
Abstract
Research on obesity- and diabetes mellitus (DM)-related carcinogenesis has expanded exponentially since these two diseases were recognized as important risk factors for cancers. The growing interest in this area is prominently actuated by the increasing obesity and DM prevalence, which is partially responsible for the slight but constant increase in pancreatic cancer (PC) occurrence. PC is a highly lethal malignancy characterized by its insidious symptoms, delayed diagnosis, and devastating prognosis. The intricate process of obesity and DM promoting pancreatic carcinogenesis involves their local impact on the pancreas and concurrent whole-body systemic changes that are suitable for cancer initiation. The main mechanisms involved in this process include the excessive accumulation of various nutrients and metabolites promoting carcinogenesis directly while also aggravating mutagenic and carcinogenic metabolic disorders by affecting multiple pathways. Detrimental alterations in gastrointestinal and sex hormone levels and microbiome dysfunction further compromise immunometabolic regulation and contribute to the establishment of an immunosuppressive tumor microenvironment (TME) for carcinogenesis, which can be exacerbated by several crucial pathophysiological processes and TME components, such as autophagy, endoplasmic reticulum stress, oxidative stress, epithelial-mesenchymal transition, and exosome secretion. This review provides a comprehensive and critical analysis of the immunometabolic mechanisms of obesity- and DM-related pancreatic carcinogenesis and dissects how metabolic disorders impair anticancer immunity and influence pathophysiological processes to favor cancer initiation.
Similar content being viewed by others


Introduction
As a multifactorial consequence of socioeconomic development, the prevalence of obesity and diabetes mellitus (DM) is booming in most parts of the world, regardless of the different landscapes among nations and regions.1,2 The detrimental outcomes and threats of obesity and DM include disability, a shortened life span, and many other critical conditions affecting both physical and mental health, whether acute, chronic, or even terminal.1,2,3,4,5,6 Some of the most vital sequelae of obesity and DM include various malignancies, including pancreatic cancer (PC).7,8,9 Despite its aggressiveness and lethality, the insidious symptoms of PC make employing applicable and sensitive screening methods difficult. However, etiological studies not only can help develop tools to detect PC at an early stage but also can provide decisive clues for effective prevention, which would undoubtedly benefit both cancer-free individuals and those with undiagnosed PC.
In this context, the accelerated rise in the prevalence of obesity and DM and the intimidating biological behaviors of PC have inspired tremendous explorations regarding their correlations. Beyond obesity and DM being causal factors of PC, PC, in turn, can also lead to an elevation of blood glucose and reduction of body weight, manifesting as the primary symptoms of occult malignancy.10 In terms of the carcinogenic effects of obesity and DM, many clinical studies and high-quality meta-analyses have confirmed the relationships of obesity and DM with PC, with basic studies focusing on the different aspects of these two most common metabolic disorders providing multitudinous insights to unveil the mechanisms behind the clinical evidence.
To date, the revealed mechanisms of obesity- and DM-related pancreatic carcinogenesis cover almost all of the immunometabolic alterations in these two diseases, which jointly reprogram systemic metabolism and remodel the local microenvironment of the pancreas, creating a perfect storm for the gradual initiation of PC. In brief, as both an endocrine and exocrine organ, the pancreas is not only the producer of hormones but also the receptor of many hormones. Thus, the changes in hormone levels and dysbiosis of the microbiome in obesity and DM inevitably lead to metabolic remodeling and pernicious accumulation of substantial nutrients and metabolites. These nutrients, metabolites, and other components within the reshaped tumor microenvironment (TME) provide precancerous and cancerous cells with mutagens, energy, hormones, and growth factors (GFs) and support the interactions between the surrounding cells and cancer cells via autocrine/paracrine signals and rewired metabolism while creating an inflammatory and immunosuppressive TME.11 As a result, the protective inflammatory/immune responses and various physiological processes collapse, and the carcinogenic microenvironment favors cancer initiation owing to continuously strengthened protumorigenic factors and compromised anticancer defense.
Although some previous studies have illustrated the correlations among obesity, DM, and PC, they were not entirely focused on the intricate mechanisms of the carcinogenic effects of obesity and DM. In addition, the exponential growth in the number of studies newly published on this topic also necessitates a comprehensive review of the key findings and questions. Beginning with a brief introduction of pancreatic carcinogenesis, we aim to provide a broad overview of the critical clinical and experimental discoveries through an in-depth look at the mechanisms of obesity- and DM-related pancreatic carcinogenesis, and we hope this overview will provide suggestions and guidance for experimental practice and research in this area.
Pancreatic carcinogenesis
PC has a variety of histological classifications that differ in terms of PC development, biological behaviors, clinical features, and response to treatments, among which pancreatic ductal adenocarcinoma (PDAC) is predominant (>90%).12 Therefore, the carcinogenic process of PDAC will be briefly introduced as a representative.
In general, it takes more than 20 years to develop clinically detectable PC,13 giving an extensive time window for obesity and DM to promote carcinogenesis. The cellular origin of PDAC is still disputable, as both acinar and ductal cells are heterogeneous in their capacity to be transformed.14,15 Nevertheless, mutation in the proto-oncogene KRAS is the most common event and the primary regulator of the initiation of PC,16,17 abetted by sequential inhibition or inactivation of tumor suppressor genes during the progression of precancerous lesions [pancreatic intraepithelial neoplasia (PanIN)], including cyclin-dependent kinase inhibitor 2A (CDKN2A), tumor suppressor p53 (TP53), and SMAD family member 4 (SMAD4).9 In other words, a single mutation in KRAS cannot cause cellular transformation without additional genetic alterations, which jointly sabotage cell identification, KRAS signaling, the cell cycle, apoptosis, senescence, DNA repair, and cellular metabolism.15 PanINs are divided into grades 1~3, and only PanIN3 (high-grade dysplasia) is the true precursor of cancer in situ.18 Within these neoplasms, the cellular heterogeneity includes metaplastic epithelia and low-grade dysplasia, with many tuft cells that mediate inflammatory responses in other glandular tissues.19 Neuroendocrine PanINs respond to neuronal signals to enhance lesion growth20 while frequently delaminating and entering the surrounding stroma,21 facilitating metastasis even in the absence of a carcinoma.
The desmoplastic stroma of PDAC harbors pancreatic stellate cells (PSCs), activated cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), other immune cells, cancer cells, and the microvasculature.15 This immunosuppressive microenvironment is characterized by a disrupted inflammatory response and an aberrant extracellular matrix (ECM), shielding cancer cells from immune surveillance and attack.22,23 As a critical part of the TME, TAMs play an essential role in the inflammatory environment that promotes pancreatic carcinogenesis. First, oncogenic KRAS drives proinflammatory signaling in precancerous lesions by activating the nuclear factor-κB (NF-κB), signal transducer and activator of transcription 3 (STAT3), and glycogen synthase kinase 3 (GSK3)/nuclear factor of activated T cells (NFAT) pathways,24,25,26 where inflammatory macrophages promote acinar cell dedifferentiation, acinar-to-ductal metaplasia (ADM), and the formation of precancerous lesions by secreting provocative mediators.27 Next, inflammatory macrophages upregulate tissue inhibitors of metalloproteinases or matrix metalloproteinases (MMPs) and remodel the acinar microenvironment by promoting ADM.27 However, the local inflammation caused by mutant KRAS alone is insufficient for pancreatic carcinogenesis, which requires additional inflammatory fuels and genetic changes.28 What are the critical drivers of fibrogenesis in the microenvironment? The inflammatory cytokines secreted in PanIN1 lesions initiate the phenotypic switch of macrophages and are vital in suppressing inflammation and promoting the growth of the lesions.29 Finally, TAMs promote the epithelial-to-mesenchymal transition (EMT), invasiveness, and metastasis of PDAC through the immunosuppression of T cells and the regulation of fibrinogenesis, vascularization, and angiogenesis.15
Other components of the TME also participate in the initiation of PDAC. CAFs from multiple origins are responsible for producing an ECM that contains various components, and PSCs are the main contributors to the desmoplastic reaction.30 In the normal pancreas, quiescent PSCs are activated during acute or chronic inflammation31 and change their morphology into myofibroblast-like cells to enhance ECM production.32 Consequently, the abundance of CAFs and a collagen- and hyaluronic-rich ECM promotes vasculature and increases tissue tension, creating a hypoxic microenvironment33 and altering tumor metabolism,34 which is essential for carcinogenesis.15 Furthermore, in contrast to the suppression of T-cell function in PDAC that leads to cancer cell proliferation, immune evasion, and metastasis, B cells enhance cell proliferation, inhibit antitumor immunity, and promote the progression and metastasis of cancer in multiple ways15 (Fig. 1).

Progression and microenvironment of PanINs that favor the formation of PDAC. Ductal cells can transdifferentiate into acinar cells under normal conditions as a compensatory regenerative process to maintain the proper function of the pancreas. Meanwhile, the highly plastic acinar cells can also be turned into ductal cells through the metaplastic process called ADM when stimulated by inflammatory macrophages via the secretion of MMPs, and they maintain their ductal phenotype in the presence of oncogenic KRAS mutation, followed by enhanced EGFR signaling and sequential inactivation of tumor-suppressive genes, such as CDKN2A, TP53, BRCA2, and SMAD4, to form a carcinoma in situ. Initially, oncogenic KRAS magnifies proinflammatory signaling in macrophages and promotes ADM by producing MMPs while secreting inflammatory cytokines into the microenvironment. Some serine/threonine-protein kinase DCLK1+ cells of acinar origin are also formed during low-grade PanIN lesions, such as PanIN1A, PanIN1B, and PanIN2, putatively serving as progenitor cells with cancer stem cell functions. Meanwhile, macrophages activate PSCs and change their morphology into CAFs, which enhance the desmoplastic reaction and ECM production, increasing tissue tension and creating a hypoxic microenvironment within the PanINs that is made up of abundant precancerous metaplastic epithelia and tuft cells. Furthermore, CAFs can activate immunosuppressive B cells, Tregs, and TH17 cells and collaboratively sabotage the anticancer immunity of CD8+ T cells with macrophages. During this process, precancerous cells are transformed by strengthened KRAS signaling. The aberrance of the cell cycle, apoptosis, senescence, DNA repair, and metabolism in this immunosuppressive microenvironment jointly favors the formation of PDAC. ADM acinar-to-ductal metaplasia, CAF cancer-associated fibroblast, CDKN2A cyclin-dependent kinase inhibitor 2A, DCLK1 doublecortin-like kinase 1, ECM extracellular matrix, EGFR epidermal growth factor receptor, KRAS Kirsten rat sarcoma viral oncogene homolog, MMPs matrix metalloproteinases, PanIN pancreatic intraepithelial neoplasia, PDAC pancreatic ductal adenocarcinoma, PSC pancreatic stellate cell, SMAD4 SMAD family member 4, TP53 tumor suppressor p53, Tregs T helper cells, TH helper T. This figure was adapted from a previous publication15
In summary, the mutation of KRAS is one of the earliest events in pancreatic carcinogenesis, which simultaneously activates intrinsic pathways by inducing inflammation and promoting interactions among acinar cells, ductal cells, immune cells, and fibroblasts that jointly favor an immunosuppressive and fibroinflammatory microenvironment suitable for the promotion of the plasticity of neoplastic cells at all stages of tumor progression.27 Regardless of the differences in pathological characteristics, microenvironmental aberrance of the inflammatory response, immune abnormalities, and fibrosis are commonly present in obesity, DM, and PC. Thus, it can be assumed that these similarities could be the main drivers of pancreatic carcinogenesis. For further reference, the carcinogenic process of PDAC is reviewed in more detail here.15
Mechanisms of obesity- and DM-related pancreatic carcinogenesis
Cancer cells are persistently influenced by the TME, which is predominantly shaped by the metabolic abnormalities of the host, providing beneficial hormones, GFs, nutrients, and metabolites and supporting the interactions between the surrounding cells and cancer cells via autocrine and paracrine signals while creating an inflammatory and immunosuppressive TME in the context of obesity and DM. The systemic immunometabolic abnormalities caused by obesity and DM are extremely complicated. Reprogrammed metabolism affected by internal or external factors and rewired glucose, amino acid, and lipid metabolism and metabolic crosstalk within the TME is critical in pancreatic carcinogenesis. As an endocrine and exocrine organ, the pancreas is not only the producer of hormones but also the receptor of many hormones. The changes in hormone levels and dysbiosis of the microbiome in obesity and DM inevitably lead to metabolic remodeling and pernicious accumulation of nutritional metabolites. At the same time, the reshaped metabolism reprograms inflammatory/immune responses and various physiological processes that are supposed to be anticarcinogenic. In this context, the aberrant microenvironment breeds pancreatic carcinogenesis owing to continuously strengthened cancer-promoting factors and the collapse of anticancer defense.
Nutrients and metabolites
High-fat diet and lipids
The contributive impact of a high-fat diet (HFD) on pancreatic carcinogenesis has been known for over 20 years,35 and not only can an HFD contribute to pancreatic carcinogenesis through the induction of obesity or DM, but it also has been shown to affect the carcinogenetic processes directly in different animal models. It was shown in P48+/Cre; LSL-KRASG12D (KC) mice that an HFD significantly increased the incidence and progression of precancerous lesions of PC via sustained inflammation and dysregulated autophagy.36 An HFD can also contribute to pancreatic carcinogenesis by augmenting pancreatic fatty infiltration through its obesogenic effect in Syrian golden hamsters treated with N-nitrosobis (2-oxopropyl) amine (BOP).37 In addition, it was demonstrated in C57BL/6 mice fed an HFD that this dietary pattern can be carcinogenic by stimulating inflammation via gut microbiome (GM) alteration, which occurs before the potential influence of circulating inflammatory cytokines.38 The effects of an HFD on inflammation and GM composition can also enhance the progression of carcinogen-induced PC in C57BL/6 mice.39 In three studies using different genetically engineered mouse models (GEMMs), an HFD was shown to increase inflammation, fibrosis, and PanIN lesions while promoting the transformation of precancerous lesions into more aggressive PDAC through enhanced cyclooxygenase 2 (COX2)-activated KRAS signaling,40 aerobic glycolysis,41 RAS activity, and reduced expression of fibroblast growth factor 21 (FGF21).42 In addition to promoting pancreatic carcinogenesis via the dysregulation of autophagy, increased genetic alterations in PanINs36 and reduced DNA repair in precancerous cells,43 an HFD also exacerbates tumor growth, angiogenesis, and EMT while decreasing apoptosis.44 In particular, the tumor-promoting effect of an HFD was suggested to be regulated by endogenous cholecystokinin (CCK),45,46 and this effect could not be ameliorated by physical exercise.47 Other physiological impacts of an HFD on PC include enhanced lipid metabolism, altered oxidative stress, extensive central necrosis, and lipid accumulation.48 In addition, diet and obesity, in a setting of an HFD, were demonstrated to promote pancreatic carcinogenesis via phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling.49
Some lipids are also involved in pancreatic carcinogenesis related to obesity and DM. Obesity and DM are common risk factors for dyslipidemia characterized by elevated circulating levels of very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and triglycerides (TAGs). In contrast, the level of high-density lipoprotein (HDL) is decreased.50 Dyslipidemia has long been recognized as a risk factor for PC,51,52 and high dietary cholesterol, which contributes to dyslipidemia, also increases the risk of PC.53 In addition, previous research has demonstrated that dyslipidemia contributes to pancreatic carcinogenesis by deteriorating pancreatic fatty infiltration.54 However, while conflicting results exist regarding the effects of pharmacological treatment of dyslipidemia (mainly statins) on the risk of PC,55,56,57 animal studies have suggested a detrimental role of statins in the development of PC.58 In contrast, atorvastatin, another 3-hydroxy-3-methyl glutaryl coenzyme A (HMG-CoA) reductase inhibitor, was demonstrated to suppress pancreatic carcinogenesis and prolong the survival of rodents with PC.59
Cholesterol is an essential molecule that maintains the normal function of cellular membranes, and it is a precursor for synthesizing steroid hormones, oxysterols, and bile acids (BAs), acting as a signaling molecule regulating the cell cycle and the modification and synthesis of proteins.60 In contrast to the speculation that cholesterol induces a higher PC risk, total serum cholesterol was inversely related to the risk of PC independent of statin use.61 Notably, LDL promotes the proliferation of PC cells by activating the STAT3 pathway while upregulating the expression of multiple oncogenic genes.62 This can be partly explained by the role of interleukin 6 (IL-6)/Janus kinase (JAK)/STAT3 signaling in cancer,63 as IL-6 is a proinflammatory and protumorigenic cytokine capable of reducing the level of total serum cholesterol.64 PC cells are highly dependent on profoundly activated cholesterol uptake, which results in an increased influx of cholesterol and overexpression of the LDL receptor (LDLR),51 a major site that transports LDL, VLDL, and VLDL into the cells,65 increasing cell proliferation and activating the extracellular signal-regulated kinase (ERK) 1/2 pathway.66 In TP53-mutant PDAC cells, sterol O-acyltransferase 1 (SOAT1) sustains the mevalonate pathway by converting cholesterol to inert cholesterol esters, thereby preventing the negative feedback elicited by unesterified cholesterol, which promotes cell proliferation in vitro and tumor progression in vivo.67 Moreover, acyl-CoA cholesterol acyltransferase-1 (ACAT-1) was found to enhance the esterification and accumulation of cholesterol in human PC specimens and cell lines, suppressing apoptosis and supporting tumor growth.68
As mentioned above, cholesterol is a precursor for progesterone, estrogen, and androgen synthesis, which implies that cholesterol may contribute to pancreatic carcinogenesis by influencing the levels of sex hormones (which will be addressed later). In addition to its direct effects on the synthesis of steroid hormones, cholesterol is also metabolized into biologically active oxysterols. Oxysterols also have multiple functions, such as affecting membrane fluidity, regulating the sterol regulatory element-binding protein (SREBP) signaling pathway, and activating several nuclear receptors, such as retinoic acid receptor-related orphan receptors (RORs), farnesoid X receptor (FXR), pregnane X receptor (PXR), estrogen receptors (ESRs), and liver X receptors (LXRs).69,70 Among them, cholesterol metabolism is under the strict regulation of SREBPs71,72 and LXRs,73 which decrease cholesterol uptake via LDLR and increase cholesterol efflux.74 SREBPs are transcription factors that activate the transcription of genes enhancing cholesterol synthesis and uptake. Despite the primary regulator of cholesterol homeostasis being SREBP-2,75 the SREBP-1 pathway is essential for the growth, viability, and proliferation of PC cells.76,77 LXRs, members of a nuclear receptor family that regulate insulin secretion, cholesterol homeostasis, lipid metabolism, and inflammation, were shown to be dramatically elevated in PDAC.78 In contrast, LXR agonists can disrupt the proliferation, cell cycle progression, and colony formation of PDAC cells.79,80 Similarly, inhibiting the transcriptional activity of LXR with synthetic ligands reduces the proliferation of PDAC cells and tumor formation.81 Furthermore, as defects in DNA repair, increased DNA strand breaks, genomic instability, and gene mutagenesis are known to induce carcinogenesis, defective LXR/SREBP-1/polynucleotide kinase/phosphatase (PNKP) signaling was demonstrated to cause a reduction in both DNA repair and apoptosis in vivo and in vitro.82
In addition to mediating SREBPs and LXRs, other mechanisms are also involved in cholesterol-related pancreatic carcinogenesis. First, oxysterols have also been shown to increase inflammatory cytokines in macrophages,83 and tumor necrosis factor α (TNF-α) can affect the lipogenesis and inflammatory status of PDAC cells by regulating SREBP-1 and acetyl-CoA carboxylase (ACC),84 suggesting that cholesterol may also affect carcinogenesis via the inflammatory response. In addition, oxy186, a semisynthetic oxysterol analog as an inhibitor of Hedgehog (Hh) signaling acting downstream of Smoothened (Smo), was illustrated to suppress Hh signaling and the proliferation of PANC-1 cells.85 Finally, oxysterol binding protein-related protein 5 (ORP5) induces the expression of SREBP-2 to enhance the cholesterol synthesis pathway and activates histone deacetylase 5 (HDAC5) to promote the growth of PC cells.86
Fatty acids
Some fatty acids (FAs) are essential for mammals, and different FAs have distinct impacts on tumor growth. For example, omega-3 FAs and omega-6 FAs can be oxidized to acetyl-CoA, while omega-3 FAs have an anti-inflammatory effect both in vivo and in vitro, and omega-6 FAs have proinflammatory and protumorigenic properties in obesity.87 Consistent with epidemiological data suggesting an anticancer effect of diets high in omega-3 FAs, a preclinical study showed that an omega-3-FA-enriched diet suppressed pancreatic carcinogenesis via reduced phosphorylated AKT (pAKT), whereas an omega-6-FA-enriched diet augmented tumor formation.88 In patients with obesity, high levels of free fatty acids (FFAs) can activate preadipocytes and inflammatory cells by inducing Toll-like receptor 4 (TLR4) signaling.89 It was also shown that lipid metabolism and fatty acid oxidation (FAO) are implicated in pancreatic carcinogenesis initiated from intraductal papillary mucinous neoplasms (IPMNs).90
Glucose
Given the long period of pancreatic carcinogenesis, patients with obesity and DM are often asymptomatic for decades. Nevertheless, many of these patients suffer from the gradual development of glucose intolerance and hyperglycemia before cancer diagnosis. Many epidemiological studies have concluded that type 1 DM (T1DM) and type 2 DM (T2DM) increase the risk of PC in both sexes.8,91 Epidemiological data have also showed that hyperglycemia in the first few years, commonly known as new-onset DM, induces a higher PC risk than long-standing DM,92 whereas studies on LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre (KPC) mice did not show a relationship of paraneoplastic DM and pancreatic carcinogenesis.93 Hyperglycemia, as a hallmark of DM, provides cancer cells with excessive energy to stimulate their proliferation and accelerate the progression of carcinogenesis. Interestingly, cancer cells tend to use glycolysis instead of efficient ATP production for their expansion, the so-called Warburg effect,94 enabling cancer cells to survive in nutrient-deficient conditions.95 Metabolically, mechanisms connecting hyperglycemia and cancer include lipotoxicity and glucose-associated pathways such as autoxidation, oxidative phosphorylation, glycosylation, the glycosamine pathway, and the Hippo-Yes-associated protein (YAP) pathways,96 with the dysfunction of these pathways increasing reactive oxygen species (ROS) and weakening DNA stability in β-cells.97
Beyond directly accelerating PC development by providing excessive glucose to cancer cells, hyperglycemia also promotes cell proliferation via the induction of epidermal growth factors (EGFs) and their receptors (EGFRs) while causing endothelial dysfunction and promoting angiogenesis.98 In addition, multiple signaling pathways can be aberrantly activated in hyperglycemia. Activated NF-кB and p38 MAPK signaling in response to cellular stress and chronic inflammation under hyperglycemic conditions augments the proliferation and apoptosis of PC cells by enhancing the secretion of proinflammatory cytokines and the paracrine effects of vascular endothelial growth factor (VEGF), promoting EMT, cell growth and PC development.98 In addition to ECM remodeling and angiogenesis,97 hyperglycemia also promotes EMT99,100 and the stemness of precancerous cells to promote pancreatic carcinogenesis through the activation of transforming growth factor β (TGF-β) signaling.101 Moreover, under hyperglycemic conditions, cellular O-GlcNAcylation can be significantly elevated in pancreatic cells that exhibit lower phosphofructokinase (PFK) activity, which compromises ribonucleotide reductase (RNR) activity and leads to deficiency in dNTP pools, enhancing genomic DNA alterations with concurrent KRAS mutations and cellular transformation. All these changes induce the initial oncogenic KRAS mutations in pancreatic cells to trigger carcinogenesis.102
Advanced glycation end products and their receptors
Referring to a heterogeneous class of molecules resulting from a nonenzymatic reaction of the oxo group of carbohydrates and the free amino group of amino acids, lipids, nucleic acids, or their combinations, advanced glycation end products (AGEs) are excessively produced and accumulate in hyperlipidemic and hyperglycemic conditions such as obesity, DM, and their comorbidities.103,104 Receptors of AGEs (RAGEs) belong to the immunoglobulin superfamily and are multiligand transmembrane receptors present on various cells.105,106 To date, ample evidence has demonstrated the potential contribution of AGE/RAGE crosstalk to pancreatic carcinogenesis through different mechanisms (Fig. 2). First, RAGEs prevent cell death and apoptosis by suppressing TP53 transcription and autophagy to improve the proliferation and survival of PC cells.107,108 Second, RAGEs promote the recruitment and retention of myeloid-derived suppressor cells (MDSCs) in the TME to protect pancreatic neoplasms from the antitumor immune response.109,110 In addition, since NF-κB is essential for inflammatory signaling in PanINs,111 the binding of NF-κB to RAGEs maintains the longstanding inflammatory state preferable for carcinogenesis.112 Meanwhile, as hypoxia induces NF-κB-dependent and hypoxia-inducible factor 1 subunit α (HIF-1α)-independent RAGE expression in PC cells, along with enhanced interaction between RAGEs and mutant KRAS facilitating the transcriptional activity of HIF-1α, the activation of NF-κB signaling deteriorates hypoxia by enhancing HIF-1α activation.113 Finally, in an NF-κB-dependent manner, RAGEs prevent PC cells from H2O2-induced oxidative injury during oxidative stress.114 Overall, RAGEs support carcinogenesis by creating an immunosuppressive TME while promoting the survival of PC cells. Interestingly, while dietary consumption of AGEs was suggested to modestly increase the risk of PC in men,115 others failed to confirm the association between AGEs/RAGEs and PC risk.116

The roles of AGEs and RAGEs in pancreatic carcinogenesis. The production of AGEs is drastically increased in obesity and DM, and the binding of AGEs to RAGEs activates MAPK and NF-κB signaling and increases the transcription of HIF-1α and NF-κB, which prevents cell death from oxidative stress and creates a hypoxic microenvironment while promoting proinflammatory signaling to exacerbate inflammatory reactions and recruit immunosuppressive MDSCs to diminish anticancer immunity. In addition to the decreased apoptosis due to the decline in the transcription of TP53 following the activation of KRAS signaling, the enhanced PI3K-AKT signaling and the direct activation of mTOR by RAGEs mitigate autophagy to improve the proliferation and survival of cancer cells, thereby promoting pancreatic carcinogenesis. AGEs advanced glycation end products, AKT protein kinase B, DM diabetes mellitus, ERK extracellular signal-regulated kinase, HIF-1α hypoxia-inducible factor 1 subunit α, IKKβ inhibitor of nuclear factor-κB (NF-κB) kinase subunit β, Ikβ inhibitor of NF-κB subunit β, KRAS Kirsten rat sarcoma viral oncogene homolog, MAPK mitogen-activated protein kinase, MDSCs myeloid-derived suppressor cells, MEK mitogen extracellular kinase, mTOR mammalian target of rapamycin, NF-κB nuclear factor-κB, P phosphorylation, PI3K phosphatidylinositol-3-kinase, RAF Raf proto-oncogene, RAGE receptor of AGEs, TP53 tumor suppressor p53
Bile and bile acids
Being influenced by lifestyle factors such as smoking, alcohol consumption, and dietary habits, bile and BAs are also closely related to the pathogenesis of obesity and DM.117,118 While heavy alcohol consumption alters the levels of BAs in the blood and intestines, which affects the GM, influences intestinal permeability, and induces systemic inflammation, intracellular signaling pathways are also activated in pancreatic epithelial cells owing to low-dose exposure to BAs.117 As a mutagen associated with PC, cigarette smoke stimulates the activation of mutated KRAS as well as that of other mutated proteins, such as those encoded by TP53, COX2, SMAD4, and p16INK4A.117 Mechanistically, nicotine promotes pancreatic carcinogenesis by increasing the secretion of gastric acid while disrupting the secretion of BAs.117 Concerning the effects of dietary habits, the physiological function of BAs is to promote the absorption of dietary fat and fat-soluble vitamins as a mediator of cholesterol metabolism. Thus, the levels of BAs are drastically elevated in individuals with an HFD, as dietary fat significantly stimulates the secretion of BAs. Although the pancreas does not make direct contact with BAs, the fact that nearly 60% of PC tumors occur in the head of the pancreas adjacent to the bile tracts implies a probability that BAs may play a role in pancreatic carcinogenesis, as previous studies have confirmed the association between BAs and cancers of multiple sites.117
FXR is a critical mediator of BA synthesis and metabolic control, and various preclinical studies have concluded that FXR is involved in the initiation of multiple cancers.119,120,121 FXR was significantly increased in PC cell lines and was found to be the regulator of the focal adhesion kinase (FAK)/JUN N-terminal kinase (JNK)/Mucin (MUC) 4 signaling pathway.122 Likewise, in both PDAC cell lines and human samples, it was found that bile accelerates carcinogenesis through the overexpression of MUC4.123 In addition to their contribution to pancreatic carcinogenesis via insulin resistance [or elevated insulin-like growth factor 1 (IGF-1) signaling], hyperinsulinemia, and the disruption of the GM in obesity and DM, BAs also elevate the risk of PC via gallstones, pancreatobiliary maljunction, and chronic pancreatitis.117,124 Moreover, BAs can have much more direct and local effects on carcinogenesis. For example, BAs induce cell membrane perturbations by disrupting the redistribution of membrane cholesterol and promoting cell proliferation with their mitogenic impact while reducing apoptosis.117 They also enhance inflammatory reactions and activate signaling pathways closely related to pancreatic carcinogenesis, such as Erb-B2 EGFR, mitogen-activated protein kinase (MAPK), and STAT3 signaling.117
Amino acids
Apart from being involved in the pathogenesis of obesity and DM,125 amino acids are vital for the survival of all cells and rewired metabolism in cancers, and they play distinct roles within the carcinogenic TME, serving as energy sources, regulators of epigenetics and immune responses, and therapeutic targets.126 Preclinical research has demonstrated that macropinocytosis, a highly conserved endocytic process transporting extracellular fluid and its contents into oncogenic Ras-transformed cells, supports the growth of these cells through the internalization of amino acids, including glutamine, that are translated into proteins.127 Emerging evidence has suggested that different amino acids participate in pancreatic carcinogenesis. It was shown in patients undergoing pancreatic resection that the circulating levels of branched-chain amino acids (BCAAs) were correlated with the dysplastic grades of IPNM, a high-risk precancerous lesion.128 A recent study indicated that BCAA uptake promotes PDAC development, while BCAA catabolism is impeded in PDAC tissue, indicating that BCAA uptake could be a promising therapeutic target for the treatment of PDAC.129 Isoleucine, one of the BCAAs, was associated with an increased risk of PC in women with long-term obesity.130 It was demonstrated in KC mice that KRAS stabilizes BCAA transaminase 2 (BCAT2) via the regulation of spleen tyrosine kinase (SYK) and E3 ligase tripartite-motif-containing protein 21 (TRIM21) to enhance BCAA uptake and mitochondrial respiration, which fosters the progression of PanIN.131 Similarly, TRIM2 was shown to promote PC progression by activating ROS-related nuclear factor (erythroid-derived 2)-like 2 (NRF2)/antioxidant response element (ARE) signaling and the integrin/FAK pathway.132
The synthesis of amino acids and proteins can also fuel pancreatic carcinogenesis. The enhanced mTOR-dependent serine synthesis and upregulation of DNA methylation due to the loss of liver kinase B1 (LKB1, also known as STK11) synergize with KRAS activation to promote pancreatic carcinogenesis in GEMMs and primary pancreatic epithelial cells.133 Similarly, protein synthesis is also involved in pancreatic carcinogenesis. In Ras-driven cancers such as PC, the guanosine triphosphatase (GTPase) activity of eukaryotic elongation factor 1A (eEF1A) catalytically increased by methyltransferase-like 13 (METTL13) augments protein production in vitro, and METTL13 dimethylation of eEF1A lysine 55 (eEF1AK55me2) enhances translation and protein synthesis to promote carcinogenesis in vivo.134
Additionally, amino acid modification can also contribute to pancreatic carcinogenesis. For example, the deregulation of lysine methylation signaling has been shown to be a common pathogenic factor in cancers, making inhibitors of several histone lysine methyltransferases (KMTs) ideal chemotherapeutics.135 Among these KMTs, SET and MYND domain-containing protein 3 (SMYD3) was suggested to promote carcinogenesis in mouse models of PDAC via the methylation of MAP kinase MAP3K2 at lysine 260 and subsequently activate RAS signaling.135
Acetyl-coenzyme A
Acetyl-coenzyme A (acetyl-CoA) is a central metabolic intermediate of the tricarboxylic acid (TCA) cycle and the primary regulator of cellular metabolism. Acetyl-CoA affects the activity and specificity of enzymes and the acetylation profile of proteins, thereby controlling vital cellular processes such as energy balance, mitosis, and autophagy that are implicated in the development of obesity and DM.136 Recent studies have also illustrated the roles of acetyl-CoA in pancreatic carcinogenesis. It was shown that the elevated levels of acetyl-CoA induced by adenosine triphosphate (ATP)-citrate lyase (ACLY) in KRAS-mutant acinar cells promoted ADM and tumor formation via histone acetylation and the mevalonate pathway.137 Fueled by the phosphorylation of acyl-CoA thioesterase (ACOT) at S392 by AKT, the accumulation of ACOT catalyzes the hydrolysis of acyl-CoA thioesters and produces nonesterified FAs and coenzyme A (CoA), which provides excessive CoA to promote the proliferation and tumor formation of PDAC cells.138
As mentioned above, many dysregulated nutrients and metabolites in obesity and DM can promote pancreatic carcinogenesis. However, most of these findings were based on observations in different animal models, and the scarcity of clinical evidence warrants more future studies to validate these impacts in humans.
Endocrine and exocrine factors
Long known as being vital for the normal functioning of the pancreas, the exocrine-endocrine axis is responsible for the extensive regulation of physiological and pathophysiological processes. The pancreas is a hormone-producing organ and a target of many hormones itself. Various gastrointestinal (GI) hormones/peptides and sex hormones have been suggested to be involved in pancreatic carcinogenesis.
Gastrin and CCK
Gastrin, a peptide released by G cells in the pyloric antrum of the stomach, duodenum, and pancreas, stimulates the secretion of gastric acid (HCl) by the parietal cells of the stomach and aids in gastric motility; gastrin also plays a critical role in the development of the GI tract and the regulation of satiety. Usually, gastrin is not expressed in the adult pancreas but surprisingly reappears in PanINs.139 Patients with pernicious anemia and elevated serum gastrin levels have an increased incidence of pancreatic neoplasia.140 It was also shown that gastrin promotes the growth of several human PC cell lines in an autocrine manner141,142 as a ligand binding to CCK B receptor (CCK-RB) to participate in pancreatic carcinogenesis.
Secreted by a unique species of enteroendocrine cells (EECs) called I cells, CCK responds to meal digestion, regulates satiety, and controls blood glucose by affecting hepatic glucose production and gastric emptying,143 and the dysregulation of CCK signaling can contribute to the pathogenesis of obesity and T2DM.143 In addition, despite neither gastrin nor CCK being mutagenic, they can accelerate the progression of existing KRAS mutations and PanIN lesions.144 Gastrin and CCK were found to significantly enhance the proliferation of PC cells in vitro,142,145 and the high level of CCK in the blood induced by dietary fat was suggested to promote the growth of an established PC tumor in animal models.146
The carcinogenic effect of gastrin and CCK lies in the autocrine mechanism of gastrin sustaining tumor growth through enhanced transcription in cancer cells by activating CCK-RB,147 and the expression of gastrin is ubiquitous and essential for carcinogenesis and cancer progression in PC.148 In contrast, CCK is not thought to be expressed in the pancreas.149 However, it has been shown that the aberrant expression of Cck in pancreatic β-cells in response to obesity enhances the proliferation and ductal transformation of acinar cells to promote Kras-driven pancreatic carcinogenesis, indicating that obesity-associated changes in the TME implicate endocrine-exocrine signaling in PDAC development.150 Nevertheless, the expression of gastrin and CCK is detectable in PC tissues,151 although CCK produced by the tumor is likely to be inefficient in influencing the growth of PC.152 Therefore, it can be hypothesized that the carcinogenic effect of gastrin and CCK along with their presence in PC result from the re-expression of endogenous gastrin through an autocrine mechanism.153
There are two classic types of CCK receptors, named CCK-RA and CCK-RB,154 that are predominant in the normal pancreas of mice and humans, respectively.155 Regardless of its low abundance, the increase in CCK-RB is significantly related to the development of PC.156 In addition, a mutant of CCK-RB called CCK-RC (CCK-cancer receptor) is related to higher aggressiveness and shortened survival.157 For the intracellular signaling of CCK-RB in PC, the activation of CCK-RB or the splice variant CCK-RC triggers a conformational change in receptors and leads to the activation of various secondary messenger molecules responsible for the regulation of cell growth, proliferation, differentiation, migration and invasion, angiogenesis, and cell survival.153 In more detail, gastrin stimulation activates AKT phosphorylation, MAPK (including the four subgroups ERK1/2, JNKs, ERK5, and p38-MAPK) pathways, and cyclins through CCK-RB.153
As introduced, the inflammatory TME of obesity and DM is important in carcinogenic progression. It has been demonstrated that CCK receptors and CCK are essential in accelerating PanIN progression under inflammatory conditions.144,155 Furthermore, CCK receptors were found on PSCs,158 the nonepithelial component of the TME, with the activation of these receptors being suggested to promote desmoplasia in PC.159 Given the nonnegligible roles of gastrin and CCK in carcinogenesis, massive efforts have been made to target CCK/gastrin signaling pathways, and selective CCK-RB antagonist blockade and downregulation as well as the neutralization of the potent trophic effects of gastrin through nanotechnology and immunotherapy have been shown to be promising in several types of malignancies.153
Insulin resistance/hyperinsulinemia and IGF-1 axis disruption
Since insulin resistance is positively correlated with obesity and DM, elevated fasting serum insulin levels and insulin resistance are also associated with a higher risk of PC through a combined effect of IGFs.160,161,162 It was found that rather than hyperglycemia or pancreatic β-cell dysfunction, circulating markers of peripheral insulin resistance were independently associated with PC risk.163 In addition, nonfasting C-peptide levels were also shown to be associated with this risk.164 Synthesized by almost every organism tissue, IGFs consist of the insulin receptor (IR), IGF-1 receptor (IGF-1R), and IGF-2R, along with the ligands of insulin, IGF-1, and IGF-2 and the IGF-binding proteins (IGFBPs) that bind to IGF-1 and IGF-2, jointly regulating the growth, development, and survival of cells. IRs and IGFRs all belong to the receptor tyrosine kinase (RTK) family, which includes two different IRs and IGFRs, IR-A/IR-B and IGF-1R/IGF-2R, respectively. While IGF-1R is expressed in nearly all tissues, with the majority found to be IGF-1R/IR hybrids,165 IGF-2R is ubiquitously expressed and does not induce activation of the insulin-IGF signaling axis.166 Furthermore, the IGF signaling pathway consists of six IGFBPs and ten IGFBP-related proteins (IGFBP-RPs),167 and the complexity of this signaling pathway endows the insulin-IGF signaling axis with numerous modes of activation and intricate roles in pancreatic carcinogenesis.
Proinsulin can contribute to pancreatic carcinogenesis by inducing cell proliferation and migration through the ERK/p70S6K pathway.168 Insulin/IGF signaling regulates the development and function of the endocrine pancreas by controlling the function of β-cells, stimulating cell proliferation, and increasing cell mass and basal insulin production.162 Due to the dysregulation of IGFs in obesity and DM,169 as well as the overexpression of IGFs or IGF-1R in cancer cells, stromal cells exert neoplastic actions by promoting cell cycle progression and inhibiting apoptosis either directly or indirectly through preacquired oncogenic drivers.162 Mutations in KRAS and elevated insulin and IGF-1 levels can activate PSCs and thereby increase stromal fibrosis within the islets162 and peri-islet tissue.170 On the one hand, elevated insulin can increase IGFs by reducing IGFBPs.171 On the other hand, insulin and IGFs induce a variety of carcinogenetic effects on target cells and influence cell proliferation, apoptosis, angiogenesis, and lymphangiogenesis,172 which also implicates the PI3K/AKT/mTOR pathway in regulating cell growth and differentiation and the MAPK pathway in enhancing cell proliferation in obesity.173 Overall, IGF-1- and IGF-1R-mediated signaling promote cell proliferation and expression of angiogenic factors and decrease apoptosis in obesity-related pancreatic carcinogenesis.174
Other mechanisms of insulin contributing to DM-related PC include the upregulation of the expression of transgelin-2, which binds with SREBP-1 to alter lipid metabolism.175 In addition, insulin regulates glucose uptake in target tissues while acting as a mitogen on PC cells. Beyond its mitogenic effects, IGF-1 promotes PC growth by enhancing angiogenesis and EMT while inhibiting apoptosis.98
Sex hormones
The latest cancer statistics suggest that men suffer from a higher incidence of PC than women,176 indicating that there could be an impact of sex hormones on pancreatic carcinogenesis. However, some have proposed that this higher incidence is a consequence of the many environmental factors that men are more likely to be exposed to, such as smoking and alcohol. So, do sex hormones have nothing to do with the discrepancies in the incidence of PC between the sexes?
Before answering that question, we should keep in mind that a vicious cyclical relationship exists between obesity and sex hormones (Fig. 3). Obesity can cause hypogonadism, which in turn can result in or exacerbate obesity and other metabolic disorders, such as insulin resistance and DM.177,178,179 In short, the impact of obesity on gonadal function involves insulin resistance at the hypothalamic and pituitary levels. The inhibitory effect on gonadotropin secretion of inflammatory mediators secreted by adipose tissue leads to sexual dimorphism with androgen deficiency, causing male obesity-associated secondary hypogonadism (MOSH). In contrast, excessive androgen leads to polycystic ovary syndrome (PCOS) and idiopathic hyperandrogenism in women, jointly contributing to metabolic disorders and the dysfunction of other organs.179 Recently, the correlation between PCOS and the risk of PC has been confirmed by a case‒control study.180 Conversely, intentional weight loss and other weight-lowering interventions effectively ameliorated obesity-related hypogonadism.179 Consistently, a recent study suggested that hormone therapy is also an ideal way to prevent or reverse T2DM in patients with obesity.181

Cyclical relationships between obesity and dysregulated sex hormone levels in both sexes. The excessive accumulation and expansion of adipose tissue disrupts the secretion of metabolic and inflammatory adipokines and cytokines, eventually causing systemic inflammation, insulin resistance, and hyperglycemia. a In men, metabolic disorders along with increased aromatase activity and estrogen levels induce an inhibitory effect on the secretion of gonadotropin from the hypothalamus and pituitary gland, suppressing the production of androgen from the testes and resulting in MOSH and the exacerbation of obesity. b In contrast, the inhibitory effect of the hypothalamus and pituitary gland on the ovaries and the influence of systemic inflammation and metabolic disorders on the adrenals and ovaries lead to the elevation of androgen, resulting in PCOS and hyperandrogenism in women. MOSH male obesity-associated secondary hypogonadism, PCOS polycystic ovary syndrome
At first glance, the pancreas is certainly not one of the target organs of sex hormones. However, estrogen was found to be predominantly localized in the cytoplasm of acinar cells,182 which seems essential for the synthesis of pancreatic digestive enzymes. The increase in estrogen levels can also lead to elevated TAG and total lipid levels in the pancreas,183,184 suggesting that increased estrogen can contribute to fatty infiltration in the pancreas (Fig. 4). In contrast to promoting digestive enzyme synthesis, estrogen seems to have an inhibitory effect on pancreatic growth due to the reduced cell numbers.185 Moreover, estrogen treatment was shown to significantly suppress the progression of precancerous lesions in vivo.186,187

Sex hormones and pancreatic carcinogenesis and cancer progression. a In the cytoplasm of pancreatic acinar cells, the increased estrogen levels lead to the elevation of TAGs and total lipids in the pancreas, contributing to fatty infiltration. b Tamoxifen can play an anticancer role by antagonizing estrogen receptors and agonizing GPER, and the latter can mitigate fibrosis and hypoxia in the TME by targeting PSCs, while it also ameliorates the immunosuppressive infiltration of macrophages and hinders cancer progression. c According to the description of Kanda et al. 195, the tumorigenic cytokine IL-6 can activate both STAT3 and MAPK signaling in PC cells, while extracellular androgen and oncogenic c-Src can also enhance AR and MAPK signaling and trigger the transactivation of nuclear ARs. Meanwhile, AHR, ARNT, and ARE interact with AR in a testosterone-dependent manner and translocate into the nucleus to increase the transcription of ADAM10, MMP9, TGFβ, and VEGF. ADAM10 and MMP-9 increase the expression of MICA and MICB and hamper the immune response of NK cells and T cells against cancer cells. In combination with the enhanced cell proliferation and invasion favored by the activation of EGF and MMP-9, TGF-β and VEGF also jointly promote angiogenesis and cell proliferation. ADAM10 a disintegrin and metalloprotease 10, AHR aryl hydrocarbon (or dioxin) receptor, AR androgen receptor, ARE androgen-responsive element, ARNT AHR nuclear translocator, ECM extracellular matrix, EGF epidermal growth factor, ERK extracellular signal-regulated kinase, GPER G-protein-coupled estrogen receptor, IL-6 interleukin 6, MAPK mitogen-activated protein kinase, MEK mitogen extracellular kinase, MICA/B major histocompatibility complex class I chain-related gene A/B, MMP-9 matrix metalloprotease 9, NK natural killer, PC pancreatic cancer, PSC pancreatic stellate cell, RAF Raf proto-oncogene, STAT3 signal transducer and activator of transcription 3, TAGs triglycerides, TAM tumor-associated macrophage, TCR T-cell receptor, TGF-β transforming growth factor β, TME tumor microenvironment, VEGF vascular endothelial growth factor. Panel c in this figure was adapted from a previous publication195
When discussing the role of estrogen in cancer, we must talk about the nuclear antagonist of estrogen receptors, the nonsteroidal drug tamoxifen, which has been used as standard endocrine therapy against breast cancer for decades. In addition to antagonizing estrogen receptors, tamoxifen also acts as an agonist of the G-protein-coupled estrogen receptor (GPER) expressed by many normal and malignant cells, commonly localized at intracellular membranes, regulating vascular tone and cell growth as well as lipid and glucose homeostasis. Hence, GPER is also implicated in obesity and DM.188 In terms of the roles of tamoxifen and GPER in PC, recent studies suggest that through the activation of GPER, tamoxifen reduces fibrosis and desmoplastic tissues by targeting PSCs and ameliorates the infiltration of macrophages by lowering the stiffness of the ECM while mitigating hypoxia and angiogenesis in the TME, which promotes apoptosis, inhibits cell proliferation, and prevents cancer progression189,190 (Fig. 4). Likewise, other agonists of GPER also showed satisfactory results in inhibiting cell proliferation and disrupting the cell cycle in PC.191 Together, these results indicate that GPER is a promising therapeutic target in the estrogen-related treatment of PC.
Androgen receptors (ARs) also exist in the normal pancreas and PC cells in humans.192 The overexpression of IL-6 in PC increases the phosphorylation of STAT3 and MAPK, which increases the activation of ARs in PC cells, promoting the progression of pancreatic carcinogenesis193 (Fig. 4). Furthermore, it was suggested that ARs might contribute to the progression of PC via the disruption of the circadian rhythm, a factor known to be associated with PC risk.194 Although it is assumed that ARs rather than androgen are involved in pancreatic carcinogenesis and progression,195 testosterone has been shown to vigorously promote experimental PC growth. In contrast, antiandrogen therapy has been shown to effectively prolong the survival of patients with unresectable PC.185
Based on the evidence above, along with the fact that men with obesity suffer from androgen deficiency with a mild increase in estrogen, while women with obesity have hyperandrogenemia and a prevalence of severe obesity approximately twice as high as that of men,178 it seems that women should be at greater risk of PC, which is obviously in contrast to the epidemiological data. Hypothetically, the best translation of these results would be, or likely be, the indirect role of sex hormones in exacerbating the abnormalities within the TME that contribute to metabolic dysfunction, inflammation, and carcinogenesis. Owing to these questions, more studies are warranted in the future to determine the roles of sex hormones in pancreatic carcinogenesis.
Microbiomes
Microbiomes can interact with the immunometabolic and endocrine systems, and it has long been known that viruses and other microbes take part in carcinogenesis. Worldwide, nearly one-fifth of malignant conditions are associated with microbial infections,196 and this percentage might have increased in recent years owing to the rising prevalence of metabolic disorders and cancers. Emerging evidence suggests that changes in the diversity, composition, and dominant organisms of the microbiome are correlated with the occurrence and development of PC and impair chemosensitivity and antitumor immunity in patients with PC.196 Animal studies identified a time-dependent gut dysbiosis associated with KRAS activation in pancreatic tumors.197 Although most studies have focused only on the carcinogenic effects of the intestinal microbiome, microbes that inhabit other parts of the digestive tract are also indispensable and unneglectable, as they can all be carcinogenic in different ways (Fig. 5).

Microbes and pancreatic carcinogenesis. Upper left panel: Beyond their distant impact and transfer of their carcinogenic products, microbes from different regions of the GI tract may migrate to the pancreas via retrograde transfer through the opening of the sphincter of Oddi and contribute to pancreatic carcinogenesis. Marks a, b, and b (corresponding to panels a, b, and c, respectively) indicate the possible distant influence and transfer of oral, gastric, and GM carcinogenic products or their migration to the pancreas. a Distinct effect of different oral microbiome species on the risk of PC. b Two hypothetical theories on pancreatic carcinogenesis related to Hp infection. c Some metabolites of the GM, such as lipopolysaccharide (LPS), can enhance chronic inflammation by activating multiple carcinogenic pathways and increasing the secretion of proinflammatory components. In contrast, altered levels of carcinogenic metabolites (e.g., BAs) can promote pancreatic carcinogenesis by accelerating the senescence-associated secretory phenotype and increasing DNA damage and genomic instability. Some viruses from the GM community (e.g., HBV and HCV) are also suggested to increase inflammation-induced DNA damage and carcinogenesis. Later, microbe-induced inflammation can be carcinogenic and initiate the activation of KRAS, which also exacerbates inflammatory reactions in return. The enhancement of oncogenic signaling subsequently triggers other factors that promote the progression of carcinogenesis, such as oxidative stress, cell cycle disruption, suppressed apoptosis, and the immune response. BAs bile acids, GI gastrointestinal, GM gut microbiome, HB (C) V hepatitis B (C) virus, Hp Helicobacter pylori, KRAS Kirsten rat sarcoma viral oncogene homolog, PC pancreatic cancer
Oral microbiome
The oral cavity contains a vast diversity of bacteria, viruses, and fungi,198 and these commensal microbes can be pathogenic and even carcinogenic under certain conditions.199 Periodontal disease, for example, an inflammation caused by oral microbes, has been suggested to elevate the risk of PC200 owing to alterations in microbial composition201 and the immune response.200,202 Specifically, high amounts of Porphyromonas gingivalis were shown to increase the PC risk up to 2-fold, while high levels of antibodies against nonpathogenic oral bacteria were shown to reduce this risk.203 Likewise, carriers of P. gingivalis share the same higher risk, regardless of the abundance of the bacteria, which indicates that P. gingivalis may serve as a biomarker for PC screening.203 Moreover, some periodontal Fusobacterium species were also detected in PC samples, but their roles remain elusive. In contrast to the increased risk of PC due to the carriage of the periodontal pathogen Aggregatibacter actinomycetemcomitans, Tannerella forsythia, Prevotella intermedia, and the genus Leptotrichia are associated with a decreased risk203 (Fig. 5). Of note, Fusobacterium, Haemophilus, Leptotrichia, and Porphyromonas are also suggested to be sensitive in distinguishing patients with PC.203 Mechanistically, the oral microbiome can promote pancreatic carcinogenesis by migrating to the pancreas through the natural digestive tract or the circulation during bacteremia, disrupting the pancreatic microenvironment.200 One of the most studied oral microbes, P. gingivalis, was speculated to increase p53 and Kras mutations following degradation through peptidyl-arginine deiminase enzyme secretion.204
Helicobacter pylori
As the only bacterium colonizing the stomach, the relationship between Helicobacter pylori (Hp) and gastric cancer has been recognized previously. Other studies in recent years have also connected Hp with PC.200 Accordingly, PC patients are more likely to test positive for Hp.205 However, there is no widely accepted explanation for the causality of Hp in pancreatic carcinogenesis. Some have proposed that pancreatic hyperplasia resulting from hyperchlorhydria and excessive release of secretin following Hp infection could be a possible answer, whereas others argued that atrophic gastritis and hypochlorhydria causing bacterial overgrowth and N-nitrosamine overproduction are the culprits200 (Fig. 5). Both of these findings coincide with the theory that the carcinogenic effects of Hp lie in chronic mucosal inflammation and aberrant cell proliferation and differentiation.206
Pancreatic microbiome
Contradictory to the obsolete views that the pancreas is sterile, previous findings proved that not only does the pancreas have its own microbiome, but there are also considerable differences in microbe abundance and composition between normal and cancerous pancreatic tissue.207 A previous study analyzing the microbes in pancreatic cyst fluid illustrated their unique microbial ecosystem and detrimental influence on the pancreatic neoplastic process, with this correlation being found with microbiome composition rather than microbe abundance.208 Later, the discovery of bile transporting some gut microbes to the pancreas209 further demonstrated the direct contact with and impact of the GM on the pancreatic microenvironment.207
The gut microbiome
Influenced by age, dietary habits, antibiotics, and other internal and external environmental factors, the GM is the most studied microbiome with a crucial impact on obesity, DM, and carcinogenesis210 (Fig. 5). However, only a few species are recognized as being carcinogenic due to their extensive colonization of the GI tract, manifesting vast complex interactions among the GM, environmental factors, and cancer initiation.211 Undoubtedly, obesity, DM, and carcinogenesis are subsequent chain reactions resulting from dysbiosis and the subsequent generation of toxic metabolites.212 As mentioned above, GM dysfunction leads to alterations in the levels of GI hormones, glucose hemostasis and energy balance. Moreover, some metabolites, such as LPS, enhance chronic inflammation, while BAs promote carcinogenesis by accelerating the senescence-associated secretory phenotype, increasing DNA damage and genomic instability and activating carcinogenic signaling pathways or inducing direct tumorigenic effects.211 For instance, Fusobacteria was shown to provoke NF-κB signaling and increase the production of proinflammatory cytokines, such as IL-1, IL-6, IL-8, TNF-α, MMP-3, and COX2.173
Fungi and viruses
Less-studied fungi and viruses are also associated with pancreatic carcinogenesis. While clinical trials in distinct populations found that Candida infection increases the risk of PC,200 preclinical research showed exponential growth of pathogenic fungi and altered composition of the mycobiome in PDAC in both humans and rodents, which promote disease development by driving the complement cascade with the cleavage of C3 into C3a and C3b through the activation of mannose-binding lectin (MBL).213 Notably, intrapancreatic fungi can increase more than 3000 times in number in PDAC compared with the normal pancreas, and the latest study also demonstrated that translocated fungi are capable of augmenting the production of IL-33 from PDAC cells to enhance the recruitment and activation of immunosuppressive T helper 2 (TH2) cells and type 2 innate lymphoid cells (ILC2s), thereby promoting the progression of PDAC.214 In addition, hepatitis viruses B and C (HBV and HCV, respectively) are also suggested to be associated with PC through inflammation-induced DNA damage and carcinogenesis.200,203
The main mechanisms by which microbes promote pancreatic carcinogenesis
Systemic and pancreatic inflammation
Microbial infections can cause carcinogenic inflammation in the pancreas, whether locally or systemically, since the constant stimulation of inflammation driven by the microbes was suggested to initiate the activation of KRAS,215 which also involves several other cancer-related inflammatory signaling pathways.
First, macropinocytosis is regulated by Wingless/Integrated (Wnt) signaling. Macropinocytosis is an endocytic process of antigen capture, presentation, and subsequent activation of the inflammatory reaction.216 Wnt signaling mediates the proliferation and differentiation of cells and tumor growth during pancreatic carcinogenesis.200 Consistently, a previous study demonstrated that fat mass and obesity-associated protein (FTO), an essential regulator of obesogenesis, mitigated pancreatic carcinogenesis by demethylating PJA2 and diminishing Wnt signaling.217 The next step is the stimulation of TLRs by LPS. TLR4 activates several downstream pathways that are carcinogenic in inflammatory conditions. mTOR signaling, for example, not only reshapes the composition of the GM but also participates in pancreatic carcinogenesis by promoting tumor growth through ERK/mTOR signaling.200 In addition, the interactions between LPS and TLR4 can activate NF-κB and STAT3 signaling and accelerate carcinogenesis by amplifying RAS signals and enhancing the progression of tumors.200,211,218 Similarly, the increased levels of inflammatory cytokines in obesity and DM, such as IL-1, IL-6, and TNF-α, also participate in the carcinogenic process via the activation of the NF-κB pathway.219 Furthermore, the activation of other pathways, such as the JNK/AKT/STAT3 and cyclin/cyclin-dependent kinase (CDK) pathways, increases oxidative stress, disrupts the cell cycle, suppresses apoptosis, and induces DNA mutations.203
Apart from the inflammation- and damage-induced metaplasia resulting from HBV and HCV infection in the pancreas, these two viruses can also promote carcinogenesis by causing a high level of mutations in the TP53 and CTNNB1 genes, activating numerous oncogenic processes, including telomere maintenance, Wnt signaling, cell cycle regulation, oxidative stress, epigenetic modifications, JAK/STAT signaling, immune response suppression and apoptosis.203
Diminished immune response
Interacting bidirectionally with each other, the microbiome and the immune system collaboratively maintain the symbiosis between the human body and microorganisms, while the immune system influences the composition and evolution of the microbiome, which in turn affects the maturation and adaptation of the immune system as well as the carcinogenesis caused by immune dysregulation.211 Different studies have emphasized the two almost opposite effects of the microbiome: the promotion of immune maturation and the suppression of antitumor immunity.
On the one hand, the gut microbiome and the immune system can affect each other in the gut lamina propria and extraintestinal sites. Microbes can act as antigens activating the immune system to promote its maturation and maintain its functional integrity.200 Some specific species, such as Bacteroides fragilis and Bifidobacterium species, were deemed essential for the maturation of the immune system.200 On the other hand, microbe-mediated immune suppression is associated with pattern recognition receptors (PRRs), which are also called pathogen-associated molecular patterns (PAMPs) and are capable of directly recognizing pathogens of microorganisms. The TLR and nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) families are two major sets of receptors that induce carcinogenic effects in GM-related inflammation.211 As introduced, TLRs can recognize microbial pathogens (e.g., LPS, lipoproteins, lipopeptides, flagellin, single- or double-stranded DNA, and CpG DNA) and trigger the inflammatory response and carcinogenesis.211 In addition, the activation of the NF-κB and MAPK signaling pathways following the stimulation of TLRs initiates the production of proinflammatory cytokines and the recruitment of inflammatory entities, accelerating the development of cancer.220 NLRs, likewise, can promote carcinogenesis by increasing the release of inflammasomes and ILs after recognizing microbial signals while activating NF-κB, p38 MAPK, and interferon signaling to modulate bacterial clearance and augment the formation of autophagosomes.211 The inhibition of these receptors diminishes tumor development, whereas several other TLRs (e.g., TLR2, TLR4, TLR5, and TLR7) suppress innate and adaptive immunity to promote the development of PC by disrupting the interactions between macrophages and lymphocytes.200
Different microbiomes play distinct roles in immunity or other tumor models. With advancing techniques in sample analysis and sequencing [e.g., single-cell analysis of host-microbiome interactions (SAHMI)221], the future combination of these methods with other multiomic data will reveal an increasing number of roles of the microbiomes in immune dysfunction in pancreatic carcinogenesis resulting from metabolic disorders.
Changes in metabolism
As mentioned above, microbes are one of the main regulators of energy balance. Given that obesity is characterized by altered microbial diversity, the excessive release of LPS from the GM in obesity often leads to endotoxemia, with this low-grade chronic inflammation increasing the risk of PC by augmenting the secretion of various proinflammatory cytokines and activating the NF-κB pathway.200 Regarding the roles of the microbiome in DM, in addition to the insulin resistance caused by GM dysfunction, it was suggested that alterations in the levels of metabolites, such as acetate and butyrate, also increase the risk of PC by enhancing chronic inflammation through endotoxemia due to impaired epithelial tight junctions in the intestinal mucosa.200
The TME and cellular perturbations
The TME comprises infiltrating immune cells, such as lymphocytes, TAMs, mast cells, antigen-presenting cells (APCs), and granulocytes, as well as CAFs, endothelial cells, ECM, and other stromal components.222 As the hallmark and most frequently mutated oncogene in PC, mutated KRAS cooperates with existing metabolic abnormalities to further influence the different components of the pancreatic TME.223 Mutated TP53, another commonly mutated gene, has been suggested to deteriorate fibrosis and immunosuppression within the TME of PDAC.224 TME aberrance results in epithelial dysfunction, carcinogenesis, and tumor promotion. Specifically, the ectopic expansion of adipose tissue fuels energy imbalance and inflammatory disruption in the TME through excessive production of proinflammatory chemokines and cytokines and dysregulated secretion of adipokines. Meanwhile, cancer-associated adipocytes (CAAs) further scatter the TME and provide crucial support for the progression of carcinogenesis, with hyperactive CAFs, ECM deposition, and hypoxia promoting fibroinflammatory desmoplastic reaction, EMT, and immunosuppression to promote tumor formation.
Ectopic adipose tissue expansion and the adipose tissue microenvironment
Adipose tissue microenvironment
In obesity, the excessively expanded adipose tissue, including subcutaneous and visceral adipose tissue and the adipose tissue surrounding many organs, is a vital organic system capable of modulating the production of adipokines, inflammatory cytokines, and other enzymes that potentially contribute to carcinogenesis and tumor growth while exerting an essential impact on cancer cells in the adipose tissue microenvironment (ATME). Weight gain is accompanied by anti-inflammatory to proinflammatory status elevation in the ATME owing to hypertrophy and adipocyte death, increasing the production and release of multiple proinflammatory cytokines into the ATME [e.g., TNF-α, interferon γ (IFN-γ), IL-1β, and IL-6)]225 and thus exacerbating chronic fibrosis and vascular inflammation, which in turn disrupts ATME homeostasis226 (Fig. 6). Specifically, the different types of adipocyte death in obesity result in the release of cellular contents such as lipids, cytokines, and other signaling molecules into the ATME, promoting the recruitment and proliferation of phagocytic macrophages.227,228 Macrophages scavenge lipids and cellular debris by encircling dying adipocytes and forming crown-like structures (CLSs) and sometimes evolve into other cell types.229 However, the direct outcomes of these processes are simple: the activation of several inflammatory pathways, including the NLR and TLR pathways, and downstream signaling through inflammasome activation, an inhibitor of NF-κB kinase subunit β (IKKβ)-NF-κB 47 and c-JNK1 (also known as MAPK8), which are all correlated with insulin resistance and metabolic abnormalities in obesity and DM. Hence, the formation of CLSs in the ATME is regarded as a principal lesion and biomarker of adipose tissue inflammation,228 which is also implicated in obesity-induced DM.230

Correlations between the ATME and pancreatic carcinogenesis. Beyond an immediate increase in the development of low-grade chronic inflammation via the enhanced secretion of proinflammatory cytokines, the excessively expanded adipose tissue exacerbates metabolic disorders and magnifies the negative impact of adipokines. Meanwhile, this hypertrophic expansion inflicts stress on adipocytes and increases the production and release of substantial proinflammatory cytokines into the ATME, promoting the recruitment and proliferation of inflammatory cells and exacerbating oxidative stress, fibrosis, hypoxia, and lipolysis, which collaboratively have toxic, diabetogenic, and carcinogenic influences on the pancreas. ATMs adipose tissue macrophages, ATME adipose tissue microenvironment, NK natural killer, ROS reactive oxygen species
To date, most of the evidence regarding the roles of CLSs in cancer is based on cancer types other than PC, such as breast cancer. CLSs are more frequently detected among obese than nonobese breast cancer patients, are associated with an increased risk of breast cancer and contribute to the development and progression of cancer as metabolic and inflammatory factors.231 Similarly, in prostate cancer, CLSs and concurrent inflammation in periprostatic fat were shown to be associated with a higher body mass index (BMI) and tumor grade in men,232 and the results from rodent studies suggest that supplementation with estrogen and caloric restriction could be an ideal anti-inflammatory therapeutic option in the treatment of obesity.233 In the context of the consistent results from studies of other nonhormone-driven cancers234 and the connections between inflammation and pancreatic carcinogenesis in obesity and DM, we can assume that CLSs could also be essential drivers of pancreatic carcinogenesis, and further studies are warranted for confirmation.
Fatty infiltration in the pancreas
Ectopic visceral adipose tissue synthesizes various adipocytokines involved in metabolic processes, inflammation, appetite regulation, immunity, hematopoiesis, angiogenesis, and diseases such as obesity and cancer.235,236 Obesity causes intrapancreatic fatty infiltration associated with PanIN.174 Consistently, previous studies have indicated that intrapancreatic adipose tissue could be toxic, diabetogenic237 and carcinogenic238 since adipocytes are also endocrinologically capable of producing many molecules, including hormones, GFs, and adipokines, to reshape the local environment,239,240 making it conducive the progression of PanIN and consequent PC development (Fig. 6).241
With the gradual increase in pancreatic fat until the age of 60 years and the slow decrease in the volume of the pancreatic parenchyma after the age of 30 years, the fat/parenchymal ratio increases with age and results in fatty infiltration in the pancreas.242 When combined with metabolic dysregulation, such as the dyslipidemia and excessive visceral adipose accumulation in obesity and T2DM, fatty infiltration can be even more severe, and it is positively associated with the risk of PC.243 Unlike in hepatosteatosis, human samples have demonstrated that adipocytes infiltrate the pancreatic parenchyma in a scattered pattern (intralobular fat) and accumulate in the perilobular space, mainly around large vessels (interlobular fat).244 While intralobular fat is speculated to be produced by transforming fibroblasts or acinar cells to fill in the spaces created by the loss of damaged acinar cells, interlobular fat is seemingly related to obesity and T2DM.244 Clinical sample analysis demonstrated that fatty infiltration is more common in the peritumoral tissues of PC patients, where the infiltration fraction correlates with BMI and HbA1c levels in both groups.241 Even in the precancerous stage, pancreatic fatty infiltration correlates with the progression of PanINs in obesity.245 The potential mechanisms of these carcinogenic processes include the reprogramming and remodeling of the immunosuppressive TME, abnormalities in inflammation via the aberrant secretion of proinflammatory cytokines, and the disruption of growth factor signals (which will be introduced in detail below).
White adipose tissue inflammation
Generally, adipose tissue in patients with obesity is characterized by hypertrophy, hyperplasia, and an increased number of preadipocytes.246 Preadipocytes, by producing inflammatory cytokines and chemokines, as well as attracting and activating macrophages and endothelial precursors, create a proinflammatory microenvironment by increasing the levels of proinflammatory cytokines such as leptin, TNF-α, retinol-binding protein 4 (RBP-4), VEGF, IL-6, IL-8, IL-17, C-C motif chemokine ligand (CCL) 2, and CCL5,247,248 while infiltrated macrophages produce IL-6, IL-8, CCL2, and CCL5 at the same time.247,248,249 As a result, the established inflammatory microenvironment supports the proliferation of preadipocytes but impairs their differentiation.250
The excessively accumulated and expanded white adipose tissue (WAT) in patients with obesity is infiltrated by immune cells (mostly macrophages and lymphocytes), which secrete proinflammatory mediators to foster tumor growth.251 In addition, the expanded WAT outgrows its blood supply, leading to hypoxia, which causes adipocyte stress and death.252 Apart from the proinflammatory mediators secreted by enlarged adipocytes, the FFAs released from adipocytes and other cells jointly activate TLR4 in macrophages, leading to the increased expression of proinflammatory genes dependent on NF-κB, such as TNFα, IL1β, and COX2.253 Conversely, TNF-α and other cytokines sustain WAT inflammation by stimulating lipolysis and the release of FFAs.
The impact of adipose tissue dysfunction extends far beyond the TME; adipose tissue dysfunction also elicits a systemic effect that may synergistically fuel tumor growth. WAT inflammation is also associated with elevated circulating levels of various adipokines, C-reactive protein (CRP), and IL-6 in patients with obesity and DM, which have all been shown to promote pancreatic carcinogenesis.254,255,256 Therefore, it is likely that both the local and systemic environments are reprogrammed to promote carcinogenesis under conditions of WAT dysfunction and inflammation.
Adipokines
Adipokines are secreted by adipocytes, while cytokines are mainly produced by immune cells infiltrating adipose tissues, including macrophages and lymphocytes. Many studies have revealed the multifaceted roles of these signaling molecules in obesity- and DM-related carcinogenesis, and some of the most studied adipokines and cytokines will be addressed below.
-
(1)
Adiponectin. The most abundant adipokine in circulation, adiponectin, was also reported to be the first dysregulated hormone in metabolic disorders.257 Although adiponectin is primarily produced by adipose tissues, the levels of circulating adiponectin are inversely decreased in patients with obesity and DM. Adiponectin functions as an insulin-sensitizing, antidiabetic, anti-inflammatory, antiangiogenic, and anticancer adipokine. Adiponectin abolition in mice resulted in increased expression of proinflammatory genes, whereas adiponectin treatment reversed these effects. Moreover, adiponectin participates in the maturation of preadipocytes,258 and its signaling increases the phosphorylation of AMP-activated protein kinase (AMPK) and antagonizes leptin signaling.259
Previous research indicated that hypoadiponectinemia is associated with a higher risk of PC.260,261 Consistently, adiponectin has been shown to restrain tumor development and growth by affecting several intracellular signaling pathways, including the MAPK, mTOR, PI3K/AKT, STAT3, NF-κB, and sphingolipid metabolic pathways.262,263 Adiponectin also significantly inhibits the proliferation of PC cells by blocking the phosphorylation/inactivation of GSK-3β and suppressing the intracellular accumulation of β-catenin, reducing the expression of cyclin D1 and causing cell cycle arrest.264 Adiponectin has been shown to have indirect antitumor activity through its insulin-sensitizing and anti-inflammatory effects as an antineoplastic therapy,265 and it has been deemed to be an ideal diagnostic and prognostic biomarker in cancer.257 Nevertheless, a Mendelian randomization study with a large sample size did not find any correlation between circulating adiponectin or leptin and PC,266 and one study even suggested that adiponectin could be cancer-promoting in a tissue-dependent manner.267 Therefore, the roles of adiponectin in obesity- and DM-related pancreatic carcinogenesis are still controversial.
-
(2)
Leptin. Leptin is an adipokine involved in the regulation of energy balance, and its receptors are widely expressed in peripheral tissues, including pancreatic β-cells,268 with their activation reducing insulin secretion and modulating the proliferation, apoptosis, and size of β-cells.269 Notably, the adipogenic insulin overproduced in patients with obesity and T2DM stimulates the production and secretion of leptin, which in turn suppresses insulin secretion through both a central effect and its direct action on β-cells, establishing a hormonal regulatory feedback loop, a so-called adipo-insular axis.270 In addition, an elevated level of leptin is common in individuals with obesity and DM due to leptin resistance without any reduction in appetite.248 In contrast to adiponectin, which has antiproliferative and anti-inflammatory functions, leptin has mitogenic and proinflammatory effects by stimulating the production of IL-1, IL-6, IL-12, TNF-α, leukotriene B4 (LTB-4), and COX2.271 Leptin is also involved in the differentiation of monocytes and promotes T-cell proliferation and the TH1 phenotype while suppressing regulatory T cells (Tregs).271
Consistent with the evidence mentioned above, an increased leptin level is correlated with PC risk.255,272 However, while some researchers have also proposed a correlation between decreased leptin levels and PC,273 none of these correlations were reported in a previous Mendelian randomization study.266 By binding to its full-length receptor, leptin activates numerous intracellular signaling pathways that promote cancer cell growth and PC progression, including the leptin-Notch/RBP/JNK, JAK/STAT, MAPK, PI3K/AKT, and suppression of cytokine signaling (SOCS) pathways.274,275,276 Additionally, classic histone deacetylase (HDAC)-microRNA-leptin signaling can be oncogenic and increase proliferation, cellular stemness, and angiogenesis in PC.277 For further details, the roles of leptin in carcinogenesis have been reviewed elsewhere.278
-
(3)
Lipocalin-2 (LCN2). LCN2, also known as neutrophil gelatinase-associated lipocalin (NGAL), siderocalin, or 24p3, belongs to the lipocalin superfamily as a pleiotropic mediator of various inflammatory processes.279 Consistently, circulating levels of LCN2 are increased in patients with obesity, insulin resistance, and DM, with a parallel elevation in inflammatory markers, such as TNF-α, IL-1β, IL-6, and IFN-γ, exerting a proinflammatory effect.280 Since LCN2 can be secreted by adipocytes, neutrophils, macrophages, and cancer cells, elevated levels of LCN2 can be detected in obesity, DM, and PC.281,282,283 Overall, the increased level of LCN2 is correlated with carcinogenesis and progression to PC, in which LC2 levels begin to rise early in PanIN development and increase constantly during malignant progression to PDAC.284 Although the changes in the levels of LCN2 in animals led to inconclusive findings,284 the elevated level of LCN2 has been shown to contribute to the establishment of an inflammatory TME in PC via increased secretion of proinflammatory cytokines, such as IL-1β, IL-6, IL-8, stromal cell-derived factor 1 (SDF-1), intercellular adhesion molecule 1 (ICAM-1), MMP-1, MMP-3, MMP-9, and α-smooth muscle actin (α-SMA) from PSCs,281,285,286 which exacerbates the infiltration of TAMs285 and collaboratively promotes pancreatic carcinogenesis. In addition to its roles in modulating the TME, the elevation of LCN2 was also reported to be an early biomarker of PDAC in human samples of urine, serum, bile, pancreatic fluid/juice, and pancreatic cyst fluid.284 However, since there are also speculations about LCN2 being a tumor suppressor gene depending on the specific type of cancer, LCN2 is of great interest for further investigations.
-
(4)
Resistin. Resistin is primarily secreted by macrophages via the induction of IL-1, IL-6, and TNF-α. Similar to leptin, resistin is a proinflammatory adipokine responsible for insulin resistance in obesity and T2DM through SOCS3.287 Given that increased levels of resistin are associated with PC risk,288 it was reported that resistin activates STAT3 in an adenylyl cyclase-associated protein 1 (CAP1)- and TLR4-dependent manner to support the proliferation of PC cells and thereby contribute to tumor progression.289 In addition, resistin functions as a regulator of inflammation in the signaling of general transcription factor II-I repeat domain-containing protein 1 (GTF2IRD1), promoting the progression of PC.290
-
(5)
Nicotinamide phosphoribosyltransferase (NAMPT)/visfatin. NAMPT is secreted by adipose tissue and many other tissues in humans, acting as an enzyme, adipocytokine, and growth factor.236 Among the two forms of NAMPT, intracellular NAMPT (iNAMPT) participates in nicotinamide adenine dinucleotide (NAD) biosynthesis, a crucial part of cellular metabolism. The other form, extracellular NAMPT (eNAMPT), is implicated in several pathologies.236 In detail, circulating levels of NAMPT are increased in patients with obesity and T2DM,291 and NAMPT was also demonstrated to increase the expression of proinflammatory cytokines and inhibit apoptosis in response to endoplasmic reticulum (ER) stress.291 Additionally, eNAMPT exerts proinflammatory, proproliferative, antiapoptotic, and proangiogenic effects through the activation of the NF-κB, Notch-1, cyclin D1, CDK 2, MAPK, ERK1/2, and p38 signaling pathways.236 Furthermore, NAMPT is the rate-limiting enzyme of the NAD salvage pathway that is vital for the proliferation and survival of PC cells.292 Hence, inhibiting the NAD salvage pathway often causes metabolic collapse and cell death. In addition to reduced viability and colony formation in different PDAC cell lines, specific NAMPT inhibitors also decreased glucose uptake, lactate excretion, and ATP levels, resulting in metabolic collapse through activated AMPK and inhibited mTOR pathways.293 Likewise, a clinical trial also validated the efficacy of NAMPT inhibition.294 Given the roles of NAMPT in cellular metabolism and that IL-1 can stimulate the production of NAMPT,295 it is likely that inflammatory conditions in obesity and DM increase the levels of NAMPT initially and contribute to pancreatic carcinogenesis by promoting glycolysis and inflammation.
-
(6)
Chemerin and omentin-1. Expressed in multiple sites in humans, chemerin and omentin-1 (also known as intelectin-1) are both produced by adipose tissue and related to metabolic disorders in obesity and DM. Chemerin is involved in innate and adaptive immunity, adipogenesis, and adipocyte metabolism, while omentin-1 exerts its various functions in the regulation of metabolic processes in endocrine, autocrine, and paracrine manners.236 The levels of chemerin and omentin-1 are similarly increased in patients with PC, with the former showing a better discriminant ability.296 Chemerin can promote carcinogenesis through the inflammation, angiogenesis, and recruitment of mesenchymal stromal cells (MSCs),296,297 while omentin-1 can also promote cancer cell proliferation via enhanced inflammation.297,298 However, the effects of chemerin and omentin-1 differ among cancers.
-
(7)
Retinol-binding protein 4 (RBP-4). RBP-4 is an adipocytokine that acts by binding to cell surface receptors or through retinoic acid and retinoic acid-X receptors and is associated with TAG, total cholesterol, and LDL-cholesterol dysregulation and high blood pressure.236 Although some have questioned the diagnostic specificity of RBP4 in PC,299 others have shown that increased RBP-4 is effective in discriminating PC in patients with T2DM.300 Consistently, RBP-4 was suggested to promote JAK/STAT signaling via its receptor stimulated by retinoic acid 6 (STRA6) in carcinogenesis.236
-
(8)
Osteopontin (OPN). OPN, a protein of the bone calcified matrix, is a proinflammatory adipocytokine expressed in different cell types301 and is involved in biomineralization, remodeling and metabolic disorders such as obesity and DM.302 In cancer, OPN impacts cell proliferation, survival, drug resistance, invasion, and cell stemness by mediating cellular crosstalk and influencing the TME.303,304 OPN was shown to be tumorigenic a decade ago,305 and a higher OPN level is a diagnostic biomarker of PC.306,307 Concerning the roles of OPN isoforms in obesity, DM, and PC, OPNc was closely associated with obesity and DM, whereas the overexpression of OPNb and OPNc in PDAC cells increased the formation of colonies and the transcription of IL6.308 In addition, in a cell model replicating DM-related PC, the increased oxidative stress fueled by high glucose levels was reported to accelerate cell proliferation and mRNA expression of OPN in human pancreatic duct epithelial cells.309 Similarly, in a ROS-dependent manner, OPN secreted from activated PSCs in hypoxia interacts with the transmembrane receptor integrin αvβ3 on PC cells to upregulate the expression of forkhead box protein M1 (FOXM1), which induces malignant phenotypes of PC cells by enhancing EMT and cancer stem cell (CSC)-like properties through the AKT and ERK pathways.310 Upregulated OPN also takes part in the subtype conversion from classic to basal-like in PDAC through the modulation of the transcription factor GLI family zinc finger 2 (GLI2), which is a driver of PDAC cell plasticity that is critical for metastatic growth and adaptation to oncogenic KRAS ablation.311 Beyond that, OPN induces the activation of MMP-2 and MMP-9,236 which are known to promote carcinogenesis in PDAC.312
-
(9)
Oncostatin M (OSM). As a part of the inflammatory response in obesity and DM, OSM belongs to the IL-6 family and is secreted by activated T cells and macrophages in WAT.313 Despite being initially introduced as an anticancer agent, OSM can promote carcinogenesis and cancer progression in some cases, and the overexpression of OSM and OSM receptor (OSMR) has been detected in various cancers, including PC.314 OSM is elevated in the serum of PDAC patients,315 with TAMs in murine PDAC exerting increased secretion of OSM as well.316 OSM activates several carcinogenic signaling pathways through the formation of a complex composed of OSM and the cell signaling molecule gp130, including JAK/STAT3, MAPK, and PI3K.317 Among them, STAT3 is the main downstream signaling molecule of OSM regulating cell growth, invasion, survival, and all other hallmarks of cancer cells.314 In line with its unique efficacy compared to other members of the IL-6 family, OSM was shown to be one of the strongest drivers of EMT and CSC plasticity fueled by the induction of ZEB1, Snail (SNAI1), and OSMR, leading to enhanced pancreatic tumorigenicity through the JAK/STAT3 pathway.318,319 In addition, the cooperative signaling among OSM, hepatocyte growth factor (HGF), and TGF-β improves EMT in PC.320
In summary, adipocytes support tumor metabolism, and adipocytokines mediate carcinogenesis and cancer progression via their paracrine and endocrine actions. In obesity and DM, adipocytokines present independent and joint effects on the activation of major intracellular signaling pathways implicated in the carcinogenesis, proliferation, expansion, and survival of cancer cells. Treatments targeting obesity and DM have been shown to alter the plasma levels of adipocytokines and influence cancer risk. Therefore, many studies are being conducted to validate the potential of adipokines as therapeutic targets for the treatment of obesity and DM and diagnostic biomarkers of cancers. For now, more translational research is essential considering the conflicting results of the roles of some adipokines (e.g., adiponectin and leptin) in pancreatic carcinogenesis.
Cancer-associated adipocytes
Peritumoral adipocytes that are capable of interacting with cancer cells and contributing to tumor formation and growth by providing energy sources are called CAAs.251 The crosstalk between CAAs and cancer cells is important in determining the biological behavior of a tumor. Characterized by a strange fibroblast morphology, smaller and scattered lipid droplets, enhanced active energy metabolism and secretion, and higher expression of adipokines and chemokines, the concept of CAAs was proposed in 2010.321 Adjacent adipocytes also undergo similar phenotypic changes orchestrated with the remodeling of the TME. Currently, it is widely accepted that adipocytes in the inflammatory TME can dedifferentiate into CAAs that are fibroblast-like and capable of promoting ECM remodeling, tumor growth, and tumor progression.322 Meanwhile, PC cells can induce the dedifferentiation of adjacent adipocytes to CAAs, reducing their lipid droplets and significantly enhancing the EMT, migration/invasion capability, and chemoresistance of cancer cells.323 In addition to the pancreatic fatty infiltration mentioned above, enhanced Wnt5α signaling, a noncanonical Wnt pathway, plays a key role in the dedifferentiation of adipocytes in PC by favoring an inflammatory TME via the production of IL-6 and STAT3.324 In addition to the effects of adipocytes promoting carcinogenesis via adipokine and chemokine secretion, CAAs support pancreatic carcinogenesis via remodeled metabolism and crosstalk with other cells within the TME.
Cancer cells must rewire their metabolism to survive in a hypoxic TME due to fibrosis, the lack of vasculature, and poor perfusion. Thus, reprogramming lipid metabolism would be an ideal solution. Lipid metabolism is upregulated in the hypoxic TME.325 Apart from synthesizing FAs on their own, cancer cells also take up lipoproteins, chyme particles, and FFAs from adipocytes to sustain their constant proliferation.326 Unlike the storage of lipid droplets in hepatocytes in the liver, the lipids in the pancreas are mainly stored in infiltrated adipocytes in a much smaller amount,327,328 implying the potential active lipid supply from adipocytes to PC cells. However, it was also confirmed that exosomes secreted by PC cells increase lipolysis in adipose tissue.329 As a result, elevated FAs resulting from lipolysis act as a powerful driver of carcinogenesis, as addressed above.
As introduced in the last section, adipocytes can produce a wide range of adipokines and chemokines to promote carcinogenesis. These signaling molecules enable crosstalk between CAAs and other cell types in the TME, such as PSCs, tumor-associated neutrophils (TANs), Tregs, and CD8+ T cells, to fuel immunosuppression as well.246,330 Therefore, targeting CAAs has now been deemed a therapeutic strategy against PC. As a vital part of the TME, the roles of adipocytes in carcinogenesis are worth further investigation, which would be key to dissecting the impacts of adipose tissue on obesity- and DM-related pancreatic carcinogenesis.
CAFs, CAF-induced immunosuppression, disrupted ECM deposition, and hypoxia
CAFs
As a solid tumor, PC is characterized by a fibrotic and highly desmoplastic TME, highlighting the extensive distribution and significance of fibroblasts and PSCs in its pathogenesis. Resident fibroblasts and PSCs maintain the connective tissue architecture of the normal pancreas. Nevertheless, in collaboration with CAFs, PSCs induce excessive production of inflammatory cytokines and tissue-healing proangiogenic GFs that enhance the recruitment of both adaptive and innate immune cells, angiogenesis, and the deposition of ECM in chronic inflammation.331 Likewise, a recent study has also demonstrated that DM can activate PSCs through RAGEs in PDAC.332 Later, following the transformation of PSCs induced by various GFs and cytokines, such as pigment epithelium-derived factor (PEDF), endothelin 1 (ET-1), IGF-1, trefoil factor 1 (TFF2), and platelet‐derived growth factors (PDGFs), both PSCs and CAFs can change their phenotypes, resulting in excessive secretion of ECM during carcinogenesis, promoting cancer cell proliferation and reducing apoptosis.331 Although more than ten types of cells have been recognized as precursors of CAFs, there are only three well-defined subtypes of CAFs in PDAC, with considerable biological heterogeneity in spatial distribution and functional properties rooted in their diverse cell of origin: 1) profibrotic, immunosuppressive, and tumor restraining myofibroblastic CAFs (myCAFs); 2) secretory and inflammatory CAFs (iCAFs), which are functionally similar to senescent fibroblasts and deemed to be immunosuppressive and tumorigenic; and 3) immunoregulatory and tumorigenic antigen-presenting CAFs (apCAFs).333
Regardless of the differences in methods and standards used in discriminating the subtypes of CAFs, the correlations between their distinct features and PC are definite.334 Moreover, since each subtype has specific markers, combining subtype-specific lineage tracing models and in vivo imaging methods is ideal for revealing the cellular origin of distinct CAF populations. Owing to their adjacent residence, the myCAFs marked by the high expression of α-SMA respond to the increased levels of local TGF-β secreted by cancer cells and promote the production of ECM via the upregulated expression of α-SMA and collagen genes. In contrast, iCAFs, with much lower α-SMA levels, are distantly located from cancer cells and carry various inflammatory markers, such as IL-1, IL-6, IL-21, C3, CXC-chemokine ligand (CXCL) 1~3, CXCL12, and other inflammatory mediators.335 IL-1-mediated JAK/STAT signaling is suggested to regulate the transdifferentiation of CAFs or quiescent PSCs into iCAFs.335 With no sign of expressing costimulatory molecules such as CD80 or CD86, apCAFs carrying distinct markers such as major histocompatibility complex (MHC) II, CD74, serum amyloid A3 (SAA3), and secretory leukocyte peptidase inhibitor (SLPI) were inferred to lessen the T-cell response as a decoy receptor activating CD4+ T cells.333,335 Although it was indicated that myCAFs are profibrotic and immunosuppressive like the other two subtypes in PDAC, studies designed to target the Hh pathway by genetically depleting α-SMA-expressing cells, deleting sonic hedgehog (Shh), or pharmacologically inhibiting Smo did somehow incur some speculation about the tumor-restraining nature of myCAFs, as these manipulations resulted in reduced ECM deposition and α-SMA+ cells but surprisingly shortened survival in both preclinical and clinical studies.333 However, these observations are skeptical and not entirely indicative of the tumor-restraining roles of myCAFs due to the possibility of other α-SMA-expressing cells accidentally being involved during this pathway-targeted intervention.
Apart from the functional differences among CAF subtypes, emerging evidence from a single-cell RNA sequencing (scRNA-seq) study has illustrated an evolutionary transition of the identity and proportion of distinct CAF subtypes across normal, inflammatory, and premalignant/malignant states.333 From the inflammatory to premalignant/malignant state, a vital turning point of carcinogenesis, elevated α-SMA expression, a secretory phenotype, ECM deposition, proliferation, contractility, morphological activation, and deteriorated immunosuppression was observed in fibroblasts.333 Likewise, it was demonstrated in KPC mice that stromal fibroblasts, defined by positive α-SMA staining, are visible in PanINs and expand during the progression of pancreatic carcinogenesis.336 In contrast to normal fibroblasts, which restrain carcinogenesis and cancer progression, CAFs can be adverse via the following mechanisms: 1) increased ECM deposition; 2) enhanced inflammation and angiogenesis; 3) an increased number of tumor-initiating cells (TICs) and secretion of carcinogenic molecules; 4) altered cancer cell signaling, metabolism, and epigenome; 5) the establishment of an immunosuppressive TME; 6) the promotion of cancer cell EMT and stemness; and 10) the supplementation of cancer cells with substantial vital metabolites.333
CAF-induced immunosuppression
CAFs contribute to carcinogenesis through immunosuppression via their interactions with infiltrating immune cells within the TME. However, further partitioning and analysis of various populations of CAFs indicated the existence of nonnegligible and capricious differences in the function and effect of CAFs during this process.336 A recent study also identified CD105 as a robust marker distinguishing two lineages of CAFs with opposite effects on anticancer immunity in PDAC using mass cytometry.337 Despite their heterogeneity in biological properties and functions, all subtypes of CAFs can singly participate in immunosuppression. Overall, CAFs not only diminish the recruitment and cytotoxic activity of T cells by secreting immunosuppressive molecules such as TGF-β, IL-1α/β, IL-6, CXCL12, FGFs, EGFs, and TNF-α but also recruit immunosuppressive cells such as MDSCs and TANs that further activate CAFs in turn, and CAFs are also implicated in the recruitment, differentiation, and polarization of TAMs.336 In addition to a tumor-restraining phenotype of CAFs, the knockout of Il17a favored ECM remodeling and increased the recruitment and activation of CD8+ T cells and TH1 cells while lowering the numbers of immunosuppressive cells.338
Primarily induced by TGF-β, myCAFs impede the infiltration of cytotoxic lymphocytes and reduce the efficacy of anticancer immunity by excessively producing collagens and other components of the ECM, leading to aberrantly enhanced tissue stiffness and increased interstitial fluid pressure (see next section). Furthermore, these physical barriers built up by myCAFs seem to disturb only the activity and functioning of cytotoxic immune cells, such as CD8+ T cells, while facilitating the infiltration of immunosuppressive Tregs, MDSCs, and M2-TAMs.335 Inflammatory iCAFs, on the other hand, are the primary producers of immunosuppressive cytochemokines and impair anticancer immunity in PDAC.333 In addition, inflammatory signals, such as the activation of TLR4, can trigger the transdifferentiation of CAFs into iCAFs and promote the M2 polarization of TAMs and the recruitment of MDSCs, TANs, regulatory B cells (Bregs), and TH17 cells while expelling cytotoxic CD8+ T cells and hindering NK cells and dendritic cells (DCs), jointly undermining immune surveillance and deteriorating immune evasion.335
Beyond the direct regulatory effects of CAFs on immune cells, some other newly found mechanisms are also involved in CAF-induced immunosuppression.335 With more direct cell-to-cell contact, the antigens carried by CAFs engage with tumor-infiltrating CD8+ T cells and induce programmed cell death due to the subsequent upregulation of programmed cell death ligand (PD-L) 2 and Fas ligand (FasL) on CAFs.335 Furthermore, the snatching of nutrients from immune cells by predominantly expanded CAFs might also oppress the normal functioning of immune cells via compromised metabolism, which can be epitomized as the reverse Warburg effect.335 In brief, the expansion of CAFs consumes most of the extracellular glucose and produces more lactate, a fuel for anabolic processes for cancer cells, which causes the scarcity of glucose in the TME, damaging the glycolytic activity essential for the competent functioning of T cells in anticancer immunity. Given the immunosuppressive roles of CAFs, the depletion of CAFs expressing fibroblast activation protein (FAP) increased the antitumor efficacy of cytotoxic T cells, diminished the activity of the CAF-secreted cytokine CXCL12, enhanced T-cell recruitment in tumors and acted synergistically with PD-L1 to eradicate cancer cells.98 In summary, the gradually increasing and aberrantly expanded CAFs serve as regulators and mediators of the complex crosstalk among various cell types in the TME, most of which, however, promote carcinogenesis.
Disrupted ECM deposition
PDAC has more extensive ECM deposition than any other cancer, implying the crucial roles of the composition and function of ECM in initiating PC. Beyond providing structural support for resident cells as a noncellular component within all tissues, the ECM comprises proteins and polysaccharides, maintaining tissue homeostasis through dynamic and reciprocal biochemical and biomechanical interactions among various cellular constituents within the microenvironment.339 Structurally, the ECM is divided into the basement membrane (BM) and interstitial matrix (IM). The BM is responsible for supporting the polarity and differentiation of cells, and the IM, in contrast, is the mechanical regulator of tissue homeostasis.339 The dynamic physical force and chemical stress within the microenvironment can inevitably have complex effects on the fate of cells, such as inducing pathological conditions related to inflammation, which contributes to pancreatic carcinogenesis by damaging the structure and function of tissues; thus, the dynamic physical force and chemical stress are the major factors of the TME influencing the initiation of cancer.339 This process results from the fact that collagen and various ECM components stored in the dense, linearized, and cross-connected tumor-associated stroma (TAS) can stimulate cancer cell proliferation and angiogenesis while suppressing antitumor immunity.98,339 Similarly, a previous study also confirmed that ECM components are mainly produced by fibroblasts, and their composition in inflammatory conditions has considerable overlap with that in premalignant and malignant states, accompanied by parallel alterations in accordance with the proportion and function of CAF subtypes.333
Unlike normal wound healing, the expansion of CAFs fueled by chronic inflammation results in the accumulation of ECM proteins and the stiffening of the matrix, along with the elevation of cytokines and GFs promoting the proliferation of cancer cells, indicating that force-mediated biochemical signaling can also remodel the surrounding microenvironment and reinforce mechanosignaling.339 CAFs are the primary producers of ECM components (e.g., fibrillar collagen, fibronectin, laminin, and proteoglycans such as hyaluronic acid). Fibrosis resulting from chronic and excessive accumulation of ECM components builds a stiff and dense barrier that augments interstitial pressure, collapses blood vessels and reduces the accessibility of oxygen and the infiltration of cytotoxic immune cells while enhancing EMT and supporting tumor growth with biochemical and mechanical fuels in an immunosuppressive TME.333,336
The fibrotic and inflammatory microenvironment in obesity and DM leads to momentous mechanical changes in the ECM that promote EMT and tumor growth.336,340 Specifically, the TME, rich in collagen and hyaluronic acid, increases the density and stiffness of the PC stroma, shielding cancer cells while reenforcing the inflammatory reaction.340 In mice with mutant Kras, increased severity of inflammation-induced pancreatic injury and fibrosis were suggested to accelerate carcinogenesis due to the loss of tuft cells and decreased production of prostaglandin D2.341 Furthermore, obesity is associated with enhanced signaling of angiotensin II type 1 (AT1), which is expressed on both adipocytes and PSCs, with the former also having a high density of AT1 receptor (AT1R) related to the profibrotic pathways and the development of obesity and insulin resistance.342,343,344 As an antifibrotic treatment, the inhibition of AT1 signaling was demonstrated to ameliorate hypoxia and EMT by decreasing the production of IL-1β in adipocytes and PSCs.246
Hypoxia
Hypoxia is a condition of insufficient O2 availability concurrently caused by a reduced vascular density due to the expansion of the desmoplastic stroma and increased demand for O2 supply resulting from unbounded oncogene-driven proliferation of cancer cells, which is a common environmental phenomenon in many solid tumors, such as PC. As mentioned above, the extensive desmoplasia and ECM deposition fueled by activation of PSCs causes the compression of blood vessels with relative vessel collapse, decreased blood flow, and consequent hypoxia.322 Obesity and T2DM also induce local hypoxia via the overexpression of VEGF, and elevated levels of proangiogenic VEGF-A in patients with obesity and T2DM are correlated with increased vascular density in PDAC and faster disease progression.98 In the presence of hyperproliferative cancer cells, the local nutrients and oxygen provided by the vasculature are rapidly exhausted, and the cellular response is consequently activated to withstand hypoxic stress. In contrast to the necrosis of cells distant from the blood supply, substantial changes in transcriptional programs orchestrated by HIF-1 are made as adaptive responses in other cells. As a master transcription regulator, HIF-1 is a heterodimer composed of an α-subunit (HIF-1α) and β-subunit (HIF-1β), with the former being primarily in charge of its activity and stability. Despite its instability and immediate proteasomal degradation under normal levels of oxygen, the stabilization and accumulation of HIF-1α under hypoxic conditions can induce enhanced transcription of numerous genes involved in the modulation of angiogenesis, cell proliferation, survival, inflammation, anaerobic metabolism, cancer initiation and progression.345
Consistent with some reports indicating that HIF-1α is not entirely detrimental in all malignancies, pancreas-specific Hif1a deletion profoundly accelerated the progression of PanINs with a significant increase in the numbers of intrapancreatic “B1b” B-cell subtypes in KrasG12D-driven murine models,346 regardless of the early emergence of HIF-1α during the preinvasive stages of PDAC in both rodents and humans. Nevertheless, the carcinogenic effects of HIF-1α were also observed in PC. In addition to its potential correlations with inflammatory TLRs in PanIN lesions,347 HIF-1α can also promote pancreatic carcinogenesis by regulating the expression of EMT-related genes336 and its downstream target retention in endoplasmic reticulum sorting receptor 1 (RER1).348 Additionally, the transcriptional activity of HIF-1α has also been shown to be correlated with the number of TICs in PC.349
In summary, the deviant TME in the context of obesity and DM provides a very favorable growth soil for carcinogenesis. If the final formation of PC is metaphorized into the fruits, then as another decisive factor in carcinogenesis, the alteration of the immune response is undoubtedly the fertilizer for fruit ripening. Next, we will meticulously dissect the contribution of inflammation and the immune response to pancreatic carcinogenesis.
Inflammation and the immune response
Obesity and DM are characterized by an inflammatory microenvironment, a central and reversible mechanism promoting cancer risk and progression.251 Epidemiologically, various inflammatory stimuli or conditions in addition to obesity and DM, such as smoking and chronic pancreatitis, are other risk factors for PC, as both chronic low-grade inflammation and acute or chronic inflammation can be carcinogenic to the pancreas.350 All cells in the pancreas are maintained by self-renewal instead of stem cell replenishment owing to the lack of a stem cell population,351 with the natural regenerative capacity of the pancreas also endowing a greater chance of slow accumulation of lethal mutations and epigenetic changes in resident cells under inflammatory conditions, eventually leading to carcinogenesis initiated by malignantly dedifferentiated cells.352 Despite being protective against tissue damage, it was recently demonstrated that recurrent inflammation in normal pancreatic epithelial cells could cooperate with oncogenic Kras to trigger carcinogenesis by inducing irreversible ADM through MAPK signaling, which could be a universal event in the process of pancreatic carcinogenesis.353 In addition to ADM,352 similar to many other malignancies, the occurrence and progression of PC in the context of obesity and DM implicate insulin resistance, inflammation, and the infiltration of immunosuppressive cells.354
Generally, the inflammatory TME of PC comprises stromal cells (mainly fibroblasts and PSCs), endothelial cells, innate immune cells (e.g., macrophages, neutrophils, DCs, and NK cells), adaptive immune cells (T and B lymphocytes), and substantial cytokines and chemokines produced by these cells, jointly triggering tumor initiation and progression in an autocrine and/or paracrine manner. As a member of the inflammatory TME, no component can stay out of the carcinogenic process, although their roles may differ regarding their numbers, distribution, and physiological effects or even playing dual or multiple roles. In the presence of chronic inflammation fueled by the anfractuous interactions among these cells and the secretion of substantial proinflammatory cytokines and GFs, such as ILs, TNF-α, SHh, and TGF-β, along with the activation of the oncogenic networks facilitating the establishment of PanINs and their eventual progression to PC. As a solution, the application of nonsteroid anti-inflammatory drugs has been shown to be protective against PC.355
Regulation of inflammation-induced pancreatic carcinogenesis
Transcription factors
-
(1)
NF‑κB. NF-κB is a master regulator of innate immunity and inflammation, serving as a molecular bridge between chronic inflammation and cancer development through the modulation of cell proliferation and differentiation as well as the immune response.356,357 There are two distinct pathways involved in NF‑κB activation: the canonical and noncanonical pathways. The former is controlled by the IκB kinase (IKK) complex composed of IKKa, IKKb, and IKKc, while the latter is regulated by IKKa and has been reviewed previously regarding its roles in orchestrating inflammation in obesity, DM, and cancer.358,359 Despite functioning as a tumor suppressor in precancerous cells by maintaining cellular senescence, NF‑κB acts as a tumor promoter by diminishing immune surveillance during cancer initiation360,361 and magnifying RAS activity to fuel the formation of PanINs in the presence of oncogenic Kras.362 For example, the increased expression of CXCL12 resulting from elevated NF-κB activity in PSCs prevents cytotoxic T cells from infiltrating the tumor and eliminating cancer cells.363 Meanwhile, the expression and secretion of many inflammatory cytokines responsive to NF-κB are elevated upon its activation, and the interaction between oncogenic KRAS and NF-κB pathways can promote pancreatic carcinogenesis. Overall, the NF-κB signaling pathway is essential to the development of PDAC induced by mutant KRAS.364 Once activated, the transcription of growth-promoting genes (e.g., cyclin D1, cyclin E, cdk2, and c-myc) and cytokines is increased, and IKKb is also suggested to be necessary for the induction of PanINs and PDAC formation.364
Additionally, other proinflammatory factors regulated by NF-κB, such as IL-1 and TNF-α, also impact NF-κB activity and are strongly associated with most types of PDAC.365 IL-1, for instance, induces the constitutive activation of NF-κB, which is capable of promoting cellular transformation,365,366 subsequent formation of PanINs,345,367 and the determination of the PC phenotype during carcinogenesis.368 The regulation of NF-κB in inflammation-related carcinogenesis is at least partially achieved in cooperation with other factors, such as STAT3, and the expression of STAT3 is mediated by NF-κB, which also modulates cytokines and GFs in the TME, controlling the carcinogenic process, cancer cell proliferation and survival.369 Notably, various ILs have been reported to be closely associated with STAT3 activation in pancreatic carcinogenesis (discussed later). Similarly, p21-activated kinase 1 (Pak1) acts as an oncogene enhancing tumor formation by binding to fibronectin and interacting with the NF-κB-p65 complex in the Kras intact model.370 Furthermore, NF‑κB also modulates inflammatory macrophages through the direct regulation of growth and differentiation factor 15 (GDF‑15)/macrophage inhibitory cytokine 1 (MIC‑1), which is highly expressed in PC,371 serving as a promoter of early carcinogenesis. Together, these observations support the critical roles of NF-κB in pancreatic carcinogenesis (Fig. 7).
Fig. 7 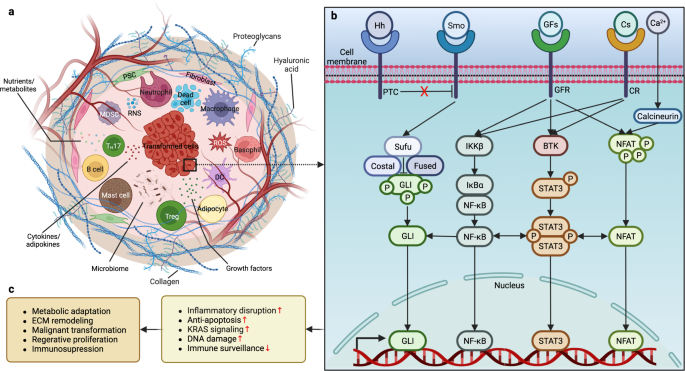
The impacts of the inflammatory microenvironment on pancreatic carcinogenesis. a The desmoplastic and inflammatory pancreatic microenvironment in obesity and DM during carcinogenesis comprises transformed cells, stromal cells (mainly CAFs and PSCs), adipocytes, diverse immune cells, dead cells, microbes, metabolites, ROS, RNS, and substantial proinflammatory cytokines/adipokines and GFs, etc., jointly fueling carcinogenesis via constantly activated inflammatory reactions. b Similar to other cells in this microenvironment, inflammatory stimuli activate various proinflammatory and oncogenic transcription factors in precancerous cells undergoing malignant transformation. The inhibitory effect of the transmembrane protein PTC on Smo is reversed after its binding to Hh, which increases the release of GLI from their inhibitory complex and favors nuclear translocation. Furthermore, the binding of GFs (e.g., EGF, etc.) and cytokines (IL-1, etc.) to calcineurin can enhance the nuclear translocation and transcriptional activity of NF-κB, STAT3, and NFAT. Bidirectional arrows indicate the interactions among transcription factors. c The activation of these transcription factors upregulates the expression of numerous target genes mediating inflammation, cell death, KRAS signaling, DNA damage, and immune surveillance, thereby jointly magnifying the impacts of inflammatory disruption on pancreatic carcinogenesis. BTK Bruton’s tyrosine kinase, Cs cytokines, CR cytokine receptor, DC dendritic cell, ECM extracellular matrix, EGF epidermal growth factor, GFR growth factor receptor, GFs growth factors, GLI glioma-associated oncogene, Hh Hedgehog, IκBα inhibitor of NF-κB subunit α, IKKβ inhibitor of nuclear factor-κB (NF-κB) kinase subunit β, IL-1 interleukin 1, MDSC myeloid-derived suppressor cell, NFAT nuclear factor of activated T cells, NF-κB nuclear factor-κB, P phosphorylation, PSCs pancreatic stellate cells, PTC patched, RNS reactive nitrogen species, ROS reactive oxygen species, Smo Smoothened, STAT3 signal transducer and activator of transcription 3, Sufu suppressor of fused, TH T helper, Treg regulatory T cell. Panel b in this figure was adapted from a previous publication369
-
(2)
Nuclear factor of activated T cells (NFAT). Capable of activating STAT3-like NF-κB, NFAT is another transcription factor family member implicated in pancreatic carcinogenesis.372 Likewise, NFAT family members modulate inflammatory processes and the expression of genes controlling cell growth and differentiation.369 Therefore, the aberrant activation of NFAT members (NFATc1 and NFATc2) can cause oncogenic transformation through the Ca2+/calcineurin/NFAT signaling pathway369,373 (Fig. 7).
-
(3)
Glioma-associated oncogene (GLI) family. GLI family members (GLI1~3) participate in pancreatic carcinogenesis by regulating the expression of genes related to inflammatory reactions, cell proliferation, apoptosis, and autophagy.369 As a downstream effector activated by two multitransmembrane proteins, patched (PTC) and Smo, in the Hh signaling pathway, GLI is mainly activated in stromal cells, which indicates that its unique contribution to the TME in pancreatic carcinogenesis is paracrine signaling369 (Fig. 7). However, this contribution is not entirely Hh-dependent.374 Moreover, KRAS was shown to induce pancreatic carcinogenesis by increasing GLI1 expression and protein stability while cooperating with GLI2.369 Collectively, the increased expression of both GLI1 and GLI2 also impacts PanIN lesions through an SMO-independent mechanism regulated by TGF-β.374,375

Other less-studied transcription factors also play different roles in pancreatic carcinogenesis. For example, the homeodomain transcription factor Prox1, which regulates mouse exocrine pancreas development, was illustrated to be transiently reactivated in acinar cells undergoing dedifferentiation and ADM. However, the expression of Prox1 was absent in the neoplastic lesions and tumors in the pancreas of both mice and humans. Furthermore, Prox1 heterozygosity drastically increased the formation of ADM and early neoplasia, with concurrent enhancement of inflammation in mice carrying oncogenic Kras.376
Cytokines
A variety of cytokines are secreted into the TME in response to inflammation and the immune response, modulating tumor development and progression. While many cytokines are secreted to inhibit carcinogenesis by the host, cancer cells can use these “double agents” to promote cell growth, reduce apoptosis, and facilitate tumor formation.377
Interleukins (ILs)
Chronic inflammation can lead to the overproduction of proinflammatory cytokines, and ILs are essential molecules downstream of NF‑κB signaling, playing crucial roles in promoting pancreatic carcinogenesis. Given their diversity and complexity in physiological functions, ILs can drive pancreatic carcinogenesis by enhancing inflammatory signaling, angiogenesis, and immunosuppression; dysregulating cell proliferation, survival and death; disrupting autophagy; and promoting ER stress, EMT and cell transformation (Table 1). To date, studies connecting ILs to pancreatic carcinogenesis are still lacking in comparison with their large numbers and considerable varieties. Given the diversity of ILs in cellular sources, receptors, signaling pathways and physiological functions, their roles in pancreatic carcinogenesis need to be further explored to fully illustrate the carcinogenic effects of these critical regulators of anticancer immunity.
Growth factors (GFs)
Accumulating evidence has suggested that various types of GFs are involved in pancreatic carcinogenesis, including TGFs, EGFs, HGFs, IGFs, VEGFs, PDGFs and FGFs.378 Functionally, GFs are secreted as signaling polypeptides capable of regulating specific cellular responses (e.g., cell proliferation, survival, migration, and differentiation), metabolism, and other cell functions by binding with specific and highly compatible cell membrane receptors. Several types of GFs produced by immune cells or other cells in the TME play crucial roles in inflammation-related carcinogenesis.
Transforming growth factors (TGFs).
TGFs are members of the EGF family that act synergistically to induce anchorage-independent growth of target cells. Two major types of TGFs, TGF-α and TGF-β, are known to be correlated with pancreatic carcinogenesis. A previous study confirmed that TGF-α is involved in ADM during the early stage of pancreatic carcinogenesis and local progression of PC,379,380,381,382 and it was demonstrated in 3D explant culture of primary pancreatic acinar cells that protein kinase D1 (PKD1), a downstream target of TGF-α and KRAS, is a mediator of ADM by activating the Notch signaling pathway.383 The combination of smad4 loss and tgfα overexpression was also shown to promote inflammation, ADM, fibrosis, and the formation and progression of PanIN lesions.384
TGF-β, on the other hand, acts as a tumor suppressor at the early stage of pancreatic carcinogenesis but promotes cancer progression later on.385 In detail, the activation of TGF-β forms the SMAD2/3/4 complex, which regulates the cell cycle, apoptosis, and differentiation to prevent carcinogenesis. However, the additional mutation and loss of SMAD4 leads to the functional switch of TGF‑β and drives carcinogenesis.385
Vascular endothelial growth factor (VEGF).
Apart from targeting endothelial cells, VEGF has been shown to affect multiple cell types.386 As its name implies, VEGF is vital in angiogenesis and is also involved in tumor formation and growth through the modulation of vascular permeability, which is regulated by HIF and other hypoxia-related genes as well as other oncogenic mutations.386 In addition, partly dependent on MEK1/2 and JNK signaling, the activation of KRAS was suggested to initiate angiogenesis through paracrine epithelial secretion of CXC chemokines and VEGF,387 and MMP-9 is one of the regulators of this angiogenic switch during carcinogenesis.388 VEGF also participates in EMT, and it was suggested that VEGF has clinical potential for predicting malignant transformation in patients with IPMN,389 with its expression also being correlated with metastasis and poor prognosis.390
Fibroblast growth factors (FGFs)
FGFs were given their names because they promote the proliferation of fibroblasts, and more than 20 members of this family have been discovered thus far. FGFs modulate several downstream pathways, such as the PI3K/AKT/mTOR and MAPK pathways, to regulate cell proliferation, differentiation, survival, migration, invasion, metastasis, angiogenesis, and wound repair, thereby being implicated in pancreatic carcinogenesis.378 However, not all FGFs are carcinogenic, and FGF21, a metabolic regulator preventing obesity, was shown to be suppressed in mice fed an HFD and carrying oncogenic kras mutations, which led to extensive inflammation, pancreatic cysts, PanIN, and eventual PDAC.42
Other chemocytokines and GFs.
TNF-α, a type II transmembrane protein with signaling potential released by proteolytic cleavage, is another central regulator of inflammation with dual roles in the TME related to carcinogenesis.28 The two binding proteins of TNF‑α, TNFR1 and TNFR2, have distinctly opposite roles in inflammatory reactions. Ubiquitously expressed TNFR1 (also known as p55) activates NF‑κB, JNK, and p38‑MAPK as a promoter of inflammation. In contrast, TNFR2 (also known as p75) is mainly expressed in immune cell receptors and regulates anti‑inflammatory signaling.28,385 The pro- or antitumoral effect of TNF‑α, of note, also depends on its local concentration and expression site, as it was demonstrated that TNF‑α can be carcinogenic by inducing DNA damage via enhanced production of ROS and reactive nitrogen species (RNS), especially at a low concentration.28 More importantly, TNF‑α can also be produced by cancer cells, which further stimulates the secretion of other cytokines and chemokines in the TME to exacerbate inflammation and immunosuppression, promoting tumor growth, angiogenesis, and metastasis.385
In contrast to the abovementioned GFs with protumor effects, PEDF has a tumor-suppressing effect on PC and limits pancreatic carcinogenesis by decreasing intratumoral inflammation and fibrosis.391 In addition, other components of inflammation also participate in carcinogenesis. Necroptosis (programmed necrosis) plays a vital role in the development of PC. It was shown that the major components of the necrosome, receptor-interacting protein (RIP) 1 and RIP3, were associated with oncogenic progression and immunosuppression mediated by the chemokine attractant CXCL1 and cytoplasmic SAP130 (a subunit of the histone deacetylase complex).392
Having a functional similarity with CXCL1, CXCL12 is one of the most studied chemokines in pancreatic carcinogenesis; CXCL12 is the ligand of CXCR4 receptors and is capable of enhancing tumor growth while inhibiting immune surveillance through local autocrine and paracrine mechanisms.385 Likewise, it was demonstrated both in vivo and in vitro that NF-κB signaling in PSCs increases the expression of CXCL12, which promotes tumor growth by diminishing the infiltration of cytotoxic T cells and the eradication of cancer cells.363 In addition, CXCL12 also promotes EMT, with the activation of CXCR4 increasing the expression of Smo, GLI1, and EMT markers to establish a fibrotic and hypovascular microenvironment in PC.385
Proteins involved in inflammation and the immune response
Toll-like receptors (TLRs)
TLRs are type I membrane receptors and the PRRs of the innate immune system; they are involved in the pathogenesis of PC by maintaining an inflammatory microenvironment and mediating the interactions between environmental stimuli and innate immunity.385,393 TLR signaling has been proven to promote carcinogenesis and cancer progression. For example, the overexpression of TLR2, TLR4, and TLR9 was detected in PanIN and PDAC lesions in resected human samples, with a linear increase in the expression of TLR2 from PanIN1 to PanIN3, TLR4 expression being the highest in inflamed ducts, and TLR9 expression in PanIN1 lesions.347 In a preclinical study, the endogenous microbiome of PDAC was shown to create an immunosuppressive environment by inhibiting T cells through selective TLR ligation.207
Overall, multiple members of the TLR family participate in pancreatic carcinogenesis. Driven by microbial-dependent activation of TLR4 signaling and subsequent engagement of the NLRP3 inflammasome, IL-1β secreted by PDAC cells supports the establishment of the carcinogenic TME by promoting the activation and secretory phenotype of quiescent PSCs and suppressing the immune response mediated by M2 macrophages, MDSCs, CD1dhiCD5+ Bregs, and TH17 cells.394 TLR2 and other TLR family members also exert a protumorigenic effect on the pancreas.395 In the context of enhanced inflammation through the activation of STAT3, Notch, NF-κB, and MAPK signaling, along with elevated expression in both the epithelial and stromal compartments of PC in humans and rodents, TLR7 promotes tumor progression by leading to the loss of phosphatase and tensin homolog (PTEN), CDKN2A, and cyclin D1 and the upregulation of CDKN1A, CDKN1B, TP53, c-Myc, SHPTP1, TGF-β, PPAR-γ, and cyclin B1.393 Similarly, the activation of TLR9 augments proinflammatory signaling in transformed epithelial cells, and its ligation promotes epithelial cell proliferation and fibrosis through the regulation of CCL11 on PSCs.396 Furthermore, TLR9 can be immunosuppressive by fueling the recruitment of Tregs and the proliferation of MDSCs.396 Apart from TLRs, NLR1 and NLR2, as bacterial sensors, function against gut inflammation and carcinogenesis in obesity.397
Cyclooxygenase 2 (COX2)
COX2 is a key enzyme that responds to multiple cytokines and GFs implicated in inflammation. Various binding elements have been found within the COX2 promoter for TP53, NF‑κB, and other transcription factors.385 Regarding its roles in pancreatic carcinogenesis, it was shown in mice harboring Kras mutations that the activation of COX2 accelerates the progression of PanIN via NOTCH1 signaling.398 Diet-induced obesity in mice can activate oncogenic Kras via COX2, leading to pancreatic inflammation, fibrosis, and the development of PanINs to PDAC.40 Additionally, COX2 was suggested to be indispensable for the positive feedback loop regulated by NF-κB, which prolongs Ras signaling and fuels chronic inflammation and PanIN formation in mice expressing physiological levels of oncogenic Kras.362 The coactivation of COX2 and KRAS was shown to accelerate the progression of PanIN lesions,398 while the inhibition of COX2 was demonstrated to abolish chemically induced pancreatic carcinogenesis in hamsters.399 Moreover, COX2 can be carcinogenic by suppressing the cell-competition-mediated apical elimination of RasV12-transformed cells, which are the initiators of pancreatic carcinogenesis.400
Bromodomain and extra terminal domain (BET) proteins
BET proteins are members of a bromodomain subfamily including BRD2~4 and BRDT, which mediate histone acetylation recognition, chromatin remodeling, and transcription regulation, therefore regulating inflammation, apoptosis, cell proliferation, the cell cycle, and cancer.98 In addition to enhancing the transcription of NF-κB-dependent proinflammatory cytokine genes, BET proteins also regulate the STAT signaling pathway.98
Immune cells
Disrupted metabolism and constantly activated inflammatory pathways provide preferable support for cancer initiation. During this process, substantial metabolic, endocrine, and inflammatory mediators as well as the intricate interactions among cells fuel malignant transformation, while the original metabolic and functional characteristics of immune cells are also reshaped. Metabolic disorders stimulate the expansion of immunosuppressive and protumorigenic immune cells, and the accumulation of these cells establishes a TME perfectly suitable for tumor formation.401
Leukocytes
Obesity and DM are associated with various immunological changes, such as leukocytosis resulting from chronic inflammation in adipose tissue,402 and the infiltration of different leukocytes has distinct effects on pancreatic carcinogenesis and progression.200
-
(1)
Lymphocytes. The impact of obesity and DM on the immune system is quite complicated. This impact not only alters the production of proinflammatory cytokines in the TME but also involves many other parts of the immune response, such as the adaptive immune response. Various cell types, including MDSCs, macrophages, TH17 cells and other CD8+ T cells, are present in the TME of Kras-mutant PDAC mice during the early stage of pancreatic carcinogenesis.98 In particular, peripancreatic leukocytes can either be protective against carcinogenesis by restraining tumor growth via antigen-restricted tumoricidal immune responses or, conversely, promote tumor progression through the induction of immunosuppression.403
-
(a)
T cells. Both obesity and DM have been shown to induce negative impacts on the population and function of T cells.404 As a vital part of the adaptive immune response, T cells are the pioneers of anticancer immunity and are supposed to be on the front lines preventing carcinogenesis. However, the incompetence of immune surveillance and evasion of precancerous cells indicate that there might be suspicious “traitors” among this defensive line. Indeed, the infiltration of distinct subpopulations of T cells induces different impacts on the carcinogenic process depending on their types, spatial distribution, and accompanying macrophage infiltration.335 In detail, as the predominant population of T cells in humans and a group of non-MHC-restricted lymphocyte subsets closely related to innate immunity, γδT cells, accounting for approximately 40% of the tumor-infiltrating T cells in human PDAC, have been shown to promote pancreatic carcinogenesis by mitigating the immune infiltration, activation, and TH1 polarization of αβT cells.403 In inflammation-driven pancreatic carcinogenesis, it was revealed that CD4+ T cells promote the establishment of an immunosuppressive TME and favor PanIN formation by mitigating the antitumor effects of CD8+ T cells, including the production of the cytotoxic molecules IFN-γ, TNF, perforin, and granzymes.335,405
The subpopulations of CD4+ T cells are made up of immune-activating and IFN-producing TH1 (T-BET+) cells and immunosuppressive TH2 (GATA3+) cells.335 While the former enhances immunity against intracellular pathogens and tumors by producing IFN-γ and other cytotoxic molecules to activate and recruit cytotoxic T cells, M1 macrophages, and NK cells, the latter secretes anti-inflammatory cytokines, such as IL-4, IL-5, and IL-13, and diminishes the antitumor response by inducing the M2 phenotype of macrophages and increasing the proliferation of PC cells via enhanced activation of TLRs and STAT3/AKT/MAPK signaling, which is jointly mediated by cancer cells, CAFs, TAMs, DCs, and B cells.335 Hence, in contrast to the inhibition of pancreatic tumor growth fueled by TH1-polarized CD4+ and CD8+ T cells, antigen-specific TH2-polarized CD4+ T cells were shown to promote PC progression in rodents.200
The main subpopulation of CD8+ T cells is cytotoxic T cells producing IFN-γ, TNF, and other cytotoxic molecules and playing critical roles in antitumor immunity. It was suggested that the functioning of cytotoxic T cells could be diminished by the increased expression of CXCL12 via the enhanced activity of NF-κB in PSCs.363 In addition, cancer cells can also sabotage the function of CD8+ T cells by reducing their survival and increasing programmed death 1 (PD-1), PD-L1, and cytotoxic T lymphocyte-associated protein 4 (CTLA-4).335 In contrast, the depletion of myeloid cells prevents mutant KrasG12D-driven pancreatic carcinogenesis, which is induced by enhancement of the antitumor activity of CD8+ T cells and elevated expression of PD-L1 in tumor cells in an EGFR/MAPK-dependent manner.406 Moreover, the oncogenic KrasG12D-dependent upregulation of granulocyte-macrophage colony-stimulating factor (GM-CSF) present in both the pancreatic ductal epithelial cells (PDECs) of mice and human PanIN lesions is essential for the recruitment of Gr1+CD11b+ myeloid cells and the establishment of an immunosuppressive TME, which causes PC cells to evade CD8+ T-cell-driven antitumor immunity.407
Through their production of multiple cytokines, including IL-17, IL-21, and IL-22, depending on the lineage-specific transcription factor RORγt, TH17 cells and Tregs are also involved in pancreatic carcinogenesis. Fueled by oncogenic KRAS, MAPK signaling induces the accumulation of TH17 cells in the TME and the subsequent upregulation of IL-17R to drive the progression of PanIN to PDAC in a cell-autonomous manner via REG3G, a member of the regenerating islet-derived gene (REG) 3 protein family that is associated with innate immunity.335 Moreover, REG3G promotes tumor growth, the differentiation of Tregs and the recruitment of MDSCs while inhibiting the maturation of DCs and hindering the activity of CD8+ T cells by interacting with PD-1/PD-L1 through the JAK2/STAT3 signaling pathway in DCs.408 Similarly, the ablation of the IFN-stimulated gene 15 (ISG15) pathway, which is highly active in PDAC, was demonstrated to suppress the expression of PD-L1 and the activity of Tregs while increasing the number of CD8+ tumor-infiltrating T cells, thereby mitigating pancreatic carcinogenesis.409 Similarly, the linear increase in the numbers of Tregs from precursor lesions to the progressed PC raised the speculation that Tregs are responsible for immunosuppression and a culprit for pancreatic carcinogenesis as a critical source of TGF-β ligands in the TME.335 However, the depletion of Tregs accelerated tumor progression by reshaping fibroblasts and increasing CCL3, CCL6, and CCL8, which augmented myeloid cell recruitment and immune suppression and promoted carcinogenesis.410 These contradictory results indicate that whether Tregs are promotive or inhibitive in pancreatic carcinogenesis may depend on a variety of factors. Based on the existing results, it would be hasty to assert the roles of Tregs, which need to be revealed by future studies.
-
(b)
B cells. As another important part of adaptive immunity, the distribution of B cells within the TME is often deemed to induce protumorigenic and immunosuppressive effects. In contrast to the perspective that HIF-1α is the main contributor to the hypoxic TME that promotes pancreatic carcinogenesis, the pancreas-specific deletion of Hif1α profoundly accelerated the development of PanINs in mice carrying mutant KrasG12D with a concurrent accumulation of intrapancreatic B lymphocytes, featured by prominent influx of a rare “B1β” B-cell subtype.346 Notably, a unique subtype of B cells, CD1dhiCD5+ Bregs, accumulate in the pancreatic parenchyma during early neoplasia and accelerate the development of PC in mice by stimulating the proliferation of cancer cells, which is mediated by the expression of IL-35.411 Later, another study showed that the inhibition of Bruton’s tyrosine kinase (BTK), a critical B-cell kinase, disrupts the differentiation of CD1dhiCD5+ Bregs and the production of IL-10 and IL-35 in the pancreatic TME, followed by an increase in stromal CD8+IFNγ+ cytotoxic T cells and the cessation of PanIN progression.412
-
(c)
NK cells. Representing 5~20% of circulating lymphocytes in humans, NK cells have long been known to participate in the antitumor immune response and reduce carcinogenesis.413 NK cells not only act as a component of anticancer immunity by nonspecifically recognizing and directly killing tumor cells but also regulate and shape the antitumor immune landscape through crosstalk with other cells, including DCs, macrophages, T cells, and endothelial cells.335 Emerging evidence has indicated that some signaling molecules within the TME can favor the induction of immune evasion from NK cells while inducing a tumor-promoting phenotype.335 However, unlike the decreased CD8+ T cells, the circulating levels of NK cells (CD56+CD3-) are elevated in patients with PDAC, which is positively correlated with a longer survival, although the cytotoxicity of NK cells in PDAC patients is weaker than that in healthy individuals owing to the reduction in cytotoxic mediators and increase in immunosuppressive signaling molecules, such as TGF-β, IL-10, IL-18, indoleamine 2,3-dioxygenase (IDO), and MMPs.335,414
Overall, apart from impaired cell recognition, the recruitment and infiltration of NK cells within the TME are reduced in PC.414 The activated PSCs in the desmoplastic stroma of PC exile NK cells from the local TME, partially accounting for the extremely low frequency (<0.5%) of NK cell infiltration in PDAC tumors, while NK cells isolated from tumor samples exert their intrinsic anticancer effects.335 Similar exciting findings inspired researchers to come up with the concept called “NK cell restoration”, aiming to reactivate the suppressed cytotoxic NK cells within the TME as a strategy of immune therapy.335 However, the extremely low numbers of NK cells in the TME of PC make it harder to depict the roles of NK cells during the early stage. Therefore, the gap in knowledge in this field awaits future efforts to reveal the roles that NK cells play and the changes they undergo during the carcinogenesis and progression of PC.
-
(d)
Dendritic cells. DCs (or S100+ cells) are clusters of specialized APCs capable of inducing antigen-specific immune responses and participating in antitumor immunity, especially myeloid DCs (mDCs, also known as CD11b+ cells).415 Quantitatively, DCs are rare in the TME of PDAC but are mainly located in the stroma adjacent to the tumor. It was suggested that a higher number of DCs, whether in the circulation or within the tumor, is correlated with longer survival, regardless of the stage of PC at diagnosis.415 Of note, the number of DCs infiltrated within the tumor is parallel to the number of CD4+ and CD8+ T cells in patients with PDAC, implying the presence of all key components in the antitumor immune response, and the decreased number and function of DCs in PDAC is responsible for the immune tolerance resulting from compromised antigen presentation.415
-
(a)
Other studies also confirmed the participation of DCs in pancreatic carcinogenesis. For example, the systemic and progressive dysfunction of type 1 conventional DCs (cDC1s) was found to occur in the earliest stages of preinvasive PanIN in KPC mice, which was fueled by increased apoptosis and production of IL-6 in the TME that subsequently leads to the disruption of cDC1 maturation.416 During pancreatic carcinogenesis in IPMN and intraepithelial precursor lesions that progress from adenoma to carcinoma, the switch from an antitumor immune response to immune tolerance occurs between the stages of intraductal papillary mucinous adenoma (IPMA) and intraductal papillary mucinous carcinoma (IPMC), where the increased expression levels of CXCL17 and intercellular adhesion molecule 2 (ICAM2) first enhance the infiltration and accumulation of immature mDCs while promoting the susceptibility of the tumor cells to cytotoxic T-cell-mediated cytolysis, which then disappears in IPMC.417
Granulocytes
-
(1)
Neutrophils. As the most abundant circulating leukocytes in humans, neutrophils are increased in WAT and produce the serine protease neutrophil elastase that cleaves insulin receptor substrate 1 (IRS1), preventing IRS1 from binding to PI3K and leading to insulin resistance,418 and the activation of the PI3K pathway is also important in pancreatic carcinogenesis in obesity and DM. Beyond that, accounting for an essential part of the innate immune system, the enormous population of neutrophils is inevitably implicated in the pathogenesis of various diseases, including the development and progression of cancer, such as PC.419,420,421 Characterized by their versatile phenotypes and functionality, TANs infiltrating tumors play crucial and decisive roles in carcinogenesis, and clinical research has shown a correlation between the elevation of the neutrophil-lymphocyte ratio and the stage and/or aggressiveness of cancer.420 TANs can be classified into two types based on polarization states, antitumor N1 neutrophils and protumor N2 neutrophils, and the transformation of both types of TANs with unique markers can be induced by multiple kinds of signaling cytokines and chemokines.421 It was also confirmed that the transformation of TANs from N1 to N2 can promote carcinogenesis in a hypoxic and immunosuppressive TME.420,422
Additionally, emerging evidence has shown that neutrophils have a longer life cycle and are capable of producing various cytokines and chemokines, affecting the TME by acting on both cancer cells and stromal cells.420 Some of these biologically active molecules can be mutagens or be in other forms to promote carcinogenesis through augmented cell proliferation, survival, migration, and angiogenesis in various cancers.420 As mentioned above, the complex and changeable phenotypes and functionality of neutrophils also have a two-sided influence on antitumor immunity. On the one hand, despite not being able to kill cancer cells solely or directly, neutrophils show an antitumor effect when stimulated by various cytokines and chemokines or kill cancer cells with indirect cytotoxicity, while they also interact with other immune cells, such as T cells and DCs, to inhibit carcinogenesis by enhancing the immune response.420 On the other hand, neutrophils also contribute to immunosuppression by recruiting immunosuppressive cells, such as Tregs and MDSCs, and diminishing the activity of T cells and NK cells via the formation of neutrophil extracellular traps (NETs).420
Mechanistically, neutrophils can be recruited to the TME by a substantial number cytokines and chemokines via various signaling pathways, and the abolition of these pathways has shown a significant antitumor effect in different animal models.421 Moreover, neutrophils can interact with other components of the TME, such as T cells, fibroblasts, PSCs, and macrophages, to participate in carcinogenesis and the subsequent development of PC by influencing the immune status.421 Finally, the formation of NETs has been implicated in the progression and metastasis of different types of cancer, yet its roles in pancreatic carcinogenesis have rarely been reported. Thus, research in this field will undoubtedly bring many interesting discoveries in the future.
-
(2)
Basophils. The number of basophils is far less than that of neutrophils, accounting for only <1% of human peripheral leukocytes. Like neutrophils, basophils also develop from hematopoietic stem cells in bone marrow via the stimulation of IL-3, the most important growth factor for the development of basophils in both humans and mice.423 Except for this common phenotypic marker IL-3, basophils of humans and mice express a variety of unique markers of their own, which basically modulate the inflammatory and immune response through the secretion of cytokines and chemokines, promoting or mitigating carcinogenesis.423 Accumulating evidence has suggested that not only do basophils reside in tissues rather than circulating in peripheral blood, but tissue-resident basophils also possess a specific phenotype shaped by the tissue microenvironment that is vital for the roles they play and the impacts they have on the development of cancer.423
Basophils are also present in the TME of PC, and beyond producing a variety of angiogenic factors, the receptors expressed on the surface of basophils also allow them to be regulated by some of these factors and participate in angiogenesis during carcinogenesis.423 Using an orthotopic model of PC, it was shown that basophils are indispensable for the formation of PC, and the interaction between basophils and Tregs contributes to immune tolerance in the TME of PC via the mediation of several proinflammatory cytokines, such as IL-3, IL-4, IL-8, and IL-13.423
-
(3)
Macrophages. Macrophages are the most abundant cell type in the TME, and they affect tumor biology in different ways. As macrophages are highly plastic, the macrophages in the ATME can be phenotypically, spatially, and functionally heterogeneous, playing a vital role in maintaining tissue homeostasis and mediating immunometabolism. Although disputable, it is clear that macrophages are a group of multisource cells originating from hematopoietic stem cells, yolk sac cells, adult monocytes, and fetal monocytes.424 Originating from embryonic development as the main source of pancreas-resident macrophages, a vast number of TAMs are recruited during PDAC progression, when they expand and exhibit a profibrotic profile fueling cancer-related inflammation, immune evasion, and matrix remodeling.336
A previous study confirmed that the infiltration and activation of macrophages are required for pancreatic carcinogenesis.425 The coculture of PDECs and macrophages supplemented with high-level glucose showed a significantly promoted malignant transformation of PDECs via enhanced EMT, suggesting a supporting role of macrophages in DM-related pancreatic carcinogenesis.426 Similar to TANs, TAMs are capable of making adaptations by switching their specific phenotype and differentiating into the M1 or M2 phenotype in response to various microenvironmental stimuli, with the latter commonly being deemed tumorigenic by contributing to the establishment of a protumor TME and promoting inflammation, angiogenesis, and EMT, especially in obesity-related PC.424,427 Moreover, it was shown that inflammatory macrophages can be converted into Ym1+ alternatively activated macrophages at ADM/PanIN lesions in the presence of IL-13 and oncogenic Kras and then drive pancreatic fibrogenesis and carcinogenesis by releasing IL-1Rα and CCL2.27 Furthermore, mutant KRAS in organoids was demonstrated to induce a protumorigenic phenotype in macrophages, with these transformed macrophages reducing epithelial PEDF expression and favoring a cancerous phenotype of epithelial cells during the early stage of pancreatic carcinogenesis, thereby promoting neoplastic growth.428 Likewise, the extracellular KRASG12D protein packed into exosomes can be released from cancer cells via autophagy-dependent ferroptosis under oxidative stress and then taken up by macrophages through an AGER-dependent mechanism, leading to the switch of TAMs into an M2-like phenotype via STAT3-dependent FAO.429
Communication between macrophages and adipocytes plays an important role in the pathogenesis of many diseases related to obesity, including DM and PC.424 TAMs in patients with obesity and DM can produce substantial proinflammatory cytokines to favor a hypoxic and immunosuppressive TME suitable for pancreatic carcinogenesis, such as IL-1β, IL-6, and TNF-α, activating NF-κB and Hh signaling.424 In addition, TAMs are involved in immunoregulation by reshaping the metabolism of the TME. While proinflammatory M2 macrophages fuel the enhancement of glycolysis and FA biosynthesis and the disruption of the TCA cycle, M2 macrophages increase oxidative phosphorylation, FAO, and arginine metabolism.424 Numerous preclinical studies have confirmed that TAMs can promote the Warburg effect in the TME of PC via different pathways and contribute to carcinogenesis and cancer progression.424
Apart from being implicated in the production of proinflammatory cytokines and chemokines, the infiltration of macrophages in adipose tissue was also suggested to promote PanIN development and PC initiation.424 In detail, the release of proinflammatory cytokines from macrophages increases the levels of FAs in the microenvironment owing to lipolysis, and this process can accelerate pancreatic carcinogenesis. In the conditional KrasG12D mouse model, mice with diet-induced obesity developed hyperinsulinemia, hyperglycemia, hyperleptinemia, and elevated levels of IGF-1. The pancreas of these animals showed important signs of inflammation with increased numbers of infiltrating inflammatory macrophages and T cells, elevated levels of several cytokines and chemokines, increased stromal fibrosis, and more advanced PanIN lesions.430
Since CAFs in the TME are capable of inducing the polarization of TAMs from the antitumorigenic M1 to the protumorigenic M2 phenotype through the secretion of proinflammatory cytokines and chemokines, polarized M2 TAMs also produce a variety of cytokines to promote EMT, which is beneficial for the spreading of PC cells during the early stage of PC formation.336,424 Through the activation of the transcription factor NRF2 and subsequent upregulation of CD163 and Arg1 and downregulation of IL-1β and IL-6, cancer cells induce an M2-like phenotype in TAMs and promote VEGF expression to augment EMT.431 Expressing multiple proangiogenic factors (e.g., VEGF-A, TGF-β1, TNF-α, IL-1β, IL-8, IL-10, IL-35, PDGF, and FGF-2) and enhancing the activity of MMPs and thymidine phosphorylase, macrophages function as one of the most important regulators of angiogenesis via the regulation of HIF-1α and participate in the entire angiogenic process, including the initiation, anastomosis, remodeling, maturation and plexus formation of vessels.424 As a major source of VEGF, a main driver of angiogenesis, macrophages facilitate angiogenesis in a VEGF-dependent manner in this process. TAMs and VEGFs all have a synergistic effect on enhanced angiogenic activity, which supplies the newly formed PC tumor with adequate blood and oxygen. Moreover, the infiltration of TAMs in the hypoxic TME can also elevate the expression of HIF-1α and VEGF-A.424
-
(4)
Mast cells. Being ubiquitous in the interstitial spaces of the body and residing in the connective tissue close to blood vessels, lymphatic capillaries, nerves, and epithelia, mast cells are known for their roles in allergic and anaphylactic reactions, with their involvement in acute or chronic inflammatory responses endowing them with the ability to participate in carcinogenesis.432 It was demonstrated in β-cell tumor models that the activation of MYC in vivo triggers fast recruitment of mast cells to the tumor site, which is necessary for macroscopic tumor expansion, while the inhibition of mast cells induces hypoxia and the death of cancer cells and endothelial cells.433 Like macrophages, mast cells can make selective adaptations to secrete pro- or anti-inflammatory molecules according to microenvironmental stimuli. Moreover, mast cells can change the microenvironment by producing cytokines and chemokines that impact fibrogenesis, angiogenesis, and ECM composition,432 while they can also be counteractivated by PC cells.434
Mast cells facilitate the invasion of cancer cells, as their infiltration in tumors can promote proliferation and immunoregulation by remodeling the TME.432 All parts of the mast cell-mediated inflammatory response participate in the inflammatory response against cancer. In contrast, the epigenetic modifications in response to environmental changes induced by the inflammatory response drive the development of cancer.432 In addition to regulating vascular permeability and enhancing tissue healing during inflammation, mast cells also modulate innate and adaptive immunity by tuning the functions and activities of other immune cells.432 Mast cells not only take part in innate immunity with neutrophils, macrophages, and NK cells but also regulate the migration of T cells and the growth and differentiation of B cells.432 In the case of low-grade chronic inflammatory conditions such as obesity and DM, mast cells synthesize extra proinflammatory signaling molecules to recruit and activate multiple types of leukocytes, including macrophages and lymphocytes, further promoting tissue destruction and inflammation, and the interactions between mast cells and fibroblasts are essential for fibrogenesis in the TME.432
Mast cells can be quite stable in the hypoxic TME, surviving via the release of IL-6, supporting tumor progression and elevating the level of ROS.432 Under this condition, cancer cells use mast cells as mediators to regulate the immune response of other immune cells via various cytokines and chemokines [e.g., TNF-α, macrophage inflammatory protein-1 (MIP-1), and MCP]. Nevertheless, similar to many other immune cells, mast cells either positively or negatively regulate the immune response against cancer, enhancing or restraining tumor growth. Accordingly, the actual impact of mast cells may differ depending on the type and phase of the tumor, and it is now generally accepted that different subsets of mast cells infiltrate tumors at different stages.432
More importantly, mast cells contribute to pancreatic carcinogenesis by enhancing angiogenesis435 and releasing a broad range of MMPs.436 In addition, via interactions with CAFs, mast cells promote fibrosis and angiogenesis by releasing substantial amounts of proangiogenic molecules, including VEGFs, angiopoietin-1, heparin, TNF, and FGF, in PC.432 Furthermore, mast cells promote EMT and create the morphogenetic basis favorable for the subsequent evolution of the tumoral interstitium by producing signaling molecules to sustain crosstalk with cancer cells,432 through which mast cells facilitate the invasion of CAFs in the tumoral interstitium and the development of a chaotic vasculature (granulation tissue), breeding tumor cells with the resources vital for their growth and metastatic invasion.432
In summary, mast cells support carcinogenesis via a complementary function to maintain the evolution of the tumor, which includes the creation of a tissue interstitium suitable for the implantation and survival of CSCs and the disruption of the immune response in the TME to ensure the proliferation and invasiveness of cancer cells. Finally, a chaotic vascular and fibrotic stroma is established to promote angiogenesis and EMT.432 However, at least for now, the roles of mast cells in pancreatic carcinogenesis remain largely unknown.
-
(5)
MDSCs. As a heterogeneous population of immature myeloid cells (CD15+CD11b+) accumulating during chronic inflammatory conditions such as cancer, MDSCs can be commonly divided into granulocytic or polymorphonuclear MDSCs (PMN-MDSCs, CD15+CD66b+) and monocytic MDSCs (M-MDSCs, CD66b-), the former are phenotypically and morphologically similar to neutrophils and represent more than 80% of all tumor-associated MDSCs, whereas the latter are phenotypically and morphologically similar to monocytes.335 Compared to the absence of MDSC infiltration in normal pancreatic tissues, the number of MDSCs is drastically increased in both the circulation and the TME, which is associated with the cancer stage in human PDAC.335,415 Moreover, human and animal studies have shown that GM-CSF and several other chemokines from the CXC family are the principal stimulators of MDSC recruitment and differentiation. At the same time, RAGEs are also likely to be a part of this process and promote pancreatic carcinogenesis and progression.335,415,437 Additionally, YAP, which is involved in obesity-related pancreatic carcinogenesis, can also deteriorate immunosuppression by elevating the expression and secretion of cytokines and chemokines that enhance the differentiation and accumulation of MDSCs.335,438
The immunosuppressive effects of MDSCs involve their various interactions with other cells within the TME, impairing innate and adaptive immunity against cancer. In detail, MDSCs have been shown to diminish the cytotoxicity of NK cells and promote the polarization of macrophages toward an M2 phenotype. MDSCs also have a suppressive impact on both CD4+ and CD8+ T cells through direct cell-to-cell contact by upregulating PD-L1 and stimulating the expansion of immunosuppressive Tregs via the IL-10-dependent secretion of TGF-β and IFN-γ,335 where a similar effect was also observed in IL-6.415 In contrast, the targeted depletion of MDSCs has been shown to reactivate the endogenous antitumor T-cell response and regress KrasG12D-driven PC initiation at early stages.335 Recently, a clinical study recruiting patients with premalignant polyps, colon cancer, premalignant IPMN, and PC confirmed a linear increase in the numbers of MDSCs from normal status to premalignancy and cancer. In contrast, no difference in their subpopulation composition or immunosuppressive capacity was noted, suggesting that the external microenvironment rather than the constant phenotypes and properties of MDSCs may play a more significant role in immunosuppression.439
Although there are many antitumor immune cells in the TME, their number is far less than that of immunosuppressive cells, which surround the antitumor cells and strongly impair their killing effect, favoring carcinogenesis and cancer progression (Fig. 8).

Interactions among cells within the tumor-killing (a) and immunosuppressive (b) TME of PC. Promotive effects are indicated with black arrows, and the suppressive impacts of cells on anticancer immunity are marked with lines in color. Some signaling components are shown near the arrows and lines. CAF cancer-associated fibroblast, CCL C-C motif chemokine ligand, CTLA4 cytotoxic T lymphocyte-associated protein 4, CXCL CXC-chemokine ligand, DC dendritic cell, EVs extracellular vesicles, FGF fibroblast growth factor, GM-CSF granulocyte-macrophage colony-stimulating factor, HGF hepatocyte growth factor, IDO indoleamine 2,3-dioxygenase, IFN interferon, IL interleukin, M1(2) M1(2) macrophage, MCP monocyte chemoattractant protein, MDSC myeloid-derived suppressor cell, MIP-1 macrophage inflammatory protein 1, MMPs matrix metalloproteinases, N2 N2 neutrophil, NE neutrophil elastase, NET neutrophil extracellular trap, NK natural killer, PC pancreatic cancer, PCCs pancreatic cancer cells, PD(-L) programmed death (ligand), PDGF platelet‐derived growth factor, PSC pancreatic stellate cell, RAGE receptor of advanced glycation end products, TGF transforming growth factor, TH T helper (cell), TNF tumor necrosis factor, Treg regulatory T cell, VEGF vascular endothelial growth factor
Physiological/pathophysiological processes
Obesity and DM are often accompanied by systemic alterations involving almost every aspect of human physiology. In addition to the abovementioned abnormalities, many vital physiological and pathological processes can also participate in pancreatic carcinogenesis. Next, we will summarize several key points that have been intensively studied in recent years.
Autophagy
What is mainly regulated by the mechanistic target of rapamycin (mTOR) kinase and the autophagy-related protein (ATG) family is autophagy, a conserved homeostatic process that maintains cellular quality and organ function through the disposal and recycling of cellular components while eliminating cells containing toxic proteins, lipids and organelles.440 Given its critical roles in mediating the homeostatic balance, detrimental alterations in autophagy are associated with metabolic disorders and other diseases, including obesity, DM, and cancer.440,441 As a unpredictable and dynamic physiological process, autophagy is highly sensitive to changes in nutrients, energy status, the microenvironment (hypoxia, oxidative stress, DNA damage, and protein aggregates), cellular metabolism, inflammatory status, and intracellular pathogens,442 and observations of the changes in autophagy in obesity can be highly dependent on the conditions of the models used.440 Therefore, it is still controversial whether alterations in autophagy are the cause or the consequence of metabolic disorders in obesity and DM.440 Nevertheless, most of the previous findings suggest that autophagy is suppressed under overnutrition conditions such as obesity and DM, although others suggest that autophagy is enhanced in adipose tissue as a compensatory anti-inflammatory response.440,443 In addition, the nature of autophagy being influenced by multiple factors results in an interesting scenario in which autophagy can be enhanced and suppressed in the same organ in different models of obesity,440 making its roles in metabolic disorders unclear.
Regarding the roles of autophagy in pancreatic carcinogenesis, more than 30 autophagy-related genes and numerous genes with similar functions have been implicated in carcinogenesis,444 and previous studies have confirmed that autophagy is vital for the initiation and progression of precancerous lesions in PC. Similar to the proinflammatory effect of suppressed autophagy in obesity,440 it was shown that inhibited autophagy could fuel inflammation445 and that autophagy participates in AGER-dependent macrophage polarization driven by oxidative stress in PDAC via ferroptosis.429 In another study, RAGEs were illustrated to promote cancer cell proliferation during the earliest stages of PC development via the regulation of autophagy, which enhances the IL-6-induced phosphorylation of STAT3.446 As one of the regulators of autophagy, ATG5 was also shown to determine the progression and metastasis of PC,447 and the knockout of Atg5 or Atg7 hindered the progression of PanINs, suggesting that autophagy is necessary for the initiation of malignant transformation.448 Likewise, although the decreased tumor growth following the inhibition of autophagy seems to prove its protumorigenic effect, the genetic ablation of autophagy in the pancreas increased tumor initiation but compromised the conversion of these premalignant lesions to invasive cancer and favored prolonged survival.449
Additionally, the dual effects of enhanced autophagy on pancreatic carcinogenesis have also been reported. Oncogenic RAS could stimulate autophagy via the BH3-protein Noxa and the autophagy regulator Beclin 1 to improve the survival of Ras-transformed cells by inducing proliferative arrest or premature senescence triggered by autophagy protein ULK3 during oncogene-induced senescence (OIS).445 However, it was also suggested that autophagy could prevent carcinogenesis through the clearance of p62, a multidomain signaling adapter protein functioning as a signaling hub in the mediation of cell survival and apoptosis, which can also augment ER stress, oxidative stress, DNA damage, and NF-kB activation.445 Similarly, it has been demonstrated that autophagy is protective against carcinogenesis by eliminating excessive ROS.450 Although it is easy to reason that suppressed autophagy in obesity and DM may contribute to pancreatic carcinogenesis based on the evidence above, we still cannot draw this conclusion considering the variable characteristics of autophagy and how it is influenced by various factors because it is very likely that different results will be observed using different models under different conditions in vivo and in vitro. Thus, determining the main factors and mechanisms that determine the roles of autophagy in the pathogenesis of diseases should be the focus of future research.
ER stress
The ER is involved in the process of protein folding, and ER stress refers to a basic cellular stress response that maintains protein homeostasis under several stressful conditions, such as the accumulation of unfolded proteins in the lumen of the ER. The increased FFA levels in individuals with obesity are associated with ER stress in adipocytes,451 which is caused by the increased number of unfolded proteins in the ER owing to the accumulation of ROS, resulting in cell apoptosis eliciting an inflammatory response.251 Moreover, WAT inflammation related to ER stress has also been noted in glucose intolerance.452 The recognition of ER stress as a factor related to carcinogenesis and cancer development involves the “unfolded protein response (UPR)”. Usually, cells adapt to ER stress by activating the UPR signaling pathway to retain ER homeostasis. Nevertheless, prolonged or severe stress often switches cellular signaling from prosurvival to ER stress-induced apoptosis.453 The high demand for protein synthesis in the exocrine pancreas requires the constitutive activation of the UPR to maintain the homeostasis of acinar cells,454 which may inevitably induce carcinogenic side effects on these cells of PC origin. In addition, ER stress can induce cellular inflammation and has been demonstrated to be related to pancreatic carcinogenesis. Accompanied by the later increase in UPR proteins, anterior gradient-2 (AGR2), a pro-oncogenic member of the protein disulfide isomerase family of ER-resident proteins, was suggested to be essential for PC initiation following inflammation induced by ER stress.455 Driven by ER stress, hypoxia, and ROS, ER oxidoreductase-1α (ERO1L), an ER luminal glycoprotein, is overexpressed in PanINs and PDAC, which promotes tumor growth via the enhanced Warburg effect.456 Nevertheless, the cellular response to ER stress is not always carcinogenic,457 and the impact of ER stress on tumor growth and the antitumor immune response also makes it a promising target in immunotherapy.458
Epithelial-mesenchymal transition
The highly fibroinflammatory (or desmoplastic) stroma consisting of a dense ECM is vital for establishing a hypoxic and nutrition-poor TME, promoting the final formation of PC. EMT is a process of epithelial cell differentiation to a mesenchymal phenotype through the loss of epithelial markers such as E-cadherin, certain cytokeratins, occludin, and claudin but the gain of mesenchymal markers such as vimentin, N-cadherin and fibronectin. EMT increases cell motility by lessening cell adhesion to other cells and the matrix to promote the development of PC.249,336
Initially, triggered by inflammatory reactions and regulated by numerous signaling pathways, EMT occurs much earlier than the final formation of pancreatic tumors but at the very early stage of carcinogenesis. As a dynamic and reversible process shaped by the TME, EMT endows cancer cells with morphological changes and a higher migratory capability. However, in response to the TME, with its considerable complexity, most cancer cells only undergo “partial” EMT and phenotypic versions instead of a thorough EMT to make adaptations and improve their survival.336 However, as mentioned above, EMT is reversible under certain conditions, as cancer cells can revert to an epithelial phenotype. Following the progression of precancerous low-grade neoplastic lesions, various protumoral factors are assembled into the TME, including cytokines and GFs that promote inflammation. At the same time, desmoplastic fibroblasts increase the intertumoral pressure and cause abnormalities in blood vessels, creating a severely hypoxic TME336 (detailed in the section Tumor microenvironment (TME) and cellular perturbations).
A microenvironment favorable for carcinogenesis exists in animal models of diet-induced obesity characterized by changes in hormones, GFs, cytokines, and adipocytes and alterations in EMT, which can be reversed by calorie restriction (CR), and EMT components may also serve as novel targets for cancer prevention or therapy.459 Indeed, enhanced EMT in PC cells is mediated by proinflammatory cytokines secreted in response to obesity,218 which is identical for DM, with a desmoplastic stroma and preexisting chronic inflammation promoting neoplastic progression.336 As we introduced above, the inflammatory response is a strong promoter of pancreatic carcinogenesis. In turn, precancerous or neoplastic cells can also trigger inflammation in the host immune system, disrupting innate and adaptive inflammatory responses to exacerbate the stromal reaction and further contribute to carcinogenesis via enhanced EMT.336
Exosome secretion
Transported by a variety of body fluids to distal tissues and organs to activate signaling pathways in target cells, exosomes are a class of nanoextracellular vesicles that function in an autocrine and paracrine manner460 and are also produced by stromal and transformed cells in the TME.461 Hence, ample evidence has suggested that exosomes play crucial roles in the pathogenesis of multiple physiological disorders and diseases, such as obesity, DM, and PC.462 During pancreatic carcinogenesis, exosomes promote the transformation of various precancerous lesions while augmenting angiogenesis, cell migration, and EMT and inhibiting apoptosis.462
Sharing similar structural proteins, exosomes secreted by different cell types contain proteins, nucleic acids, lipid molecules, and other inorganic substances. The surface of exosomes contains a variety of lipid raft microdomains that can transduce essential signals, such as signals for apoptosis and cell cycle arrest, via lipid molecules or proteins.462 Like exosomal lipids consisting of various components, exosomal nucleic acids consist of almost all kinds of RNAs and several types of DNAs.462 Exosomes are released into body fluids upon stimulation by specific signals. Then, they activate the corresponding signaling pathways in target cells, with their production and function varying according to the types and needs of cells.
The validation of targeting oncogenic KRAS using exosomes in PC models provided solid proof that exosomes participate in pancreatic carcinogenesis.463 Since exosomes play critical roles in the pathogenesis of obesity and DM,464 they can also promote carcinogenesis through various metabolic disorders. A previous study showed that exosomes derived from the adipose tissue of obese rodents can induce the differentiation of peripheral blood monocytes into activated macrophages and stimulate the secretion of proinflammatory IL-6 and TNF-α in a TLR4-dependent manner to fuel insulin resistance.465 In addition, obstructive sleep apnea (OSA), a comorbidity commonly seen in patients with obesity as a potential cause of intermittent hypoxia (IH),462 was suggested to increase the risk and metastasis of PC.466,467 In detail, IH promotes the proliferation and migration of cancer cells and angiogenesis by regulating the exosome content and increasing the production of tumor-promoting exosomes.462 In addition, exosomes also promote pancreatic carcinogenesis by mediating the occurrence of both T1DM and T2DM.462 In T1DM, apart from enhancing the autoimmune response via the contained proteins functioning as antigens, miRNAs transported by exosomes have also been shown to influence the pathogenesis of T1DM by influencing the apoptosis of β-cells. Similarly, various miRNAs in exosomes can modulate the apoptosis of β-cells in T2DM and control insulin secretion and the formation of pancreatic islets, thereby regulating glucose homeostasis.
In addition to the abovementioned metabolic disorders, exosomes are implicated in the inflammatory reactions of pancreatic carcinogenesis. With an elevated level in the circulation under inflammatory conditions, exosomes lead to pancreatic damage through an action similar to inflammatory factors, mainly characterized by irreversible damage in interstitial fibrosis and parenchymal calcification, where the proinflammatory effects of exosomes originating from different sources vary according to their components.462 As a result, pancreatic tissue destruction and exocrine/endocrine deficiency may turn quiescent PSCs with a high proliferative capacity into myofibroblasts, promoting the progression of PanINs.468 In particular, exosomal miRNAs can cause extensive fibrosis by promoting desmoplasia,462 a common aberrance in the TME that promotes PC development.
Previous studies have shown that exosomes secreted by PC cells can promote the malignant transformation and tumorigenesis of human pancreatic duct epithelial (HPDE) cells via the inhibition of SCL/TAL1 interrupting locus (STIL)469 and initiate malignant transformation and carcinogenesis in vivo by inducing random mutations in recipient cells.470 Comparisons among human PC cell lines and normal pancreatic epithelial cells confirmed that vesicles from PC cells with enriched proteins contribute to oncogenic cellular transformation. In contrast, vesicles from normal pancreatic cells are abundant in proteins related to the immune response.471 To facilitate and maintain PC development, cancer cells produce exosomes to favor an immunosuppressive TME to escape immune surveillance.472 DCs are the most critical APCs in the human body, acting through TLRs and producing various ILs, among which TLR4 is particularly indispensable for the antitumor effect of DCs.462 However, exosomes produced by PC cells have been shown to suppress the immune response of DCs by increasing intracellular levels of miR-203 and inhibiting the expression of TLR-4, TNF-α, and IL-12.473 Moreover, the miR-212-3p+ exosomes produced by PC cells were demonstrated to result in the failure of DCs.474 PC cells also diminish adaptive and innate antitumor responses by preventing B lymphocytes from recognizing cancer cells and triggering the cytotoxic killing effects of other immune cells.475 To create an immunosuppressive TME, exosomes produced by SMAD4-deficient PC cells carrying miR-1260a and miR-494-3p augment cell proliferation and glycolysis.476 Exosomes secreted by PC cells in rats can also reduce the proliferation and antiapoptotic capacity of leukocytes and abolish the chemotactic migration of leukocytes to tumor sites,477 contributing to tumor formation.
Oxidative stress and ROS
Reduction‒oxidation (redox) chemical reactions are a principal constituent of all life. With no exceptions, all normal and neoplastic transformed cells have a redox balance essential for maintaining cell functions. However, incomplete oxygen reduction leads to ROS formation, which is associated with the principle of oxidative stress mediating pathology, as ROS are damaging agents that can structurally and functionally compromise macromolecules, such as nucleic acids, proteins, and lipids. While normal cells have only a limited capacity to maintain the redox balance and nucleotide synthesis, oncogenic transformation can occur once this limit is overcome.478 Increased ROS is the direct cause of oxidative stress, and it has been implicated in various conditions such as obesity, DM, and the initiation and progression of PC.345,479,480
As a defense against oxidative stress, cells orchestrate a complex network of antioxidants to maintain proper cellular function. Several transcription factors strictly regulate the activity and production of antioxidant equivalents to protect macromolecules from the indiscriminate damage incurred by free radicals.481 In particular, nuclear factor erythroid 2-like 2 (NFE2L2/NRF2) is a master regulator of this network and can elevate the expression of genes involved in cytoprotective activities, maintaining redox homeostasis in response to oxidative stress, implicating xenobiotic metabolism, regulating proteasomal subunits and inflammatory response.481 In addition, the well-known transcription factor TP53 suppresses ROS accumulation by directly regulating the expression of a variety of antioxidant genes and indirectly inducing the metabolic gene TP53-inducible glycolysis and apoptosis regulator (TIGAR).481
ROS represent a double-edged sword in pancreatic carcinogenesis. Even though a low to moderate concentration of ROS in the TME could be adjusted by the antioxidant defense system, they still act as signaling molecules promoting genomic DNA mutation and the proliferation of cancer cells, which initiates neoplastic transformation by activating oncogenes as well as altering gene expression.345 Interestingly, an ROS level exceeding the optimum concentration beyond the capacity of the antioxidant defense system leads to irreversible oxidative damage, enhancing cell death via apoptosis, necrosis, and autophagy.345 High levels of ROS can cause damage to nucleic acids, thus being carcinogenic as a DNA mutagen or a promoter of genomic instability via the activation of topoisomerase II.482,483 On the other hand, cancer cells can enhance ROS generation,484 indicating the existence of a vicious cycle in ROS-promoting tumorigenesis. In addition to tumorigenesis, ROS have also been shown to promote cancer cell proliferation, survival, and metastasis in murine models and human cell lines.485 Mechanistically, it was suggested that carcinogenesis and its progression driven by ROS are achieved through sustained H2O2-dependent activation of the PI3K/AKT/mTOR and MAPK/ERK signaling cascades.481 Surprisingly, enhanced ROS production also plays a tumor-suppressing role by inducing cell cycle arrest, senescence, and cancer cell death.481
Given the biological features of oxidative stress, antioxidant strategies are seemingly a good option for the prevention and treatment of cancer. However, endogenous or exogenous antioxidants all have a dual effect of promoting and suppressing carcinogenesis and progression, similar to oxidative stress. For example, dietary antioxidants are considered cancer-promotive rather than prophylactic in experimental animal models.481 Moreover, as two maintainers of redox homeostasis, the elevated expression of nrf2 and increased intracellular levels of glutathione (GSH) in mice contribute to the initiation and progression of PC.486 Similarly, while the high generation of ROS caused by the oncogene KRAS increased the proliferation of PC cells and was reversed by manganese superoxide dismutase (MnSOD),487 the oxidizing agents, ironically, have been shown to inactivate mitogenic signaling cascades driven by AKT in pancreatic ductal cells.486
These self-contradictory results make the experimental evidence regarding the contribution of ROS to carcinogenesis difficult to interpret. Many questions are pending regarding the role of ROS in carcinogenesis, mainly because of the elusive effect of ROS that depends on their origin and cellular location as well as the stage of cancer progression.481 This is the main obstacle in implementing antioxidants in cancer prevention and treatment, owing to the topology and temporality of ROS regulation.488 In line with this concept, clinical trials have confirmed that antioxidants do more harm than good in cancer prevention.489 In this context, with both increased ROS and antioxidants promoting carcinogenesis, we have a long way ahead of us to reveal the mechanisms regulating the distinct response of different cells to oxidative stress, and only when we start to explore the spatial, temporal, and chemical specificity of individual redox couples will we be headed in the right direction to prevent pancreatic carcinogenesis by tackling oxidative stress.
Closing remarks and perspective
As the two most common and closely related metabolic diseases, obesity and DM are risk factors for many cancers. As the prevalence of obesity and DM is growing rapidly in most parts of the world, increasing attention is being paid to these two diseases and their effects on other diseases. Metabolism is a realm of great significance in cancer research; therefore, the impact of metabolic disorders on carcinogenesis and the progression of cancer has been the focus of scientific research for a long time. Benefitting from these efforts, the great number of publications and emerging novel findings in recent years have deepened our understanding of the relationship between metabolic disorders and carcinogenesis. In this article, we introduced the most studied mechanisms of obesity- and DM-related pancreatic carcinogenesis from several aspects. Therefore, beyond a broader and deeper understanding of the pathogenesis of PC in metabolic disorders, how should we reflect on these findings and what should they bring to patients? We would like to talk about the three most important values of this research field to answer these questions.
The first is the idea of PC prevention. There is no doubt that, if possible, prevention is always the best treatment for every disease. From an epidemiological perspective, the constant rise in the prevalence of obesity and DM is likely to favor a further increase in the occurrence of PC. Currently, the therapeutic options for obesity and DM vary, and their efficacy is promising. Numerous studies have also shown that most treatments for obesity and DM can significantly reduce the risk of PC.10 Hence, it is possible that effective prevention or treatment of obesity and DM can benefit patients at a higher risk, which not only will reduce the number of individuals with obesity and DM but also will lighten the future cancer burden. However, if the measures taken to tackle obesity and DM keep failing as they have been, the potentially growing future cancer burden of PC necessitates the translation of the research findings into clinical practice, whether developing sensitive methods to identify PC at an early stage in patients with obesity and DM or at least improving the efficacy of current obesity and DM treatments to prevent long-term lethal outcomes at best. At least for now, detecting early-stage PC in these patients and starting timely interventions to maximize the efficacy of treatments and prolong overall survival seems more ideal and practical than simply attempting to lighten the future cancer burden by tackling obesity and DM.
More in-depth exploration of the correlations of obesity, DM, and PC to favor a broader understanding of the mechanisms behind obesity- and DM-related pancreatic carcinogenesis is obviously essential to achieve this goal. Herein, we summarized and introduced the relevant research findings from six aspects (Fig. 9). Considering both the systemic and local influence of obesity and DM on the whole body and the pancreas, pancreatic carcinogenesis could be a multifactorial outcome involving joint input from different culprits. Due to the limited space, it is difficult to be fully comprehensive even if we have included as much varying content as possible, and we must admit that there are some other contributors to obesity- and DM-related pancreatic carcinogenesis that have been missed in this article. Nevertheless, now that we have learned the critical mutations initiating pancreatic carcinogenesis and some of the subsequent genomic alterations,490 identifying the enhanced triggering factors that result in modifications of these genes and the maintainers of the premalignant progression in metabolic disorders such as obesity and DM will be a stepstone for future strategies for PC prevention. Therefore, the ultimate challenge in this long journey will be, if it is possible and truly exists, looking for the genuine fuse of this timed bomb or finding the scissor to cut it off.

The intricate relationships among obesity, DM, and pancreatic carcinogenesis and the three most important values of relevant research. DM diabetes mellitus
Finally, and most importantly and realistically, efforts should be made to translate these findings into clinical practice for earlier PC detection. The timing of therapeutic intervention is the decisive factor in the treatment efficacy and prognosis of PC. Even if our abovementioned vision sounds like a daydream, we cannot stop searching for sensitive markers and physiological and biochemical indicators that can be used for surveillance and screening for PC, helping physicians detect early-stage PC in high-risk patients with obesity and DM. In recent years, emerging multiomic studies and advances in sequencing techniques have provided solid evidence on the metabolic landscape of obesity, DM, and other potential triggering factors of pancreatic carcinogenesis from multiple dimensions. The analysis of different clinical samples also hinted at promising directions for experimental practice. In light of these inspirational discoveries, future efforts will breed fruitful results in developing sensitive and cost-effective biomarkers by establishing modified disease models that highly replicate the original and natural pathogenesis through multidisciplinary cooperation and maximum utilization of the available techniques, which we believe serves as the starting point to developing reliable early screening and early diagnostic strategies for PC in populations with obesity and DM in the future.
References
World Obesity Federation. World Obesity Atlas 2022 (World Obesity Federation, 2022).
International Diabetes Federation. IDF Diabetes Atlas (International Diabetes Federation, Brussels, Belgium, 2021).
Tsai, A. G. & Bessesen, D. H. Obesity. Ann. Intern. Med. 170, Itc33–itc48 (2019).
Kumanyika, S. & Dietz, W. H. Solving population-wide obesity - progress and future prospects. N. Engl. J. Med. 383, 2197–2200 (2020).
Blüher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 15, 288–298 (2019).
Chan, J. C. N. et al. The Lancet Commission on diabetes: using data to transform diabetes care and patient lives. Lancet 396, 2019–2082 (2021).
Jain, T. & Dudeja, V. The war against pancreatic cancer in 2020 - advances on all fronts. Nat. Rev. Gastroenterol. Hepatol. 18, 99–100 (2021).
Carstensen, B. et al. Cancer incidence in persons with type 1 diabetes: a five-country study of 9,000 cancers in type 1 diabetic individuals. Diabetologia 59, 980–988 (2016).
Mizrahi, J. D., Surana, R., Valle, J. W. & Shroff, R. T. Pancreatic cancer. Lancet 395, 2008–2020 (2020).
Ruze, R. et al. Obesity, diabetes mellitus, and pancreatic carcinogenesis: correlations, prevention, and diagnostic implications. Biochim. Biophys. Acta Rev. Cancer 1878, 188844 (2022).
Ho, W. J., Jaffee, E. M. & Zheng, L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 17, 527–540 (2020).
Collisson, E. A., Bailey, P., Chang, D. K. & Biankin, A. V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 16, 207–220 (2019).
Yachida, S. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010).
Neuhöfer, P. et al. Acinar cell clonal expansion in pancreas homeostasis and carcinogenesis. Nature 597, 715–719 (2021).
Storz, P. & Crawford, H. C. Carcinogenesis of pancreatic ductal adenocarcinoma. Gastroenterology 158, 2072–2081 (2020).
Buscail, L., Bournet, B. & Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nature reviews. Nat. Rev. Gastroenterol. Hepatol. 17, 153–168 (2020).
Assi, M. et al. Dynamic regulation of expression of KRAS and its effectors determines the ability to initiate tumorigenesis in pancreatic acinar cells. Cancer Res. 81, 2679–2689 (2021).
Basturk, O. et al. A revised classification system and recommendations from the baltimore consensus meeting for neoplastic precursor lesions in the pancreas. Am. J. Surg. Pathol. 39, 1730–1741 (2015).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016).
Sinha, S. et al. PanIN neuroendocrine cells promote tumorigenesis via neuronal cross-talk. Cancer Res. 77, 1868–1879 (2017).
Farrell, A. S. et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun. 8, 1728 (2017).
Michl, P. & Gress, T. M. Improving drug delivery to pancreatic cancer: breaching the stromal fortress by targeting hyaluronic acid. Gut 61, 1377–1379 (2012).
Zeligs, K. P., Neuman, M. K. & Annunziata, C. M. Molecular pathways: the balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-κB signaling for cancer treatment. Clin. Cancer Res. 22, 4302–4308 (2016).
Tandon, M. et al. Prolactin promotes fibrosis and pancreatic cancer progression. Cancer Res. 79, 5316–5327 (2019).
Liou, G.-Y. et al. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14, 2325–2336 (2016).
Baumgart, S. et al. GSK-3β governs inflammation-induced NFATc2 signaling hubs to promote pancreatic cancer progression. Mol. Cancer Ther. 15, 491–502 (2016).
Liou, G.-Y. et al. The presence of interleukin-13 at pancreatic ADM/PanIN lesions alters macrophage populations and mediates pancreatic tumorigenesis. Cell Rep. 19, 1322–1333 (2017).
Padoan, A., Plebani, M. & Basso, D. Inflammation and pancreatic cancer: focus on metabolism, cytokines, and immunity. Int. J. Mol. Sci. 20, 676 (2019).
Bastea, L. I. et al. Pomalidomide alters pancreatic macrophage populations to generate an immune-responsive environment at precancerous and cancerous lesions. Cancer Res. 79, 1535–1548 (2019).
Bachem, M. G. et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921 (2005).
Masamune, A., Watanabe, T., Kikuta, K. & Shimosegawa, T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin. Gastroenterol. Hepatol. 7, S48–S54 (2009).
Kraman, M. et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830 (2010).
Jacobetz, M. A. et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120 (2013).
Auciello, F. R. et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Disco. 9, 617–627 (2019).
Birt, D. F., Julius, A. D., White, L. T. & Pour, P. M. Enhancement of pancreatic carcinogenesis in hamsters fed a high-fat diet ad libitum and at a controlled calorie intake. Cancer Res. 49, 5848–5851 (1989).
Chang, H.-H. et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS One 12, e0184455 (2017).
Hori, M. et al. Enhancement of carcinogenesis and fatty infiltration in the pancreas in N-nitrosobis(2-oxopropyl)amine-treated hamsters by high-fat diet. Pancreas 40, 1234–1240 (2011).
Guo, X. et al. High fat diet alters gut microbiota and the expression of paneth cell-antimicrobial peptides preceding changes of circulating inflammatory cytokines. Mediat. Inflamm. 2017, 9474896 (2017).
Liu, B. et al. Effects of high-fat diet on carcinogen-induced pancreatic cancer and intestinal microbiota in C57BL/6 wild-type mice. Pancreas 50, 564–570 (2021).
Philip, B. et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 145, 1449–1458 (2013).
Wang, D. et al. Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun. Signal.: CCS 17, 19 (2019).
Luo, Y. et al. Oncogenic KRAS reduces expression of FGF21 in acinar cells to promote pancreatic tumorigenesis in mice on a high-fat diet. Gastroenterology 157, 1413–1428.e11 (2019).
Kishikawa, T. et al. Satellite RNA increases DNA damage and accelerates tumor formation in mouse models of pancreatic cancer. Mol. Cancer Res. MCR 16, 1255–1262 (2018).
Garcia, D. I. et al. High-fat diet drives an aggressive pancreatic cancer phenotype. J. Surg. Res. 264, 163–172 (2021).
Smith, J. P., Kramer, S. & Bagheri, S. Effects of a high-fat diet and L364,718 on growth of human pancreas cancer. Digest. Dis. Sci. 35, 726–732 (1990).
Herrington, M. K. et al. Effects of high fat diet and cholecystokinin receptor blockade on pancreatic growth and tumor initiation in the hamster. Carcinogenesis 14, 1021–1026 (1993).
Kazakoff, K. et al. Effects of voluntary physical exercise on high-fat diet-promoted pancreatic carcinogenesis in the hamster model. Nutr. Cancer 26, 265–279 (1996).
Wang, F., Kumagai-Braesch, M., Herrington, M. K., Larsson, J. & Permert, J. Increased lipid metabolism and cell turnover of MiaPaCa2 cells induced by high-fat diet in an orthotopic system. Metabolism 58, 1131–1136 (2009).
Torres, C. et al. p110γ deficiency protects against pancreatic carcinogenesis yet predisposes to diet-induced hepatotoxicity. Proc. Natl Acad. Sci. USA 116, 14724–14733 (2019).
Kopin, L. & Lowenstein, C. Dyslipidemia. Ann. Intern. Med. 167, ITC81–ITC96 (2017).
Wang, F. et al. Dyslipidemia in Chinese pancreatic cancer patients: a two-center retrospective study. J. Cancer 12, 5338–5344 (2021).
Tseng, C.-H. New-onset diabetes with a history of dyslipidemia predicts pancreatic cancer. Pancreas 42, 42–48 (2013).
Wang, J., Wang, W.-J., Zhai, L. & Zhang, D.-F. Association of cholesterol with risk of pancreatic cancer: a meta-analysis. World J. Gastroenterol. 21, 3711–3719 (2015).
Catanzaro, R., Cuffari, B., Italia, A. & Marotta, F. Exploring the metabolic syndrome: nonalcoholic fatty pancreas disease. World J. Gastroenterol. 22, 7660–7675 (2016).
Kirkegård, J., Lund, J. L., Mortensen, F. V. & Cronin-Fenton, D. Statins and pancreatic cancer risk in patients with chronic pancreatitis: a Danish nationwide population-based cohort study. Int. J. Cancer 146, 610–616 (2020).
Chen, M.-J. et al. Statins and the risk of pancreatic cancer in Type 2 diabetic patients-A population-based cohort study. Int. J. Cancer 138, 594–603 (2016).
Hamada, T. et al. Statin use and pancreatic cancer risk in two prospective cohort studies. J. Gastroenterol. 53, 959–966 (2018).
Gabitova-Cornell, L. et al. Cholesterol pathway inhibition induces TGF-β signaling to promote basal differentiation in pancreatic cancer. Cancer Cell 38, 567–583.e11 (2020).
Liao, J. et al. Atorvastatin inhibits pancreatic carcinogenesis and increases survival in LSL-KrasG12D-LSL-Trp53R172H-Pdx1-Cre mice. Mol. Carcinog. 52, 739–750 (2013).
Simons, K. & Ikonen, E. How cells handle cholesterol. Science 290, 1721–1726 (2000).
Chen, W. C.-Y., Boursi, B., Mamtani, R. & Yang, Y.-X. Total serum cholesterol and pancreatic cancer: a nested case-control study. Cancer Epidemiol. Biomark. Prev. 28, 363–369 (2019).
Jung, Y. Y. et al. LDL cholesterol promotes the proliferation of prostate and pancreatic cancer cells by activating the STAT3 pathway. J. Cell. Physiol. 236, 5253–5264 (2021).
Johnson, D. E., O’Keefe, R. A. & Grandis, J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nature reviews. Clin. Oncol. 15, 234–248 (2018).
Hashizume, M. & Mihara, M. IL-6 and lipid metabolism. Jpn. Soc. Inflamm. Regen. 31, 325–333 (2011).
Chappell, D. A., Fry, G. L., Waknitz, M. A., Muhonen, L. E. & Pladet, M. W. Low density lipoprotein receptors bind and mediate cellular catabolism of normal very low density lipoproteins in vitro. J. Biol. Chem. 268, 25487–25493 (1993).
Guillaumond, F. et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl Acad. Sci. USA 112, 2473–2478 (2015).
Oni, T. E. et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J. Exp. Med. 217, e20192389 (2020).
Li, J. et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 35, 6378–6388 (2016).
De Boussac, H. et al. Oxysterol receptors and their therapeutic applications in cancer conditions. Expert Opin. Ther. Targets 17, 1029–1038 (2013).
Björkhem, I. & Diczfalusy, U. Oxysterols. Arterioscler. Thromb. Vasc. Biol. 22, 734–742 (2002).
Brown, M. S. & Goldstein, J. L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 (1997).
Radhakrishnan, A., Ikeda, Y., Kwon, H. J., Brown, M. S. & Goldstein, J. L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl Acad. Sci. 104, 6511–6518 (2007).
Zelcer, N., Hong, C., Boyadjian, R. & Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325, 100–104 (2009).
Jo, Y. & Debose-Boyd, R. A. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit. Rev. Biochem. Mol. Biol. 45, 185–198 (2010).
Shimano, H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 40, 439–452 (2001).
Sun, Y. et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumor Biol. 36, 4133–4141 (2015).
Siqingaowa, S. S., Gopalakrishnan, V. & Taghibiglou, C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem. Biophys. Res. Commun. 488, 136–140 (2017).
Kashiwagi, K. et al. Expression of liver X receptors in normal and refractory carcinoma tissues of the human lung and pancreas. Histol. Histopathol. 33, 497–505 (2018).
Candelaria, N. R. et al. Antiproliferative effects and mechanisms of liver X receptor ligands in pancreatic ductal adenocarcinoma cells. PLoS One 9, e106289 (2014).
Srivastava, S. et al. Novel liver X receptor ligand GAC0001E5 disrupts glutamine metabolism and induces oxidative stress in pancreatic cancer cells. Int. J. Mol. Sci. 21, 9622 (2020).
Karaboga, H. et al. Screening of focused compound library targeting liver X receptors in pancreatic cancer identified ligands with inverse agonist and degrader activity. ACS Chem. Biol. 15, 2916–2928 (2020).
Yang, B. et al. The lipogenic LXR-SREBF1 signaling pathway controls cancer cell DNA repair and apoptosis and is a vulnerable point of malignant tumors for cancer therapy. Cell Death Differ. 27, 2433–2450 (2020).
Kim, S.-M. et al. 27-hydroxycholesterol induces production of tumor necrosis factor-alpha from macrophages. Biochem. Biophys. Res. Commun. 430, 454–459 (2013).
Al-Zoubi, M. et al. Overexpressing TNF-alpha in pancreatic ductal adenocarcinoma cells and fibroblasts modifies cell survival and reduces fatty acid synthesis via downregulation of sterol regulatory element binding protein-1 and activation of acetyl CoA carboxylase. J. Gastrointest. Surg. 18, 257–268 (2014).
Wang, F., Stappenbeck, F. & Parhami, F. Inhibition of hedgehog signaling in fibroblasts, pancreatic, and lung tumor cells by Oxy186, an oxysterol analogue with drug-like properties. Cells 8, 509 (2019).
Ishikawa, S. et al. The role of oxysterol binding protein-related protein 5 in pancreatic cancer. Cancer Sci. 101, 898–905 (2010).
Sasaki, A. et al. Obesity suppresses cell-competition-mediated apical elimination of RasV12-transformed cells from epithelial tissues. Cell Rep. 23, 974–982 (2018).
Ding, Y. et al. Omega-3 fatty acids prevent early pancreatic carcinogenesis via repression of the AKT pathway. Nutrients 10, 1289 (2018).
Poulain-Godefroy, O. & Froguel, P. Preadipocyte response and impairment of differentiation in an inflammatory environment. Biochem. Biophys. Res. Commun. 356, 662–667 (2007).
Patra, K. C. et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat. Cell Biol. 20, 811–822 (2018).
Ling, S. et al. Association of type 2 diabetes with cancer: a meta-analysis with bias analysis for unmeasured confounding in 151 cohorts comprising 32 million people. Diabetes Care 43, 2313–2322 (2020).
Pereira, S. P. et al. Early detection of pancreatic cancer. lancet Gastroenterol. Hepatol. 5, 698–710 (2020).
Pasquale, V. et al. Glucose metabolism during tumorigenesis in the genetic mouse model of pancreatic cancer. Acta Diabetol. 56, 1013–1022 (2019).
Heiden, M. G. V., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Young, C. D. & Anderson, S. M. Sugar and fat - that’s where it’s at: metabolic changes in tumors. Breast Cancer Res. BCR 10, 202 (2008).
Peng, C. et al. Regulation of the Hippo-YAP pathway by glucose sensor O-GlcNAcylation. Mol. Cell 68, 591–604.e5 (2017).
Chang, S.-C. & Yang, W.-C. V. Hyperglycemia, tumorigenesis, and chronic inflammation. Crit. Rev. Oncol. Hematol. 108, 146–153 (2016).
Quoc Lam, B., Shrivastava, S. K., Shrivastava, A., Shankar, S. & Srivastava, R. K. The Impact of obesity and diabetes mellitus on pancreatic cancer: molecular mechanisms and clinical perspectives. J. Cell. Mol. Med. 24, 7706–7716 (2020).
Li, W. et al. Hyperglycemia promotes the epithelial-mesenchymal transition of pancreatic cancer via hydrogen peroxide. Oxid. Med. Cell. Longev. 2016, 5190314 (2016).
Wang, L. et al. Diabetes mellitus stimulates pancreatic cancer growth and epithelial-mesenchymal transition-mediated metastasis via a p38 MAPK pathway. Oncotarget 7, 38539–38550 (2016).
Rahn, S. et al. Diabetes as risk factor for pancreatic cancer: Hyperglycemia promotes epithelial-mesenchymal-transition and stem cell properties in pancreatic ductal epithelial cells. Cancer Lett. 415, 129–150 (2018).
Hu, C.-M. et al. High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab. 29, 1334–1349.e10 (2019).
Ruiz, H. H., Ramasamy, R. & Schmidt, A. M. Advanced glycation end products: building on the concept of the “common soil” in metabolic disease. Endocrinology 161, bqz006 (2020).
Vlassara, H. & Striker, G. E. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinol. Metab. Clin. North Am. 42, 697–719 (2013).
Bucciarelli, L. G. et al. RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell. Mol. Life Sci. 59, 1117–1128 (2002).
Hoppmann, S., Steinbach, J. & Pietzsch, J. Scavenger receptors are associated with cellular interactions of S100A12 in vitro and in vivo. Int. J. Biochem. Cell Biol. 42, 651–661 (2010).
Kang, R. et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 17, 666–676 (2010).
Jung, C. H., Ro, S.-H., Cao, J., Otto, N. M. & Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295 (2010).
Vernon, P. J., Zeh Iii, H. J. & Lotze, M. T. The myeloid response to pancreatic carcinogenesis is regulated by the receptor for advanced glycation end-products. Oncoimmunology 2, e24184 (2013).
Bayne, L. J. et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835 (2012).
Nomura, A. et al. Inhibition of NF-kappa B pathway leads to deregulation of epithelial–mesenchymal transition and neural invasion in pancreatic cancer. Lab. Invest. 96, 1268–1278 (2016).
Hoesel, B. & Schmid, J. A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 12, 86 (2013).
Kang, R. et al. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 5, e1480–e1480 (2014).
Kang, R. The receptor for advanced glycation end-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid. Redox Signal. 15, 2175–2184 (2011).
Jiao, L. et al. Dietary consumption of advanced glycation end products and pancreatic cancer in the prospective NIH-AARP Diet and Health Study. Am. J. Clin. Nutr. 101, 126–134 (2015).
Grote, V. A. et al. The associations of advanced glycation end products and its soluble receptor with pancreatic cancer risk: a case-control study within the prospective EPIC Cohort. Cancer Epidemiol. Biomark. Prev. 21, 619–628 (2012).
Feng, H.-Y. & Chen, Y.-C. Role of bile acids in carcinogenesis of pancreatic cancer: an old topic with new perspective. World J. Gastroenterol. 22, 7463–7477 (2016).
Chávez-Talavera, O., Tailleux, A., Lefebvre, P. & Staels, B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 152, 1679–1694.e3 (2017).
Yu, J. et al. Farnesoid X receptor antagonizes Wnt/β-catenin signaling in colorectal tumorigenesis. Cell Death Dis. 11, 640 (2020).
Zhou, M. et al. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J. Hepatol. 66, 1182–1192 (2017).
Liu, T. et al. The gut microbiota at the intersection of bile acids and intestinal carcinogenesis: an old story, yet mesmerizing. Int. J. Cancer 146, 1780–1790 (2020).
Joshi, S. et al. Bile acids-mediated overexpression of MUC4 via FAK-dependent c-Jun activation in pancreatic cancer. Mol. Oncol. 10, 1063–1077 (2016).
Gál, E. et al. Bile accelerates carcinogenic processes in pancreatic ductal adenocarcinoma cells through the overexpression of MUC4. Sci. Rep. 10, 22088 (2020).
MORI, H. et al. Bile metabolites and risk of carcinogenesis in patients with pancreaticobiliary maljunction: a pilot study. Anticancer Res. 41, 327–334 (2021).
Yang, Q., Vijayakumar, A. & Kahn, B. B. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 19, 654–672 (2018).
Lieu, E. L., Nguyen, T., Rhyne, S. & Kim, J. Amino acids in cancer. Exp. Mol. Med. 52, 15–30 (2020).
Commisso, C. et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013).
Yip-Schneider, M. T. et al. Circulating leptin and branched chain amino acids-correlation with intraductal papillary mucinous neoplasm dysplastic grade. J. Gastrointestinal. Surg. 23, 966–974 (2019).
Li, J.-Y. et al. GEO data mining and TCGA analysis reveal altered branched chain amino acid metabolism in pancreatic cancer patients. Aging 13, 11907–11918 (2021).
Tobias, D. K. et al. Circulating branched-chain amino acids and long-term risk of obesity-related cancers in women. Sci. Rep. 10, 16534 (2020).
Li, J.-T. et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 22, 167–174 (2020).
Sun, Q. et al. Oncogenic function of TRIM2 in pancreatic cancer by activating ROS-related NRF2/ITGB7/FAK axis. Oncogene 39, 6572–6588 (2020).
Kottakis, F. et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395 (2016).
Liu, S. et al. METTL13 methylation of eEF1A increases translational output to promote tumorigenesis. Cell 176, 491–504.e21 (2019).
Mazur, P. K. et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287 (2014).
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F. & Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 (2015).
Carrer, A. et al. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Disco. 9, 416–435 (2019).
Ni, C. et al. ACOT4 accumulation via AKT-mediated phosphorylation promotes pancreatic tumourigenesis. Cancer Lett. 498, 19–30 (2021).
Prasad, N. B. et al. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 65, 1619–1626 (2005).
Borch, K., Kullman, E., Hallhagen, S., Ledin, T. & Ihse, I. Increased incidence of pancreatic neoplasia in pernicious anemia. World J. Surg. 12, 866–870 (1988).
Smith, J. P., Shih, A., Wu, Y., McLaughlin, P. J. & Zagon, I. S. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am. J. Physiol. 270, R1078–R1084 (1996).
Smith, J. P., Fantaskey, A. P., Liu, G. & Zagon, I. S. Identification of gastrin as a growth peptide in human pancreatic cancer. Am. J. Physiol. 268, R135–R141 (1995).
Steinert, R. E. et al. Ghrelin, CCK, GLP-1, and PYY(3-36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol. Rev. 97, 411–463 (2017).
Carrière, C., Young, A. L., Gunn, J. R., Longnecker, D. S. & Korc, M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem. Biophys. Res. Commun. 382, 561–565 (2009).
Smith, J. P., Kramer, S. T. & Solomon, T. E. CCK stimulates growth of six human pancreatic cancer cell lines in serum-free medium. Regul. Pept. 32, 341–349 (1991).
Matters, G. L. et al. Cholecystokinin mediates progression and metastasis of pancreatic cancer associated with dietary fat. Dig. Dis. Sci. 59, 1180–1191 (2014).
Kovac, S., Xiao, L., Shulkes, A., Patel, O. & Baldwin, G. S. Gastrin increases its own synthesis in gastrointestinal cancer cells via the CCK2 receptor. FEBS Lett. 584, 4413–4418 (2010).
Matters, G. L. et al. Growth of human pancreatic cancer is inhibited by down-regulation of gastrin gene expression. Pancreas 38, e151–e161 (2009).
Dockray, G. J. Cholecystokinin. Curr. Opin. Endocrinol. Diabetes Obes. 19, 8–12 (2012).
Chung, K. M. et al. Endocrine-exocrine signaling drives obesity-associated pancreatic ductal adenocarcinoma. Cell 181, 832–847.e18 (2020).
Goetze, J. P., Nielsen, F. C., Burcharth, F. & Rehfeld, J. F. Closing the gastrin loop in pancreatic carcinoma: coexpression of gastrin and its receptor in solid human pancreatic adenocarcinoma. Cancer 88, 2487–2494 (2000).
Matters, G. L. et al. Role of endogenous cholecystokinin on growth of human pancreatic cancer. Int. J. Oncol. 38, 593–601 (2011).
Smith, J. P., Fonkoua, L. K. & Moody, T. W. The role of gastrin and CCK receptors in pancreatic cancer and other malignancies. Int. J. Biol. Sci. 12, 283–291 (2016).
Wank, S. A., Pisegna, J. R. & de Weerth, A. Cholecystokinin receptor family. Molecular cloning, structure, and functional expression in rat, guinea pig, and human. Ann. N. Y. Acad. Sci. 713, 49–66 (1994).
Smith, J. P. & Solomon, T. E. Cholecystokinin and pancreatic cancer: the chicken or the egg? Am. J. Physiol. Gastrointest. Liver Physiol. 306, G91–G101 (2014).
Smith, J. P., Hamory, M. W., Verderame, M. F. & Zagon, I. S. Quantitative analysis of gastrin mRNA and peptide in normal and cancerous human pancreas. Int. J. Mol. Med. 2, 309–315 (1998).
Smith, J. P. et al. Distribution of cholecystokinin-B receptor genotype between patients with pancreatic cancer and controls and its impact on survival. Pancreas 44, 236–242 (2015).
Berna, M. J. et al. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J. Biol. Chem. 285, 38905–38914 (2010).
Schnittert, J., Bansal, R. & Prakash, J. Targeting pancreatic stellate cells in cancer. Trends cancer 5, 128–142 (2019).
Carreras-Torres, R. et al. The role of obesity, type 2 diabetes, and metabolic factors in pancreatic cancer: a mendelian randomization study. J. Natl Cancer Inst. 109, djx012 (2017).
Stolzenberg-Solomon, R. Z. et al. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA 294, 2872–2878 (2005).
Mutgan, A. C. et al. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol. Cancer 17, 66 (2018).
Wolpin, B. M. et al. Hyperglycemia, insulin resistance, impaired pancreatic β-cell function, and risk of pancreatic cancer. J. Natl Cancer Inst. 105, 1027–1035 (2013).
Michaud, D. S. et al. Prediagnostic plasma C-peptide and pancreatic cancer risk in men and women. Cancer Epidemiol. Biomark. Prev. 16, 2101–2109 (2007).
Bailyes, E. M. et al. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochemical J. 327, 209–215 (1997).
Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928 (2008).
Brahmkhatri, V. P., Prasanna, C. & Atreya, H. S. Insulin-like growth factor system in cancer: novel targeted therapies. BioMed. Res. Int. 2015, 538019 (2015).
Malaguarnera, R. et al. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology 153, 2152–2163 (2012).
Haywood, N. J., Slater, T. A., Matthews, C. J. & Wheatcroft, S. B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol. Metab. 19, 86–96 (2019).
Yang, J. et al. Insulin promotes proliferation and fibrosing responses in activated pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G675–G687 (2016).
Douglas, J. B. et al. Serum IGF-I, IGF-II, IGFBP-3, and IGF-I/IGFBP-3 molar ratio and risk of pancreatic cancer in the prostate, lung, colorectal, and ovarian cancer screening trial. Cancer Epidemiol. Biomark. Prev. 19, 2298–2306 (2010).
Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat. Rev. Cancer 12, 159–169 (2012).
Berger, N. A. Obesity and cancer pathogenesis. Ann. N. Y. Acad. Sci. 1311, 57–76 (2014).
Majumder, K., Gupta, A., Arora, N., Singh, P. P. & Singh, S. Premorbid obesity and mortality in patients with pancreatic cancer: a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 14, 355–368.e (2016). quiz e332.
Sun, Y. et al. Role of transgelin-2 in diabetes-associated pancreatic ductal adenocarcinoma. Oncotarget 8, 49592–49604 (2017).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Kelly, D. M. & Jones, T. H. Testosterone and obesity. Obes. Rev. 16, 581–606 (2015).
Leeners, B., Geary, N., Tobler, P. N. & Asarian, L. Ovarian hormones and obesity. Hum. Reprod. Update 23, 300–321 (2017).
Escobar-Morreale, H. F., Santacruz, E., Luque-Ramírez, M. & Botella Carretero, J. I. Prevalence of ‘obesity-associated gonadal dysfunction’ in severely obese men and women and its resolution after bariatric surgery: a systematic review and meta-analysis. Hum. Reprod. Update 23, 390–408 (2017).
Peeri, N. C. et al. Association between polycystic ovary syndrome and risk of pancreatic cancer. JAMA Oncol. 8, 1845–1847 (2022).
Wittert, G. et al. Testosterone treatment to prevent or revert type 2 diabetes in men enrolled in a lifestyle programme (T4DM): a randomised, double-blind, placebo-controlled, 2-year, phase 3b trial. Lancet Diabetes Endocrinol. 9, 32–45 (2021).
Sandberg, A. A. & Rosenthal, H. E. Estrogen receptors in the pancreas. J. Steroid Biochem. 5, 969–975 (1974).
Grossman, A., Boctor, A. M., Band, P. & Lane, B. Role of steroids in secretion-modulating effect of triamcinolone and estradiol on protein synthesis and secretion from the rat exocrine pancreas. J. Steroid Biochem. 19, 1069–1081 (1983).
Tiscornia, O. M., Cresta, M. A., de Lehmann, E. S., Belardi, G. & Dreiling, D. A. Estrogen effects on exocrine pancreatic secretion in menopausal women: a hypothesis for menopause-induced chronic pancreatitis. Mt. Sinai J. Med. 53, 356–360 (1986).
Andrén-Sandberg, A., Hoem, D. & Bäckman, P. L. Other risk factors for pancreatic cancer: hormonal aspects. Ann. Oncol. 10, 131–135 (1999).
C Sumi, D. L. & Roebuck, B. D. Is pancreatic tumor growth inhibited by sex steroid hormones? An experimental study in hamster. Dig. Dis. Sci. 31, 868–870 (1986).
Sumi, C., Longnecker, D. S., Roebuck, B. D. & Brinck-Johnsen, T. Inhibitory effects of estrogen and castration on the early stage of pancreatic carcinogenesis in Fischer rats treated with azaserine. Cancer Res. 49, 2332–2336 (1989).
Barton, M. & Prossnitz, E. R. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol. Metab. 26, 185–192 (2015).
Cortes, E. et al. GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. 20, e46556 (2019).
Cortes, E. et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 20, e46557 (2019).
Natale, C. A. et al. Pharmacologic activation of the G protein–coupled estrogen receptor inhibits pancreatic ductal adenocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 10, 868–880.e861 (2020).
Corbishley, T. P., Iqbal, M. J., Wilkinson, M. L. & Williams, R. Androgen receptor in human normal and malignant pancreatic tissue and cell lines. Cancer 57, 1992–1995 (1986).
Okitsu, K. et al. Involvement of interleukin-6 and androgen receptor signaling in pancreatic cancer. Genes Cancer 1, 859–867 (2010).
Relles, D. et al. Circadian gene expression and clinicopathologic correlates in pancreatic cancer. J. Gastrointest. Surg. 17, 443–450 (2013).
Kanda, T., Jiang, X. & Yokosuka, O. Androgen receptor signaling in hepatocellular carcinoma and pancreatic cancers. World J. Gastroenterol. 20, 9229–9236 (2014).
Armstrong, H., Bording-Jorgensen, M., Dijk, S. & Wine, E. The complex interplay between chronic inflammation, the microbiome, and cancer: understanding disease progression and what we can do to prevent it. Cancers 10, 83 (2018).
McAllister, F., Khan, M. A. W., Helmink, B. & Wargo, J. A. The tumor microbiome in pancreatic cancer: bacteria and beyond. Cancer Cell 36, 577–579 (2019).
Lamont, R. J., Koo, H. & Hajishengallis, G. The oral microbiota: dynamic communities and host interactions. Nat. Rev. Microbiol. 16, 745–759 (2018).
Karpiński, T. M. Role of oral microbiota in cancer development. Microorganisms 7, 20 (2019).
Wang, Y. et al. Role of the microbiome in occurrence, development and treatment of pancreatic cancer. Mol. Cancer 18, 173 (2019).
Farrell, J. J. et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 61, 582–588 (2012).
Michaud, D. S. et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 62, 1764–1770 (2013).
Karpiński, T. M. The microbiota and pancreatic cancer. Gastroenterol. Clin. North Am. 48, 447–464 (2019).
Öğrendik, M. Periodontal pathogens in the etiology of pancreatic cancer. Gastrointest. Tumors 3, 125–127 (2017).
Mei, Q.-X. et al. Characterization of the duodenal bacterial microbiota in patients with pancreatic head cancer vs. healthy controls. Pancreatology 18, 438–445 (2018).
Ding, S.-Z., Goldberg, J. B. & Hatakeyama, M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 6, 851–862 (2010).
Pushalkar, S. et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Disco. 8, 403–416 (2018).
Li, S. et al. Pancreatic cyst fluid harbors a unique microbiome. Microbiome 5, 147 (2017).
Maekawa, T. et al. Possible involvement of Enterococcus infection in the pathogenesis of chronic pancreatitis and cancer. Biochem. Biophys. Res. Commun. 506, 962–969 (2018).
Thomas, R. M. et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 39, 1068–1078 (2018).
Li, Q., Jin, M., Liu, Y. & Jin, L. Gut microbiota: its potential roles in pancreatic cancer. Front. Cell. Infect. Microbiol. 10, 572492 (2020).
Mendez, R. et al. Microbial dysbiosis and polyamine metabolism as predictive markers for early detection of pancreatic cancer. Carcinogenesis 41, 561–570 (2020).
Aykut, B. et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574, 264–267 (2019).
Alam, A. et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell 40, 153–167.e11 (2022).
Daniluk, J. et al. An NF-κB pathway–mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Investig. 122, 1519–1528 (2012).
Redelman-Sidi, G. et al. The canonical Wnt pathway drives macropinocytosis in cancer. Cancer Res. 78, 4658–4670 (2018).
Azzam, S. K., Alsafar, H. & Sajini, A. A. FTO m6A demethylase in obesity and cancer: implications and underlying molecular mechanisms. Int. J. Mol. Sci. 23, 3800 (2022).
Cascetta, P. et al. Pancreatic cancer and obesity: molecular mechanisms of cell transformation and chemoresistance. Int. J. Mol. Sci. 19, 3331 (2018).
Pagliari, D. et al. Gut microbiota-immune system crosstalk and pancreatic disorders. Mediators Inflamm. 2018, 7946431 (2018).
Rakoff-Nahoum, S. & Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 9, 57–63 (2009).
Ghaddar, B. et al. Tumor microbiome links cellular programs and immunity in pancreatic cancer. Cancer Cell 40, 1240–1253.e5 (2022).
Rozenblatt-Rosen, O. et al. The human tumor atlas network: charting tumor transitions across space and time at single-cell resolution. Cell 181, 236–249 (2020).
Hafezi, S., Saber-Ayad, M. & Abdel-Rahman, W. M. Highlights on the role of mutations in reshaping the microenvironment of pancreatic adenocarcinoma. Int. J. Mol. Sci. 22, 10219 (2021).
Maddalena, M. et al. missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Natl Acad. Sci. USA 118, e2025631118 (2021).
Brestoff, J. R. & Artis, D. Immune regulation of metabolic homeostasis in health and disease. Cell 161, 146–160 (2015).
Crewe, C., An, Y. A. & Scherer, P. E. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 127, 74–82 (2017).
Kuroda, M. & Sakaue, H. Adipocyte death and chronic inflammation in obesity. J. Med. Investig. 64, 193–196 (2017).
Quail, D. F. & Dannenberg, A. J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 15, 139–154 (2019).
Haka, A. S. et al. Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J. Lipid Res. 57, 980–992 (2016).
van Diepen, J. A. et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 60, 1304–1313 (2017).
Maliniak, M. L. et al. Crown-like structures in breast adipose tissue: early evidence and current issues in breast cancer. Cancers 13, 2222 (2021).
Gucalp, A. et al. Periprostatic adipose inflammation is associated with high-grade prostate cancer. Prostate Cancer Prostatic Dis. 20, 418–423 (2017).
Bhardwaj, P. et al. Supplemental estrogen and caloric restriction reduce obesity-induced periprostatic white adipose inflammation in mice. Carcinogenesis 40, 914–923 (2019).
Iyengar, N. M. et al. White adipose tissue inflammation and cancer-specific survival in patients with squamous cell carcinoma of the oral tongue. Cancer 122, 3794–3802 (2016).
Cabia, B., Andrade, S., Carreira, M. C., Casanueva, F. F. & Crujeiras, A. B. A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes. Rev. 17, 361–376 (2016).
Spyrou, N., Avgerinos, K. I., Mantzoros, C. S. & Dalamaga, M. Classic and novel adipocytokines at the intersection of obesity and cancer: diagnostic and therapeutic strategies. Curr. Obes. Rep. 7, 260–275 (2018).
Chan, T. T. et al. Fatty pancreas is independently associated with subsequent diabetes mellitus development: a 10-year prospective cohort study. Clin. Gastroenterol. Hepatol. 20, 2014–2022.e4 (2021).
Renehan, A. G., Zwahlen, M. & Egger, M. Adiposity and cancer risk: new mechanistic insights from epidemiology. Nat. Rev. Cancer 15, 484–498 (2015).
Coelho, M., Oliveira, T. & Fernandes, R. Biochemistry of adipose tissue: an endocrine organ. Arch. Med. Sci. 9, 191–200 (2013).
Parida, S., Siddharth, S. & Sharma, D. Adiponectin, obesity, and cancer: clash of the bigwigs in health and disease. Int. J. Mol. Sci. 20, 2519 (2019).
Hori, M. et al. Association of pancreatic Fatty infiltration with pancreatic ductal adenocarcinoma. Clin. Transl. Gastroenterol. 5, e53 (2014).
Saisho, Y. Pancreas volume and fat deposition in diabetes and normal physiology: consideration of the interplay between endocrine and exocrine pancreas. Rev. Diabet. Stud. 13, 132–147 (2016).
Hoogenboom, S. A. et al. Pancreatic steatosis on computed tomography is an early imaging feature of pre-diagnostic pancreatic cancer: a preliminary study in overweight patients. Pancreatology 21, 428–433 (2021).
Takahashi, M. et al. Fatty pancreas: a possible risk factor for pancreatic cancer in animals and humans. Cancer Sci. 109, 3013–3023 (2018).
Rebours, V. et al. Obesity and fatty pancreatic infiltration are risk factors for pancreatic precancerous lesions (PanIN). Clin. Cancer Res. 21, 3522–3528 (2015).
Incio, J. et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Disco. 6, 852–869 (2016).
Carbone, C. et al. An angiopoietin-like protein 2 autocrine signaling promotes EMT during pancreatic ductal carcinogenesis. Oncotarget 6, 13822–13834 (2015).
Ouchi, N., Parker, J. L., Lugus, J. J. & Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97 (2011).
Gilbert, C. A. & Slingerland, J. M. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu. Rev. Med. 64, 45–57 (2013).
Naugler, W. E. & Karin, M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 14, 109–119 (2008).
Iyengar, N. M., Gucalp, A., Dannenberg, A. J. & Hudis, C. A. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J. Clin. Oncol. 34, 4270–4276 (2016).
Rosen, E. D. & Spiegelman, B. M. What we talk about when we talk about fat. Cell 156, 20–44 (2014).
Lee, J. Y., Sohn, K. H., Rhee, S. H. & Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 276, 16683–16689 (2001).
Xia, B. et al. Metabolic syndrome and risk of pancreatic cancer: a population-based prospective cohort study. Int. J. Cancer 147, 3384–3393 (2020).
Stolzenberg-Solomon, R. Z. et al. Circulating leptin and risk of pancreatic cancer: a pooled analysis from 3 cohorts. Am. J. Epidemiol. 182, 187–197 (2015).
Mace, T. A. et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 67, 320–332 (2018).
Avgerinos, K. I., Spyrou, N., Mantzoros, C. S. & Dalamaga, M. Obesity and cancer risk: emerging biological mechanisms and perspectives. Metab. Clin. Exp. 92, 121–135 (2019).
Berg, A. H. & Scherer, P. E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 96, 939–949 (2005).
Simons, P. J., van den Pangaart, P. S., van Roomen, C. P. A. A., Aerts, J. M. F. G. & Boon, L. Cytokine-mediated modulation of leptin and adiponectin secretion during in vitro adipogenesis: evidence that tumor necrosis factor-alpha- and interleukin-1beta-treated human preadipocytes are potent leptin producers. Cytokine 32, 94–103 (2005).
Stolzenberg-Solomon, R. Z. et al. Prediagnostic adiponectin concentrations and pancreatic cancer risk in male smokers. Am. J. Epidemiol. 168, 1047–1055 (2008).
Bao, Y. et al. A prospective study of plasma adiponectin and pancreatic cancer risk in five US cohorts. J. Natl Cancer Inst. 105, 95–103 (2013).
Dalamaga, M., Diakopoulos, K. N. & Mantzoros, C. S. The role of adiponectin in cancer: a review of current evidence. Endocr. Rev. 33, 547–594 (2012).
Hopkins, B. D., Goncalves, M. D. & Cantley, L. C. Obesity and cancer mechanisms: cancer metabolism. J. Clin. Oncol. 34, 4277–4283 (2016).
Jiang, J. et al. Adiponectin suppresses human pancreatic cancer growth through attenuating the β-catenin signaling pathway. Int. J. Biol. Sci. 15, 253–264 (2019).
Akimoto, M., Maruyama, R., Kawabata, Y., Tajima, Y. & Takenaga, K. Antidiabetic adiponectin receptor agonist AdipoRon suppresses tumour growth of pancreatic cancer by inducing RIPK1/ERK-dependent necroptosis. Cell Death Dis. 9, 804 (2018).
Dimou, N. L. et al. Circulating adipokine concentrations and risk of five obesity-related cancers: a Mendelian randomization study. Int. J. Cancer 148, 1625–1636 (2021).
Huang, B. et al. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1α signaling. Oncotarget 5, 4732–4745 (2014).
Denroche, H. C., Huynh, F. K. & Kieffer, T. J. The role of leptin in glucose homeostasis. J. Diabetes Investig. 3, 115–129 (2012).
Marroquí, L. et al. Role of leptin in the pancreatic β-cell: effects and signaling pathways. J. Mol. Endocrinol. 49, R9–R17 (2012).
Kieffer, T. J. & Habener, J. F. The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 278, E1–E14 (2000).
Carbone, F., La Rocca, C. & Matarese, G. Immunological functions of leptin and adiponectin. Biochimie 94, 2082–2088 (2012).
Babic, A. et al. Pancreatic cancer risk associated with prediagnostic plasma levels of leptin and leptin receptor genetic polymorphisms. Cancer Res. 76, 7160–7167 (2016).
Kadri Colakoglu, M. et al. Roles of adiponectin and leptin as diagnostic markers in pancreatic cancer. Bratisl. Lek. Listy 118, 394–398 (2017).
Gallagher, E. J. & LeRoith, D. Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol. Rev. 95, 727–748 (2015).
Harbuzariu, A. et al. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 8, 7740–7752 (2017).
Harbuzariu, A., Oprea-Ilies, G. M. & Gonzalez-Perez, R. R. The role of notch signaling and leptin-notch crosstalk in pancreatic cancer. Medicines 5, 68 (2018).
Tchio Mantho, C. I., Harbuzariu, A. & Gonzalez-Perez, R. R. Histone deacetylases, microRNA and leptin crosstalk in pancreatic cancer. World J. Clin. Oncol. 8, 178–189 (2017).
Lin, T.-C. & Hsiao, M. Leptin and cancer: updated functional roles in carcinogenesis, therapeutic niches, and developments. Int. J. Mol. Sci. 22, 2870 (2021).
Kjeldsen, L., Cowland, J. B. & Borregaard, N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim. Biophys. Acta 1482, 272–283 (2000).
Moschen, A. R., Adolph, T. E., Gerner, R. R., Wieser, V. & Tilg, H. Lipocalin-2: a master mediator of intestinal and metabolic inflammation. Trends Endocrinol. Metab. 28, 388–397 (2017).
Auguet, T. et al. Upregulation of lipocalin 2 in adipose tissues of severely obese women: positive relationship with proinflammatory cytokines. Obesity 19, 2295–2300 (2011).
Mosialou, I. et al. Lipocalin-2 counteracts metabolic dysregulation in obesity and diabetes. J. Exp. Med. 217, e20191261 (2020).
Kaur, S. et al. MUC4-mediated regulation of acute phase protein lipocalin 2 through HER2/AKT/NF-κB signaling in pancreatic cancer. Clin. Cancer Res. 20, 688–700 (2014).
Gumpper, K. et al. Lipocalin-2 expression and function in pancreatic diseases. Pancreatology 20, 419–424 (2020).
Gomez-Chou, S. B. et al. Lipocalin-2 promotes pancreatic ductal adenocarcinoma by regulating inflammation in the tumor microenvironment. Cancer Res. 77, 2647–2660 (2017).
Catalán, V. et al. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J. Mol. Med. 87, 803–813 (2009).
Steppan, C. M. et al. The hormone resistin links obesity to diabetes. Nature 409, 307–312 (2001).
Gąsiorowska, A. et al. Role of adipocytokines and its correlation with endocrine pancreatic function in patients with pancreatic cancer. Pancreatology 13, 409–414 (2013).
Zhang, M., Yan, L., Wang, G.-J. & Jin, R. Resistin effects on pancreatic cancer progression and chemoresistance are mediated through its receptors CAP1 and TLR4. J. Cell. Physiol. 234, 9457–9466 (2019).
Zhuang, H., Zhang, C. & Hou, B. GTF2IRD1 overexpression promotes tumor progression and correlates with less CD8+T cells infiltration in pancreatic cancer. Biosci. Rep. 40, BSR20202150 (2020).
Garten, A., Petzold, S., Körner, A., Imai, S.-I. & Kiess, W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol. Metab. 20, 130–138 (2009).
Chini, C. C. S. et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin. Cancer Res. 20, 120–130 (2014).
Espindola-Netto, J. M. et al. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget 8, 85054–85067 (2017).
Davis, K. et al. Nicotinamide phosphoribosyltransferase expression and clinical outcome of resected stage I/II pancreatic ductal adenocarcinoma. PLoS One 14, e0213576 (2019).
Bauer, L., Venz, S., Junker, H., Brandt, R. & Radons, J. Nicotinamide phosphoribosyltransferase and prostaglandin H2 synthase 2 are up-regulated in human pancreatic adenocarcinoma cells after stimulation with interleukin-1. Int. J. Oncol. 35, 97–107 (2009).
Kiczmer, P. et al. Serum omentin-1 and chemerin concentrations in pancreatic cancer and chronic pancreatitis. Folia Med. Cracov. 58, 77–87 (2018).
Reizes, O. & Berger, N. A. Adipocytokines, energy balance, and cancer, 1st ed., 2199–2622 (Springer, Cham, 2017).
Karabulut, S. et al. Clinical significance of serum omentin-1 levels in patients with pancreatic adenocarcinoma. BBA Clin. 6, 138–142 (2016).
Wlodarczyk, B., Gasiorowska, A., Borkowska, A. & Malecka-Panas, E. Evaluation of insulin-like growth factor (IGF-1) and retinol binding protein (RBP-4) levels in patients with newly diagnosed pancreatic adenocarcinoma (PDAC). Pancreatology 17, 623–628 (2017).
El-Mesallamy, H. O., Hamdy, N. M., Zaghloul, A. S. & Sallam, A. M. Clinical value of circulating lipocalins and insulin-like growth factor axis in pancreatic cancer diagnosis. Pancreas 42, 149–154 (2013).
Cymbaluk-Płoska, A. et al. Evaluation of biologically active substances promoting the development of or protecting against endometrial cancer. Onco Targets Ther. 11, 1363–1372 (2018).
Kahles, F., Findeisen, H. M. & Bruemmer, D. Osteopontin: a novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 3, 384–393 (2014).
Shevde, L. A. & Samant, R. S. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. J. Int. Soc. Matrix Biol. 37, 131–141 (2014).
Kaleağasıoğlu, F. & Berger, M. R. SIBLINGs and SPARC families: their emerging roles in pancreatic cancer. World J. Gastroenterol. 20, 14747–14759 (2014).
Zhivkova-Galunska, M. et al. Osteopontin but not osteonectin favors the metastatic growth of pancreatic cancer cell lines. Cancer Biol. Ther. 10, 54–64 (2010).
Rychlíková, J. et al. Osteopontin as a discriminating marker for pancreatic cancer and chronic pancreatitis. Cancer Biomarkers Sect. A Dis. Markers 17, 55–65 (2016).
Poruk, K. E. et al. Serum osteopontin and tissue inhibitor of metalloproteinase 1 as diagnostic and prognostic biomarkers for pancreatic adenocarcinoma. Pancreas 42, 193–197 (2013).
Sarosiek, K. et al. Osteopontin (OPN) isoforms, diabetes, obesity, and cancer; what is one got to do with the other? A new role for OPN. J. Gastrointest. Surg. 19, 639–650 (2015).
Ito, M. et al. High Glucose Accelerates Cell Proliferation and Increases the Secretion and mRNA Expression of Osteopontin in Human Pancreatic Duct Epithelial Cells. Int. J. Mol. Sci. 18, 807 (2017).
Cao, J. et al. Hypoxia-driven paracrine osteopontin/integrin αvβ3 signaling promotes pancreatic cancer cell epithelial-mesenchymal transition and cancer stem cell-like properties by modulating forkhead box protein M1. Mol. Oncol. 13, 228–245 (2019).
Adams, C. R. et al. Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer. eLife 8, e45313 (2019).
Jakubowska, K. et al. Expressions of matrix metalloproteinases 2, 7, and 9 in carcinogenesis of pancreatic ductal adenocarcinoma. Dis. Markers 2016, 9895721 (2016).
Sanchez-Infantes, D. et al. Oncostatin M is produced in adipose tissue and is regulated in conditions of obesity and type 2 diabetes. J. Clin. Endocrinol. Metab. 99, E217–E225 (2014).
Masjedi, A. et al. Oncostatin M: a mysterious cytokine in cancers. Int. Immunopharmacol. 90, 107158 (2021).
Torres, C. et al. Serum cytokine profile in patients with pancreatic cancer. Pancreas 43, 1042–1049 (2014).
Benson, D. D. et al. Activation state of stromal inflammatory cells in murine metastatic pancreatic adenocarcinoma. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R1067–R1075 (2012).
Richards, C. D. The enigmatic cytokine oncostatin M and roles in disease. ISRN Inflamm. 2013, 512103 (2013).
Junk, D. J. et al. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 36, 4001–4013 (2017).
Smigiel, J. M., Parameswaran, N. & Jackson, M. W. Potent EMT and CSC phenotypes are induced by oncostatin-M in pancreatic cancer. Mol. Cancer Res. 15, 478–488 (2017).
Argast, G. M. et al. Cooperative signaling between oncostatin M, hepatocyte growth factor and transforming growth factor-β enhances epithelial to mesenchymal transition in lung and pancreatic tumor models. Cells Tissues Organs 193, 114–132 (2011).
Dirat, B. A., Bochet, L., Escourrou, G., Valet, P. & Muller, C. Unraveling the obesity and breast cancer links: a role for cancer-associated adipocytes? Endocr. Dev. 19, 45–52 (2010).
Lunardi, S., Muschel, R. J. & Brunner, T. B. The stromal compartments in pancreatic cancer: are there any therapeutic targets? Cancer Lett. 343, 147–155 (2014).
Takehara, M. et al. Cancer-associated adipocytes promote pancreatic cancer progression through SAA1 expression. Cancer Sci. 111, 2883–2894 (2020).
Zoico, E. et al. Adipocytes WNT5a mediated dedifferentiation: a possible target in pancreatic cancer microenvironment. Oncotarget 7, 20223–20235 (2016).
Palm, W. & Thompson, C. B. Nutrient acquisition strategies of mammalian cells. Nature 546, 234–242 (2017).
Kuemmerle, N. B. et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 10, 427–436 (2011).
Pinnick, K. E. et al. Pancreatic ectopic fat is characterized by adipocyte infiltration and altered lipid composition. Obesity 16, 522–530 (2008).
Cai, Z. et al. Cancer‑associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 42, 2537–2549 (2019).
Sagar, G. et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 65, 1165–1174 (2016).
Nov, O. et al. Interleukin-1β regulates fat-liver crosstalk in obesity by auto-paracrine modulation of adipose tissue inflammation and expandability. PLoS One 8, e53626 (2013).
Melstrom, L. G., Salazar, M. D. & Diamond, D. J. The pancreatic cancer microenvironment: a true double agent. J. Surg. Oncol. 116, 7–15 (2017).
Uchida, C. et al. Diabetes in humans activates pancreatic stellate cells via RAGE in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 22, 11716 (2021).
Biffi, G. & Tuveson, D. A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 101, 147–176 (2021).
Ogawa, Y. et al. Three distinct stroma types in human pancreatic cancer identified by image analysis of fibroblast subpopulations and collagen. Clin. Cancer Res. 27, 107–119 (2021).
Huber, M. et al. The immune microenvironment in pancreatic cancer. Int. J. Mol. Sci. 21, 7307 (2020).
Bulle, A. & Lim, K.-H. Beyond just a tight fortress: contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct. Target. Ther. 5, 249 (2020).
Hutton, C. et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 39, 1227–1244.e20 (2021).
Mucciolo, G. et al. IL17A critically shapes the transcriptional program of fibroblasts in pancreatic cancer and switches on their protumorigenic functions. Proc. Natl Acad. Sci. 118, e2020395118 (2021).
Piersma, B., Hayward, M. K. & Weaver, V. M. Fibrosis and cancer: a strained relationship. Biochim. Biophys. Acta Rev. Cancer 1873, 188356 (2020).
Hosein, A. N., Brekken, R. A. & Maitra, A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 17, 487–505 (2020).
DelGiorno, K. E. et al. Tuft cells inhibit pancreatic tumorigenesis in mice by producing prostaglandin D. Gastroenterology 159, 1866–1881.e8 (2020).
Sun, K., Tordjman, J., Clément, K. & Scherer, P. E. Fibrosis and adipose tissue dysfunction. Cell Metab. 18, 470–477 (2013).
Hosogai, N. et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56, 901–911 (2007).
Azushima, K. et al. Adipocyte-specific enhancement of angiotensin II type 1 receptor-associated protein ameliorates diet-induced visceral obesity and insulin resistance. J. Am. Heart Assoc. 6, e004488 (2017).
Shahab, U. et al. The receptor for advanced glycation end products: a fuel to pancreatic cancer. Semin. Cancer Biol. 49, 37–43 (2018).
Lee, K. E. et al. Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Disco. 6, 256–269 (2016).
Leppänen, J. et al. Toll-like receptors 2, 4 and 9 and hypoxia markers HIF-1alpha and CAIX in pancreatic intraepithelial neoplasia. APMIS 126, 852–863 (2018).
Chen, S. et al. RER1 enhances carcinogenesis and stemness of pancreatic cancer under hypoxic environment. J. Exp. Clin. Cancer Res. 38, 15 (2019).
McGinn, O. et al. Inhibition of hypoxic response decreases stemness and reduces tumorigenic signaling due to impaired assembly of HIF1 transcription complex in pancreatic cancer. Sci. Rep. 7, 7872 (2017).
Forsmark, C. E. Incretins, diabetes, pancreatitis and pancreatic cancer: what the GI specialist needs to know. Pancreatology 16, 10–13 (2016).
Kopp, J. L., Grompe, M. & Sander, M. Stem cells versus plasticity in liver and pancreas regeneration. Nat. Cell Biol. 18, 238–245 (2016).
Krah, N. M. & Murtaugh, L. C. Differentiation and inflammation: ‘best enemies’ in gastrointestinal. Carcinog. Trends Cancer 2, 723–735 (2016).
Poggetto, E. D. et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 373, eabj0486 (2021).
Jain, R. K. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J. Clin. Oncol. 31, 2205–2218 (2013).
Shebl, F. M. et al. Non-steroidal anti-inflammatory drugs use is associated with reduced risk of inflammation-associated cancers: NIH-AARP study. PLoS One 9, e114633 (2014).
Melisi, D. & Chiao, P. J. NF-kappa B as a target for cancer therapy. Expert Opin. Ther. Targets 11, 133–144 (2007).
Dai, J.-J., Jiang, M.-J., Wang, X.-P. & Tian, L. Inflammation-related pancreatic carcinogenesis: mechanisms and clinical potentials in advances. Pancreas 46, 973–985 (2017).
Colotta, F., Allavena, P., Sica, A., Garlanda, C. & Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081 (2009).
Shin, C. H. & Choi, D.-S. Essential roles for the non-canonical IκB kinases in linking inflammation to cancer, obesity, and diabetes. Cells 8, 178 (2019).
Wang, D. J., Ratnam, N. M., Byrd, J. C. & Guttridge, D. C. NF-κB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 9, 90–103 (2014).
Lesina, M. et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J. Clin. Investig. 126, 2919–2932 (2016).
Daniluk, J. et al. An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Investig. 122, 1519–1528 (2012).
Garg, B. et al. NFκB in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via up-regulation of CXCL12. Gastroenterology 155, 880–891.e8 (2018).
Ling, J. et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 21, 105–120 (2012).
Niu, J., Li, Z., Peng, B. & Chiao, P. J. Identification of an autoregulatory feedback pathway involving interleukin-1alpha in induction of constitutive NF-kappaB activation in pancreatic cancer cells. J. Biol. Chem. 279, 16452–16462 (2004).
Hayden, M. S. & Ghosh, S. Shared principles in NF-kappaB signaling. Cell 132, 344–362 (2008).
Qian, W. et al. Resveratrol slows the tumourigenesis of pancreatic cancer by inhibiting NFκB activation. Biomed. Pharmacother. 127, 110116 (2020).
Mueller, S. et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 554, 62–68 (2018).
Baumgart, S., Ellenrieder, V. & Fernandez-Zapico, M. E. Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis. Gut 62, 310–316 (2013).
Jagadeeshan, S. et al. Transcriptional regulation of fibronectin by p21-activated kinase-1 modulates pancreatic tumorigenesis. Oncogene 34, 455–464 (2015).
Wang, X. et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) as a novel diagnostic serum biomarker in pancreatic ductal adenocarcinoma. BMC Cancer 14, 578 (2014).
Lagunas, L. & Clipstone, N. A. Deregulated NFATc1 activity transforms murine fibroblasts via an autocrine growth factor-mediated Stat3-dependent pathway. J. Cell. Biochem. 108, 237–248 (2009).
König, A., Fernandez-Zapico, M. E. & Ellenrieder, V. Primers on molecular pathways-the NFAT transcription pathway in pancreatic cancer. Pancreatology 10, 416–422 (2010).
Nolan-Stevaux, O. et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 23, 24–36 (2009).
Dennler, S. et al. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 67, 6981–6986 (2007).
Drosos, Y. et al. Prox1-heterozygosis sensitizes the pancreas to oncogenic Kras-induced neoplastic transformation. Neoplasia 18, 172–184 (2016).
Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 4, 11–22 (2004).
Kang, X., Lin, Z., Xu, M., Pan, J. & Wang, Z. W. Deciphering role of FGFR signalling pathway in pancreatic cancer. Cell Prolif. 52, e12605 (2019).
Visser, C. J. et al. Transforming growth factor-alpha and epidermal growth factor expression in the exocrine pancreas of azaserine-treated rats: modulation by cholecystokinin or a low fat, high fiber (caloric restricted) diet. Carcinogenesis 16, 2075–2082 (1995).
Shen, J. et al. GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, signaling, and mutant Kras-driven pancreatic tumorigenesis in mice. Proc. Natl Acad. Sci. USA 114, E4020–e4029 (2017).
Kibe, S. et al. Cancer-associated acinar-to-ductal metaplasia within the invasive front of pancreatic cancer contributes to local invasion. Cancer Lett. 444, 70–81 (2019).
Wagner, M., Lührs, H., Klöppel, G., Adler, G. & Schmid, R. M. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology 115, 1254–1262 (1998).
Liou, G. Y. et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 6, 6200 (2015).
Garcia-Carracedo, D. et al. Smad4 loss synergizes with TGFα overexpression in promoting pancreatic metaplasia, PanIN development, and fibrosis. PLoS One 10, e0120851 (2015).
Shadhu, K. & Xi, C. Inflammation and pancreatic cancer: an updated review. Saudi J. Gastroenterol. 25, 3–13 (2019).
Apte, R. S., Chen, D. S. & Ferrara, N. VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264 (2019).
Matsuo, Y. et al. K-Ras promotes angiogenesis mediated by immortalized human pancreatic epithelial cells through mitogen-activated protein kinase signaling pathways. Mol. Cancer Res. 7, 799–808 (2009).
Bergers, G. et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744 (2000).
Yabusaki, N. et al. A vascular endothelial growth factor gene polymorphism predicts malignant potential in intraductal papillary mucinous neoplasm. Pancreas 44, 608–614 (2015).
Zhang, Q. et al. Expression of the PTEN/FOXO3a/PLZF signalling pathway in pancreatic cancer and its significance in tumourigenesis and progression. Invest. N. Drugs 38, 321–328 (2020).
Principe, D. R. et al. PEDF inhibits pancreatic tumorigenesis by attenuating the fibro-inflammatory reaction. Oncotarget 7, 28218–28234 (2016).
Seifert, L. et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249 (2016).
Ochi, A. et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J. Clin. Investig. 122, 4118–4129 (2012).
Das, S., Shapiro, B., Vucic, E. A., Vogt, S. & Bar-Sagi, D. Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 80, 1088–1101 (2020).
Pradere, J. P., Dapito, D. H. & Schwabe, R. F. The Yin and Yang of Toll-like receptors in cancer. Oncogene 33, 3485–3495 (2014).
Zambirinis, C. P. et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 212, 2077–2094 (2015).
Denou, E. et al. Defective NOD2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol. Med. 7, 259–274 (2015).
Chiblak, S. et al. K-Ras and cyclooxygenase-2 coactivation augments intraductal papillary mucinous neoplasm and Notch1 mimicking human pancreas lesions. Sci. Rep. 6, 29455 (2016).
Furukawa, F. et al. A cyclooxygenase-2 inhibitor, nimesulide, inhibits postinitiation phase of N-nitrosobis(2-oxopropyl)amine-induced pancreatic carcinogenesis in hamsters. Int. J. Cancer 104, 269–273 (2003).
Sato, N. et al. The COX-2/PGE pathway suppresses apical elimination of RasV12-transformed cells from epithelia. Commun. Biol. 3, 132 (2020).
Sanchez-Pino, M. D., Gilmore, L. A., Ochoa, A. C. & Brown, J. C. Obesity-associated myeloid immunosuppressive cells, key players in cancer risk and response to immunotherapy. Obesity 29, 944–953 (2021).
Grant, R. W. & Dixit, V. D. Adipose tissue as an immunological organ. Obesity 23, 512–518 (2015).
Daley, D. et al. γδ T cells support pancreatic oncogenesis by restraining αβ T cell activation. Cell 166, 1485–1499.e15 (2016).
Touch, S., Clément, K. & André, S. T cell populations and functions are altered in human obesity and type 2 diabetes. Curr. Diab. Rep. 17, 81 (2017).
Zhang, Y. et al. CD4+T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol. Res. 2, 423–435 (2014).
Zhang, Y. et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 66, 124–136 (2017).
Pylayeva-Gupta, Y., Lee, K. E., Hajdu, C. H., Miller, G. & Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847 (2012).
Liu, X. et al. Acceleration of pancreatic tumorigenesis under immunosuppressive microenvironment induced by Reg3g overexpression. Cell Death Dis. 8, e3033–e3033 (2017).
Burks, J., Fleury, A., Livingston, S. & Smith, J. P. ISG15 pathway knockdown reverses pancreatic cancer cell transformation and decreases murine pancreatic tumor growth via downregulation of PDL-1 expression. Cancer Immunol. Immunother. 68, 2029–2039 (2019).
Zhang, Y. et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Disco. 10, 422–439 (2020).
Pylayeva-Gupta, Y. et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Disco. 6, 247–255 (2016).
Das, S. & Bar-Sagi, D. BTK signaling drives CD1dhiCD5+regulatory B-cell differentiation to promote pancreatic carcinogenesis. Oncogene 38, 3316–3324 (2019).
Yoshida, Y. et al. Impaired tumorigenicity of human pancreatic cancer cells retrovirally transduced with interleukin-12 or interleukin-15 gene. Cancer Gene Ther. 7, 324–331 (2000).
Melzer, M. K. et al. An immunological glance on pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 21, 3345 (2020).
Muller, M. et al. The immune landscape of human pancreatic ductal carcinoma: key players, clinical implications, and challenges. Cancers 14, 995 (2022).
Lin, J. H. et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J. Exp. Med. 217, e20190673 (2020).
Hiraoka, N. et al. CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology 140, 310–321 (2011).
Talukdar, S. et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 18, 1407–1412 (2012).
Coffelt, S. B., Wellenstein, M. D. & de Visser, K. E. Neutrophils in cancer: neutral no more. Nat. Rev. Cancer 16, 431–446 (2016).
Mukaida, N., Sasaki, S.-i & Baba, T. Two-faced roles of tumor-associated neutrophils in cancer development and progression. Int. J. Mol. Sci. 21, 3457 (2020).
Jin, L., Kim, H. S. & Shi, J. Neutrophil in the pancreatic tumor microenvironment. Biomolecules 11, 1170 (2021).
Munir, H. et al. Stromal-driven and Amyloid β-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat. Commun. 12, 683 (2021).
Marone, G. et al. Is there a role for basophils in cancer? Front. Immunol. 11, 2103 (2020).
Yang, J., Li, Y., Sun, Z. & Zhan, H. Macrophages in pancreatic cancer: an immunometabolic perspective. Cancer Lett. 498, 188–200 (2021).
Dai, E. et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat. Commun. 11, 6339 (2020).
Otto, L. et al. Initiation of pancreatic cancer: the interplay of hyperglycemia and macrophages promotes the acquisition of malignancy-associated properties in pancreatic ductal epithelial cells. Int. J. Mol. Sci. 22, 5086 (2021).
Helm, O. et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 135, 843–861 (2014).
Bishehsari, F. et al. KRAS mutation and epithelial-macrophage interplay in pancreatic neoplastic transformation. Int. J. Cancer 143, 1994–2007 (2018).
Dai, E. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 16, 2069–2083 (2020).
Dawson, D. W. et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev. Res. 6, 1064–1073 (2013).
Feng, R. et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal 16, 54 (2018).
Aller, M.-A., Arias, A., Arias, J.-I. & Arias, J. Carcinogenesis: the cancer cell–mast cell connection. Inflamm. Res. 68, 103–116 (2019).
Soucek, L. et al. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 13, 1211–1218 (2007).
Theoharides, T. C. Mast cells and pancreatic cancer. N. Engl. J. Med. 358, 1860–1861 (2008).
Longo, V. et al. Mast cells and angiogenesis in pancreatic ductal adenocarcinoma. Clin. Exp. Med. 18, 319–323 (2018).
Komi, D. E. A. & Redegeld, F. A. Role of mast cells in shaping the tumor microenvironment. Clin. Rev. Allergy Immunol. 58, 313–325 (2020).
Hoffman, M. T. et al. The gustatory sensory G-protein GNAT3 suppresses pancreatic cancer progression in mice. Cell Mol. Gastroenterol. Hepatol. 11, 349–369 (2021).
Eibl, G. & Rozengurt, E. KRAS, YAP, and obesity in pancreatic cancer: a signaling network with multiple loops. Semin. Cancer Biol. 54, 50–62 (2019).
Ma, P. et al. Circulating myeloid derived suppressor cells (MDSC) that accumulate in premalignancy share phenotypic and functional characteristics with MDSC in cancer. Front. Immunol. 10, 1401 (2019).
Zhang, Y., Sowers, J. R. & Ren, J. Targeting autophagy in obesity: from pathophysiology to management. Nat. Rev. Endocrinol. 14, 356–376 (2018).
Kim, K. H. & Lee, M. S. Autophagy-a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 10, 322–337 (2014).
Cheng, Y., Ren, X., Hait, W. N. & Yang, J. M. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol. Rev. 65, 1162–1197 (2013).
Jansen, H. J. et al. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 153, 5866–5874 (2012).
Usman, R. M. et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac. J. Clin. Oncol. 17, 193–208 (2021).
Aghajan, M., Li, N. & Karin, M. Obesity, autophagy and the pathogenesis of liver and pancreatic cancers. J. Gastroenterol. Hepatol. 27, 10–14 (2012).
Kang, R., Tang, D., Lotze, M. T. & Zeh, H. J. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 8, 989–991 (2012).
Görgülü, K. et al. Levels of the autophagy-related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology 156, 203–217.e220 (2019).
Wang, Y., Qin, C., Yang, G., Zhao, B. & Wang, W. The role of autophagy in pancreatic cancer progression. Biochim. Biophys. Acta 1876, 188592 (2021).
Yang, A. et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Disco. 4, 905–913 (2014).
Manent, J. et al. Autophagy suppresses Ras-driven epithelial tumourigenesis by limiting the accumulation of reactive oxygen species. Oncogene 36, 5576–5592 (2017).
Balkwill, F., Charles, K. A. & Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7, 211–217 (2005).
Yang, L. et al. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506 (2015).
Hetz, C., Chevet, E. & Oakes, S. A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838 (2015).
Hess, D. A. et al. Extensive pancreas regeneration following acinar-specific disruption of Xbp1 in mice. Gastroenterology 141, 1463–1472 (2011).
Dumartin, L. et al. ER stress protein AGR2 precedes and is involved in the regulation of pancreatic cancer initiation. Oncogene 36, 3094–3103 (2017).
Zhang, J. et al. Endoplasmic Reticulum stress-dependent expression of ERO1L promotes aerobic glycolysis in pancreatic cancer. Theranostics 10, 8400–8414 (2020).
Clarke, H. J., Chambers, Joseph, E., Liniker, E. & Marciniak, S. J. Endoplasmic reticulum stress in malignancy. Cancer Cell 25, 563–573 (2014).
Mohamed, E., Cao, Y. & Rodriguez, P. C. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: a promising opportunity for cancer immunotherapy. Cancer Immunol. immunotherapy CII 66, 1069–1078 (2017).
Hursting, S. D. & Dunlap, S. M. Obesity, metabolic dysregulation, and cancer: a growing concern and an inflammatory (and microenvironmental) issue. Ann. N. Y. Acad. Sci. 1271, 82–87 (2012).
Zhang, W. et al. Liquid biopsy for cancer: circulating tumor cells, circulating free DNA or exosomes? Cell. Physiol. Biochem. 41, 755–768 (2017).
Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 126, 1208–1215 (2016).
Sun, W., Ren, Y., Lu, Z. & Zhao, X. The potential roles of exosomes in pancreatic cancer initiation and metastasis. Mol. Cancer 19, 135 (2020).
Kamerkar, S. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503 (2017).
Pardo, F., Villalobos-Labra, R., Sobrevia, B., Toledo, F. & Sobrevia, L. Extracellular vesicles in obesity and diabetes mellitus. Mol. Asp. Med. 60, 81–91 (2018).
Deng, Z.-b et al. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58, 2498–2505 (2009).
Gozal, D., Ham, S. A. & Mokhlesi, B. Sleep apnea and cancer: analysis of a nationwide population sample. Sleep 39, 1493–1500 (2016).
Dal Molin, M. et al. Obstructive sleep apnea and pathological characteristics of resected pancreatic ductal adenocarcinoma. PLoS One 11, e0164195 (2016).
Jin, G., Hong, W., Guo, Y., Bai, Y. & Chen, B. Molecular mechanism of pancreatic stellate cells activation in chronic pancreatitis and pancreatic cancer. J. Cancer 11, 1505–1515 (2020).
Ye, Z. et al. Hsa_circ_0000069 knockdown inhibits tumorigenesis and exosomes with downregulated hsa_circ_0000069 suppress malignant transformation via inhibition of STIL in pancreatic cancer. Int. J. Nanomed. 15, 9859–9873 (2020).
Stefanius, K. et al. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. eLife 8, e40226 (2019).
Servage, K. A., Stefanius, K., Gray, H. F. & Orth, K. Proteomic profiling of small extracellular vesicles secreted by human pancreatic cancer cells implicated in cellular transformation. Sci. Rep. 10, 7713 (2020).
Whiteside, T. L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 126, 1216–1223 (2016).
Zhou, M. et al. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell. Immunol. 292, 65–69 (2014).
Ding, G. et al. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget 6, 29877–29888 (2015).
Capello, M. et al. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat. Commun. 10, 254 (2019).
Basso, D. et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a -dependent manner through a new calcium related axis. Oncotarget 8, 84928–84944 (2017).
Zech, D., Rana, S., Büchler, M. W. & Zöller, M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun. Signal. CCS 10, 37 (2012).
Zhang, Y. et al. Upregulation of antioxidant capacity and nucleotide precursor availability suffices for oncogenic transformation. Cell Metab. 33, 94–109.e8 (2021).
Cross, C. E. Oxygen radicals and human disease. Ann. Intern. Med. 107, 526–545 (1987).
Cheung, E. C. et al. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 37, 168–182.e4 (2020).
Chio, I. I. C. & Tuveson, D. A. ROS in cancer: the burning question. Trends Mol. Med. 23, 411–429 (2017).
Ames, B. N., Shigenaga, M. K. & Hagen, T. M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl Acad. Sci. USA 90, 7915–7922 (1993).
Shibutani, S., Takeshita, M. & Grollman, A. P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349, 431–434 (1991).
Szatrowski, T. P. & Nathan, C. F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 51, 794–798 (1991).
Sabharwal, S. S. & Schumacker, P. T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 14, 709–721 (2014).
Chio, I. I. C. et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 166, 963–976 (2016).
Cullen, J. J. et al. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res. 63, 1297–1303 (2003).
Chandel, N. S. & Tuveson, D. A. The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 371, 177–178 (2014).
Bjelakovic, G., Nikolova, D., Gluud, L. L., Simonetti, R. G. & Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA 297, 842–857 (2007).
Baslan, T. et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 608, 795–802 (2022).
Tjomsland, V. et al. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 13, 664–675 (2011).
Skaug, B., Jiang, X. & Chen, Z. J. The role of ubiquitin in NF-kappaB regulatory pathways. Annu. Rev. Biochem. 78, 769–796 (2009).
Apte, R. N. et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 25, 387–408 (2006).
Melisi, D. et al. Secreted interleukin-1alpha induces a metastatic phenotype in pancreatic cancer by sustaining a constitutive activation of nuclear factor-kappaB. Mol. cancer Res. MCR 7, 624–633 (2009).
Briukhovetska, D. et al. Interleukins in cancer: from biology to therapy. Nat. Rev. Cancer 21, 481–499 (2021).
Dinarello, C. A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27 (2018).
O’Neill, C. M. et al. Circulating levels of IL-1B+IL-6 cause ER stress and dysfunction in islets from prediabetic male mice. Endocrinology 154, 3077–3088 (2013).
Levine, B., Mizushima, N. & Virgin, H. W. Autophagy in immunity and inflammation. Nature 469, 323–335 (2011).
Ochi, A. et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 209, 1671–1687 (2012).
Daley, D. et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 214, 1711–1724 (2017).
Bent, R., Moll, L., Grabbe, S. & Bros, M. Interleukin-1 Beta-A friend or foe in malignancies? Int. J. Mol. Sci. 19, 2155 (2018).
Saijo, Y. et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. J. Immunol. 169, 469–475 (2002).
Bunt, S. K., Sinha, P., Clements, V. K., Leips, J. & Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 176, 284–290 (2006).
Mantovani, A., Barajon, I. & Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 281, 57–61 (2018).
Mantovani, A., Dinarello, C. A., Molgora, M. & Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 50, 778–795 (2019).
Alonso-Curbelo, D. et al. A gene-environment-induced epigenetic program initiates tumorigenesis. Nature 590, 642–648 (2021).
Liew, F. Y., Girard, J.-P. & Turnquist, H. R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 16, 676–689 (2016).
Todaro, M. et al. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 15, 762–772 (2008).
Prokopchuk, O., Liu, Y., Henne-Bruns, D. & Kornmann, M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: evidence for autocrine and paracrine actions. Br. J. Cancer 92, 921–928 (2005).
Traub, B. et al. Endogenously expressed IL-4Rα promotes the malignant phenotype of human pancreatic cancer in vitro and in vivo. Int. J. Mol. Sci. 18, 716 (2017).
Akdis, M. et al. Interleukins, from 1 to 37, and interferon-γ: receptors, functions, and roles in diseases. J. allergy Clin. Immunol. 127, 701–21.e1-70 (2011).
Shi, J., Song, X., Traub, B., Luxenhofer, M. & Kornmann, M. Involvement of IL-4, IL-13 and their receptors in pancreatic cancer. Int. J. Mol. Sci. 22, 2998 (2021).
Junttila, I. S. Tuning the cytokine responses: an update on Interleukin (IL)-4 and IL-13 receptor complexes. Front. Immunol. 9, 888 (2018).
Opal, S. M. & DePalo, V. A. Anti-inflammatory cytokines. Chest 117, 1162–1172 (2000).
Suzuki, A., Leland, P., Joshi, B. H. & Puri, R. K. Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine 75, 79–88 (2015).
Gitto, S. B. et al. Identification of a novel IL-5 signaling pathway in chronic pancreatitis and crosstalk with pancreatic tumor cells. Cell Commun. Signal. CCS 18, 95 (2020).
Dougan, M., Dranoff, G. & Dougan, S. K. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity 50, 796–811 (2019).
Grisaru-Tal, S., Itan, M., Klion, A. D. & Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 20, 594–607 (2020).
Kang, R. et al. Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Res. 27, 916–932 (2017).
Huang, L. et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J. Exp. Clin. Cancer Res. CR 35, 27 (2016).
Mills, L. D. et al. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem. 288, 11786–11794 (2013).
Xing, H.-B. et al. Suppression of gene by shRNA augments gemcitabine chemosensitization in pancreatic adenocarcinoma cells. BioMed. Res. Int. 2018, 3195025 (2018).
Lesina, M. et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19, 456–469 (2011).
Holmer, R., Goumas, F. A., Waetzig, G. H., Rose-John, S. & Kalthoff, H. Interleukin-6: a villain in the drama of pancreatic cancer development and progression. Hepatobiliary Pancreat. Dis. INT. HBPD INT 13, 371–380 (2014).
Guo, Y., Xu, F., Lu, T., Duan, Z. & Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 38, 904–910 (2012).
Rose-John, S. Interleukin-6 family cytokines. Cold Spring Harb. Perspect. Biol. 10, a028415 (2018).
Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 33, 127–148 (2021).
Perusina Lanfranca, M. et al. Interleukin 22 signaling regulates acinar cell plasticity to promote pancreatic tumor development in mice. Gastroenterology 158, 1417–1432.e11 (2020).
He, W. et al. IL22RA1/STAT3 signaling promotes stemness and tumorigenicity in pancreatic cancer. Cancer Res 78, 3293–3305 (2018).
Sabat, R., Ouyang, W. & Wolk, K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat. Rev. Drug Discov. 13, 21–38 (2014).
Markota, A. & Endres, S. & Kobold, S. Targeting interleukin-22 for cancer therapy. Hum. Vaccin. Immunother. 14, 2012–2015 (2018).
Lim, C. & Savan, R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev. 25, 257–271 (2014).
Huber, S. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263 (2012).
Ouyang, W. & O’Garra, A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity 50, 871–891 (2019).
Ouyang, W., Rutz, S., Crellin, N. K., Valdez, P. A. & Hymowitz, S. G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 29, 71–109 (2011).
Yazdani, Z. et al. IL-35, a double-edged sword in cancer. J. Cell. Biochem. 121, 2064–2076 (2020).
Das, S. & Bar-Sagi, D. BTK signaling drives CD1dCD5 regulatory B-cell differentiation to promote pancreatic carcinogenesis. Oncogene 38, 3316–3324 (2019).
Loncle, C. et al. IL17 functions through the novel REG3β-JAK2-STAT3 inflammatory pathway to promote the transition from chronic pancreatitis to pancreatic cancer. Cancer Res. 75, 4852–4862 (2015).
McAllister, F. et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25, 621–637 (2014).
Gu, C., Wu, L. & Li, X. IL-17 family: cytokines, receptors and signaling. Cytokine 64, 477–485 (2013).
Cua, D. J. & Tato, C. M. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10, 479–489 (2010).
Amatya, N., Garg, A. V. & Gaffen, S. L. IL-17 signaling: the Yin and the Yang. Trends Immunol. 38, 310–322 (2017).
McGeachy, M. J., Cua, D. J. & Gaffen, S. L. The IL-17 family of cytokines in health and disease. Immunity 50, 892–906 (2019).
Hill, K. S. et al. Met receptor tyrosine kinase signaling induces secretion of the angiogenic chemokine interleukin-8/CXCL8 in pancreatic cancer. PLoS One 7, e40420 (2012).
Matsuo, Y. et al. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 124, 853–861 (2009).
Petreaca, M. L., Yao, M., Liu, Y., Defea, K. & Martins-Green, M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol. Biol. Cell 18, 5014–5023 (2007).
Waugh, D. J. J. & Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 14, 6735–6741 (2008).
Russo, R. C., Garcia, C. C., Teixeira, M. M. & Amaral, F. A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 10, 593–619 (2014).
Liu, Q. et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 31, 61–71 (2016).
Acknowledgements
We regretfully failed to cite many critical and excellent original studies related to the topic of this article due to format constraints. Beyond our cordial apology, we sincerely hope the reviews we cited can make it easier for these studies to be discovered by interested readers. All figures were created on BioRender.com with permission for publication.
Funding
Research work related to the topic of this article that is currently being carried out in the authors’ laboratories is supported in whole or in part by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS, 2021-I2M-1-002), National High Level Hospital Clinical Research Funding (2022-PUMCH-D-001), the National Natural Science Foundation of China (NSFC, 81970763), the Foundation Project for Young Scientists of NSFC (82102810), the Fellowship of China Postdoctoral Science Foundation (2021M700501), the Postdoctoral Science Foundation of Peking Union Medical College Hospital (2022T150067), and a Nonprofit Central Research Institute Fund of CAMS (2018PT32014).
Author information
Authors and Affiliations
Contributions
R.R. and C.W. conceptualized and designed the article. R.R., J.S., and X.Y. prepared the initial manuscript. Y.C. and R.X. performed literature categorization and helped with the illustrations. C.W. and Y.Z. critically reviewed and supervised the revision of the manuscript. All authors have read and approved the final version of the submitted and published article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors agree to publish the article.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruze, R., Song, J., Yin, X. et al. Mechanisms of obesity- and diabetes mellitus-related pancreatic carcinogenesis: a comprehensive and systematic review. Sig Transduct Target Ther 8, 139 (2023). https://doi.org/10.1038/s41392-023-01376-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01376-w
- Springer Nature Limited
This article is cited by
-
Emerging mechanisms and promising approaches in pancreatic cancer metabolism
Cell Death & Disease (2024)
-
Evaluation of pancreatic iodine uptake and related influential factors in multiphase dual-energy CT
European Radiology (2024)