
Abstract
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by clonal expansion of myeloid blasts in the bone marrow (BM). Despite advances in therapy, the prognosis for AML patients remains poor, and there is a need to identify novel molecular pathways regulating tumor cell survival and proliferation. F-box ubiquitin E3 ligase, FBXO21, has low expression in AML, but expression correlates with survival in AML patients and patients with higher expression have poorer outcomes. Silencing FBXO21 in human-derived AML cell lines and primary patient samples leads to differentiation, inhibition of tumor progression, and sensitization to chemotherapy agents. Additionally, knockdown of FBXO21 leads to up-regulation of cytokine signaling pathways. Through a mass spectrometry-based proteomic analysis of FBXO21 in AML, we identified that FBXO21 ubiquitylates p85α, a regulatory subunit of the phosphoinositide 3-kinase (PI3K) pathway, for degradation resulting in decreased PI3K signaling, dimerization of free p85α and ERK activation. These findings reveal the ubiquitin E3 ligase, FBXO21, plays a critical role in regulating AML pathogenesis, specifically through alterations in PI3K via regulation of p85α protein stability.
Similar content being viewed by others


Introduction
Post-translational regulation of hematopoietic differentiation by the ubiquitin proteasome system, specifically the substrate-recognizing ubiquitin E3 ligases is an important direction in unraveling molecular mechanisms regulating cell fate decisions in normal and malignant hematopoiesis [1, 2]. The FBOX family of proteins are a group of approximately 69 ubiquitin E3 ligases that function as the substrate recognition component of the SKP1-CUL1-FBOX (SCF) complex [3, 4]. Of the FBOX family of proteins only 15 of the 69 have known roles in normal and malignant hematopoiesis [5]. Analysis of AML expression datasets revealed one of the FBOX E3 ubiquitin ligases, FBXO21, is differentially expressed in leukemia compared to normal bone marrow (BM). Interestingly, low expression of FBXO21 correlates with better overall survival in AML patients [6]. Little is known about the molecular mechanism of FBXO21, and it has not yet been studied within the context of malignant hematopoiesis.
FBXO21 has two known substrates: EP300-inhibitor of differentiation 1 (EID1) and Apoptosis signal-regulating kinase 1 (ASK1), which were identified in 293 T and RAW264.7 cells, respectively [7,8,9,10]. EID1 interacts with RB1 and EP300 leading to maintenance of pluripotent stem cells, and inhibition of differentiation [11, 12]. Additionally, FBXO21 has been shown regulate the response to immune stimuli via proinflammatory cytokine mediated pathways through ubiquitination of ASK1 via K29 linkage [10]. FBXO21 is highly expressed in stem and progenitor (HSPC) population, the tumor initiating population in AML, but has low to no expression in mature myeloid populations [13]. To determine the role of FBXO21 in hematopoietic development we generated a conditional knockout model and crossed it to Vav1-CRE, which deletes during early hematopoiesis. Deletion of Fbxo21 revealed minimal change in the hematopoietic development [13]. However, knockdown (KD) of FBXO21 in AML patient samples and patient derived cells lines led to apoptosis and decreased proliferation at a greater degree compared to normal human CD34 + HSPC. Silencing of FBXO21 in AML increased expression of inflammatory cytokines and chemokines, including CXCL10. Further, we utilized a mass spectrometry based proteomic approach to identify p85α as a substrate of FBXO21. p85α is targeted for ubiquitination by FBXO21, and stabilization of p85α leads to apoptosis, differentiation, decreased canonical PI3K signaling, and ERK activation. Taken together, the data suggest that FBXO21 plays a key role leukemia progression and maintenance but has a limited role in normal hematopoiesis suggesting FBXO21 as a potential therapeutic target for drug discovery.
Results
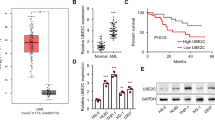
Low ubiquitin E3 ligase FBXO21 expression correlates with improved survival in AML
Utilizing AML primary patient expression data from the Microarray Innovations in Leukemia study [14], we determined that the expression of the E3 ligase FBXO21 is significantly downregulated in AML compared to healthy BM (Fig. 1A). The most pronounced decrease in FBXO21 expression was in patients with mixed lineage leukemia (MLL) and t(8;21) translocations (Fig. 1A). Interestingly, lower expression of FBXO21 is associated with improved survival (Fig. 1B) [6]. Protein expression analysis in mononuclear cells from peripheral blood (PB) of nine AML patients with French-American-British classifications ranging from M0–M4 showed an increase in the levels of FBXO21 compared to two healthy human total BM samples (Fig. 1C). These patients had AML blast counts ranging from 36 to 95%, although only two representative patients relapsed samples had high FBXO21 protein similarly to hematopoietic stem and progenitor population (HPSC) characterized by CD34+ expression. In addition, protein expression in patient-derived AML (MOLM-13, THP-1, HL-60, and KASUMI-1) and acute lymphoblastic leukemia (MOLT-4, CCRF-CEM, and RS4(11)) cell lines exhibited high level of FBXO21 compared to two independent human BM samples (Fig. 1D). These findings suggest FBXO21 expression is associated with prognosis and may contribute to progression leukemia.

A FBXO21 gene expression analysis of patient samples with different karyotypes from the Leukemia MILE study. B Overall patient survival from TCGA AML patient dataset. C Western blot of FBXO21 from two independent human BM (HBM) samples, two healthy CD34+ samples isolated from mobilized PB, and mononuclear cells from PB of nine AML patients (Supplementary Table 1). D Western blot probing for FBXO21 in two independent HBM samples compared to patient-derived AML (MOLM-13, THP-1, HL-60, and KASUMI-1) and ALL (MOLT-4, CCRF-CEM, and RS4(11)) cell lines. (ns non-significant, *p ≤ 0.05, ****p ≤ 0.0001).
Silencing of FBXO21 in AML alters proliferation, differentiation, and tumor progression
To understand the role of FBXO21 in AML, we generated FBXO21 knockdown (KD) AML cells. Here we utilized four patient derived AML cell lines (MOLM-13 (FLT3-IDT/MLL-AF9), THP-1 (MLL-AF9/TP53), Kasumi-1 (AML-ETO/TP53/KIT), and HL-60 (TP53/NRAS)) and 4 independent samples from primary mononuclear PB of AML patients (2 de novo, 2 relapse; Supplementary Table 1), and infected them with lentivirus expressing shRNAs against either a non-targeting control (shNTC) or shFBXO21 to silence FBXO21 expression (Fig. 2 and Supplementary Fig. 1). Knockdown yielded a 75–93% decrease in FBXO21 protein levels (Fig. 2A, B) and led to a decrease in the cell’s ability to proliferate (Fig. 2C, and Supplementary Fig. 1A, B) and an increase in cells undergoing early (Annexin V+/PI-) and late (Annexin V+/PI+) apoptosis (Fig. 2D, E, and Supplementary Fig. 1C). Knocking down FBXO21 also promoted cell differentiation, indicated by increased expression of CD11b in MOLM-13 cells (Fig. 2F) and CD15 in primary AML cells and AML cell lines, both mature myeloid cell surface markers (Fig. 2G, and Supplementary Fig. 1D). Colony forming potential was decreased following silencing of FBXO21 when both MOLM-13 cells and primary AML cells were plated in methylcellulose (Fig. 2H, I, and Supplementary Fig. 1E). Importantly, silencing FBXO21 delayed disease onset of NSG mice following transplantation in comparison to mice transplanted with the shNTC in patient derived cell lines, consistent with survival in human AML patients with low FBXO21 expression (Fig. 2J, and Supplementary Fig. 1F). Combining these findings with previous data showing that patients with low expression of FBXO21 yields better prognosis suggests that FBXO21 may act as an oncogene in AML.

A–J (MOLM-13: n = 3 biological replicates, AML Patients: n = 3 technical replicates) MOLM-13 cells and 4 AML primary samples (2 de novo, 2 relapse) were infected with lentiviral shRNAs against shFBXO21 and shNTC were analyzed at 72 h post puromycin selection by (A, B) western blot for knockdown in (A) AML patient derived cell line, MOLM13, and (B) 4 AML primary samples (2 de novo, 2 relapse), (C) proliferative ability of MOLM-13 cells by cell count. Cells were stained with (D, E) (left) representative flowcytometry plot and (right) bar graph of Annexin V and propidium iodide (PI) for percent of (1) AnnexinV+/PI− and (2) AnnexinV+/PI+ apoptotic cells in (D) MOLM-13 and (E) AML primary cells. Cells were analyzed by flowcytometry for (F) (right) representative flowcytometry plot and (left) bar graph of CD11b (MOLM-13) and (G) (right) representative flowcytometry plot and (left) bar graph of CD15 (AML primary cells) expression. Colony forming ability by CFU assay in (H) MOLM-13 and (I) AML primary cells. J Survival of sub-lethally irradiated NSG mice transplanted with 5 × 105 MOLM-13 cells infected with shRNAs against shFBXO21 and shNTC. (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Overexpression of FBXO21 leads to increased proliferation and colony formation
Expression data suggest that higher expression of FBXO21 is correlated with poor prognosis and relapse. To determine if overexpression leads to increased proliferation, we retrovirally expressed empty vector or flag-tagged FBXO21 to overexpress FBXO21 in tandem with GFP in MOLM-13 cells (Fig. 3A). In addition, we overexpressed FBXO21 with the FBOX domain deleted (ΔFBXO21), a catalytically dead mutant of FBXO21 due to FBOX domain required for binding to SKP1. Overexpression of FBXO21 led to increased proliferation and colony formation, whereas ΔFBXO21 overexpression did not alter proliferation or colony formation potential (Fig. 3B, C). Increased expression did not alter apoptosis but led to decreased cell surface expression of CD15 and increased disease onset in NSG mice (Fig. 3D–F). These findings of FBXO21 overexpression displayed an inverse effect from cells having FBXO21 knocked down, where KD had led to increased expression of CD15, decreased colony formation, and a delay in disease onset in NSG mice (Fig. 2G, J). The decrease in survival of NSG mice that received FBXO21 overexpression AML cells correlates with expression data linking patients with higher expression of FBXO21 have poorer survival.

A–F (n = 6, 2 biological, 3 technical replicates) MOLM-13 cells were infected with retrovirus expressing FBXO21, ∆FBXO21, and Empty control were analyzed after sorting via FACS for (A) protein expression by western blot, (B) proliferative ability by cell count, (C) colony forming ability by CFU assay, (D) for percent of Annexin V+/PI- and Annexin V+/PI+ apoptotic cells, (E) (left) representative flowcytometry plot and (right) bar graph of CD15 expression by flow cytometry, and (F) survival of sub-lethally irradiated NSG mice transplanted with 5 × 105 cells infected with an Empty control or a plasmid overexpressing FBXO21. G MTT assay in MOLM-13 NTC and shFBXO21.55 following treatment with between 0.1-1000 nM cytarabine for 48 h. H MOLM-13, and (I) AML primary cells with FBXO21 KD and shNTC, and (J) MOLM-13 FBXO21 and Empty were treated with 50 nM cytarabine for 48 h, stained with Annexin V and PI for Annexin V+/PI− and Annexin V+/PI+ apoptotic cells, and analyzed by flow cytometry. (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Typically, the first line of treatment for AML is administrating intense induction chemotherapy—generally a combination of cytarabine and an anthracycline such as daunorubicin, with the occasional addition of all trans retinoic acid for certain subtypes [15,16,17,18]. To determine whether FBXO21 expression has an impact on cytarabine sensitivity, we treated FBXO21 KD or overexpression AML cells with cytarabine. Silencing of FBXO21 led to an additive effect of cytarabine decreasing the IC:50 from 42 nM to 23 nM (Fig. 3G). Following treatment of 50 nM of cytarabine for 48 h, we measured apoptosis of the live cell population by flow cytometry for annexin V. Silencing of FBXO21 led to increased sensitivity to cytarabine (Fig. 3H, I), whereas cells overexpressing FBXO21 had no changes in apoptosis and cell death (Fig. 3J). Together these finding demonstrate that levels of FBXO21 impact survival, differentiation, and sensitivity to current therapeutics.
Silencing of FBXO21 leads to cytokine and chemokine response
To understand transcriptional changes due to depletion of FBXO21, we performed RNA-sequencing on MOLM-13 shNTC and shFBXO21 targeted cells. RNA-sequencing data showed that silencing of FBXO21 led to a dramatic increase in the expression of inflammatory cytokine/chemokine related genes, including CXCL10, CXCL11, IFIT1, IFIT2, IFIT3, IL1β, STAT1, and STAT2 (Fig. 4A). These genes are associated to pathways including inflammatory response, signal transduction, and positive regulation of the ERK1 and ERK2 cascade (Fig. 4B). Differentially expressed RNAs were found in all cellular compartments with ~12% associated with the extracellular space and ~25% associated with the plasma membrane (Fig. 4C). Cytokine arrays confirmed that CCL5 and CXCL10 proteins are also found to be upregulated in the media following silencing of FBXO21 (Fig. 4D). We identified a 14-fold change increase of CXCL10 expression at the RNA level and confirmed up-regulation of the protein in both shFBXO21 KD AML cell lines and primary patient samples (Fig. 4E, F). Interestingly, overexpression of FBXO21 in MOLM-13 led to a decrease in the amount of CXCL10 in the media (Fig. 4G). These findings suggest that CXCL10 is regulated downstream of FBXO21. Although we have observed an increase in CXCL10 at both RNA and protein levels, we did not observe a change in CXCL10 receptor, CXCR3 at the RNA level (data not shown). CXCL10 is known to be regulated through various MAP kinase pathways, including through the activation of transcription factor NFκB via JNK, p38, ERK1/2, and JAK/STAT [19]. These findings suggest that silencing of FBXO21 in AML alters cytokine signaling.

A Volcano plot showing fold change of expressed genes from MOLM-13 cells with silenced shFBXO21 and shNTC. B Gene ontology analysis showing pathways known to be associated with significantly upregulated (p < 0.05, ≥1-fold change) and downregulated (p < 0.05, ≤1-fold change) genes using DAVID bioinformatics database. C Localization of significantly upregulated genes (405). D Cytokine array for supernatant of MOLM-13 cells with shFBXO21 KD and shNTC, membranes showing the change in inflammatory cytokines and quantified intensities relative to internal standards. CXCL10 secretion was evaluated by enzyme-linked immunosorbent assay (ELISA) in (E) MOLM-13 cells with silenced shFBXO21 and shNTC, (F) AML primary cells with silenced shFBXO21 and shNTC, and (G) MOLM-13 cells expressing FBXO21 and Empty control.
FBXO21 substrate identification in AML
Since FBXO21 is a substrate recognition component of the SKP1-Cullin-FBOX (SCF)-type E3 ligase complex, we performed a combination of mass spectrometry (MS) approaches to identify the substrate of FBXO21 in AML. First, we performed tandem mass tag (TMT) MS, which allowed us to quantitatively identify changes in protein abundance between our MOLM-13 shFBXO21 KD, and its respective shNTC cells (Fig. 5A). In addition, we performed K-ε-GG immunoprecipitation followed by MS on our shNTC and shFBXO21 KD cell lines, which allowed for identification of unique ubiquitination sites through enrichment of Ub-reminant diglycyl-lysine (K-ε-GG) and protein identification by tandem MS (Fig. 5A). Here we identified 260 proteins upregulated following silencing of FBXO21 in TMT MS, including proteins associated with cytokine-mediated signaling pathways and 1297 ubiquitinated peptides in K-ε-GG MS, of which 50 were found either more abundant or only present in the MOLM-13 shNTC cell line, which would be predicted for a substrate targeted by polyubiquitination followed for proteasomal degradation (Fig. 5B, C; Supplementary Tables 2, 3).

A Schematic of TMT MS and K-ε-GG IP/MS using MOLM-13 cells infected with shRNAs against shFBXO21 and shNTC. B Volcano plots showing fold change of expressed proteins from shFBXO21 compared to shNTC cells. C Gene ontology analysis showing pathways known to be associated with significantly upregulated (p < 0.05, ≥1.3-fold change) proteins using DAVID bioinformatics database. Western blot for previously known FBXO21 substrates (ASK1, EID1) and other MAPK pathway proteins (D) in MOLM-13 cells and (E) AML primary cells. F Venn Diagram of combined TMT MS, K-ε-GG IP/MS, cytosolic proteins, proteins involved in cytokine signaling pathways, and RNA-seq data comparing overlap of differentially expressed proteins and genes. G Western blot in MOLM-13 cells for validation of upregulated proteins of interest identified via the combination of proteomic and genomic analysis.
FBXO21 has only two known substrates in other cell types, EP300-interacting inhibitor of differentiation 1 (EID1) and apoptosis signaling kinase 1 (ASK1, also known as MAP3K5) [7,8,9,10]. EID1 has a role in inhibiting differentiation and maintaining the pluripotency of stem cells through interacting with RB1 and EP300 [11, 12]. FBXO21 has also been shown to target ASK1 for K29 ubiquitination in vitro in cell lines regulating response to immune stimuli. However, neither ASK1 nor EID1 were identified in MS approaches, and western blot analysis of shNTC and shFBXO21 KD cells revealed no alterations in EID1, ASK1 activation, or ASK1’s down-stream target, p38; suggesting EID1 and ASK1 are not substrates of FBXO21 in AML (Fig. 5D). In addition, we treated MOLM-13 cells with Selonsertib, a small molecular inhibitor of ASK1. ASK1 inhibition showed that depletion of phosphorylated ASK1 led to diminished p38 phosphorylation, which was not seen following FBXO21 silencing (Supplementary Fig. 2A). Similar to shFBXO21 KD, Selonsertib treatment led to decreased proliferation, increased apoptosis, but did not demonstrate the same cell surface phenotype as shFBXO21 KD further supporting ASK1 is not likely a substrate of FBXO21 in the context of AML (Supplementary Fig. 2B, F).
Interestingly, no changes in total or phospho-p38 was observed, however another MAPK pathway, phospho-ERK1/2 was highly upregulated (Fig. 5D, E). Although, no changes in total ERK1/2 were observed suggesting FBXO21 does not target ERK for poly-ubiquitination, it does not rule out that FBXO21 targets the ERK pathway upstream or an activator of the ERK pathway (Fig. 5D, E). Further supporting that ERK1/2 is not the direct substrate, inhibition of ERK with SCH772984 did not rescue the decrease in colony formation or proliferation in shFBXO21 KD cells, and CXCL10 levels did not decrease to levels seen in shNTC cells (Supplementary Fig. 3). Contrary to previously published worked suggesting ERK activation promotes proliferation, FBXO21 KD led to decreased proliferation supporting that ERK itself is not a substrate of FBXO21 [20].
Since EID1 and ASK1, the known substrates of FBXO21, showed no alterations in protein abundance, we used a combinational approach to identify potential substrates. Criteria included 1) upregulated at the protein level by TMT MS in FBXO21 KD cell line (unable to be ubiquitinated and sent to proteasome for degradation), 2) ubiquitin modified as identified in the K-ε-GG mass spectrometry, 3) cytoplasmic proteins since FBXO21 localizes to the cytoplasm, 4) unaltered at the RNA level (as we are identifying modification at the posttranslational level), and 5) proteins associated with cytokine signaling (Fig. 5F, Supplementary Tables 2–5). Crossing the datasets revealed two proteins that meet all five criteria, ARF6 and p85α, both of which were confirmed by western blot to be upregulated following silencing of FBXO21 in the MOLM-13 cell line (Fig. 5G). In summary, the combination of proteomic and genomic analysis yielded two potential novel FBXO21 substrates in AML.
FBXO21 targets p85α for ubiquitination and degradation
To determine if either ARF6 and/or p85α are substrates of FBXO21, we performed endogenous IP of FBXO21 in MOLM-13 cells to determine protein interaction. Of the two potential substrates identified through MS approaches, only p85α was found to interact with FBXO21 (Fig. 6A). To further confirm binding of p85α and FBXO21, we transiently expressed p85α tagged with GFP with either wild-type FBXO21 or FBOX domain deleted FBXO21 (ΔFBXO21) tagged with HA in HEK293T, and immunoprecipitated GFP or HA. We found p85α interacted with both full-length and FBOX domain deleted FBXO21 suggesting a direct interaction with FBXO21, and that the interaction is not through the SCF complex (Fig. 6B). To investigate whether p85α is targeted for degradation by the proteasome and whether depletion of FBXO21 blocks degradation, we treated both shNTC and shFBXO21 KD MOLM-13 cells with the proteasome inhibitor MG132. Immunoblotting revealed that in shNTC MOLM-13s p85α accumulated in the presence of proteasome inhibitor (lane 1 and 2); however, silencing of FBXO21 inhibited accumulation of p85α protein following MG132 treatment (lanes 3 and 4) (Fig. 6C). In contrast, ARF6 protein abundance was unaltered in either shNTC or shFBXO21 KD following MG132 suggesting it is likely not regulated in AML by proteasomal degradation and further confirming ARF6 is not a substrate of FBXO21. cMYC was used as a positive control due to known to be targeted by the proteasome in leukemia, and shows accumulation in both shNTC or shFBXO21 KD following MG132 [2].

A Western blot of endogenous immunoprecipitation of FBXO21 in MOLM-13 cell line. B Western blot of GFP and HA immunoprecipitation in HEK293T cells transiently transfected with plasmids expressing GFP-tagged p85α and/or HA-tagged FBXO21/∆FBXO21. C Western blot of shNTC/shFBXO21.55 MOLM-13 cells treated with 20 μM MG132 or DMSO. D Western blot of Ubiquitin immunoprecipitation in shNTC/shFBXO21.55 HEK293T cells transiently transfected with p85α and 2, 5, or 10 μg Ubiquitin. E Western blot of Ubiquitin immunoprecipitation in Empty/FBXO21/∆FBXO21 HEK293T cells transiently transfected with p85α and 5 or 10 μg Ubiquitin. F HEK293T cells were transfected with GFP-tagged p85α, HA-tagged FBXO21, HA-tagged ΔFBXO21 as indicated. After immunopurification with anti-GFP/anti-HA, in vitro ubiquitylation of p85α was performed in the presence of E1, E2, and ubiquitin (Ub). Samples were analyzed by western blot with the indicated antibodies. n = 3 for all experiments.
To further confirm FBXO21 regulates the degradation of p85α, we immunoprecipitated ubiquitin in HEK293T cells stably expressing either shNTC or shFBXO21, and transiently expressing GFP-tagged p85α, along with increasing concentrations of HA-tagged ubiquitin. Increased ubiquitinated p85α protein was only found in cells expressing FBXO21 (lane 4–6), whereas ubiquitinated p85α was not immunoprecipitated in cells depleted for FBOX21 (lanes 10–12) (Fig. 6D). Similarly, we preformed ubiquitin immunoprecipitation in HEK293T cells stably overexpressing FBXO21 or ΔFBXO21. Overexpression of FBXO21 led to increased immunoprecipitation of ubiquitinated p85α (lane 6–8) compared to endogenous expression of FBXO21 (lanes 2–4) (Fig. 6E). Deletion of the FBOX domain, due to being catalytically dead and unable to bind the SCF complex, inhibited p85α ubiquitination (lanes 10–12) (Fig. 6E). Finally, we reconstituted the ubiquitination of p85α in vitro. Immunopurified FBXO21, but not FBOX deficient FBXO21 ubiquitinated p85α in vitro (Fig. 6F). Together, these results demonstrate that FBXO21 directly mediates the ubiquitylation and degradation of p85α in AML.
p85α regulates CXCL10 promoting apoptosis and differentiation of AML
p85α is part of the PI3K pathway and is a central signaling pathway for hematopoietic cells, and regulates crucial functions such as proliferation, differentiation, and survival [21, 22]. p85α (PIK3R1) and p85β (PIK3R2) are the main regulatory subunits of PI3K which mediate the catalytic activity of p110 [23], however mass spectrometry approaches only found p85α differentially expression and p85β isoform was not found among the proteins expressed (Supplementary Table 2). p85α is significantly upregulated in AML patients and increased expression correlates with a worse survival rate [24]. However, we found high levels of p85α due to silencing of FBXO21 led to an increase in differentiation, and promoted cell death in AML. This suggests that p85α could work in a dose dependent manner. To determine if increased expression of p85α in MOLM-13 cells contributed to the increased cell death, and decreased proliferation, we stably overexpressed flag-tagged p85α in MOLM-13 cells. Overexpression of p85α increased ERK activation, similarly, to silencing of FBXO21, as well as, decreased colony formation, proliferation, and promoted apoptosis (Fig. 7A–D). Corresponding to what was previously shown in our FBXO21 KD, ERK activation due to p85α overexpression led to elevated CXCL10 (Fig. 7E).

(n = 6, 2 biological, 3 technical replicates) MOLM-13 cells were infected with retrovirus expressing p85α and Empty control were analyzed after sorting via FACS by (A) western blot, (B) proliferative ability by cell count, (C) for percent of Annexin V+/PI- and Annexin V+/PI+ apoptotic cells, (D) colony forming ability by CFU assay, and (E) CXCL10 secretion by ELISA. Western blot in MOLM-13 cells with silenced shFBXO21 and shNTC for (F) PI3K pathway proteins and (G) native gel for PI3K complex proteins. H Schematic highlighting FBXO21 mediated alterations within PI3K signaling pathway.
Both silencing of FBXO21 and overexpression of p85α induced ERK activation and CXCL10 expression. CXCL10 protein expression in shFBXO21 MOLM-13 cells could be partially rescued by inhibition of ERK suggesting high levels of CXCL10 affected AML survival and proliferation (Supplemental Fig. 3E). Addition of increasing concentration of CXCL10 at dosages corresponding to levels seen with elevated p85α, similarly decreased colony formation and proliferation, and led to increased apoptosis in MOLM-13 cells (Supplemental Fig. 4A–D). Together these finding suggest that p85α overexpression directly leads to ERK activation and elevated CXCL10 expression, and elevated CXCL10 has a negative impact on AML cells.
In canonical PI3K signaling, the catalytic reaction depends on the activity of p110 binding p85α leading to downstream activation of mTOR and AKT [25,26,27,28]. However previous studies have revealed free p85 can dimerize, negatively impacting PI3K signaling and activating MAPK pathways [29,30,31]. shFBXO21 KD AML cell lines revealed no alterations in mTOR activation following p85α stabilization, however decreased AKT activation was seen with a slightly decreased interaction was seen between p110 and p85α, and dimerization of p85α is found (Fig. 7G). This suggests that loss of FBXO21 leads to decreased canonical PI3K signaling and promotes dimerization of p85α leading to cell death and differentiation of AML cells by elevated CXCL10 via ERK activation.
Discussion
Collectively, our data suggest a novel role of ubiquitin E3 ligase FBXO21 in mediating AML survival and cytokine signaling pathways via p85α ubiquitylation. Although FBXO21 RNA is expressed at lower levels of RNA overall, patients with normal or complex karyotypes and MLL translocations had higher expression FBXO21 protein than healthy BM. Higher expression of FBXO21 correlates with poor survival, which fits with patients with both complex karyotypes and MLL translocations being associated with poor survival [32,33,34]. Silencing in both primary patient samples and AML derived cell lines revealed FBXO21 is required for proliferation, and survival of AML cells. We utilized 4 different AML cell lines, including MOLM-13, which contains a MLL translocation and by protein showed the highest expression of FBXO21. Although MOLM-13 showed significant alterations in vivo, the other cell lines similarly demonstrated that loss of FBXO21 led to decreased proliferation, and increased apoptosis/cell death suggesting FBXO21 could not only be a target in patients with high expression of FBXO21 but all patients. We also showed previously that alterations to FBXO21 minimally affects steady-state hematopoiesis. Silencing of FBXO21 in primary human CD34+ cells showed only ~50% reduction in colony-formation with no induction of early apoptosis, whereas in primary AML cells there was ~90% reduction in colony formation and up to 40% induction of early apoptosis suggesting a therapeutic window for targeting FBXO21 in the context of AML. These findings suggest targeting FBXO21 could solely affect the AML tumor cells with minimal toxicity to the remaining healthy hematopoietic cells [13].
Tight regulation of cytokine signaling within the BM is vital for normal hematopoiesis, and disruptions in cytokine signaling exert profound effects on disease progression and survival of AML. Cytokine and chemokine production in AML is known to be induced by the activation of MAP kinase pathways, and through silencing of FBXO21 in AML, we have observed modulation of the MAP kinase pathway, ERK1/2. The ERK pathway has been previously found to play a major role in the differentiation and proliferation of myeloid cells by regulating inflammatory cytokines and chemokines [35, 36]. Total ERK protein expression was unchanged following silencing of FBXO21 suggesting ERK itself is not the substrate of FBXO21. However mass spectrometry combined with our genomic dataset revealed p85α (PIK3R1) as the ubiquitination target of FBXO21. p85α (PIK3R1) expression is significantly upregulated in AML and increased expression correlates with a worse survival rate, and PI3K pathway is among one of the most frequently upregulated intracellular pathways [6, 37, 38]. Our studies revealed overexpression of p85α was detrimental to the AML cells suggesting that too much p85α also impacts the survival and proliferation of AML. In previous studies we demonstrated that in chronic myeloid leukemia (CML) the oncogene cMYC was regulated by FBXW7 and in a similar fashion silencing of FBXW7 led to stabilization of cMYC which was toxic to the CML cells suggesting specific dosage of oncogenes may be required to drive the disease [2].
Although p85α does not directly bind ERK, it has been suggested that there is crosstalk between the two pathways and that they are not linear signaling cascades (Fig. 7H) [39,40,41]. Overexpression of p85α in MOLM-13 cells, similar to silencing of FBXO21, led to activation of ERK and increased CXCL10 suggesting that p85α is either indirectly or directly activating ERK. In addition, we found that silencing of FBXO21 leads to decreased canonical PI3K signaling with decreased activation of AKT, as well as decreased interaction with the catalytic sub-unit p110. Excess free p85α due to overexpression and stabilization of p85α protein promoted dimerization of p85α. Future studies are needed to decipher the signaling cascade altered by FBXO21 silencing and p85α overexpression. It is unknown whether p85α dimers can activate ERK or decreased AKT phosphorylation promotes ERK activation. Addition of CXCL10 to the media led to an anti-proliferative effect, however previous studies have shown inhibition of AKT signaling can also inhibit suggesting multiple players in the signaling cascade maybe contributing to the anti-proliferative effects. These findings identify a novel role of FBXO21 in regulating PI3K signaling in AML.
These findings identify a novel role for FBXO21 in PI3K signaling by ubiquitination of p85α. Although p85α is a regulatory sub-unit of PI3K signaling and lacks kinase activity, excess free p85α following silencing leads to dimerization and decreased interaction with its catalytic sub-unit p110. Taken together, the data suggest targeting FBXO21 could inhibit canonical PI3K signaling and impedes growth of AML, but may also impede growth of other cancer sub-types dependent on canonical PI3K signaling.
Materials and methods
Cell culture/transplantation
HEK293T, PHOENIX-Amphos, HL-60, KASUMI-1, THP-1, and MOLM-13 cells were purchased from ATCC and DSMZ. Human AML cells (Cureline Translational Cro) were cultured in StemSpan SFEM II media with CD34+ Expansion Supplement (StemCell Technologies). The shRNA plasmids were purchased from Sigma-Aldrich. FBXO21 and ∆FBXO21 were subcloned from pCDNA (kind gift from Dr. Yukiko Yoshida, Tokyo Metropolitan Institute of Medical Science) into pMIGR1 with tandem Strep and Flag tags, and retrovirus was produced as previously described [8, 42]. Lentivirus was produced according to manufacturer instructions. Cells were treated with 1 μg/ml of puromycin 48 h post infection. For cytarabine treatment, cells were treated for 48 h with 50 nM cytarabine, and for MG132 treatment, cells were treated for 4 h with 20 µM MG132. For CFC assay cells were plated 100 cells/well of a 24-well plate in Methocult (H4434, StemCellTechnologies, Vancouver, BC, Canada), and counted between day 7-10.
For transplants, 5 × 105 MOLM-13 cells were transplanted into sub-lethally (250 cGy) irradiated 6–10 week old NSG mice (#005557, Jackson Labs). All experiments performed were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center in accordance with NIH guidelines.
Flow cytometry analysis
For apoptosis, staining was performed following BioLegend apoptosis staining protocol. Antibodies listed in supplemental materials and methods.
RNA-sequencing
Total RNA was harvested from cells using the Monarch Total RNA Miniprep Kit (New England Biolabs, Ipswich, MA, USA). RNA sequencing and analysis was performed by Novogene.
Cytokine and ELISA assays
Cytokine arrays were performed in accordance with the manufacturer’s protocol (R&D Systems, Proteome Profiler Array: Human Cytokine Array Kit). CXCL10 ELISA assays were performed per manufacturer’s protocol (R&D Systems, Human CXCL10/IP-10 DuoSet ELISA).
TMT labeling and mass spectrometry
Samples were prepared and TMT-labeled per manufacturer’s protocol (ThermoFisher TMT16plex Mass Tag Labeling Kits). Following TMT labeling, acetonitrile was removed by speedvac, and samples were resuspended in 0.1% trifluoroacetic acid. Sample concentrations were re-quantified (Pierce Quantitative Colorimetric Peptide Assay kit) and combined in equal concentration. Following combination, samples were fractionated by ThermoFisher high pH reverse phase fractionation kit following manufacturer’s protocol. Fractions were dried and resuspended in 0.1% Formic Acid for MS analysis as previously described [43, 44]. K-ε-GG immunoprecipitation followed by mass spectrometry was performed according to manufacturer’s protocol (Cell Signaling, PTMScan® Ubiquitin Remnant Motif (K-ε-GG) Kit #5562) [45].
In vitro ubiquitination
HEK293T cells were transfected with plasmids encoding HA-FBXO21, HA-ΔFBXO21, or GFP-p85α. 48 h post transfection, HA-FBXO21, HA-ΔFBXO21, or GFP-p85α were immunopurified from the whole cell extracts using Anti-HA (Sigma) or Anti-GFP (MBL International) beads overnight at 4 °C. The immunopurified HA-FBXO21 or HA-ΔFBXO21 (0.5 µg) proteins were incubated with immunopurified GFP-p85α (0.5 µg), E1 (500 ng), E2-UbcH5a (500 ng), FLAG-ubiquitin (0.5 µg) (BostonBiochem), and ATP (10 mM). Ubiquitylation reactions were performed in 100 mM NaCl, 1 mM DTT, 5 mM MgCl2, 25 mM Tris-Cl (pH 7.5), incubated at 30 °C for 2 h, and stopped with 2× laemmli buffer (10 min at 95 °C).
Statistical analysis
All experiments were performed in triplicate unless noted and statistical analyses were performed using unpaired two-tailed Student’s t test assuming samples of equal variance. Error bars depict the standard deviation ± mean. For survival curve, the p value was calculated using a Log-rank (Mantel-cox) test.
Data availability
The datasets generated during and/or analyzed during the current study are available in the ProteomeXchange repository, PXD04419246 [46]. The remaining data needed to evaluate all conclusions are available within the Article and/or Supplementary Information.
References
King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552–66.
Reavie L, Buckley SM, Loizou E, Takeishi S, Aranda-Orgilles B, Ndiaye-Lobry D, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23:362–75.
Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–51.
Kipreos ET, Pagano M. The F-box protein family. Genome Biol. 2000;1:REVIEWS3002.
Hynes-Smith RW, Wittorf KJ, Buckley SM. Regulation of normal and malignant hematopoiesis by FBOX ubiquitin E3 ligases. Trends Immunol. 2020;41:1128–40.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Watanabe K, Yumimoto K, Nakayama KI. FBXO21 mediates the ubiquitylation and proteasomal degradation of EID1. Genes Cells Devoted Mol Cell Mech. 2015;20:667–74.
Yoshida Y, Saeki Y, Murakami A, Kawawaki J, Tsuchiya H, Yoshihara H, et al. A comprehensive method for detecting ubiquitinated substrates using TR-TUBE. Proc Natl Acad Sci USA 2015;112:4630–5.
Zhang C, Li X, Adelmant G, Dobbins J, Geisen C, Oser MG, et al. Peptidic degron in EID1 is recognized by an SCF E3 ligase complex containing the orphan F-box protein FBXO21. Proc Natl Acad Sci USA 2015;112:15372–7.
Yu Z, Chen T, Li X, Yang M, Tang S, Zhu X, et al. Lys29-linkage of ASK1 by Skp1-cullin 1-Fbxo21 ubiquitin ligase complex is required for antiviral innate response. Elife. 2016;5:e14087.
Ye B, Dai Z, Liu B, Wang R, Li C, Huang G, et al. Pcid2 inactivates developmental genes in human and mouse embryonic stem cells to sustain their pluripotency by modulation of EID1 stability. Stem Cells. 2014;32:623–35.
Randle SJ, Laman H. F-box protein interactions with the hallmark pathways in cancer. Semin Cancer Biol. 2016;36:3–17.
Wittorf KJ, Weber KK, Swenson SA, Buckley SM. Ubiquitin E3 ligase FBXO21 regulates cytokine-mediated signaling pathways, but is dispensable for steady-state hematopoiesis. Exp Hematol. 2022;114:33–42.e33.
Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37.
Armand P, Kim HT, Logan BR, Wang Z, Alyea EP, Kalaycio ME, et al. Validation and refinement of the Disease Risk Index for allogeneic stem cell transplantation. Blood. 2014;123:3664–71.
Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74.
Gupta V, Tallman MS, Weisdorf DJ. Allogeneic hematopoietic cell transplantation for adults with acute myeloid leukemia: myths, controversies, and unknowns. Blood. 2011;117:2307–18.
Sorror ML, Storb RF, Sandmaier BM, Maziarz RT, Pulsipher MA, Maris MB, et al. Comorbidity-age index: a clinical measure of biologic age before allogeneic hematopoietic cell transplantation. J Clin Oncol. 2014;32:3249–56.
Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review). Oncol Lett. 2011;2:583–9.
Lunghi P, Tabilio A, Dall’Aglio PP, Ridolo E, Carlo-Stella C, Pelicci PG, et al. Downmodulation of ERK activity inhibits the proliferation and induces the apoptosis of primary acute myelogenous leukemia blasts. Leukemia. 2003;17:1783–93.
Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102:972–80.
Hemmati S, Sinclair T, Tong M, Bartholdy B, Okabe RO, Ames K, et al. PI3 kinase alpha and delta promote hematopoietic stem cell activation. JCI Insight. 2019;5:pii:125832.
Ito Y, Hart JR, Ueno L, Vogt PK. Oncogenic activity of the regulatory subunit p85β of phosphatidylinositol 3-kinase (PI3K). Proc Natl Acad Sci USA 2014;111:16826–9.
Liu Y, Wang D, Li Z, Li X, Jin M, Jia N, et al. Pan-cancer analysis on the role of PIK3R1 and PIK3R2 in human tumors. Sci Rep. 2022;12:5924.
Fox M, Mott HR, Owen D. Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem Soc Trans. 2020;48:1397–417.
Li X, Mak VCY, Zhou Y, Wang C, Wong ESY, Sharma R, et al. Deregulated Gab2 phosphorylation mediates aberrant AKT and STAT3 signaling upon PIK3R1 loss in ovarian cancer. Nat Commun. 2019;10:716.
Hsu J-H, Shi Y, Frost P, Yan H, Hoang B, Sharma S, et al. Interleukin-6 activates phosphoinositol-3′ kinase in multiple myeloma tumor cells by signaling through RAS-dependent and, separately, through p85-dependent pathways. Oncogene. 2004;23:3368–75.
Zhou C, Du J, Zhao L, Liu W, Zhao T, Liang H, et al. GLI1 reduces drug sensitivity by regulating cell cycle through PI3K/AKT/GSK3/CDK pathway in acute myeloid leukemia. Cell Death Dis. 2021;12:231.
Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002;22:965–77.
Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–85.
Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N, et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell. 2014;26:479–94.
Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–84. 2018/02/01
Mrozek K, Bloomfield CD. Chromosome aberrations, gene mutations and expression changes, and prognosis in adult acute myeloid leukemia. Hematol Am Soc Hematol Educ Program. 2006: 169–77.
Dimartino JF, Cleary ML. Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol. 1999;106:614–26.
Kamezaki K, Shimoda K, Numata A, Haro T, Kakumitsu H, Yoshie M, et al. Roles of Stat3 and ERK in G-CSF signaling. Stem Cells. 2005;23:252–63.
Kurosawa M, Numazawa S, Tani Y, Yoshida T. ERK signaling mediates the induction of inflammatory cytokines by bufalin in human monocytic cells. Am J Physiol Cell Physiol. 2000;278:C500–8.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research. 2017;45:W98–102.
Nepstad I, Hatfield KJ, Grønningsæter IS, Reikvam H. The PI3K-Akt-mTOR signaling pathway in human acute myeloid leukemia (AML) cells. Int J Mol Sci. 2020;21:2907.
Ebi H, Costa C, Faber AC, Nishtala M, Kotani H, Juric D, et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc Natl Acad Sci USA 2013;110:21124–9.
Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320–8.
Won JK, Yang HW, Shin SY, Lee JH, Heo WD, Cho KH. The crossregulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. J Mol Cell Biol. 2012;4:153–63.
Caplan M, Wittorf KJ, Weber KK, Swenson SA, Gilbreath TJ, Willow Hynes-Smith R, et al. Multi-omics reveals mitochondrial metabolism proteins susceptible for drug discovery in AML. Leukemia. 2022;36:1296–305.
Hynes-Smith RW, Swenson SA, Vahle H, Wittorf KJ, Caplan M, Amador C, et al. Loss of FBXO9 enhances proteasome activity and promotes aggressiveness in acute myeloid leukemia. Cancers. 2019;11:1717.
Swenson SA, Gilbreath TJ, Vahle H, Hynes-Smith RW, Graham JH, Law HC, et al. UBR5 HECT domain mutations identified in mantle cell lymphoma control maturation of B cells. Blood. 2020;136:299–312.
Buckley SM, Aranda-Orgilles B, Strikoudis A, Apostolou E, Loizou E, Moran-Crusio K, et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell. 2012;11:783–98.
Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45(D1):D1100-D1106.
Acknowledgements
We would like to thank the University of Utah Flow Cytometry Shared Resource Laboratory, Division of Hematology Biorepository at Huntsman Cancer Institute, UNMC Flow Cytometry Research Facility and UNMC Mass Spectrometry and Proteomics Core Facility for expert assistance. The UNMC core facilities are administrated through the Office of the Vice Chancellor for Research and supported by state funds from the Nebraska Research Initiative (NRI) and The Fred and Pamela Buffett Cancer Center’s National Cancer Institute Cancer Support Grant. Human CD34+ cells were available with the support of Cooperative Centers of Excellence in Hematology NIDDK Grant # DK106829. KJW was supported by the UNMC Internal Fellowship. SMB is supported by the National Institutes of Health P20GM121316, 1R37CA262635-01, and 1R01AI53090-01A1. SAS, RKH and SMB are supported by the UNMC Pediatric Cancer Group. This publication was supported by the Huntsman Cancer Institute at the University of Utah, supported by the National Cancer Institute of the National Institutes of Health (NIH) under award number P30CA042014 and Fred & Pamela Buffett Cancer Center Support Grant from the National Cancer Institute under award number P30CA036727.
Author information
Authors and Affiliations
Contributions
KKD, KJW, and SMB conceived and designed the experiments. KKD, KJW, SAS, DCB, CMG and SMB performed experiments and analysis. KKD, KJW, and SMB wrote the manuscript. RKH, GG, NTW provided technical and/or material support. All authors reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dobish, K.K., Wittorf, K.J., Swenson, S.A. et al. FBXO21 mediated degradation of p85α regulates proliferation and survival of acute myeloid leukemia. Leukemia 37, 2197–2208 (2023). https://doi.org/10.1038/s41375-023-02020-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-023-02020-w
- Springer Nature Limited