
Abstract
N6-methyladenosine (m6A), an internal modification in mRNA, plays a critical role in regulating gene expression. Dysregulation of m6A modifiers promotes oncogenesis through enzymatic functions that disrupt the balance between the deposition and removal of m6A modification on critical transcripts. However, the roles of mRNA m6A in multiple myeloma (MM) are poorly understood. The present study showed that RNA demethylase ALKBH5 was overexpressed in MM and associated with a poor prognosis in MM patients. Knocking down ALKBH5 induced apoptosis and inhibited the growth of MM cells in vitro. Xenograft models and gene set enrichment analysis with patient transcriptome datasets also supported the oncogenic role of ALKBH5 in MM. Mechanistic studies showed that ALKBH5 exerted tumorigenic effects in myeloma in an m6A-dependent manner, and TNF receptor-associated factor 1 (TRAF1) was a critical target of ALKBH5. Specifically, ALKBH5 regulated TRAF1 expression via decreasing m6A abundance in the 3'-untranslated region (3'-UTR) of TRAF1 transcripts and enhancing TRAF1 mRNA stability. As a result, ALKBH5 promoted MM cell growth and survival through TRAF1-mediated activation of NF-κB and MAPK signaling pathways. Collectively, our data demonstrated that ALKBH5 played a critical role in MM tumorigenesis and suggested that ALKBH5 could be a novel therapeutic target in MM.
Similar content being viewed by others


Introduction
Multiple myeloma (MM) is a hematological malignancy characterized by uncontrolled expansion of monoclonal plasma cells in the bone marrow (BM) [1]. The malignant development of MM is associated with the accumulation of acquired genetic events and additional epigenetic alterations [2,3,4,5]. The disease remains incurable despite the progress made in MM treatments (for example, proteasome inhibitors, immunomodulatory drugs, and autologous stem cell transplantation) [6, 7]. New therapies with novel mechanisms of action are needed for patients with MM, especially those with refractory and high-risk myeloma [1, 6, 7]. Thus, exploring distinct molecular mechanisms underlying myeloma tumorigenesis is essential to develop potentially effective therapeutic agents.
N6-methyladenosine (m6A) is a prevalent and abundant chemical modification in messenger RNA (mRNA) in eukaryotes [8, 9]. The methylation of N6-adenosine is mainly catalyzed by m6A methyltransferase (writer) complex composed of methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and Wilms’s tumor 1-associating protein (WTAP) [10, 11]. As a reversible modification, m6A methylation can be removed by m6A demethylases (erasers), including fat mass and obesity-associated protein (FTO) and α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) [12, 13]. The dynamic m6A modification of mRNAs can be recognized by various m6A-binding proteins (readers), leading to diverse alterations in mRNA metabolisms, such as mRNA stability [14,15,16], translation efficiency [17, 18], and alternative splicing [19]. Accumulating evidence indicated that the m6A modification and its modulators (writers, erasers, and readers) are involved in the pathological processes in various cancers [20, 21]. In MM, the m6A reader protein heterogeneous nuclear ribonucleoproteins A2/B1 (HNRNPA2B1) plays a pivotal role in promoting MM progression by upregulating AKT3 expression through m6A-dependent stabilization of ILF3 mRNA [22]. However, the functions of mRNA m6A methylation in MM have not yet been investigated in depth. Especially, the roles of m6A writers and erasers in MM development remain largely unknown.
ALKBH5 is a 2-oxoglutarate (2OG) and ferrous iron-dependent nucleic acid oxygenase [23, 24]. Soon after its identification as an m6A eraser [12], ALKBH5 was found to be involved in the initiation and progression of various cancers. Zhang et al. demonstrated that high ALKBH5 expression in glioblastoma (GBM) promotes the proliferation of GBM stem-like cells by sustaining FOXM1 expression [25]. In acute myeloid leukemia (AML), ALKBH5 was required to maintain leukemia stem cell self-renewal and promote AML tumorigenesis by regulating the expression of a set of critical genes (AXL and TACC3) at the posttranscriptional level [26, 27]. Conversely, ALKBH5 also served as a tumor suppressor in some malignancies. For example, Chen et al. found that ALKBH5 exhibited a tumor-suppressive role by inhibiting the expression of LYPD1 in hepatocellular carcinoma cells [28]. These studies suggested that the aberrant expression of key genes caused by the dysregulation of ALKBH5 led to a significant phenotypic change in specific cancers. However, the roles and underlying mechanisms of ALKBH5 in MM are not yet reported. Hence, we conducted a series of functional and mechanistic studies, which indicated a critical role of ALKBH5 in MM tumorigenesis as an m6A demethylase, thereby suggesting that ALKBH5 is a promising therapeutic target in MM.
Results
Increased ALKBH5 expression was associated with poor prognosis in MM
The publicly available MM datasets were queried to examine the expression profile of m6A modifiers in MM. The Amazonia! (amazonia.transcriptome.eu) MM atlas showed that the expression of m6A demethylase ALKBH5, but not of other m6A modifiers (METTL3, METTL14, WTAP, or FTO), was significantly higher in MM cells compared with the normal BM plasma cells (nBMPCs) (Figs. 1A and S1A). Quantitative PCR (qPCR) analyses also showed that ALKBH5 mRNA levels were significantly higher in purified primary myeloma cells and established human myeloma cell lines (HMCLs) compared with peripheral blood mononuclear cells (PBMCs) from healthy donors (Fig. 1B). Gene Expression Omnibus (GEO) dataset analysis demonstrated that the elevated ALKBH5 expression was correlated with myeloma disease progression from normal to smoldering myeloma (SMM) (Fig. 1C) [29] and from MM to plasma cell leukemia (PCL) (Fig. 1D) [30]. Moreover, according to Amazonia! Atlas, ALKBH5 expression was significantly higher in MM compared with other hematological malignancies (Fig. 1E). The Cancer Cell Line Encyclopedia (CCLE) dataset also revealed that ALKBH5 was highly expressed in MM compared with other cancer cell lines (Fig. 1F). In addition, we observed that the top-ranked tumors with high ALKBH5 expression were largely lymphocyte malignancies (Fig. 1E, F).

A Normalized expression signal of ALKBH5 in nBMPCs and MM cells according to Amazonia! Atlas. B qRT-PCR analysis of ALKBH5 expression in normal peripheral blood mononuclear cells (PBMCs) from healthy donors (black), primary cells derived from patients with MM (red), and human myeloma cell lines (blue), using GAPDH as the reference. C ALKBH5 expression in samples from healthy donors (HD) or patients diagnosed with monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM (SMM) in Zhan et al. [29] myeloma dataset according to Oncomine database. D ALKBH5 expression in samples from patients diagnosed with MM and PCL in the dataset GSE39925 [30]. E Normalized expression signal of ALKBH5 in various hematological malignancies according to the Amazonia! Atlas. CLL Chronic lymphocytic leukemia; T-ALL T-cell acute lymphoblastic leukemia; B-ALL B-cell acute lymphoblastic leukemia; CML Chronic myeloid leukemia; AML Acute myeloid leukemia; B-NHL B-cell non-Hodgkin lymphoma; T-NHL T-cell non-Hodgkin lymphoma; MDS Myelodysplastic syndromes. F ALKBH5 mRNA expression in MM and other indicated cancer cell lines, according to the Cancer Cell Line Encyclopedia dataset. T-cell ALL T-cell acute lymphoblastic leukemia; DLBCL Diffuse large B-cell lymphoma; B-cell ALL B-cell acute lymphoblastic leukemia; lung NSC Non-small cell lung cancer; AML Acute myeloid leukemia; CML Chronic myeloid leukemia. G, H Kaplan–Meier survival analysis in indicated myeloma datasets [31, 32]. Patients were divided into two groups based on ALKBH5 expression levels (high and low). Cutoff values were determined by the maximum standardized log-rank statistic. P-value was calculated by the log-rank test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (t-test). ns No significance. Error bars denote mean ± SD.
Next, we investigated the prognostic value of these five canonical m6A modifiers in MM. According to the Arkansas myeloma cohort (GEO accession number GSE4581) [31], the elevated expression of two m6A demethylases (ALKBH5 and FTO) was correlated with poor patient survival (Figs. 1G and S1E). Conversely, the high expression of WTAP predicted improved patient survival, while the expression of METTL3 and METTL14 was not significantly associated with prognosis in MM (Fig. S1B–D). We also validated that patients with high ALKBH5 or FTO expression had significantly shorter overall survival compared with those with low expression in Mulligan et al. [32] myeloma cohort (GSE9782) (Figs. 1H and S1F). Next, we knocked down all the five m6A modifiers in HMCLs using short hairpin RNAs (shRNAs) to clarify their functions in MM (Fig. S1G). Similar to the results of survival analysis (Figs. 1G, H; S1B–F), the knockdown (KD) of ALKBH5 or FTO inhibited the proliferation of HMCLs (Fig. S1H). These phenomena indicated that the hypomethylation of mRNAs mediated by m6A erasers (ALKBH5 and FTO) could promote tumorigenesis in MM. Therefore, we compared the expression pattern of FTO with that of ALKBH5 in MM. We also found that FTO was not significantly overexpressed in MM cells based on another MM atlas [33] and our own samples (Fig. S1I, J). Unlike ALKBH5, FTO expression in MM was not high compared with other blood cancers and solid tumors (Fig. S1K, L). Collectively, increased ALKBH5 expression was correlated with a poor prognosis of MM. Thus, we focused on exploring the role of ALKBH5 in myeloma tumorigenesis.
ALKBH5 played a pro-proliferative and -survival role in MM cells in vitro
Both gain- and loss-of-function studies were performed to investigate the role of ALKBH5 in MM. We conducted shRNA-mediated KD experiments (shA5#1 and shA5#2) to decrease ALKBH5 expression in HMCLs (Fig. 2A). Consequently, the depletion of ALKBH5 had a significant inhibitory effect on cellular growth (Fig. 2B–H), and colony formation (Fig. 2I, J) compared with that in the control group. ALKBH5 KD significantly induced cell apoptosis (Fig. 2K–O) and decreased DNA synthesis (Fig. 2P–T) compared with that in the control group, as revealed by apoptosis and 5-ethynyl-2'-deoxyuridine (EdU) assays, respectively. The cell cycle analysis showed that ALKBH5 KD in MM cells increased the proportion of cells in the G0/G1 phase and decreased the proportions of cells in the S and G2/M phases (Fig. S2A, B). In contrast, the overexpression of ALKBH5 (Fig. S2C) significantly promoted the growth of HMCLs (Fig. S2D–H). Furthermore, we established doxycycline (Dox)-inducible ALKBH5 conditional KD MM cell lines (shA5_Tet-on), and conditional depletion of ALKBH5 (Fig. S2I) significantly inhibited MM cell proliferation (Fig. 2U) and colony formation (Fig. 2V, W). Together, these results indicated that ALKBH5 plays a major role in the proliferation of human MM cells in vitro.

A Immunoblotting of ALKBH5 after shRNA-mediated KD in RPMI-8226 cells. B Proliferation of MM cells RPMI-8226, C CAG, D U266, E MM1.S, F NCI-H929, G AR-P1, H JJN-3 transduced with indicated lentiviruses, as determined by CCK-8. I, J Representative colony images (I) and statistics of colony counts (J) of CAG and U266 cells after stable transduction of the indicated lentiviruses. K–O Apoptotic analysis of MM cells with shNC or shALKBH5s. Representative flow cytometry plots from RPMI-8226 and CAG cells (K) and statistics of the percentage of apoptotic (Annexin V + ) cells in RPMI-8226 (L), M CAG, N U266, and MM1.S (O) cells transduced with indicated lentiviruses. P–T EdU assay of MM cells with shNC or shALKBH5s. Representative flow cytometry plots from RPMI-8226 and CAG cells (P) and statistics of the percentage of EdU-positive cells in RPMI-8226 (Q), R CAG, S U266, T MM1.S. U Growth curves of MM cells after transduction with control or shALKBH5_Tet-on lentiviruses and treatment with Dox at indicated concentrations. V, W Representative colony images (V) and statistics of colony number (W) of CAG and U266 cells after ALKBH5 KD by Dox (1 μg/mL). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (t-test). ns No significance. Error bars denote mean ± SD.
ALKBH5 promoted MM cell growth in vivo
We generated various xenograft models via tail vein injection of the luciferase-labeled MM cell lines into immune-deficient NOD CRISPR Prkdc Il2r Gamma (NCG) mice to evaluate the role of ALKBH5 in MM cell growth in vivo (Figs. 3A–J and S3A–C). In the first xenograft assay, ALKBH5 KD of CAG cells was achieved using Dox-inducible shRNA (Fig. 3A). Chemiluminescence imaging (Fig. 3C, D) and detection of MM cells in BM (Fig. 3E, F) revealed that the bone infiltration of MM cells and subsequent malignant expansion were significantly suppressed by ALKBH5 KD induced by Dox intake (Dox vs. vehicle). As a result, shALKBH5-ON (Dox) group displayed significantly alleviated tumor burden, as revealed by the concentration of immunoglobulin kappa (Igκ) light chain in serum (Fig. 3G) and prolonged survival (Fig. 3B) compared with the shALKBH5-OFF (vehicle) group. We also generated xenograft models using MM cell lines with stable ALKBH5 KD (Fig. 3H–J) or ALKBH5 overexpression (Fig. S3A–C). As expected, shALKBH5 significantly delayed human MM cell RPMI-8226 progression and prolonged survival in recipient mice compared with shNC (Fig. 3H–J). Conversely, the overexpression of ALKBH5 significantly increased the tumorigenic potential of MM cell line U266 and had an adverse impact on the survival in xenografted models (Fig. S3A–C). Furthermore, gene set enrichment analysis (GSEA) with transcriptome datasets of primary MM samples (GSE26760 [34] and GSE19784 [35]) showed that high ALKBH5 expression was positively correlated to genes related to cell growth, anti-apoptosis signaling, cell cycle progression, cancer pathways, and genes upregulated in MM (Figs. 3K–O and S3D–M). Taken together, our MM cell xenograft models and GSEA of myeloma patient data sets supported the role of ALKBH5 in the malignant progression of myeloma in vivo.

A Experimental scheme for (B–G). B Kaplan–Meier survival curve of NCG mice xenografted with the luciferase-labeled CAG cells transduced with Dox-inducible shALKBH5 (shA5_Tet-on) subjected to the vehicle or Dox treatment (n = 10/group). C, D Representative images of chemiluminescence (C) and summary measurement of signals (D). E, F Representative flow plot (E) and statistics of the percentage of human CD138-positive cells in BM samples prepared 26 days after xenograft from the vehicle and Dox groups (n = 5/group). G Concentration of Igκ light chain detected in serum from xenografted mice using ELISA, either mock-treated (vehicle) or with ALKBH5 KD (Dox) (n = 5/group). H–J Kaplan–Meier survival curve (H), representative image (20 days post-xenograft) (I), and summary of luminescence signals (J) of mice implanted with luciferase-labeled RPMI-8226 cells with or without stable KD of ALKBH5 (n = 8/group). K–O GSEA plots show that high ALKBH5 expression is positively related to gene sets related to positive regulation of cell growth, K Negative regulation of apoptotic signaling pathway, L Positive regulation of cell cycle G1 and S phase transition, M pathways in cancer, N and Zhan multiple myeloma up (O) in 304 MM patients based on a transcriptome data set (GSE26760 [34]). The cohort was divided into ALKBH5-High (top 50%) and ALKBH5-Low (bottom 50%) groups. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (t-test). ns, No significance. Error bars denote mean ± SD.
Demethylation activity was required for ALKBH5 to promote MM cell growth
It was determined whether demethylation activity was required for ALKBH5 function. First, we found that global m6A abundance increased as a result of ALKBH5 KD in MM cells, as assessed by the dot blot assay (Fig. 4A, left panel), which confirmed the m6A demethylation activity of ALKBH5 in MM cells. Next, we restored ALKBH5 expression by forced expression of wild-type (WT) ALKBH5 or ALKBH5-H204 A mutant (a catalytically inactive mutant [12]) (Fig. 4B). The dot blot assay showed that the WT-ALKBH5, but not the H204A mutant, restored the global m6A level of ALKBH5 KD cells (Fig. 4A, right panel). Consecutively, we observed that the restoration of WT-ALKBH5 substantially rescued the proliferation (Fig. 4C) and colony formation of ALKBH5-KD HMCLs (Fig. 4D, E). However, the ALKBH5 H204A mutant failed to rescue cell growth inhibition caused by ALKBH5 deficiency (Fig. 4C–E). Similarly, the biological changes, including apoptosis induction (Fig. 4F, G) and DNA synthesis disruption (Fig. 4H, I) caused by ALKBH5 KD in MM cells, were rescued by WT-ALKBH5 but not H204A ALKBH5. Moreover, the disruption of tumor formation by shALKBH5 was rescued by the overexpression of the WT-ALKBH5 but not the catalytic inactive mutant in vivo (Fig. 4J–L). These results confirmed that intact demethylase activity was required for the oncogenic function of ALKBH5 in MM.

A m6A dot blot assays of global m6A abundance in CAG cells transduced with indicated lentiviruses (n = 2 biological replicates). Methylene blue staining (MB) was used as a loading control. B Western blots of ALKBH5 in MM cells transduced with indicated lentiviruses. GAPDH was used as a loading control. C Growth curves of MM cells transduced with indicated lentiviruses. D, E Representative colony images (D) and statistics of colony number (E) of MM cells after the transduction of the indicated lentiviruses. F, G Representative flow plots (F) and statistics of the percentage of apoptotic cells (G) in RPMI-8226 and CAG cells transduced with indicated lentiviruses. H, I Representative flow plots (H) and statistics of the percentage of EdU-positive cells (I) in CAG and U266 cells transduced with indicated lentiviruses. J–L Kaplan–Meier curve for the survival (J), representative image (K; 30 days post-xenograft), and statistics of luminescence counts (L) of mice xenografted with luciferase-labeled CAG cells that transduced with indicated lentiviruses (n = 8/group). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (t-test). ns, No significance. Error bars denote mean ± SD.
Identification of potential downstream targets of ALKBH5 in MM
RNA sequencing (RNA-seq) was conducted to compare the gene expression profile of MM cells with or without ALKBH5 KD to explore the potential mechanisms underlying ALKBH5 function in MM. A total of 2896 and 4570 genes were differentially expressed in cell lines RPMI-8226 and CAG, respectively (Fig. S4A, B). Several of these differentially expressed genes were enriched in apoptosis, cell cycle, pathways in cancer, nuclear factor-kappa B (NF-κB) signaling pathway, mitogen-activated protein kinase (MAPK) signaling pathway, and phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway, according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (Fig. 5A), which was consistent with the phenotype alterations caused by ALKBH5 KD and GSEA of patient datasets. The differentially expressed genes by at least 1.5-fold in both cell lines were overlapped as the candidates of direct targets of ALKBH5 (Fig. 5B). Then, we compared the m6A methylomes in shNC and shALKBH5 RPMI-8226 cells by m6A sequencing (m6A-seq). Consistent with previous reports [8, 9], our m6A-seq data showed that m6A peaks were mainly distributed in the coding region (CDS) and 3'-untranslated region (3'-UTR) of transcripts in MM cells, displaying the consensus motif DRACH (Fig. 5C, Fig. S4C, D). Given the demethylase activity of ALKBH5, up to 1512 genes with significantly elevated m6A peaks upon ALKBH5 KD were identified for KEGG pathway enrichment (Figs. 5D and S4E). Also, the proliferation-associated pathways detected by RNA-seq were significantly enriched in genes with m6A-hyper peaks (Fig. 5D).

A, B RNA-seq analysis of MM cells upon ALKBH5 KD. Heatmap (A) shows differential expression of indicated KEGG pathway genes in RPMI-8226 (left panel) and CAG (right panel) upon ALKBH5 KD. Venn diagram (B) shows the number of genes with significant changes in expression (fold change > 1.5) upon ALKBH5 KD in RPMI-8226 and CAG cells. C, D m6A-seq analysis of ALKBH5 KD in RPMI-8226 cells. Distribution of total m6A peaks in the indicated regions of mRNA transcripts in the control and ALKBH5 KD cells (C). Bubble plot (D) shows KEGG pathway analysis of the genes with significantly increased m6A abundance in ALKBH5 KD cells (P < 0.05). E Integrative analysis of RNA-seq and m6A-seq to identify the potential targets of ALKBH5 in MM. Up or Down indicates genes with significantly increased or decreased expression upon ALKBH5 KD in both RPMI-8226 and CAG cells as detected by RNA-seq (fold change > 1.5), respectively. Hyper indicates genes with significantly higher m6A abundance in m6A-Seq (P < 0.05). F, G Heatmap shows differential expression of overlapped genes in RMPI-8226 (F) and CAG (G) upon ALKBH5 KD. H Kaplan–Meier survival analysis in the Arkansas myeloma dataset [31]. The cutoff value was determined by the maximum standardized log-rank statistic. P-value was detected using the log-rank test. I Pearson’s correlation between ALKBH5 and TRAF1 mRNA expression in the indicated datasets [35,36,37,38]. The expression values were log2 transformed.
Integrative analysis of the RNA-seq and m6A-seq data identified 10 downregulated and 1 upregulated potential targets of ALKBH5 in MM (Fig. 5E–G). The qPCR data validated that most of these genes were significantly regulated by ALKBH5 in accordance with RNA-seq results (Fig. S5A, B). Moreover, the prognostic value of the 11 genes in MM was investigated. We found that high expression of TRAF1, CHST7 (excluded by qPCR data), PPL, and ERO1A (excluded by qPCR data and unexpected prognostic impact) was correlated with poor survival of MM patients in the Arkansas myeloma cohort [31], whereas the other potential targets of ALKBH5 did not show a prognostic significance in MM (Figs. 5H; S5C–L). Furthermore, gene expression analysis by querying GEO datasets (GSE57317 [36], GSE82307 [37], GSE31161 [38], and GSE19784 [35]) validated the significant positive correlation between ALKBH5 and TRAF1 expression in samples from MM patients (Fig. 5I) but not between ALKBH5 and PPL expression (Fig. S5M and N).
Hence, TRAF1 may be responsible for the altered phenotype following ALKBH5 intervention in MM cells. Tumor necrosis factor receptor (TNFR)-associated factor 1 (TRAF1) is an adapter molecule known for its role in TNFR-induced cell survival [39]. In normal conditions, TRAF1 expression was largely restricted to activated lymphocytes, dendritic cells, and certain epithelia [40, 41]. Several lymphoid malignancies, especially B-cell types, displayed high levels of TRAF1 expression [39, 41,42,43,44,45,46]. Besides, TRAF1 overexpressed and played a critical oncogenic role in other types of cancers [47,48,49]. We found that TRAF1 was highly expressed in MM and shared a similar expression pattern with ALKBH5 (Fig. S5O–R). Thus, we focused on TRAF1 as a potential key target of ALKBH5 in subsequent studies.
TRAF1 was a direct target of ALKBH5 in MM
In line with RNA-seq data (Fig. 6A), our qPCR results confirmed the decreased mRNA level of TRAF1 following ALKBH5 KD in MM cells (Figs. 6B and S6A). ALKBH5 KD also decreased the TRAF1 protein level in MM cell lines (Fig. 6C). On the other hand, the forced expression of the wild-type ALKBH5, but not the mutant H204A, restored TRAF1 expression in ALKBH5 KD cells (Figs. 6D, E; S6B, C). These findings indicated that ALKBH5 regulated TRAF1 expression through its demethylation activity. In addition, we found that TRAF1 expression was significantly upregulated by the KD of m6A methyltransferase METTL3 (Fig. S6F, G), which confirmed that TRAF1 was regulated by m6A modulators. Next, we performed m6A immunoprecipitation (m6A-IP), followed by TRAF1-specific qPCR to substantiate the change in TRAF1 m6A methylation levels following ALKBH5 KD. Consistent with the current m6A-seq results (Fig. 6A), TRAF1 transcripts showed an increased m6A level after ALKBH5 KD (Fig. 6F and S6H). Conversely, the forced expression of WT-ALKBH5, but not the H204A mutant ALKBH5, decreased the m6A abundance of TRAF1 mRNA in MM cells (Fig. 6G). Furthermore, we conducted RNA immunoprecipitation (RIP) of ALKBH5 in MM cells and found that TRAF1 transcripts were significantly enriched, suggesting that ALKBH5 is bound to TRAF1 transcripts directly (Figs. 6H, I; S6I). Together, these results indicated that TRAF1 was a direct downstream target of ALKBH5 in MM.

A IGV tracks displaying the mRNA (top) and m6A (bottom) abundance in TRAF1 transcripts in control and ALKBH5 KD MM cells as detected by RNA-seq and m6A-seq. B qRT-PCR analysis showing TRAF1 levels in MM cells transduced with indicated lentiviruses. C Western blots of ALKBH5 and TRAF1 in MM cells transduced with indicated lentiviruses. Cyclophilin B was used as a loading control. D, E Relative mRNA level (D) and Western blots (E) of ALKBH5 and TRAF1 in MM cells transduced with indicated lentiviruses. GAPDH was used as a loading control. F m6A-IP-qPCR analysis of m6A enrichment on TRAF1 mRNA in RPMI-8226 cells with or without ALKBH5 KD. G m6A-IP-qPCR analysis of m6A enrichment on TRAF1 mRNA in U266 cells transduced with indicated lentiviruses. H, I ALKBH5-RIP-qPCR validation of ALKBH5 binding to TRAF1 mRNA in RPMI-8226 (H) and U266 (I) cells. J, K mRNA half-life (t1/2) of TRAF1 in RPMI-8226 (H) and U266 (I) cells transduced with indicated lentiviruses. L qRT-PCR analysis of TRAF1 mRNA expression in MM cells transduced with indicated lentiviruses. M Immunoblotting for ALKBH5, YTHDF2, and TRAF1 in RPMI-8226 cells after transduction with indicated lentiviruses. Cyclophilin B was used as a loading control. N Graphical representation of dual-luciferase reporters. The wild-type or mutant (m6A motif DRACH mutated) sequence of TRAF1–3'-UTR was cloned into pmirGLO vector between a firefly and Renilla luciferase elements. O, P Relative luciferase activity of the wild-type or mutant TRAF1–3'-UTR dual-luciferase reporter in 293 T cells treated with indicated lentivirus. Relative luciferase activity was calculated using the ratio of firefly and Renilla luciferase activity values. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (t-test). ns No significance. Error bars denote mean ± SD.
ALKBH5 regulated TRAF1 mRNA stability by altering m6A modification
RNA m6A modification was reported to affect mRNA stability [14,15,16]. The loss of mRNA stability could explain the reduced TRAF1 mRNA expression in ALKBH5 KD cells. Therefore, we conducted an RNA decay assay using the transcription inhibitor actinomycin D to assess the change of TRAF1 mRNA stability following ALKBH5 KD or overexpression in MM cells. Consequently, ALKBH5 KD significantly decreased TRAF1 mRNA half-life (Fig. 6J), while the overexpression of ALKBH5 prolonged the half-life of TRAF1 transcripts (Fig. 6K). The various biological effects of mRNA m6A modification are effectuated by different m6A readers [50]. YTH domain-containing family protein 2 (YTHDF2) is the major m6A reader responsible for the degradation of its target m6A modified transcripts [14, 15]. Thus, we knocked down YTHDF2 in ALKBH5 KD MM cells and observed that TRAF1 expression was restored (Figs. 6L, M; S6J), suggesting that YTHDF2 mediated the effect of ALKBH5 on TRAF1 mRNA. In m6A-seq, ALKBH5 deficiency markedly increased m6A abundance in the 3'-UTR of TRAF1 transcripts (Fig. 6A). We performed the TRAF1 3'-UTR-reporter dual-luciferase assay to determine the role of TRAF1 3'-UTR (Fig. 6N) and found that the activity of luciferase decreased upon ALKBH5 KD, while mutations at m6A motif sites (A to C) in TRAF1 3'-UTR resulted in resistance to the effect of ALKBH5 KD (Fig. 6O). In contrast, the forced expression of WT-ALKBH5, but not H204A mutant, increased the activity of luciferase, while the mutation of m6A sites also abolished this effect (Fig. 6P). Together, these findings suggested that ALKBH5 regulated YTHDF2-mediated TRAF1 mRNA stability in MM cells via m6A modifications in the 3'-UTR of TRAF1 transcript.
TRAF1 was a major contributor to the function of ALKBH5 in MM
Since TRAF1 could be a direct target of ALKBH5, we sought to study the function of TRAF1 in MM. In line with the phenotype resulting from ALKBH5 KD, TRAF1 KD significantly inhibited cell growth and colony formation in HMCLs (Fig. 7A–D). Moreover, TRAF1 KD induced apoptosis and inhibited DNA duplication in MM cells (Figs. 7E, F; S7A, B), which mimicked the effects of ALKBH5 depletion. Cell cycle analysis of MM cells post-KD of TRAF1 also detected defects in the G1-S phase transition (Fig. S7C, D). In addition, our in vivo study also confirmed that TRAF1 KD substantially reduced MM tumor formation in xenograft mice (Fig. 7G–I). On the other hand, the in vitro study showed that the overexpression of TRAF1 promoted MM growth (Fig. S7E–J). Thus, it could be speculated that TRAF1 mediated the function of ALBKH5 in MM cells. Next, we restored TRAF1 expression in ALKBH5 KD MM cells to clarify whether TRAF1 was a significant contributor to the function of ALKBH5 in MM proliferation (Fig. S7K) and found that the growth inhibitory effects caused by ALKBH5 deficiency were largely rescued by restoration of TRAF1 expression (Figs. 7J–M; S7L–N). Overall, these data suggested that TRAF1 was a functional target of ALKBH5 in MM.

A Western blots show the KD efficiency of TRAF1 in MM cells. B Effects of TRAF1 KD on cell growth in MM cells. C, D Representative colony images (C) and statistics of colony counts (D) of CAG and U266 cells after TRAF1 KD. E Statistics of the percentage of apoptotic cells of MM cells upon TRAF1 KD (see also Fig. S7A). F Statistics of the percentage of EdU-positive cells in RPMI-8226 and CAG cells upon TRAF1 KD (see also Fig. S7B). G–I Representative image (G; 15 days post-xenograft), a summary of luminescence counts (H), and Kaplan–Meier curve for the survival (I) of mice implanted with luciferase-labeled CAG cells with or without TRAF1 KD (n = 8/group). J–M Effects of TRAF1 restoration on cell proliferation (J), colony-forming capacity (K), apoptosis (L), and DNA synthesis (revealed by EdU; M) of ALKBH5 KD MM cells (see also supplemental S7L–N). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (t-test). ns No significance. Error bars denote mean ± SD.
ALKBH5-TRAF1 regulated NF-κB and MAPK pathways in MM
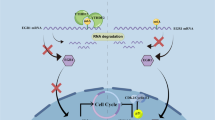
Since ALKBH5 upregulates TRAF1 expression and contributes to MM growth, we sought to explore the specific mechanism underlying their oncogenic actions. TRAF1 contributes to apoptosis resistance in the TNFR signaling pathway as part of the complex with TRAF2, where it promotes NF-κB activation through recruitment of cellular inhibitor of apoptosis protein (cIAP) [39, 51]. Reportedly, TRAF1 enhances the MAPK pathway by affecting TRAF2-mediated Lys48-linked ubiquitination of the serine/threonine-protein kinase B-raf (BRAF) [48]. TRAF1 KD led to decreased BRAF protein expression, decreased downstream MEK/ERK activation, and inhibited cell growth in human lung cancer cell lines [48]. Consistently, we found that TRAF1 inhibition also downregulated NF-κB and MAPK pathways in MM cells (Fig. 8A). Interestingly, we observed significant enrichment of the gene sets of NF-κB and MAPK pathways in ALKBH5 high-expression cases from the cohorts of patients with MM (GSE26760 [34] and GSE19784 [35]) by GSEA (Figs. 8B; S8A, B), suggesting that ALKBH5 also positively regulated these pathways. Thus, we proposed that ALKBH5 enhances TRAF1-mediated activation of NF-κB and MAPK pathways in MM cells, followed by the promotion of cell growth. As expected, knocking down ALKBH5 in MM cells not only led to TRAF1 suppression but also reduced the activation of downstream NF-κB and MAPK signaling molecules (Fig. 8C), which was rescued by TRAF1 restoration (Fig. 8D). Collectively, these data indicated that the ALKBH5/m6A/TRAF1 axis regulated MAPK and NF-κB pathways to promote survival and cell growth in MM cells (Fig. 8E).

A Western blots of TRAF1, NF-κB p105, P-NF-κB p105 (Ser932), NF-κB p50, NF-κB p65, BRAF, p-BRAF (Ser445), p-MEK1/2 (Ser221), p-ERK1/2 (Thr202/Tyr204), c-Myc, and p-c-Fos (Ser32) in RPMI-8226 cells with or without TRAF1 KD. Cyclophilin B was used as a loading control. B GSEA plots show enriched gene sets of NF-κB and MAPK signaling pathways and MAPK downstream targets in ALKBH5-High (top 50%) vs. ALKBH5-Low (bottom 50%) groups in a cohort of patients with MM (GSE26760 [34]). C, D Western blots of NF-κB and MAPK pathway molecules in MM cells upon ALKBH5 KD (C) and TRAF1 restoration (D). Cyclophilin B was used as a loading control. E Proposed model showing the role of ALKBH5 in MM pathogenesis.
ALKBH5 expression in MM subgroups with various genetic features
We analyzed ALKBH5 expression in several MM cohorts containing different genetic subgroups, most of which were defined via immunoglobulin heavy-chain locus (IgH; at 14q32) translocations and cyclin D overexpression (TC classification) [31, 52], to determine whether ALKBH5 overexpression was associated with recurrent genetic changes in MM. ALKBH5 expression levels did not show a significant difference between patients with TC populations and unclassified group (None) in Mulligan et al. [32] and HOVON65/GMMG-HD4 [35] cohort (Fig. S9A, B). Interestingly, ALKBH5 expression was significantly lower in the hyperdiploid group compared with other clusters with IgH translocations in both HOVON65/GMMG-HD4 [35] and Zhan et al. [31] cohorts (Fig. S9B, C). Previous studies suggested that hyperdiploidy conferred a relatively favorable prognosis in MM [53,54,55,56], which was consistent with our finding that lower ALKBH5 expression predicted a better prognosis. Moreover, in line with the activation of the NF-κB pathway by ALKBH5, we also observed high ALKBH5 expression in a subgroup (NFκB) characterized by the high expression of genes involved in the NF-κB pathway in the HOVON65/GMMG-HD4 [35] cohort (Fig. S9B). In addition, ALKBH5 was highly expressed in the MMSET group in all cohorts (Fig. S9A–C), indicating that ALKBH5 might be regulated by dimethylation of histone H3 at lysine 36 (H3K36me2) mediated by histone methyltransferase MMSET [3]. However, ALKBH5 expression was not altered significantly upon MMSET KD nor alterations of MMSET translocation in MM cell line KMS11 according to GEO datasets (GSE29147 [57], GSE29148 [57], GSE57863 [58], and GSE24746 [3]) (Fig. S9D–G). Furthermore, compared with patients with no 1q21 amplification (two copies), patients with three copies showed a significant increase in ALKBH5 expression in the cohort by Zhan et al. [31] (Fig. S9H). However, ALKBH5 expression did not show a significant difference between two copies and more than four copies (Fig. S9H). Overall, MM with IgH translocations showed higher ALKBH5 expression compared with the hyperdiploid group, and ALKBH5 expression was not likely to be regulated by MMSET in MM cells.
Discussion
RNA m6A represents a new layer of the regulatory mechanism controlling gene expression [19]. Accumulating data suggested that m6A modulators are dysregulated in various cancers [19, 20]. In this study, we found that m6A eraser ALKBH5 was highly expressed in MM, and increased ALKBH5 expression was correlated with a poor prognosis in MM patients. Using various HMCLs and xenograft models, we demonstrated that ALKBH5 and its demethylation activity were required for myeloma cell growth and survival. RNA-seq, m6A-seq, and ALKBH5-RIP assay identified TRAF1 as a direct target of ALKBH5. We showed that ALKBH5 positively regulated TRAF1 mRNA stability in an m6A-dependent manner. Further functional studies validated that TRAF1 was a pivotal contributor to the oncogenic effect of ALKBH5 in MM. Of course, other potential targets of ALKBH5 may also contribute to the overall function of ALBKH5 in MM, which needs to be investigated systematically. Additionally, we examined the mechanisms underlying the MM-promoting effects of the ALKBH5-TRAF1 axis. Activation of NF-κB and MAPK signaling pathways plays a critical role in tumorigenesis in MM [59]. We demonstrated that as downstream targets of TRAF1, the key components of NF-κB and MAPK pathways could also be indirectly regulated by ALKBH5 in MM cells. Besides these mechanisms, other undiscovered tumor-promoting targets of TRAF1 might exist in MM cells, which requires systematic exploration.
The current study also provides insights into the expression profile of the other canonical m6A writers and erasers (METTL3, METTL14, WTAP, and FTO) and their impacts on MM proliferation. Several other reported m6A regulators were not covered in this study. However, we found YTHDF2 was a crucial reader protein that mediates the effect of ALKBH5 on TRAF1 transcripts. YTHDF2 was dysregulated in various human malignancies and played an oncogenic or tumor-suppressive role by facilitating the degradation of m6A-modified transcripts [60,61,62,63,64,65,66]. The role of YTHDF2 in MM has not yet been elucidated. Therefore, it needs to be investigated in the future. The current study mainly focused on the effects of ALKBH5 on myeloma cell growth and survival. RNA m6A modification was also found to be involved in the self-renewal of cancer stem cells, tumor metastasis, and drug response/resistance [20, 21], which encourages us to study other effects of ALKBH5 (or other regulators) on MM for a comprehensive understanding of the roles of m6A modification in MM.
Recent studies have linked ALKBH5 overexpression to epigenetic changes in some cancers [26, 67, 68]. For example, Wang et al. demonstrated that histone demethylase KDM4C reduced the H3K9me3 (trimethylation of lysine 9 on histone H3) level and increased the chromatin accessibility of ALKBH5 locus, that in turn promoted the transcription of ALKBH5 in AML cells [26]. Thus, we sought to investigate the upstream regulators of ALKBH5 in MM by studying the correlation between ALKBH5 expression and primary/secondary genetic events in MM. Although ALKBH5 was expressed at a high level in MM cluster with MMSET translocation, it might not be regulated by methyltransferase MMSET. Also, the expression of ALKBH5 was significantly higher in patients with IgH translocations compared with those with hyperdiploidy. However, tumor-promoting effects of ALKBH5 existed across our MM cell lines with various genetic characteristics (Fig. S9I), including RPMI-8226 that had no IgH translocation but displayed the highest ALKBH5 expression. Thus, the correlation between ALKBH5 expression and IgH translocation warrants an in-depth investigation. Furthermore, the dataset GSE39925 [30] showed that ALKBH5 expression was higher in the PCL group than the MM group, but we found that ALKBH5 deletion had a relatively moderate inhibitory effect on the cellular growth of JJN-3, a PCL cell line, compared with the MM cell lines, suggesting that ALKBH5 overexpression might not be the key driver for MM progression to PCL. Therefore, further investigation is needed into other potential upstream regulators of ALKBH5 in MM and the exact correlation between ALKBH5 expression and MM disease evolution.
In summary, the current study uncovered the importance of ALKBH5-mediated mRNA m6A demethylation in MM pathogenesis. ALKBH5 promoted MM cell survival and proliferation via posttranscriptional regulation of TRAF1 expression, which activated NF-κB and MAPK signaling pathways. Given the critical role of ALKBH5 in MM tumorigenesis, targeting ALKBH5 is a promising therapeutic strategy for treating MM patients.
Methods
Primary samples
All samples from newly diagnosed MM patients were collected from BM aspirations after obtaining informed consent. CD138 + MM cells were isolated using CD138 beads (Miltenyi Biotec, Germany). PBMCs from healthy donors were purified using Ficoll (Basal Media, Shanghai, China). All experiments using primary human samples were conducted in accordance with the ethical regulations and were approved by the ethics committees of the First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China.
Human MM cell line xenograft models
CAG, RPMI-8226, and U266 cells were transduced with indicated lentiviruses and selected using 2 μg/mL puromycin for 2 days. 5 × 105 selected cells were injected via the tail vein into 4- to 5-week-old NCG mice (GemPharmatech, Nanjing, China). In vivo MM growth was monitored via chemiluminescence imaging of mice following intraperitoneal injection with luciferin (Promega, Madison, WA, USA) every 5 days. The mice were randomly assigned to the experimental groups. All animal experiments were approved by and conducted in accordance with the guidelines of the Animal Experimental Ethical Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University.
Quantification and statistical analysis
The quantification data were presented as mean ± standard deviation (SD) of three independent experiments. The log-rank (Mantel-Cox) test was performed for Kaplan–Meier survival analysis. All the other data were analyzed using the two-tailed Student’s t-test. Statistical analyses were performed using GraphPad Prism 9.0.
Additional methods are provided in supplemental Methods.
References
van de Donk NWCJ, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397:410–27.
Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol. 2018;15:409–21.
Martinez-Garcia E, Popovic R, Min DJ, Sweet SMM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011;117:211–20.
Ren Z, Ahn JH, Liu H, Tsai YH, Bhanu NV, Koss B, et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 2019;134:1176–89.
Alzrigat M, Parraga AA, Jernberg-Wiklund H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin Cancer Biol. 2018;51:101–15.
Yang Y, Li Y, Gu H, Dong M, Cai Z. Emerging agents and regimens for multiple myeloma. J Hematol Oncol. 2020;13:150.
Pinto V, Bergantim R, Caires HR, Seca H, Guimaraes JE, Vasconcelos MH. Multiple myeloma: available therapies and causes of drug resistance. Cancers 2020;12:407.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012;485:201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012;149:1635–46.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014;505:117–20.
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5’ UTR m(6)A promotes cap-independent translation. Cell 2015;163:999–1010.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015;161:1388–99.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19.
Huang H, Weng H, Chen J. m6A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37:270–88.
Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol. 2018;11:48.
Jiang F, Tang X, Tang C, Hua Z, Ke M, Wang C, et al. HNRNPA2B1 promotes multiple myeloma progression by increasing AKT3 expression via m6A-dependent stabilization of ILF3 mRNA. J Hematol Oncol. 2021;14:54.
Thalhammer A, Bencokova Z, Poole R, Loenarz C, Adam J, O’Flaherty L, et al. Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1alpha (HIF-1alpha). PLoS One. 2011;6:e16210.
Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, et al. Structure of human RNA N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014;42:4741–54.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606 e596.
Wang J, Li Y, Wang P, Han G, Zhang T, Chang J, et al. Leukemogenic chromatin alterations promote AML leukemia stem cells via a KDM4C-ALKBH5-AXL signaling axis. Cell Stem Cell. 2020;27:81–97.e88.
Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. 2020;27:64–80 e69.
Chen Y, Zhao Y, Chen J, Peng C, Zhang Y, Tong R, et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m6A-guided epigenetic inhibition of LYPD1. Mol Cancer. 2020;19:123.
Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2006;109:1692–1700.
Todoerti K, Agnelli L, Fabris S, Lionetti M, Tuana G, Mosca L, et al. Transcriptional characterization of a prospective series of primary plasma cell leukemia revealed signatures associated with tumor progression and poorer outcome. Clin Cancer Res. 2013;19:3247–58.
Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood 2006;108:2020–8.
Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2006;109:3177–88.
Tarte K, Zhan F, De Vos J, Klein B, Shaughnessy J Jr. Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B-cell differentiation. Blood 2003;102:592–600.
Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011;471:467–72.
Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, Jauch A, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 2010;116:2543–53.
Heuck CJ, Qu P, van Rhee F, Waheed S, Usmani SZ, Epstein J, et al. Five gene probes carry most of the discriminatory power of the 70-gene risk model in multiple myeloma. Leukemia 2014;28:2410–3.
Weinhold N, Ashby C, Rasche L, Chavan SS, Stein C, Stephens OW, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016;128:1735–44.
Mitchell JS, Li N, Weinhold N, Forsti A, Ali M, van Duin M, et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat Commun. 2016;7:12050.
Edilova MI, Abdul-Sater AA, Watts TH. TRAF1 signaling in human health and disease. Front Immunol. 2018;9:2969.
Zapata JM, Reed JC. TRAF1: Lord without a RING. Sci STKE. 2002;2002:pe27.
Zapata JM, Krajewska M, Krajewski S, Kitada S, Welsh K, Monks A, et al. TNFR-associated factor family protein expression in normal tissues and lymphoid malignancies. J Immunol. 2000;165:5084–96.
Zhu S, Jin J, Gokhale S, Lu AM, Shan H, Feng J, et al. Genetic alterations of TRAF proteins in human cancers. Front Immunol. 2018;9:2111.
Durkop H, Hirsch B, Hahn C, Foss HD, Stein H. Differential expression and function of A20 and TRAF1 in Hodgkin lymphoma and anaplastic large cell lymphoma and their induction by CD30 stimulation. J Pathol. 2003;200:229–39.
Feldman AL, Vasmatzis G, Asmann YW, Davila J, Middha S, Eckloff BW, et al. Novel TRAF1-ALK fusion identified by deep RNA sequencing of anaplastic large cell lymphoma. Genes Chromosomes Cancer. 2013;52:1097–102.
Abate F, Todaro M, van der Krogt JA, Boi M, Landra I, Machiorlatti R, et al. A novel patient-derived tumorgraft model with TRAF1-ALK anaplastic large-cell lymphoma translocation. Leukemia 2015;29:1390–401.
Munzert G, Kirchner D, Stobbe H, Bergmann L, Schmid RM, Dohner H, et al. Tumor necrosis factor receptor-associated factor 1 gene overexpression in B-cell chronic lymphocytic leukemia: Analysis of NF-kappa B/Rel-regulated inhibitors of apoptosis. Blood 2002;100:3749–56.
Wu TF, Li YC, Ma SR, Bing L, Zhang WF, Sun ZJ. Expression and associations of TRAF1, BMI-1, ALDH1, and Lin28B in oral squamous cell carcinoma. Tumour Biol. 2017;39:1010428317695930.
Wang Q, Gao G, Zhang T, Yao K, Chen H, Park MH, et al. TRAF1 is critical for regulating the BRAF/MEK/ERK pathway in non–small cell lung carcinogenesis. Cancer Res. 2018;78:3982.
Wen X, Wang B, Feng T, Yuan W, Zhou J, Fang T. TNF receptor-associated factor 1 as a biomarker for assessment of non-small cell lung cancer metastasis and overall survival. Clin Respir J. 2018;12:2197–203.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998;281:1680–3.
Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J Jr. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 2005;106:296–303.
Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone du Myelome. Blood 2007;109:3489–95.
Chng WJ, Santana-Davila R, Van Wier SA, Ahmann GJ, Jalal SM, Bergsagel PL, et al. Prognostic factors for hyperdiploid-myeloma: effects of chromosome 13 deletions and IgH translocations. Leukemia 2006;20:807–13.
Pawlyn C, Melchor L, Murison A, Wardell CP, Brioli A, Boyle EM, et al. Coexistent hyperdiploidy does not abrogate poor prognosis in myeloma with adverse cytogenetics and may precede IGH translocations. Blood 2015;125:831–40.
Chretien ML, Corre J, Lauwers-Cances V, Magrangeas F, Cleynen A, Yon E, et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood 2015;126:2713–9.
Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44:609–20.
Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014;10:e1004566.
Hideshima T, Anderson KC. Signaling pathway mediating myeloma cell growth and survival. Cancers 2021;13:216.
Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA m6A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25:137–.e136.
Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Disco. 2021;11:480.
Li J, Xie H, Ying Y, Chen H, Yan H, He L, et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Mol Cancer. 2020;19:152.
Li Y, Sheng H, Ma F, Wu Q, Huang J, Chen Q, et al. RNA m6A reader YTHDF2 facilitates lung adenocarcinoma cell proliferation and metastasis by targeting the AXIN1/Wnt/β-catenin signaling. Cell Death Dis. 2021;12:479.
Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61.
Shen X, Zhao K, Xu L, Cheng G, Zhu J, Gan L, et al. YTHDF2 inhibits gastric cancer cell growth by regulating FOXC2 signaling pathway. Front Genet. 2020;11:592042.
Wang JY, Lu AQ. The biological function of m6A reader YTHDF2 and its role in human disease. Cancer Cell Int. 2021;21:109.
Hao L, Yin J, Yang H, Li C, Zhu L, Liu L, et al. ALKBH5-mediated m(6)A demethylation of FOXM1 mRNA promotes progression of uveal melanoma. Aging 2021;13:4045–62.
Zhang D, Ning J, Okon I, Zheng X, Satyanarayana G, Song P, et al. Suppression of m6A mRNA modification by DNA hypermethylated ALKBH5 aggravates the oncological behavior of KRAS mutation/LKB1 loss lung cancer. Cell Death Dis. 2021;12:518.
Acknowledgements
We thank the members of our laboratory for helpful discussions and assistance. This study was supported by grants from the National Natural Science Foundation of China (81800201, 81800202, and 81872322), the Zhejiang Key Research and Development Project (2020C03014), and the National Major Scientific and Technological Special Project for Significant New Drug Development (2018ZX09733-003).
Author information
Authors and Affiliations
Contributions
JQ and YH designed the project, performed experiments, analyzed results, and wrote the paper. JQ and JC performed the bioinformatics analyses of MM datasets. QC and HG performed xenograft studies. YL provided MM samples. RX, YL, WC, JZ, and LC provided critical suggestions. CZ, JH, and EZ supervised the project. All authors discussed the results and provided assistance in some experiments and data analyses.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qu, J., Hou, Y., Chen, Q. et al. RNA demethylase ALKBH5 promotes tumorigenesis in multiple myeloma via TRAF1-mediated activation of NF-κB and MAPK signaling pathways. Oncogene 41, 400–413 (2022). https://doi.org/10.1038/s41388-021-02095-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-021-02095-8
- Springer Nature Limited
This article is cited by
-
Pan-cancer analysis of the TRAF family genes and their correlation with prognosis, TME, immune and drug sensitivity
European Journal of Medical Research (2024)
-
ALKBH5 regulates chicken adipogenesis by mediating LCAT mRNA stability depending on m6A modification
BMC Genomics (2024)
-
KIAA1429 increases FOXM1 expression through YTHDF1–mediated m6A modification to promote aerobic glycolysis and tumorigenesis in multiple myeloma
Cell Biology and Toxicology (2024)
-
N6-Methyladenosine Methylation of mRNA in Cell Apoptosis
Molecular Neurobiology (2024)
-
PRMT1 methylation of WTAP promotes multiple myeloma tumorigenesis by activating oxidative phosphorylation via m6A modification of NDUFS6
Cell Death & Disease (2023)