiation of apoptosis135. However, when caspase-8 activity is suppressed, RIPK1 and RIPK3 are activated, leading to necroptosis136. During TNF-induced necroptosis, RIPK1 can recruit RIPK3 through the RIP homotypic interaction motif (RHIM) to form necrosomes, which promote the oligomerization and phosphorylation of mixed lineage kinase domain-like protein (MLKL)137. The oligomeric form of MLKL is translocated from the cytosol to the plasma membrane, leading to the formation of membrane pores and the subsequent rupture of the plasma membrane, resulting in the release of DAMPs138. The released DAMPs are recognized by PRRs on immune cells, leading to the activation of inflammatory responses (Fig. 7c)108. This inflammatory response can contribute to the clearance of dead cells and the initiation of tissue repair processes139. However, excessive or prolonged inflammation can cause tissue damage and contribute to the pathogenesis of various diseases140.
Research results suggest that necroptosis is involved in the pathogenesis of several diseases, including neurodegenerative diseases, viral infections, ischemic injury, and cancer141,142,143,144. The inhibition of necroptosis has shown therapeutic potential in some disease models, making it an attractive target for drug development145.
Cuproptosis
Cuproptosis is a form of PCD triggered by copper (Cu)146. The term “cuproptosis” is derived from the Latin word “cuprum,” which means copper, and the Greek word “ptosis,” meaning falling off. This was first described and the term was initially defined by Tsvetkov, P. et al. in 2019 (Nat Chem Biol 15, 681–689, 2019)147. Cuproptosis differs from other types of oxidative stress-related cell death, such as apoptosis and ferroptosis, and is characterized by mitochondrial stress caused by the aggregation of lipoylated mitochondrial enzymes and the loss of Fe–S cluster proteins31.
Copper is an essential trace element that plays vital roles in various biological processes, including oxygen transport, energy production, and antioxidant defense148,149. However, excess copper can be toxic to cells and tissues, leading to a condition known as copper overload or copper toxicity150. Two mitochondrial proteotoxic stress pathways mediate cuproptosis. The mitochondrial matrix reductase ferredoxin 1 (FDX1) catalyzes the reduction of ES–Cu2+ to Cu+, releasing it into mitochondria151. FDX1 has also been identified as a novel effector of lipoylation that contributes to the accumulation of toxic lipoylated dihydrolipoamide S-acetyltransferase (DLAT)152. Cu+ binds to lipoylated DLAT, promoting the disulfide bond-dependent aggregation of lipoylated DLAT, which leads to the accumulation of toxic lipoylated DLAT and subsequent cuproptotic cell death153. In addition, FDX1-dependent degradation of Fe-S cluster proteins may favor cuproptosis (Fig. 8A)31. This type of cell death depends on the amount of copper in cells and the lipoylation status of tricarboxylic acid (TCA) cycle enzymes.

A Cuproptosis is triggered by the accumulation of copper. It results in mitochondrial stress due to the aggregation of lipoylated mitochondrial enzymes and the loss of Fe–S cluster proteins, which can be mediated by ferredoxin 1 (FDX1). B Ferroptosis is characterized by the depletion of intracellular glutathione and decreased activity of glutathione peroxidase 4 (GPX4), which leads to the accumulation of unmetabolized lipid peroxides and increased ROS production. Membrane damage is also a result of lipid peroxidation.
Cuproptosis has been implicated in several pathological conditions, such as Wilson’s disease, a genetic disorder characterized by the accumulation of copper in the liver, brain, and other organs154. Therefore, understanding the mechanisms underlying coproptosis may provide insights into the pathogenesis and treatment of copper-related diseases.
Ferroptosis
Ferroptosis is an iron-dependent form of PCD that involves the accumulation of lipid peroxides and oxidative stress leading to membrane damage155. Ferroptosis was first described in 2012 by a research team led by Brent Stockwell155. The name ferroptosis comes from the Latin word “ferrum,” meaning iron, and the Greek word “ptosis,” meaning falling, which together refer to the iron-dependent process of cellular demise27. Ferroptosis is thought to play a role in various physiological processes, including ischemia, cancer, and neurodegeneration156.
Ferroptosis is regulated by several factors, such as iron metabolism, lipid peroxidation, and antioxidant systems157. Ferroptosis is initiated by the accumulation of lipid peroxides generated through the oxidation of polyunsaturated fatty acids via lipoxygenases or other enzymes158. Lipid peroxides accumulate through the oxidation of polyunsaturated fatty acids, a process that can be further amplified via the iron-catalyzed Fenton reaction, generating ROS and hydroxyl radicals that attack and damage cellular components, particularly the cell membrane, resulting in cell death (Fig. 8B)159. The cystine/glutamate antiporter system imports cystine, a precursor of the antioxidant glutathione, and thus plays a significant role in regulating the accumulation of lipid peroxides and iron160. Glutathione neutralizes free radicals and ROS and protects cells from oxidative stress and lipid peroxidation161. Ferroptosis is characterized by the depletion of intracellular glutathione and decreased activity of glutathione peroxidase 4 (GPX4), leading to the accumulation of unmetabolized lipid peroxides and the production of high levels of ROS27,162. Other factors that can regulate ferroptosis include iron metabolism; the activity of lipid metabolism enzymes, such as acyl-CoA synthetase long-chain family member 4 (ACSL4); and the expression of genes involved in cell stress response pathways, such as the p53 pathway163,164. In cancer treatment, inhibition of the cystine/glutamate antiporter system induces ferroptosis165. In addition, the use of ferroptosis-inducing agents, such as erastin and RSL3, may become a novel approach to cancer therapy166.
Ferroptosis is a unique and important form of PCD with broad implications in various physiological processes and disease states. Further studies may provide new insights into the mechanisms underlying this form of cell death and potential therapeutic interventions.
Paraptosis
Paraptosis, the name of which is derives from the combination of “para”, meaning next to or related to, and “apoptosis,” is a type of PCD that was initially discovered by Sabina Sperandio et al. in 2000167. Paraptosis and apoptosis are typically induced simultaneously in cells. In contrast to apoptosis, paraptosis does not involve caspase activation or DNA fragmentation167. Paraptosis is characterized by the swelling and vacuolization of the ER and mitochondria, resulting in the formation of large cytoplasmic vacuoles168.
Multiple mechanisms can trigger paraptosis. Impaired proteostasis due to proteasomal inhibition or altered protein thiol homeostasis, as well as unbalanced ion homeostasis, can lead to paraptosis169. Paraptosis is characterized by cytoplasmic vacuolization resulting from swelling of the ER and mitochondria. The accumulation of misfolded proteins within the ER lumen leads to the development of an osmotic force that causes water to be drawn away from the cytoplasm, causing ER distension (Fig. 9)170. ER stress and dilation can contribute to the release of Ca2+ from the ER, which can cause mitochondrial Ca2+ overload via an intracellular Ca2+ flux mechanism located at the ER-mitochondrial axis and thus mitochondrial dilatation169. Stimulation of the MEK-2 and JNK pathways by IGF-IR, as well as its inhibition mediated by AIP-1/Alix, is known to promote paraptosis171. Paraptosis is believed to play a role in various physiological and pathological processes, including embryonic development, neurodegeneration, and cancerogenesis172. In cancer cells, paraptosis is induced by various chemotherapeutic agents, including the proteasome inhibitor bortezomib and histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA)173,174.

Paraptosis is characterized by the development of large vacuoles in the endoplasmic reticulum (ER) and mitochondria, ultimately leading to the formation of large cytoplasmic vacuoles. Impaired proteostasis, altered ion homeostasis, and ER stress cause paraptosis, resulting in the discharge of Ca2+ from the ER and accumulation of Ca2+ in mitochondria. Paraptosis can be facilitated by the activation of mitogen-activated protein kinase (MAPK) signaling pathways via IGF-IR and inhibited by AIP-1/Alix.
In summary, paraptosis is a type of PCD characterized by the formation of large cytoplasmic vacuoles and activation of multiple signaling pathways. Further research is required to fully elucidate the mechanisms underlying paraptosis and its potential as a therapeutic target for various diseases.
Methuosis
Methuosis is a nonapoptotic form of cell death characterized by the accumulation of vacuoles derived from macropinosomes, which are large endocytic vesicles175. The term methuosis is derived from the Greek word for “methuo,” meaning to drink to intoxication, and refers to the fact that the vacuoles in methuotic cells appear to be filled with an unknown substance25. Methuosis was first described by Overmeyer et al. in 2008176.
Methuosis is triggered by sustained high-level expression of the activated form of Ras (G12V) and chronic stimulation of Rac1177. This stimulation increases the rate of macropinocytic, which is the process of molecules uptake into the extracellular fluid through the formation of large vesicles called macropinosomes178. However, methuosis impairs macropinosome recycling by decreasing the pool of active Arf6, which is a protein involved in vesicle trafficking25. Thus, macropinosomes accumulate and fuse to form large vacuoles that displace the nucleus and other organelles in a cell. Eventually, the vacuoles become sufficiently large to rupture the cell membrane, leading to cell death (Fig. 10)179. However, the exact molecular mechanisms underlying this form of cell death are not fully understood.

This image depicts the working model of methuosis. Methuosis is initiated by prolonged high-level expression of RAS (G12V) and chronic activation of Rac1, which leads to enhanced macropinocytic activity. Moreover, this mechanism hampers macropinosome recycling by lowering the active Arf6 pool. Nascent macropinosomes, which are created from lamellipodial membrane projections, penetrate the cell and merge to form large fluid-filled vacuoles that, in contrast to typical macropinosomes, cannot be recycled. These vacuoles grow rapidly, resulting in a stable population with certain late endosomal features (Rab7 and LAMP1).
Methuosis has been observed in various cancer cell lines and has been proposed to be a potential therapeutic target for cancer treatment180,181. However, further research is required to fully understand the molecular mechanisms underlying methuosis and the potential of these mechanisms as targets for cancer therapy.
Entosis
Entosis is a nonapoptotic form of cell death in which one living cell actively internalizes and degrades another living cell175. Entosis is derived from the Greek word “entos,” meaning inside or within, and was first described by Overholtzer et al. in 2007182. The internalization of a living cell by another cells leads to the formation of a double-membrane vesicle called the entotic vacuole183. During normal development, entosis is thought to play a role in the removal of excess cells and in shaping tissues and organs184. In cancer, entosis has been shown to contribute to tumor growth and tumor cell invasion by facilitating the engulfment of neighboring cells183.
Entosis is triggered when cells detach from the extracellular matrix (ECM) leading to the internalization of one cell by another cell. Entosis requires the activation of multiple molecular signaling pathways. One of the crucial pathways involved in entosis is the Rho/Rho-associated protein kinase (ROCK)/actomyosin pathway, which regulates actin and myosin II activities and is essential for cell engulfment185. The Rho/ROCK/actomyosin signaling pathway is involved in various processes, including cell migration, division, and shape changes175. This pathway involves the activation of the Rho family of small GTPases, which activate downstream effectors, such as ROCK186. In turn, ROCK activates myosin II, a motor protein that generates contractile forces by interacting with actin filaments186. During entosis, an invading (engulfed) entotic cell forms an actin-rich structure that protrudes into the cytoplasm of the engulfing cell183. Myosin II is recruited to this structure and contracts it, pulling the invading cell into the engulfing cell187. The internalized cells are subsequently degraded by lysosomes within the engulfing cell (Fig. 11)182.

Entosis is a biological process characterized by the internalization of one living cell into the cytoplasm of another. It is caused by adherent cell matrix separation, which results in the establishment of E-cadherin-mediated cell connections (shown in red) between the engulfing cell and the entotic cell. RhoA activity within the entotic cell causes actomyosin buildup at the cell cortex, resulting in the creation of cell-in-cell structures that mimic an active invasion-like process. Most internalized cells die as a result of entotic cell death, which is followed by lysosome fusion or apoptosis, especially when macroautophagy has been inhibited. However, certain entotic cells may divide within their hosts or even escape death.
Entosis is induced by various stimuli, such as nutrient starvation and ultraviolet radiation. Nutrient starvation, particularly glucose starvation, plays a pivotal role in inducing entosis by activating AMP-activated protein kinase (AMPK) in internal cells188, while ultraviolet radiation induces JNK and p38 stress-activated kinase signaling to activate entosis189. Entosis differs from anoikis, which is triggered by a lack of cell attachment to the ECM but not by matrix detachment182. Entosis shares more similarities to cell invasion than to a cell engulfment mechanism182. Entosis is also distinct from phagocytosis, which involves phagocyte engulfment of a dead or dying cell190.
Recent studies provided crucial insights into the mechanisms underlying entosis and its relevance in cancer development191,192. Specifically, Orai1, a Ca2+ channel protein, has emerged as a key player in entosis193. Orai1 plays a critical role in regulating intracellular Ca2+ levels during entosis, thereby influencing the activation of signaling pathway involved in cell engulfment and degradation. Dysregulation of Orai1-mediated Ca2+ signaling has been implicated in enhanced entosis, tumor growth, and invasion194. Although much is still unknown about the molecular mechanisms underlying entosis, this form of cell death has emerged as an important area of research with potential implications for both developmental biology and cancer therapeutics.
Parthanatos
Parthanatos is a form of PCD that is mediated by the activation of poly ADP-ribose polymerase (PARP)195. The name is a combination of “PAR” and “thanatos” (the Greek word for death), reflecting the role of PAR-mediated cell death in various pathological conditions. It was first discovered by Karen Kate David in 200926. This type of cell death was first identified in neurons and plays an important role in several neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases196.
Parthanatos is triggered by various agents, such as ROS, hydrogen peroxide, ionizing radiation, and alkylating agents195. When DNA damage is mild, PARP-1 recruits DNA damage repair proteins to repair damaged DNA197. Severe DNA damage leads to PARP-1 overactivation and PAR polymer formation198. Accumulated PAR polymers bind to apoptosis-inducing factor (AIF) and mediate AIF release from mitochondria199. AIF interacts with MIF to form the AIF/MIF complex, which is translocated to the nucleus, causing DNA fragmentation and leading to parthanatos200 (Fig. 12).

This diagram depicts the molecular processes underlying parthanatos. ROS, ischemia, alkylating chemicals, and radiation activate PARP-1 by activating NOS, resulting in the creation of excess NO and subsequent synthesis of peroxynitrite (ONOO−). Peroxynitrite activates PARP-1, resulting in the formation of copious amounts of PAR polymer in the nucleus. Certain poly(ADP)-ribosylated carrier proteins escape from the nucleus, prompting the outer mitochondrial membrane to release apoptosis-inducing factor (AIF). AIF then enters the cytoplasm and attaches to macrophage migration inhibitory factor (MIF). AIF and MIF enter the nucleus and cause widespread DNA degradation, ultimately resulting in cell death.
Overall, parthanatos is a complex form of PCD that plays an important role in several disease processes. Further research is needed to fully understand the mechanisms underlying parthanatos and its potential therapeutic applications.
Alkaliptosis
Alkaliptosis is a recently discovered form of regulated necrosis triggered by exposure to alkaline agents, such as ammonia, sodium hydroxide, or high-pH buffers201. Alkaliptosis was first discovered and named by Daolin Tang in 2018201. Alkaliptosis is a sequential molecular mechanism modulated by several factors.
Alkaliptosis can be activated by the upregulation of nuclear factor-kappa B (NF-κB) pathways and subsequent downregulation of carbonic anhydrase 9 (CA9) (Fig. 13)29. CA9 is a member of the carbonic anhydrase family that plays a role in regulating pH levels29. NF-κB negatively regulates CA9 activity, which in turn inhibits alkaliptosis29. Depletion of CA9 can restored the sensitivity of cancer cells that lack functional NF-κB to alkaliptosis29,202. Another study showed that ACSS2-mediated NF-κB activation promoted alkaliptosis in human pancreatic cancer cells203. ACSS2 has been found in the nucleus and cytoplasm and provides AcCoA, which is important for lipogenesis and histone acetylation204. ACSS2 plays a role in alkaliptosis by maintaining NF-κB activation and increasing the pH value via histone acetylation in human PDAC cells203.

This figure illustrates the activation mechanism of alkaliptosis, which is characterized by intracellular alkalinization and subsequent cell death. JTC801 activates the IKK protein complex, which includes CHUK (IKKα), IKBKB (IKKβ), and IKBKG (IKKγ). Then, the IKK protein complex phosphorylates and degrades NFKBIA (IκBα), leading to the nuclear translocation of NFKB1 (p50) or RELA (p65), which regulate gene expression. Furthermore, NF-κB negatively regulates the expression of CA9, a member of the carbonic anhydrase family, to inhibit alkaliptosis.
Alkaliptosis is a promising strategy for cancer therapy because cancer cells have a profoundly unbalanced pH, and their proliferation, metastasis, and metabolic adaptation are determined by their pH sensitivity205. Drugs developed to target alkaliptosis may be a new approach to cancer treatment, especially for those resistant to conventional therapies29. However, further research is required to fully understand the mechanism of alkaliptosis and its potential for cancer therapy.
Oxeiptosis
The term “oxeiptosis” was coined by Holze et al. in 2018 to describe a caspase-independent, ROS-sensitive, and noninflammatory cell death pathway that protects against inflammation induced by ROS or ROS-generating agents, such as viral pathogens30. Oxeiptosis is characterized by the activation of the KEAP1/PGAM5/AIFM1 signaling pathway206. Under oxidative stress conditions, AIFM1 is dephosphorylated, and its activity is regulated by KEAP1 and PGAM5. Dephosphorylated AIFM1 is translocated from mitochondria to the nucleus, where it induces chromatin condensation and DNA fragmentation, leading to cell death (Fig. 14)30. Activation of the KEAP1/PGAM5/AIFM1 signaling pathway is a hallmark of apoptosis and differs from other cell death pathways, such as the apoptosis and necrosis pathways. The mechanisms underlying oxeiptosis are not yet fully understood, but it is thought to involve the activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway207.

This figure illustrates the key features of oxeiptosis. Oxeiptosis is activated in response to oxidative stress induced by ROS or ROS-generating agents, such as viral pathogens. The KEAP1/PGAM5/AIFM1 signaling pathway plays a central role in oxeiptosis, in which AIFM1 is dephosphorylated under oxidative stress conditions via the regulatory action of PGAM5. Dephosphorylated AIFM1 is translocated from mitochondria to the nucleus, leading to chromatin condensation and DNA fragmentation, ultimately resulting in cell death.
Many conditions, such as allergies, autoimmune diseases, allograft rejection, cancer, and infection with pathogens, lead to ROS production, indicating that oxeiptosis may be triggered under various pathological conditions208. Notably, excessive or dysregulated oxeiptosis can lead to tissue damage and contribute to disease development. Understanding the mechanisms underlying oxeiptosis and its role in health and disease is an active area of research that may identify new therapeutic intervention targets.
Erebosis
In 2022, Sa Kan Yoo et al. reported a novel form of cell death called erebosis during the natural turnover of gut enterocytes, which are the cells that make up the gut epithelium (Fig. 15)32. The term “erebosis” is derived from the Greek word “erebos,” meaning complete darkness32. Erebotic cells undergo membrane blebbing and actin cytoskeleton changes, eventually leading to cell disintegration32. This process is marked by the loss of cell adhesion, organelles, and fluorescence emitted from labeled proteins and the accumulation of angiotensin-converting enzyme (ACE)209. In contrast to cell undergoing apoptosis, necrosis, and autophagic cell death, cells undergoing erebosis do not exhibit distinguishing features. Notably, apoptosis inhibition does not affect apoptosis or gut cell turnover209. The authors suggested that erebosis may play a vital role in maintaining gut barrier function through the natural shedding of gut enterocytes32. In contrast, abnormal erebosis may contribute to the development of gastrointestinal conditions, such as inflammatory bowel disease32. Although this phenomenon has only been discovered in Drosophila, further research is required to determine whether it also occurs in other organisms, including humans. Such investigations may provide insights into the evolutionary origins and physiological significance of erebosis and enhance our understanding of gut physiology and related disorders.

This figure depicts the process of erebosis, a novel form of cell death observed during the natural turnover of enterocytes that constitute the gut epithelium. Nuclear expansion and accumulation of angiotensin-converting enzyme (ACE) are observed in the early stages of erebosis. Subsequently, cell shrinkage and nuclear fragmentation are observed. Late erebotic cells are surrounded by stem cells that eventually undergo division to generate new epithelial cells, contributing to the replenishment of the gut epithelium.
The complexity underlying cell death
The complexity of cell death classification
The categorization system for cell death is intrinsically complexity. Initially, cell death was classified primarily based on morphological characteristics, as described below2. Type 1 cell death, known as apoptosis, is characterized by cytoplasm shrinkage, chromatin condensation, membrane blebbing, and the formation of apoptotic bodies, which are cleared through phagocytosis and lysosomal degradation by the surrounding cells210. Type 2 cell death is characterized by intense autophagy or cytoplasmic vacuolization, leading to phagocytosis by neighboring cells and degradation via lysosomes210. Type 3 cell death, or necrosis, results in cell death without triggering phagocytosis or lysosomal degradation and is not characterized by any of the features used to identify type 1 and 2 cell death modalities210. As more forms of cell death were identified, type 4 cell death modalities were classified, and these types of cell death includes those that cannot be classified into one of the three previously defined categories211. However, the current classification system of cell death, based on morphological changes, is limited because newly discovered forms of cell death cannot be integrated into it. Furthermore, the existing classification system does not account for the increasingly recognized importance of molecular pathways, specifically their greater importance than morphological changes, in identifying forms of cell death210. Thus, there is a need for more comprehensive guidelines that are based on genetic, biochemical, and functional criteria.
To address this need, the NCCD issued guidelines on the “classification of cell death” in 2005212, 2009213, and 20182. The 2018 classification system aimed to establish a more comprehensive system based on genetic, biochemical, and functional criteria, not merely morphological features (Fig. 16)2. However, this system also has several limitations. First, certain forms of cell death, namely, paraptosis, methuosis, alkaliptosis, oxeiptosis, cuproptosis, and erebosis, were not classified. Second, although pyroptosis, NETosis, and necroptosis are categorized as different types of cell death67, these modalities all involve immunogenic cell death98. Finally, the current classification system may not account for the potential interplay and crosstalk among different forms of cell death because cell death processes are mediated via a complex network of interactions contributing to cellular processes that may not be fully reflected in the classification system.

This figure illustrates the classification of the different forms of cell death and nonlethal processes based on their underlying mechanisms and morphological features. This figure was generated according to the 2018 guidelines for the classification of cell death issued by the Nomenclature Committee on Cell Death (NCCD).
Complexity of the interconnectedness among different types of cell death
The interconnectedness among the various forms of cell death is an important factor that contributes to cell death complexity (Fig. 17). Necroptosis, a regulated form of necrosis, shares similarities with necrosis and apoptosis131,133. Autophagy is required for both autosis and autophagic cell death programs, which are triggered via different molecular pathways53,75. Similarly, cells undergoing parthanatos display morphological and cytological characteristics of both apoptosis and necrosis26. Recent studies have suggested that autophagy and ferroptosis pathways interact in a complex manner. Autophagy has been suggested to regulate ferroptosis by eliminating damaged mitochondria and peroxidized lipids214. However, excessive or prolonged autophagy can lead to ferritin degradation, which can trigger ferroptosis215.

This figure illustrates the complex and interconnected nature of cell death pathways. The figure shows the mechanisms by which different types of cell death pathways interact and influence each other and the ways in which they can be regulated by various signaling pathways and environmental factors.
Cell death is influenced by various factors, including cellular organelles and environmental conditions. Mitochondria play crucial roles in different types of cell death, including apoptosis, NETosis, paraptosis, parthanatos, and oxeiptosis216. For example, apoptosis is triggered by mitochondrial outer membrane permeabilization (MOMP), which leads to the release of cytochrome c and activation of the caspase cascade216,217. In cells undergoing NETosis, mitochondria produce mitochondrial ROS (mtROS), whereas in cells undergoing paraptosis, Ca2+ released from the ER causes mitochondrial Ca2+ overload and mitochondrial dilation218,219. Both parthanatos and oxeiptosis involve mitochondrial release of AIF, which is translocated from the mitochondria to the nucleus, resulting in cell death30,195.
Lysosomes play several roles in cell death. During lysosomal cell death, lysosomal membrane permeabilization results in the release of cathepsins, which activate apoptotic pathways83. During necrosis, lysosomal membrane permeabilization causes the release of lysosomal hydrolases and ROS, which cause cell damage and inflammation [183]. In cells undergoing autophagic death, lysosomes fuse with autophagosomes to degrade cellular components, leading to cell death220. Lysosomal exocytosis has been implicated in the induction of pyroptosis, a type of inflammatory cell death221. In addition, endothelial cells undergo lysosomal degradation182.
Similar to lysosomal factors, ROS trigger cell death in various ways. For example, ROS play a crucial role in activating NADPH oxidase, which is required for the degradation of azurophilic granules that trigger NETosis116. Moreover, ROS can cause lipid peroxidation, leading to ferroptosis27. Additionally, ROS can cause ER stress by inducing the accumulation of unfolded proteins, triggering paraptosis222. ROS production is also the primary cause of oxeiptosis initiation30. Furthermore, a rapid increase in the cytosolic pH vale can induce both lysosomal cell death and alkaliptosis17,29. Some types of cell death, such as paraptosis and methuosis, are caused by the formation of large vacuoles, highlighting the importance of organelle dysfunction in regulating cell death223.
Overall, the interconnectedness among the different types of cell death and their regulation via diverse signaling pathways and environmental factors highlight the complexity of cell death. Understanding the interplay among different signaling pathways and the impact of the cell context on the cell death modality is crucial for developing new therapeutic strategies that target cell death pathways for the treatment of various diseases. Further research is needed to fully characterize and differentiate among the various forms of cell death and their roles in health and disease.
PANoptosis: an emerging and complex form of cell death
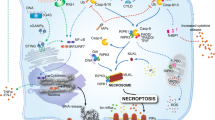
In addition to the different types of cell death that share the same molecular pathways, it has recently been shown that different types of cell death are mediated simultaneously in a single cell. This phenomenon was first described by Kanneganti et al. in 2016 when they studied inflammasome activation by influenza virus224 and named it PANoptosis in 2019 (Fig. 18)225. PANoptosis is triggered by the formation of a protein complex called the PANoptosome, which is composed of several proteins, including RIPK1, RIPK3, caspase-8, NLRP3, and ASC226. This complex activates various cell death pathways, including pyroptosis, apoptosis, and necroptosis, resulting in an inflammatory cell death response226.

PANoptosis is triggered by the formation of a protein complex called the PANoptosome, which includes several protein domains, namely, RIPK1, RIPK3, caspase-8, NLRP3, and ASC. This complex activates multiple types of cell death, including pyroptosis, apoptosis, and necroptosis, resulting in an inflammatory cell death response. During influenza A virus (IAV) infection, Z-DNA-binding protein (ZBP1) recognizes viral ribonucleoproteins and induces the formation of the ZBP1-dependent PANoptosome. TGF-β-activated kinase 1 (TAK1) is a crucial regulator of PANoptosis that negatively controls this process; however, bacterial infections can interrupt its suppression. Inhibition of TAK1 and activation of signaling through TLRs or death receptors promotes the formation of RIPK1-dependent PANoptosomes. During PANoptosis, the activation of caspase-1 or caspase-8 leads to the cleavage and activation of downstream effector proteins, such as gasdermin D and RIPK3, which drive pyroptosis and necroptosis, respectively. Activated caspase-8 subsequently cleaves and activates caspase-3, resulting in cell apoptosis.
During influenza A virus (IAV) infection, Z-DNA-binding protein (ZBP1) plays a critical role in activating PANoptosis by recognizing viral ribonucleoproteins and inducing the formation of the ZBP1-dependent PANoptosome224,227. This complex consists of ZBP1 (the sensor), RIPK3, RIPK1, NLRP3, ASC, caspase-1, caspase-8, and scaffold caspase-6228. TGF-β-activated kinase 1 (TAK1) is a crucial regulator of PANoptosis that negatively controls its initiation. Notably, bacterial infections can interrupt PANoptosis suppression via the action of the Yersinia T3SS effector YopJ, leading to PANoptosome formation225. When TAK1 was inhibited and signaling through TLRs or death receptors was activated, RIPK1-dependent PANoptosomes were formed229.
During PANoptosis, the activation of caspase-1 or caspase-8 leads to the cleavage and activation of downstream effector proteins, such as gasdermin D and RIPK3, which drive pyroptosis and necroptosis, respectively226. In addition, activated caspase-8 can cleave and activate caspase-3, leading to apoptosis230. The pathophysiological functions and importance of phagocytosis in relation to viral infections have been extensively studied during the COVID-19 pandemic; however, clear evidence of a phagocytic protein complex has yet to be established. Further research is required to identify this complex or eliminate the possibility that a complex is formed.
Conclusion and future perspectives
In the preceding sections, we discussed the many types of cell death and history of their discovery. Understanding the diverse and complex processes underlying cell death is crucial for understanding diseases and may be beneficial for the development of new therapies. The classification of cell death based on morphological features is limited because it cannot accommodate newly discovered forms of cell death and may not reflect the underlying molecular pathways that determine a form of cell death. Therefore, recent guidelines proposed by the NCCD aim to establish a more comprehensive classification scheme based on genetic, biochemical, and functional criteria2. Moreover, several new forms of cell death have been discovered that are not included in the latest NCCD classification, and some scholars have disputed the usefulness of this classification. We hope that the next NCCD consensus will produce a new cell death classification system that addresses the aforementioned issues.
Researchers have made significant strides in characterizing and distinguishing various forms of cell death, thereby advancing our understanding of the roles of these modalities in health and disease. The complex mechanisms underlying cell death are underscored by the intricate interconnections among different types of cell death and the regulation of these mechanism through diverse signaling pathways and environmental factors. In addition, the importance of crosstalk among signaling pathways and the influence of the cellular context on cell death outcomes has become increasingly evident. These findings pave the way for the development of novel therapeutic strategies targeting cell death pathways for the treatment of diverse diseases. Further research is crucial to characterize and differentiate various forms of cell death, to gain a better understanding of their roles in disease progression, and to develop targeted therapeutic strategies (Table 2). Ultimately, a comprehensive understanding of the multifaceted nature of cell death will be indispensable for the development of innovative and more efficacious treatments for a broad spectrum of diseases.
As we gain a better understanding of new types of cell death and their complexities, the study of cell death is becoming increasingly difficult. In particular, different processes of cell death are linked by molecular mechanisms and, in some cases, are potentially coactivated. Because of these connections, the specificities of molecular markers used to distinguish among different types of cell death are becoming increasingly ambiguous. Furthermore, whether the agents used to inhibit specific cell death are sufficiently specific is an ongoing concern. Developing new inhibitors with greater specificity or modulating key genes may solve the problems associated with lack of specificity among inhibitors. However, the emergence of complex forms of cell death suggests that the inhibition of only one type of cell death may not be sufficient to achieve therapeutic results. Therefore, future studies on cell death may require an integrated view of different types of cell death.
A scientist studying death of a cell can be compared with a forensic physician investigating a crime scene. However, in contrast to forensic physicians who focus on identifying the cause of a murder, scientists focused on cell death are primarily interested in understanding the mechanisms and types of PCD, that is, cell suicide. From a forensic standpoint, some may question the need for such a detailed classification and identification of cell death. However, understanding the processes and types of cell death is important to fully comprehend the manner in which cells resolve internal disharmony and maintain balance. Therefore, scientists interested only in cell suicide will play an increasingly important role in the cellular universe, similar to forensic scientists investigating crime scenes in the macroscopic world.
Change history
20 September 2023
A Correction to this paper has been published: https://doi.org/10.1038/s12276-023-01107-9
References
Majno, G. & Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 146, 3 (1995).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742–758 (2011).
Lowe, S. W. & Lin, A. W. Apoptosis in cancer. Carcinogenesis 21, 485–495 (2000).
Kroemer, G., Galluzzi, L., Kepp, O. & Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013).
Buja, L. M. The cell theory and cellular pathology: discovery, refinements and applications fundamental to advances in biology and medicine. Exp. Mol. Pathol. 121, 104660 (2021).
Clarke, P. G. & Clarke, S. Nineteenth century research on naturally occurring cell death and related phenomena. Anat. Embryol. (Berl.) 193, 81–99 (1996).
Virchow, R. Cellular-pathologie. Arch. für. Pathologische Anat. und Physiologie und für. Klinische Med. 8, 3–39 (1855).
Virchow, R. & Chance, F. N. Cellular Pathology as Based Upon Physiological and Pathological Histology: Twenty Lectures Delivered in the Pathological Institute of Berlin [luring the Months of February, March and April, 1858]. (P. Blakiston, Son & Company, 1880).
Cummings, M. C., Winterford, C. M. & Walker, N. I. Apoptosis. Am. J. Surg. Pathol. 21, 88–101 (1997).
Maghsoudi, N., Zakeri, Z. & Lockshin, R. A. Programmed cell death and apoptosis-where it came from and where it is going: from Elie Metchnikoff to the control of caspases. Exp. Oncol. 34, 146–152 (2012).
Kerr, J. F., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Kerr, J. F. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J. Pathol. Bacteriol. 90, 419–435 (1965).
Kanduc, D. et al. Cell death: apoptosis versus necrosis. Int. J. Oncol. 21, 165–170 (2002).
Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 (2007).
Kroemer, G. & Levine, B. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010 (2008).
Guicciardi, M. E., Leist, M. & Gores, G. J. Lysosomes in cell death. Oncogene 23, 2881–2890 (2004).
Jangamreddy, J. R. & Los, M. J. Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat. Monthly 12, e6159 (2012).
Lee, D., Kim, I. Y., Saha, S. & Choi, K. S. Paraptosis in the anti-cancer arsenal of natural products. Pharmacol. Ther. 162, 120–133 (2016).
Fang, Y. et al. Pyroptosis: a new frontier in cancer. Biomed. Pharmacother. 121, 109595 (2020).
Yipp, B. G. & Kubes, P. NETosis: how vital is it? Blood J. Am. Soc. Hematol. 122, 2784–2794 (2013).
Linkermann, A. & Green, D. R. Necroptosis. N. Engl. J. Med. 370, 455–465 (2014).
Galluzzi, L., Buqué, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Sun, Q. et al. Competition between human cells by entosis. Cell Res. 24, 1299–1310 (2014).
Maltese, W. A. & Overmeyer, J. H. Methuosis: nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am. J. Pathol. 184, 1630–1642 (2014).
David, K. K., Andrabi, S. A., Dawson, T. M. & Dawson, V. L. Parthanatos, a messenger of death. Front. Biosci. (Landmark Ed.) 14, 1116 (2009).
Li, J. et al. Ferroptosis: past, present and future. Cell Death Dis. 11, 88 (2020).
Liu, Y. et al. Autosis is a Na+, K+-ATPase–regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia–ischemia. Proc. Natl Acad. Sci. 110, 20364–20371 (2013).
Liu, J., Kuang, F., Kang, R. & Tang, D. Alkaliptosis: a new weapon for cancer therapy. Cancer Gene Ther. 27, 267–269 (2020).
Holze, C. et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 19, 130–140 (2018).
Tang, D., Chen, X. & Kroemer, G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 32, 417–418 (2022).
Ciesielski, H. M. et al. Erebosis, a new cell death mechanism during homeostatic turnover of gut enterocytes. PLoS Biol. 20, e3001586 (2022).
Golstein, P. & Kroemer, G. Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32, 37–43 (2007).
Zong, W. X. & Thompson, C. B. Necrotic death as a cell fate. Genes Dev. 20, 1–15 (2006).
Bousselin, M. Observations on Necrosis. Lond. Med. J. 7, 263–279 (1786).
Khalid, N. & Azimpouran, M. Necrosis Treasure Island (FL): StatPearls Publishing (2020).
Berghe, T. V., Linkermann, A., Jouan-Lanhouet, S., Walczak, H. & Vandenabeele, P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15, 135–147 (2014).
Thompson, C. B. Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456–1462 (1995).
Peter, M. E., Heufelder, A. E. & Hengartner, M. O. Advances in apoptosis research. Proc. Natl Acad. Sci. USA 94, 12736–12737 (1997).
Kerr, J. F. Shrinkage necrosis: a distinct mode of cellular death. J. Pathol. 105, 13–20 (1971).
Kerr, J. F., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Kerr, J. F. History of the events leading to the formulation of the apoptosis concept. Toxicology 181-182, 471–474 (2002).
Chen, Q., Kang, J. & Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 3, 18 (2018).
Ghobrial, I. M., Witzig, T. E. & Adjei, A. A. Targeting apoptosis pathways in cancer therapy. CA: Cancer J. Clin. 55, 178–194 (2005).
Wajant, H. The Fas signaling pathway: more than a paradigm. Science 296, 1635–1636 (2002).
Locksley, R. M., Killeen, N. & Lenardo, M. J. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501 (2001).
Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2, 420–430 (2002).
Riedl, S. J. & Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 5, 897–907 (2004).
Redza-Dutordoir, M. & Averill-Bates, D. A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 1863, 2977–2992 (2016).
Czabotar, P. E., Lessene, G., Strasser, A. & Adams, J. M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 (2014).
Kluck, R. M., Bossy-Wetzel, E., Green, D. R. & Newmeyer, D. D. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275, 1132–1136 (1997).
Ow, Y.-L. P., Green, D. R., Hao, Z. & Mak, T. W. Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542 (2008).
Galluzzi, L. et al. Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836 (2017).
Jung, S., Jeong, H. & Yu, S. W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 52, 921–930 (2020).
Klionsky, D. J. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937 (2007).
Ktistakis, N. T. In praise of M. Anselmier who first used the term "autophagie" in 1859. Autophagy 13, 2015–2017 (2017).
Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 24, 9–23 (2014).
Nevalainen, T. J. Cytotoxicity of vinblastine and vincristine to pancreatic acinar cells. Virchows Arch. B Cell Pathol. 18, 119–127 (1975).
Mizushima, N., Levine, B., Cuervo, A. M. & Klionsky, D. J. Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 (2008).
Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008).
Klionsky, D. J. & Emr, S. D. Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 (2000).
Zachari, M. & Ganley, I. G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 61, 585–596 (2017).
Jung, C. H., Ro, S.-H., Cao, J., Otto, N. M. & Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295 (2010).
Klionsky, D. J., Eskelinen, E. -L. & Deretic, V. Vol. 10 549–551 (Taylor & Francis, 2014).
Li, X., He, S. & Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19, 1–16 (2020).
Nishimura, T. & Tooze, S. A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 6, 32 (2020).
Mathew, R., Karantza-Wadsworth, V. & White, E. Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967 (2007).
Li, X. et al. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 119, 109415 (2019).
Levy, J. M. M., Towers, C. G. & Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017).
Lin, L. & Baehrecke, E. H. Autophagy, cell death, and cancer. Mol. Cell. Oncol. 2, e985913 (2015).
Chu, C. T., Zhu, J. & Dagda, R. K. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 3, 663–666 (2007).
Ma, S., Wang, Y., Chen, Y. & Cao, F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1852, 271–276 (2015).
Cursio, R., Colosetti, P. & Gugenheim, J. Autophagy and liver ischemia-reperfusion injury. BioMed. Res. Int. 2015, 417590 (2015).
Nah, J., Zablocki, D. & Sadoshima, J. The roles of the inhibitory autophagy regulator Rubicon in the heart: a new therapeutic target to prevent cardiac cell death. Exp. Mol. Med. 53, 528–536 (2021).
Liu, Y. et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl Acad. Sci. USA 110, 20364–20371 (2013).
Liu, Y. & Levine, B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 22, 367–376 (2015).
Nah, J. et al. Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J. Clin. Invest. 130, 2978–2991 (2020).
Tanaka, S. et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 64, 1994–2014 (2016).
Ikeda, S., Zablocki, D. & Sadoshima, J. The role of autophagy in death of cardiomyocytes. J. Mol. Cell Cardiol. 165, 1–8 (2022).
Nah, J., Zablocki, D. & Sadoshima, J. Autosis: a new target to prevent cell death. JACC Basic Transl. Sci. 5, 857–869 (2020).
Zheng, N. et al. Induction of tumor cell autosis by myxoma virus-infected CAR-T and TCR-T cells to overcome primary and acquired resistance. Cancer Cell 40, 973–985.e977 (2022).
Kheloufi, M., Boulanger, C. M., Codogno, P. & Rautou, P. E. Autosis occurs in the liver of patients with severe anorexia nervosa. Hepatology 62, 657–658 (2015).
Aits, S. & Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 126, 1905–1912 (2013).
de Duve, C. Lysosomes revisited. Eur. J. Biochem. 137, 391–397 (1983).
Lodish, H. et al. Molecular cell biology 4th edition. National Center for Biotechnology Information, Bookshelf 9 (2000).
Saftig, P. & Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 (2009).
Zhu, S.-y et al. Lysosomal quality control of cell fate: a novel therapeutic target for human diseases. Cell Death Dis. 11, 817 (2020).
Boya, P. & Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451 (2008).
Repnik, U., Stoka, V., Turk, V. & Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 1824, 22–33 (2012).
Wang, F., Gómez‐Sintes, R. & Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931 (2018).
Gómez-Sintes, R., Ledesma, M. D. & Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 32, 150–168 (2016).
Skulachev, V. P. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol. Asp. Med. 20, 139–184 (1999).
Wallace, D. C. Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698 (2012).
Mijaljica, D., Prescott, M. & Devenish, R. J. Mitophagy and mitoptosis in disease processes. Methods Mol. Biol. 648, 93–106 (2010).
Lyamzaev, K. G. et al. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta 1777, 817–825 (2008).
Lyamzaev, K. G., Knorre, D. A. & Chernyak, B. V. Mitoptosis, twenty years after. Biochem. (Mosc.) 85, 1484–1498 (2020).
Ding, W. X. & Yin, X. M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564 (2012).
Zhou, J. et al. Immunogenic cell death in cancer therapy: present and emerging inducers. J. Cell Mol. Med. 23, 4854–4865 (2019).
Casares, N. et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701 (2005).
Obeid, M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007).
Fucikova, J. et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 11, 1013 (2020).
Bobryshev, Y. V. Dendritic cells and their role in atherogenesis. Lab Invest. 90, 970–984 (2010).
Chijioke, O. & Münz, C. Dendritic cell derived cytokines in human natural killer cell differentiation and activation. Front. Immunol. 4, 365 (2013).
Ahmed, A. & Tait, S. W. G. Targeting immunogenic cell death in cancer. Mol. Oncol. 14, 2994–3006 (2020).
Bergsbaken, T., Fink, S. L. & Cookson, B. T. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109 (2009).
Cookson, B. T. & Brennan, M. A. Pro-inflammatory programmed cell death. Trends Microbiol. 9, 113–114 (2001).
Aachoui, Y., Sagulenko, V., Miao, E. A. & Stacey, K. J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 16, 319–326 (2013).
Amarante-Mendes, G. P. et al. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 9, 2379 (2018).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Kordes, M., Matuschewski, K. & Hafalla, J. C. Caspase-1 activation of interleukin-1β (IL-1β) and IL-18 is dispensable for induction of experimental cerebral malaria. Infect. Immun. 79, 3633–3641 (2011).
Lopez-Castejon, G. & Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 22, 189–195 (2011).
Man, S. M., Karki, R. & Kanneganti, T. D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75 (2017).
Li, Q. et al. The role of mitochondria in pyroptosis. Front. Cell Develop. Biol. 8, 630771 (2021).
Wei, Y. et al. Pyroptosis-induced inflammation and tissue damage. J. Mol. Biol. 434, 167301 (2022).
Zheng, Z. & Li, G. Mechanisms and therapeutic regulation of pyroptosis in inflammatory diseases and cancer. Int. J. Mol. Sci. 21, 1456 (2020).
Vorobjeva, N. V. & Chernyak, B. V. NETosis: molecular mechanisms, role in physiology and pathology. Biochem. (Mosc.) 85, 1178–1190 (2020).
Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147 (2018).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A. & Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 8, 883–896 (2014).
Thiam, H. R., Wong, S. L., Wagner, D. D. & Waterman, C. M. Cellular mechanisms of NETosis. Annu Rev. Cell Dev. Biol. 36, 191–218 (2020).
Ravindran, M., Khan, M. A. & Palaniyar, N. Neutrophil extracellular trap formation: physiology, pathology, and pharmacology. Biomolecules 9, 365 (2019).
Kambara, H. et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 22, 2924–2936 (2018).
Sollberger, G. et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 3, eaar6689 (2018).
Gupta, S. & Kaplan, M. J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 12, 402–413 (2016).
Takeuchi, A., Kim, B. & Matsuoka, S. The destiny of Ca(2+) released by mitochondria. J. Physiol. Sci. 65, 11–24 (2015).
Duchen, M. R. Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 529, 57–68 (2000).
Vorobjeva, N. et al. Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165664 (2020).
Chen, T. et al. Receptor-mediated NETosis on neutrophils. Front. Immunol. 12, 775267 (2021).
He, Y., Yang, F. Y. & Sun, E. W. Neutrophil extracellular traps in autoimmune diseases. Chin. Med. J. (Engl.) 131, 1513–1519 (2018).
Kwak, S. B. et al. Tumor regionalization after surgery: roles of the tumor microenvironment and neutrophil extracellular traps. Exp. Mol. Med. 54, 720–729 (2022).
Cho, Y. S. The role of necroptosis in the treatment of diseases. BMB Rep. 51, 219–224 (2018).
Chan, F. K., Luz, N. F. & Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev. Immunol. 33, 79–106 (2015).
Saeed, W. K. & Jun, D. W. Necroptosis: an emerging type of cell death in liver diseases. World J. Gastroenterol. 20, 12526–12532 (2014).
Fulda, S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol. Ther. 14, 999–1004 (2013).
Tummers, B. & Green, D. R. Caspase-8: regulating life and death. Immunol. Rev. 277, 76–89 (2017).
Han, J. H., Park, J., Kang, T. B. & Lee, K. H. Regulation of caspase-8 activity at the crossroads of pro-inflammation and anti-inflammation. Int. J. Mol. Sci. 22, 3318 (2021).
Samson, A. L. et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 11, 3151 (2020).
Kaczmarek, A., Vandenabeele, P. & Krysko, D. V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013).
Cooke, J. P. Inflammation and its role in regeneration and repair. Circ. Res. 124, 1166–1168 (2019).
Mohammed, S. et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 20, e13512 (2021).
Yuan, J., Amin, P. & Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2019).
Liu, Z. et al. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54, 247–258.e247 (2021).
Maslov, L. N. et al. The regulation of necroptosis and perspectives for the development of new drugs preventing ischemic/reperfusion of cardiac injury. Apoptosis 27, 697–719 (2022).
Gong, Y. et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 18, 100 (2019).
Seo, J., Nam, Y. W., Kim, S., Oh, D. B. & Song, J. Necroptosis molecular mechanisms: recent findings regarding novel necroptosis regulators. Exp. Mol. Med. 53, 1007–1017 (2021).
Xie, J., Yang, Y., Gao, Y. & He, J. Cuproptosis: mechanisms and links with cancers. Mol. Cancer 22, 46 (2023).
Hunsaker, E. W. & Franz, K. J. Emerging opportunities to manipulate metal trafficking for therapeutic benefit. Inorg. Chem. 58, 13528–13545 (2019).
Festa, R. A. & Thiele, D. J. Copper: an essential metal in biology. Curr. Biol. 21, R877–R883 (2011).
Drkiewicz, M., Skorzyska-Polit, E. & Krupa, Z. Copper-induced oxidative stress and antioxidant defence in Arabidopsis thaliana. Biometals 17, 379 (2004).
Gaetke, L. M. & Chow, C. K. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 189, 147–163 (2003).
Zulkifli, M. et al. FDX1-dependent and independent mechanisms of elesclomol-mediated intracellular copper delivery. Proc. Natl Acad. Sci. USA 120, e2216722120 (2023).
Li, S. R., Bu, L. L. & Cai, L. Cuproptosis: lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal Transduct. Target Ther. 7, 158 (2022).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254–1261 (2022).
Chen, L., Min, J. & Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target Ther. 7, 378 (2022).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Yan, H. F. et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct. Target Ther. 6, 49 (2021).
Chen, X., Yu, C., Kang, R. & Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 8, 590226 (2020).
Lee, J. Y., Kim, W. K., Bae, K. H., Lee, S. C. & Lee, E. W. Lipid metabolism and ferroptosis. Biol. (Basel) 10, 184 (2021).
Lyngsie, G., Krumina, L., Tunlid, A. & Persson, P. Generation of hydroxyl radicals from reactions between a dimethoxyhydroquinone and iron oxide nanoparticles. Sci. Rep. 8, 10834 (2018).
Ashraf, A., Jeandriens, J., Parkes, H. G. & So, P. W. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: evidence of ferroptosis. Redox Biol. 32, 101494 (2020).
Adeoye, O., Olawumi, J., Opeyemi, A. & Christiania, O. Review on the role of glutathione on oxidative stress and infertility. JBRA Assist. Reprod. 22, 61–66 (2018).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Jyotsana, N., Ta, K. T. & DelGiorno, K. E. The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front. Oncol. 12, 858462 (2022).
Xu, T. et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell Mol. Med. 23, 4900–4912 (2019).
Sperandio, S., de Belle, I. & Bredesen, D. E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl Acad. Sci. 97, 14376–14381 (2000).
Allen, T. C., Cagle, P. T. & Popper, H. H. Basic concepts of molecular pathology. Arch. Pathol. Lab. Med. 132, 1551–1556 (2008).
Kim, E., Lee, D. M., Seo, M. J., Lee, H. J. & Choi, K. S. Intracellular Ca(2 +) imbalance critically contributes to paraptosis. Front. Cell Dev. Biol. 8, 607844 (2020).
Hetz, C., Zhang, K. & Kaufman, R. J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438 (2020).
Sperandio, S. et al. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 11, 1066–1075 (2004).
Tardito, S. et al. The thioxotriazole copper (II) complex A0 induces endoplasmic reticulum stress and paraptotic death in human cancer cells. J. Biol. Chem. 284, 24306–24319 (2009).
Lee, D. M., Kim, I. Y., Seo, M. J., Kwon, M. R. & Choi, K. S. Nutlin-3 enhances the bortezomib sensitivity of p53-defective cancer cells by inducing paraptosis. Exp. Mol. Med. 49, e365–e365 (2017).
Ye, R.-R., Tan, C.-P., Lin, Y.-N., Ji, L.-N. & Mao, Z.-W. A phosphorescent rhenium (I) histone deacetylase inhibitor: mitochondrial targeting and paraptosis induction. Chem. Commun. 51, 8353–8356 (2015).
Kianfar, M., Balcerak, A., Chmielarczyk, M., Tarnowski, L. & Grzybowska, E. A. Cell death by entosis: triggers, molecular mechanisms and clinical significance. Int. J. Mol. Sci. 23, 4985 (2022).
Overmeyer, J. H., Kaul, A., Johnson, E. E. & Maltese, W. A. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 6, 965–977 (2008).
Bhanot, H., Young, A. M., Overmeyer, J. H. & Maltese, W. A. Induction of nonapoptotic cell death by activated Ras requires inverse regulation of Rac1 and Arf6. Mol. Cancer Res. 8, 1358–1374 (2010).
Lin, X. P., Mintern, J. D. & Gleeson, P. A. Macropinocytosis in different cell types: similarities and differences. Membr. (Basel) 10, 177 (2020).
Overmeyer, J. H., Young, A. M., Bhanot, H. & Maltese, W. A. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 10, 69 (2011).
Liu, Y. et al. Targeting VPS41 induces methuosis and inhibits autophagy in cancer cells. Cell Chem. Biol. 30, 130–143.e135 (2023).
Liu, X. et al. Epimedokoreanin C, a prenylated flavonoid isolated from Epimedium koreanum, induces non-apoptotic cell death with the characteristics of methuosis in lung cancer cells. Am. J. Cancer Res. 11, 3496–3514 (2021).
Overholtzer, M. et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131, 966–979 (2007).
Durgan, J. & Florey, O. Cancer cell cannibalism: multiple triggers emerge for entosis. Biochim. Biophys. Acta Mol. Cell Res. 1865, 831–841 (2018).
Krishna, S. & Overholtzer, M. Mechanisms and consequences of entosis. Cell Mol. Life Sci. 73, 2379–2386 (2016).
Zeng, C., Zeng, B., Dong, C., Liu, J. & Xing, F. Rho-ROCK signaling mediates entotic cell death in tumor. Cell Death Discov. 6, 4 (2020).
Spiering, D. & Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr. 5, 170–180 (2011).
Yamada, S. & Nelson, W. J. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J. Cell Biol. 178, 517–527 (2007).
Hamann, J. C. et al. Entosis is induced by glucose starvation. Cell Rep. 20, 201–210 (2017).
Chen, R., Ram, A., Albeck, J. G. & Overholtzer, M. Entosis is induced by ultraviolet radiation. iScience 24, 102902 (2021).
Arandjelovic, S. & Ravichandran, K. S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 16, 907–917 (2015).
Kroemer, G. & Perfettini, J. L. Entosis, a key player in cancer cell competition. Cell Res. 24, 1280–1281 (2014).
Mlynarczuk-Bialy, I. et al. Entosis: from cell biology to clinical cancer pathology. Cancers (Basel) 12, 2481 (2020).
Shawer, H. et al. ORAI1 Ca(2+) channel as a therapeutic target in pathological vascular remodelling. Front. Cell Dev. Biol. 9, 653812 (2021).
Lee, A. R. & Park, C. Y. Orai1 is an Entotic Ca(2+) channel for non-apoptotic cell death, entosis in cancer development. Adv. Sci. (Weinh.) 10, e2205913 (2023).
Fatokun, A. A., Dawson, V. L. & Dawson, T. M. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharm. 171, 2000–2016 (2014).
Liu, L. et al. The key players of parthanatos: opportunities for targeting multiple levels in the therapy of parthanatos-based pathogenesis. Cell Mol. Life Sci. 79, 60 (2022).
Ko, H. L. & Ren, E. C. Functional aspects of PARP1 in DNA repair and transcription. Biomolecules 2, 524–548 (2012).
Andrabi, S. A., Dawson, T. M. & Dawson, V. L. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N. Y Acad. Sci. 1147, 233–241 (2008).
Wang, Y., Dawson, V. L. & Dawson, T. M. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol. 218, 193–202 (2009).
Wang, Y. et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354, aad6872 (2016).
Song, X. et al. JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology 154, 1480–1493 (2018).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019).
Que, D., Kuang, F., Kang, R., Tang, D. & Liu, J. ACSS2-mediated NF-κB activation promotes alkaliptosis in human pancreatic cancer cells. Sci. Rep. 13, 1483 (2023).
Bulusu, V. et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 18, 647–658 (2017).
Koltai, T. Cancer: fundamentals behind pH targeting and the double-edged approach. Onco Targets Ther. 9, 6343–6360 (2016).
Kang, P. et al. Oxeiptosis: a novel pathway of melanocytes death in response to oxidative stress in vitiligo. Cell Death Discov. 8, 70 (2022).
Liu, Y. et al. NRF2 signalling pathway: new insights and progress in the field of wound healing. J. Cell Mol. Med. 25, 5857–5868 (2021).
Szatrowski, T. P. & Nathan, C. F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 51, 794–798 (1991).
Bergmann, A. Erebosis is a new type of cell death for tissue homeostasis in the Drosophila intestine. PLoS Biol. 20, e3001614 (2022).
Green, D. R. & Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 7, a006080 (2015).
Martins, I. et al. Entosis: the emerging face of non-cell-autonomous type IV programmed death. Biomed. J. 40, 133–140 (2017).
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 12, 1463–1467 (2005).
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11 (2009).
Liu, J. et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem. Biol. 27, 420–435 (2020).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Bock, F. J. & Tait, S. W. G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100 (2020).
Wang, C. & Youle, R. J. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 43, 95–118 (2009).
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Yoon, M. J. et al. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget 5, 6816–6831 (2014).
Glick, D., Barth, S. & Macleod, K. F. Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12 (2010).
Bergsbaken, T., Fink, S. L., den Hartigh, A. B., Loomis, W. P. & Cookson, B. T. Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J. Immunol. 187, 2748–2754 (2011).
Raimondi, M. et al. Ca(2+) overload- and ROS-associated mitochondrial dysfunction contributes to δ-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 26, 277–292 (2021).
Shubin, A. V., Demidyuk, I. V., Komissarov, A. A., Rafieva, L. M. & Kostrov, S. V. Cytoplasmic vacuolization in cell death and survival. Oncotarget 7, 55863–55889 (2016).
Kuriakose, T. et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1, aag2045 (2016).
Malireddi, R. K. S., Kesavardhana, S. & Kanneganti, T. D. ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front. Cell Infect. Microbiol. 9, 406 (2019).
Samir, P., Malireddi, R. K. S. & Kanneganti, T. D. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front. Cell Infect. Microbiol. 10, 238 (2020).
Kesavardhana, S. et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 214, 2217–2229 (2017).
Zheng, M., Karki, R., Vogel, P. & Kanneganti, T. D. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell 181, 674–687.e613 (2020).
Place, D. E., Lee, S. & Kanneganti, T. D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 59, 42–49 (2021).
Jiang, M. et al. Caspase-8: a key protein of cross-talk signal way in "PANoptosis" in cancer. Int. J. Cancer 149, 1408–1420 (2021).
Kam, P. C. & Ferch, N. I. Apoptosis: mechanisms and clinical implications. Anaesthesia 55, 1081–1093 (2000).
Tinari, A., Garofalo, T., Sorice, M., Esposti, M. D. & Malorni, W. Mitoptosis: different pathways for mitochondrial execution. Autophagy 3, 282–284 (2007).
Künzi, L. & Holt, G. E. Cigarette smoke activates the parthanatos pathway of cell death in human bronchial epithelial cells. Cell Death Discov. 5, 127 (2019).
Jang, K. H. et al. AIF-independent parthanatos in the pathogenesis of dry age-related macular degeneration. Cell Death Dis. 8, e2526 (2017).
Kam, T. I. et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 362, eaat8407 (2018).
Jiang, H. Y. et al. The dual role of poly(ADP-ribose) polymerase-1 in modulating parthanatos and autophagy under oxidative stress in rat cochlear marginal cells of the stria vascularis. Redox Biol. 14, 361–370 (2018).
Han, C. et al. Ferroptosis and its potential role in human diseases. Front. Pharm. 11, 239 (2020).
Acknowledgements
This study was supported by the Basic Science Research Program through National Research Foundation of Korea (NRF) grants from the Korean government (MIST; grant nos. NRF-2022R1A2C2005130 and NRF-2021R1A4A1025662). The figures in this review paper were created using BioRender.
Author information
Authors and Affiliations
Contributions
W.Y.P., as the first author, contributed to the primary writing and organization of the manuscript and conducted the literature review. W.Y.P. and S.B.W. collected and analyzed data. B.S.K. created the figures and tables. Y.C.C., S.J.B., and D.R.R. contributed to the writing and revision of the manuscript and provided critical feedback throughout the study. K.T.H., as the corresponding author, supervised the study, contributed to the interpretation of the results, and helped revise the final manuscript. All the authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, W., Wei, S., Kim, BS. et al. Diversity and complexity of cell death: a historical review. Exp Mol Med 55, 1573–1594 (2023). https://doi.org/10.1038/s12276-023-01078-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01078-x
- Springer Nature Limited
This article is cited by
-
Osteoking promotes bone formation and bone defect repair through ZBP1–STAT1–PKR–MLKL-mediated necroptosis
Chinese Medicine (2024)
-
6-Methyl-5-hepten-2-one promotes programmed cell death during superficial scald development in pear
Molecular Horticulture (2024)
-
Role of Exosomes in Cancer and Aptamer-Modified Exosomes as a Promising Platform for Cancer Targeted Therapy
Biological Procedures Online (2024)
-
Entosis: the core mechanism and crosstalk with other cell death programs
Experimental & Molecular Medicine (2024)
-
LincRNA-p21/AIF-1/CMPK2/NLRP3 pathway promoted inflammation, autophagy and apoptosis of human tubular epithelial cell induced by urate via exosomes
Scientific Reports (2024)