
Abstract
Amyloid β protein (Aβ) is the main component of neuritic plaques in Alzheimer’s disease (AD), and its accumulation has been considered as the molecular driver of Alzheimer’s pathogenesis and progression. Aβ has been the prime target for the development of AD therapy. However, the repeated failures of Aβ-targeted clinical trials have cast considerable doubt on the amyloid cascade hypothesis and whether the development of Alzheimer’s drug has followed the correct course. However, the recent successes of Aβ targeted trials have assuaged those doubts. In this review, we discussed the evolution of the amyloid cascade hypothesis over the last 30 years and summarized its application in Alzheimer’s diagnosis and modification. In particular, we extensively discussed the pitfalls, promises and important unanswered questions regarding the current anti-Aβ therapy, as well as strategies for further study and development of more feasible Aβ-targeted approaches in the optimization of AD prevention and treatment.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder leading to progressive cognitive decline with pathological hallmarks of senile plaque and neurofibrillary tangle formation in the brain. In 1984, Glenner & Wong discovered that the amyloid β protein (Aβ) is the central component of extracellular amyloid plaques in AD.1 Since then, Aβ has been considered as a driver of Alzheimer’s pathological processes and the “amyloid cascade hypothesis” has become a leading theory of AD pathogenesis.2 Over the past decades, targeting Aβ has been the main direction of developing AD treatment.3,4,5,6 However, the repetitive failures of Aβ-targeted clinical trials have cast considerable doubt on this hypothesis. Anti-Aβ therapy has now become a significant controversy in AD drug development and treatment.
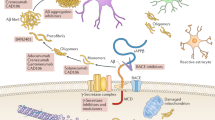
Aβ is generated from the amyloid precursor protein (APP) by sequential cleavage of β- and γ-secretase. However, the non-amyloidogenic pathway is the predominant pathway in vivo.7 APP is mostly cleaved first by α-secretase within Aβ domain at the Aβ Leu17 site in the non-amyloidogenic pathway, generating a secreted form of APP (sAPPα) and an 83-amino acid membrane-bound C-terminal fragment (CTF) C83, thus precluding Aβ production. The beta site APP cleaving enzyme 1 (BACE1), the β-secretase, and its homolog BACE2, the θ-secretase, also contribute to the non-amyloidogenic pathway.7,8 Under physiological conditions, BACE1 predominantly processes APP at the Aβ Glu11 β-secretase site to generate C89, and γ-secretase cleaves C89 to produce a truncated Aβ11-40.7,8 BACE2 cleaves APP at the Aβ Phe20 θ-secretase site to generate C80 and precludes Aβ generation.9,10,11 Two enzymatic cleavages of APP by BACE1 and γ-secretase are required to produce Aβ in the amyloidogenic pathway. BACE1 first cleaves APP at the Asp1 site to generate sAPPβ and C99. Subsequently, γ-secretase cleaves C99 to release Aβ and CTFγ. γ-secretase is a presenilins 1 (PS1)-containing macromolecular complex12,13,14,15,16 and this high molecular weight complex also requires nicastrin, anterior pharynx-defective 1, and PEN-2 for its enzymatic activity17,18 (Fig. 1).

Amyloidogenic and non-amyloidogenic processing pathways of APP. In the amyloidogenic pathway, BACE1 first cleaves APP at the Asp1 site to generate sAPPβ and a 99-amino acid membrane-bound C-terminal fragment (CTF) C99. Subsequently, γ-secretase cleaves C99 to release Aβ and CTFγ. Under physiological conditions (non-amyloidogenic pathways), APP is mostly cleaved first by α-secretase within Aβ domain at the Aβ Leu17 site, generating a secreted form of APP (sAPPα) and an 83-amino acid membrane-bound C-terminal fragment (CTF) C83, thus precluding Aβ production; BACE1 predominantly processes APP at the Aβ Glu11 β-secretase site to generate C89, and γ-secretase cleaves C89 to produce a truncated Aβ11-40; BACE2 cleaves APP at the Aβ Phe20 θ-secretase site to generate C80 and precludes Aβ generation. APP amyloid precursor protein, BACE1 β-site APP-cleaving enzyme 1, sAPP secreted APP, CTF C-terminal fragment, Aβ amyloid-β, tAβ truncated amyloid-β, BACE2 β-site APP-cleaving enzyme 2
The balance between continual Aβ generation and efficient clearance is important for Aβ homeostasis to prevent its toxic aggregation into misfolded assemblies.19 Similar to other brain metabolites, Aβ clearance depends on different pathways including enzyme degradation, crossing the blood–brain barrier (BBB), interstitial fluid (ISF) bulk-flow and CSF absorption.19,20 The BBB is composed of endothelial cells connected by tight junctions to form a selectively permeable system.21 The transport of soluble Aβ across brain endothelial cells to the peripheral circulation is mainly via low density lipoprotein receptor-related protein 1(LRP-1) and ABC transporter sub-family A and B member 1 (ABCA1 and ABCB1),22,23 while receptors for advanced glycosylation end-products (RAGE) is responsible for circulating Aβ entering into the brain.24 It has been identified that the expressions of the two blood efflux transporters LRP1 and ABCB1 were reduced during AD, whereas the expression of the blood influx transporter RAGE is elevated.21,25 The perivascular drainage pathway plays a vital role in ISF bulk-flow clearance of Aβ.26 Failure of perivascular drainage of Aβ altered Aβ homeostasis associated with synaptic dysfunction and cognitive impairment, leading to the development of AD.27 CSF absorption clearance of Aβ depends on factors including CSF production by the choroid plexus, integrity of the blood-CSF barrier, relevant transporters and CSF lymphatic absorption.28 In AD, the structural integrity of the blood-CSF barrier is destroyed, resulting in aberrant Aβ clearance.29 Enzymatic pathways for Aβ degradation include the zinc metalloendopeptidases, insulin-degrading enzyme (IDE), matrix metalloproteinase (MMPs), angiotensin converting enzyme (ACE), and endothelin-converting enzyme (ECE), serine proteases, cystein proteases, and kallikrein-related peptidase 7.30,31 In the hippocampus of AD patients, the enzymes IDE, ACE and NEP had decreased activity.30 AD model mice also showed the impaired Aβ degradation system.21,32 In GWAS, many genetic risk factors for AD (e.g. RIN3, CLU and PTK2B) are linked to Aβ degradation.33,34
Extensive genetic studies have supported the causative role of Aβ accumulation in AD pathogenesis. Down syndrome (DS) patients with trisomy-21 having extra copy of APP gene develop typical Alzheimer’s neuropathology including amyloid plaques and neurofibrillary tangles.35,36,37 Mutations in APP, presenilin 1 (PSEN1) and PSEN2 genes that increase Aβ production, elevate Aβ42/Aβ40 ratio and promote plaque formation cause autosomal dominant early-onset familial AD (FAD), implicating a role of altering APP processing in AD pathogenesis.7,38,39 In contrast, an APP mutation identified in the Icelandic population reduces Aβ production, leading to protection against cognitive decline in the elderly.40 Both genetic (e.g., ApoE4, TREM2) and non-genetic (e.g. diabetes, obesity, stroke, or physical inactivity) risk factors for late-onset sporadic Alzheimer’s disease (SAD) have also been identified to increase Aβ generation and/or reduce Aβ clearance for its accumulation.4,41,42,43,44,45 These studies suggest that Aβ accumulation drives disease progression in both FAD and SAD and thus illustrates why clinical trials involving anti-Aβ therapies have garnered so much attention in the Alzheimer’s community.
Recently, Aβ-based therapy has received encouraging results. Aducanumab, a monoclonal antibody against Aβ aggregates, has obtained the FDA’s approval as an Alzheimer’s drug for its ability to reduce the level of Aβ plaques in patients with early AD or mild cognitive impairment (MCI).46,47,48 On Nov 30 2022, Eli Lilly and Company (https://investor.lilly.com/news-releases/news-release-details/lilly-shares-positive-donanemab-data-first-active-comparator) announced the result of the first active comparator study (TRAILBLAZER-ALZ 4), which showed that donanemab, another monoclonal antibody targeting deposited plaques had outperformed aducanumab-avwa treatment in terms of brain amyloid clearance in patients with early symptomatic AD.49 At the same time, results from the highly anticipated CLARITY AD study were published, showing that 18 months of treatment with lecanemab, a humanized IgG1 monoclonal antibody targeting Aβ soluble protofibrils, reduced markers of amyloid and moderately improved cognitive decline in patients with early AD.50 Recently, the FDA approved lecanemab as the second-ever monoclonal antibody to treat AD. ANAVEX®2-73 (Blarcamesine), which targets sigma-1 and M1 muscarinic receptors, has also demonstrated its disease-modifying activity in AD transgenic mice (3xTg-AD), including reducing amyloid and tau pathologies as well as improving cognitive deficits.51,52 The results of its Phase 2B/3 study, presented at the Clinical Trials on Alzheimer’s Disease (CTAD) Congress 2022, showed that 48 weeks of blarcamesine treatment significantly reduced cognitive decline in patients with early AD. This series of positive results offers a fresh hope and indicates that Aβ-based therapy may be indeed the right direction to be followed. In this review, we summarized the history and current understanding of the “amyloid cascade hypothesis”. In particular, we discussed the pitfalls, promise and important unanswered questions about the current anti-Aβ therapy, which will provide a foundation for further studying and developing more feasible Aβ-targeted strategies to optimize AD prevention and treatment.
The history of amyloid cascade hypothesis (Fig. 2)
In 1984, Aβ was identified as the primary component of extracellular amyloid plaques in AD,1 which is the unique pathological hallmark of the disease.53 Hardy and Higgins then proposed “the amyloid cascade hypothesis” in 1992, positing that Aβ deposits in the brain are the initiating event of AD pathogenesis, resulting in subsequent tau tangle formation, neuronal loss and dysfunction as well as cognitive decline.2 Since then, many genetic and non-genetic studies have supported this hypothesis. Down syndrome with APP gene triplication or APP locus duplications produces an increase in Aβ production and the Aβ42/40 ratio, leading to plaque formation and cognitive decline. APP mutations increase total Aβ and the ratio of Aβ42/Aβ40, leading to early-onset Alzheimer’s disease (EOAD). The apolipoprotein E (APOE) and clusterin (CLU), the strongest genetic risk factors for late-onset Alzheimer’s disease (LOAD), has also been identified to influence Aβ seeding and clearance.4,41

Milestone of the amyloid cascade hypothesis and its applications. Yellow box: key research findings; blue box: the Aβ-related toxicity; green box: the diagnostic application; pink box: important drug and non-drug anti-Aβ therapies. AD Alzheimer’s disease, CSF cerebrospinal fluid, FDA food and Drug Administration, LTP long-term potentiation
Morphology of Aβ aggregates
After secretion, Aβ first aggregates into different soluble species that then change their conformation into cross-β-sheet fibrils to form plaques. There are two types of amyloid plaques: classical and diffuse ones. The classical plaques have a compact core of Aβ surrounded by an optically clear area and an outer corona.54 The corona consists of both neuronal and glial elements, including degenerative neuronal processes (neurites) along with reactive astrocytes and microglia.55,56 Diffuse plaques comprise very small, often stellate assemblies scattered about the parenchyma. It refers to the fact that the Aβ accumulation is widely spread or scattered, but not concentrated.57,58,59 Without consideration of the nature of the Aβ deposits (e.g. thread-like or punctate), “diffuse” thus denotes only the characteristics of the Aβ deposits, and not the dysmorphic neuritis or any other component of the plaques. A recent study showed that it is the classical plaques with inflammatory cells rather than diffuse plaques that correlate with the cognitive impairment during AD.60
Pathological role of Aβ aggregates (Fig. 3)
The amyloid cascade hypothesis has been the leading model of AD pathogenesis since it was proposed, and the hypothesis has being revised over time. The original hypothesis focuses on large insoluble Aβ fibrils as the key offender of neuronal damage, while growing evidence supports that the Aβ oligomers exist and exert their neurotoxicity independently of mature fibrils.61 The amyloid-β oligomer (AβO) hypothesis suggests that AD pathogenesis was instigated by soluble, ligand-like Aβ oligomers.

The generation, aggregation and pathological functions of Aβ. Aβ is generated from APP by sequential cleavage of β-secretase (beta-site APP cleaving enzyme 1, BACE1) and γ-secretase. BACE1 first cleaves APP at the Asp1 site to generate sAPPβ and C99. Subsequently, γ-secretase cleaves C99 to release Aβ (Aβ1-40/42 are the most common isoforms) and CTFγ. After secretion, Aβ peptides first oligomerize into different soluble species then convert their conformation into profibrils and cross-β-sheet fibrils, forming amyloid plaques. Aβ aggregates interact with tau proteins to exert the toxic effects. In addition, they contribute to other AD pathological features including neuroinflammation, oxidative stress and mitochondrial dysfunction, leading to neuronal death and dysfunction. Aβ amyloid β, APP amyloid precursor protein
Interact with cell membrane
Aβ aggregates can directly interact with the lipid and cholesterol components of the cell membrane, forming channels and destroying membrane integrity and permeability, which allows Ca2+ entering into the cell, leading to LTP inhibition and neuronal death.62,63 For example, AβOs bind to sialic acid-containing GM1 ganglioside on cell membrane to induce LTP impairment.64 On the other hand, cholesterol-rich lipid rafts provide an optimal environment for Aβ synthesis and enhance the interaction of Aβ with the membrane.65 Both β- and γ-secretases show increased enzymatic activity in the lipid rafts with higher cholesterol level, while non-amyloidogenic α-secretase activity is inhibited by cholesterol.66,67,68,69 In addition, It is well established that cholesteral-containing lipid membrane can influence Aβ seeding and aggregation.70,71 As a nucleation process, cholesterol and GM1-rich lipid rafts accelerate Aβ aggregation by binding with Aβ to stabilize its structure.72,73 Thus, reduction of cholesterol in endosomes or lysosomes ameliorates Aβ aggregation and its toxicity in mouse models.74
Interfere with synaptic plasticity
Impaired synaptic function is considered to be an early and key pathology of AD. Synaptic loss is also closely correlated with cognitive decline in Alzheimer’s patients.75 Aβ oligomers change the morphology and density of synapses, leading to the impairment of synaptic plasticity.76,77 As a glutamate receptor, functional NMDARs regulate the formation of synapes and synaptic plasticity.78 AβOs directly disturb the activity of NMDARs and impair NMDAR-mediated signaling pathways (e.g. Wnt/β-catenin signaling pathway), leading to synaptic loss and the reduction of spinal density.79 Furthermore, AβOs destroy Glu-recycling at the synapse by increasing glutamate release, reducing glutamate uptake and impairing glutamate transporters, which causes the overactivation of extrasyaptic NMDARs, ultimately leading to LTP suppression, LTD enhancement, and synaptic loss.80 α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) is another glutamate receptor containing four subunits GluA1-4, which makes up to 80% of the excitatory synapses in the CA1 region of hippocampus.81,82 Many studies have shown that AMPARs also take part in the modulation of synaptic plasticity.83,84 However, AβOs induce AMPAR ubiquitination and degradation, leading to the loss of AMPARs followed by the suppression of synaptic transmission.85,86 Recently, two parallel studies have further investigated the underlying mechanism of the Aβ’s detrimental effect over synaptic transmission.87,88 They found that intracellular administration of the AβOs rather than administration of the AβOs at the extracellular level altered the synaptic transmission and fast axonal transport via the casein kinase 2 (CK-2) activation. In addition, the LTP inhibtion and LTD enhancement mediated by Aβ aggregates further result in the shrinkage of dendritic spines by remodeling actin.89,90 Furthermore, Aβ aggregates and hyperphosphorylated tau protein exert synergistic effect on impairing synapse function.91,92,93 AβOs induce tau hyperphosphorylation and accumulation in dendritic spine, which further lead to synaptic loss and dysfunction.94,95 The level of pathological tau in AD patients is correlated with the severity of impaired synaptic plasticity and cognitive dysfunction.96 The pathological tau interacts with the presynaptic compartments including synapsin-1, synaptophysin, to inhibit the mobility and release of synaptic vesicles, leading to the development of AD.97,98,99 Missorted tau proteins at postsynaptic terminals interacts with the subunits of AMPARs and NMDARs, leading to the excessive activation of glutamate receptors, Ca2+ influx, impaired LTP and enhanced LTD.92,100,101 It has been demonstrated that the absence of tau proteins prevent Aβ-induced LTP impairment mouse hippocampal slices.102 Another study also indentified that reduction of tau could ameliorate Aβ-induced Ca2+ influx into neurons and AD-related excitotoxicity in vivo.103 These findings suggest that the synaptic toxicity induced by Aβ was dependent on pathological tau proteins to some extent.
Aβ-induced tauopathy
Beside Aβ plaques, neurofibrillary tangles (NFTs) containing hyperphosphorylated tau are also a hallmark of Alzheimer’s pathology.104,105,106,107 Over past dozen years, a growing number of evidence has indicated the importance of Aβ-tau interaction in Alzheimer’s pathogenesis. In the tripple transgenic mice (3xTg-AD), extracellular Aβ accumulates in the neocortex and hippocampus followed by tau seeding into fibrillar tangles.108 Injection of Aβ aggregates into brain of P301L mutant tau transgenic mice triggers a five-fold elevation in NFTs in the amygdala.109 In the clinical setting, neuroimaging of sporadic Alzheimer’s patients show the increased cortical tau-PET ligand retention only in the presence of Aβ accumulation, which is also associated with cortical atrophy in AD.110 In addition, longitudinal studies idenfied that antecedent Aβ aggregates could successfully predict the subsequent tau changes in the inferior temporal cortex.111 As the upstream factor, Aβ triggered the hyperphosphorylation of tau protiens,112,113,114 which synergistically induced neuronal impairment and cognitive deficits.111,115 Aβ accelerated the tau hyperphosphorylation by the activation of cyclin-dependent kinase 5 (CDK5) and glycogen synthase kinase 3 (GSK-3).116,117,118 GSK-3β, which is inextricably associated with Aβ production and accumulation,119 is a key trigger of tau phosphorylation and aggravates Aβ-induced tau toxicity.120 CDK5-P25 phosphorylates tau at sites of Thr181, Ser199, Ser202, Thr205, Thr212, Ser214, Ser217, Thr231, Ser235, Ser396, and Ser404.121 Thus, inhibitors of GSK-3β or CDK-5 such as AZD1080 and roscovitine, markedly reduced the levels of tau phosphorylation and prevented further tau aggregation.122,123 Further studies have found that mitogen-activated protein kinases (MAPKs) including ERK1/2, SAPKs and p38 are also involved in Aβ-induced formation of PHF-tau during AD.124,125 Cellular prion protein (PrPC) has been found as a receptor for toxic Aβ oligomers to induce LTP loss and cognitive impairment in AD model mice.126,127 PrPC has been also detected in Aβ plaques in Alzheimer’s patients,128,129,130 which activates Fyn kinase and phosphorylates tau by the GluN2B subunit of NMDARs.100,131,132,133 In addition to its stimulatory effect on tau phosphorylation, Aβ also affected tau oligomerization and tangle formation.134 Aβ triggered caspase-3 (CASP3)-induced cleavage of tau at Asp421 to yield an N-terminal product, which self-aggregated and further assembled into neurotoxic oligomers.134,135 Tau oligomers not only led to neuronal damage but also bound to astrocytes and microglia to induce neuroinflammation.136 In hippocampal neurons, Aβ also induced the activation of calpain-1 and generated a 17-kDa tau fragment, resulting in neurite degeneration and neuronal death.137
Meanwhile, the toxic state of tau proteins also influence Aβ production. Thus, knocking out the tau genes in the APP/PS1 mice inhibited the amyloidogenic pathway of APP processing, Aβ production and the amyloid plaque formation.138 Furthermore, the neurotoxicity of Aβ is tau-dependent. Absence of tau on NMDARs of spines successfully prevented the toxic effect induced by the binding of Aβ to NMDARs.92 A recent study proposed that the phosphorylation of tau at Tyr18 by Fyn kinase also blocked Aβ toxicity.139,140 Aβ promoted the phosphorylation and activation of Fyn kinase, which further migrated into dendritic spines, leading to synaptic impariment.141,142 Tau protein mediated Aβ toxicity by interacting with Fyn kinase via its amino-terminal projection domain (PD).143 Accordingly, inhibition of Fyn improved the cognitive deficits in transgenic mice with Aβ and tau depositions.144,145 PET and CSF tests also indicate the synergy between Aβ and tau, which leads to brain dysfunction and cognitive impairment.146,147,148 In contrast, Aβ and tau have antagonistic effects on neural circuit.149 Tau induces the profound silencing of circuits by blocking Aβ-dependent hyperactivity in the cortex.150
Induce inflammation
Neuroinflammation is chronic inflammation in the CNS, which is attributed to activated microglia and astrocytes to produce numerous pro-inflammatory cytokines.151 Growing evidence demonstrates that neuroinflammation plays a vital role in the neuropathological changes in AD.152,153,154 In addition, patients who are with long-term nonsteroidal anti-inflammatory drugs (NSAIDs) for treating other diseases such as rheumatoid arthritis, showed a 50% reduction in the risk for developing AD.155 It has been reported that the inflammation-associated proteins and cells were localized closely to Aβ plaques in AD brain.156 However, the possible underlying mechanisms are still unclear. One potential explanation for the activated glia cells in AD brain could be the response to Aβ produced largely by neurons.157,158 Aβ shares structural similarities with antimicrobial peptides (AMPs) and viral fusion domains, which stimulates glia cells to secrete a mass of pro-inflammatory cytokines.159 Similar to AMPs, Aβ aggregates can also induce pores in cell membranes, which allow a variety of stimuli to activate glia cells.160
Microglia comprise around 10–15% of all glial cells, which are the resident macrophages within the CNS.161 In a healthy adult brain, microglia are in a resting state and highly ramified morphology with small somas.162 These cells communicate with surrounding environments including neurons, astrocytes and blood vessels to maintain the development and homeostasis of the CNS.163,164 When microglia recognize the insults of the CNS, they respond to the injury or invasion by a morphological change, resulting in cell enlargement and migration.165 In the development of AD, it has been suggested that Aβ aggregates are the primary driver to activate microglia and set them into motion. Activated microglia migrated to the Aβ deposition and stimulated the phagocytosis of Aβ.166,167,168 Thus, factors such as CD33, which impedes Aβ phagocytosis by microglia, has been considered to increase the risk for suffering from AD.169 However, the prolonged activation of microglia become enlarged and are no longer able to exert their phagocytic function. In contrast, their capacity of pro-inflammatory cytokine production is unaffected, contributing to an exacerbation of AD pathology including Aβ accumulation and neuronal damage.170,171 To compensate the impaired clearance of Aβ, peripheral macrophages are recruited to the brain in an effort to clear Aβ plaques, which likely worsens the sustained inflammation and thus AD pathologies.172,173 Compared with microglia distal to the amyloid in AD brain tissues, there is an increased expression of TREM2 in the cells close to Aβ plaques.174,175 Increased TERM2 experession has been found in human AD blood, indicating the important role of peripheral TREM2 in Alzheimer’s pathogenesis.176,177 Using flow cytometry identified that these cells also contained high levels of CD45, Ly6c, and CD11b, which highly express in peripheral macrophages as well.174 Partial or completed deletion of TREM2 markedly reduced the number of Aβ-associated macrophages and increased cerebral Aβ plaques in AD model mice.174,175,178 The reduction of TREM2 in Aβ-associated macrophages also altered astrocytosis detected by glial fibrillary acidic protein (GFAP) and S100β.178
As the most abundant glial cells in the CNS, astrocytes play an essential role in the communication with neurons and regulation of synapse formation and function.179 Under pathological conditions, astrocytes become reactive, which are characterized by cell hypertrophy with GFAP and vimentin expressions as well as the release of cytotoxins.180,181,182 The reactive astrocytes are close to Aβ plaques in brains from AD patients and rodent models.183,184 Astrocytes response to Aβ aggregates in a TLR-dependent manner, which further activates the target genes to produce proinflammatory factors.185,186 The excessive production of proinflammatory cytokines such as TNF-α or IFN-γ modulated the APP processing in astrocytes, leading to the increased Aβ levels and toxicity.187 These studies have revealed a significant role of reactive astrocytes in the loop between inflammatory cytokines and Aβ load.188 Disturbed this cross-talk has been considered to underlay Alzheimer’s pathogenesis. Impaired astrocyte activity also increased the number of microglia surrounding Aβ plaques and altered the microglia status.189 In turn, microglia could alter the status of astrocytes. The activated microglia secreted IL-1α, TNF and C1q cytokines to further induce A1 reactive astrocytes, which are neurotoxic and increased in human AD post-mortem tissues.190 In addition, Aβ produced by neurons induced the complement protein C3a released by astrocytes via NFκB signaling, which interacted with the receptors (C3aRs) on microglia and neurons to aggravate Aβ aggregate loads and cognitive impairment.191
Mitochondrial dysfuntion and oxidative stress
Mitochondria are the major powerhouses for cells, where oxidative phosphorylation (OXPHOS) occurs to generate ATP for maintaining the optimal neuronal activities.192 Mitochondria are essential for the glutamate synthesis, synaptic transmission and calcium regulation.193,194 Disrupted energy metabolism has been found in early AD and precedes the disease development, suggesting the core role of mitochondria dysfunction in Alzheimer’s pathogenesis.195,196 Soluble Aβ oligomers disrupted the balance between mitochondrial fission and fusion, leading to significant mitochondrial dysfunction.197,198 Excessive mitocondrial fission is a key modulator of Aβ toxicity.199 Thus, restoration of mitochondrial fission rescued APP- or Aβ-induced mitochondrial abnormality and neuronal damage.200,201 Only 1% of mitochondrial proteins are synthesized in the mitochondria itself. Instead, most proteins of the mitochondria are synthesized by cytosolic ribosomes then imported into the organelle.202 APP- or Aβ-induced impairment of mitochondrial import pathway has been considered as a hallmark of AD.203,204,205 It has been demonstrated that APP blocked mitochondrial import machinery and impaired mitochondrial function in AD brain by forming a complex with translocases of the inner and outer mitochondrial membranes.204 In addition, endoplasmic reticulum (ER)-mitochondria contact sites provide a platform to regulate important cellular activities, including synthesis of phospholipids, calcium transport between ER and mitochondria, regulation of mitochondrial homeostasis, activation of inflammasome, and induction of apoptosis.206,207 Alteration of mitochondria-associated endoplasmic reticulum membrane (MAM) signaling has been implicated in neurodegenerative diseases such as AD.208,209 Overexpression of APP mutants or Aβ aggregates increased ER-mitochondria connectivity, resulting in the elevation of mitochondrial calcium.208,210,211 C99, a C-terminal fragment of APP cleaved by β-secretase, also activated sphingolipid turnover and increased ceramide to impact the ER-mitochondria contacts, leading to impaired mitochondrial respiration and metabolic disturbance.212
Mitochondria are also the major source of oxidative stress because the inevitable leakage of electrons at complex I and complex III of the electron-transport chain to produce reactive oxygen species (ROS).213,214 Mitochondria generate approximately 90% of the cellular ROS.215 The damaged mitochondria are less efficient to generate ATP but more efficient to produce ROS.216 The vulnerability of the brain to ROS is now emerging as a key detrimental factor driving AD pathogenesis. Neurons exposed to ROS stimuli are more susceptible to developing age-related neurodegenerative pathologies, as seen in AD brains. Redox active metal ions, such as Cu and Fe bind to Aβ to produce the ROS, which contributes to the oxidative damage on proteins and lipids leading to impaired membrane integrity, neuronal dysfunction and DNA damage.217,218,219,220 In addition, mitochondrion-derived ROS modulated the APP processing and triggered Aβ production to form a vicous cycle.221
Change neurochemical systems
Aβ aggregates interact with glutamatergic neurotransmission, which impairs excitatory synaptic plasticity, leading to cognitive decline.222,223,224,225 Excessive Aβ peptides induced LTD by inhibiting LTP and making a shift of the NMDAR-dependent signaling cascades.226 Thus, Aβ accumulation inhibited the synaptic transmission, resulting in early cognitive impairment.224 Aβ-induced LTD is also caused by inhibiting glutamate uptake and stimulating glutamate releasing, which evently elevates glutamate levels in the synapse cleft.222,225,227,228 An increase of glutamate activated GluN2B-bearing NMDARs, which further led to calcium-induced LTD and synaptic depression.85 Aβ oligomers also regulated the trafficking of NMDARs to change dendritic spine density.222,227,229 As with NMDARs, AMPARs are also the principal receptors mediating excitatory synaptic transmission.230 It has been identified that APP overexpression and increase of soluble Aβ oligomers are related with the downregulation of GluA1/2 subunits of AMPARs, leading to the inhibition of synaptic plasticity, spine loss, and memory deficits.231,232
The basal forebrain cholinergic system is one of the earliest brain regions vulnerable to degeneration during AD.233 The correlations between enhanced BACE1 activity, Aβ accumulation with atrophy of basal forebrain and loss of functional connectivity have been found in neuropathological and neuroimaging studies.234,235,236,237 Furthermore, such an inverse correlation seems to be intensified with the ε4 allele of the apolipoprotein E (APOE) gene, which is one of the strongest risk factors for LOAD.238
Impair brain networks
Decrease of default-mode network (DMN) functional connectivity has been found in prodromal stages of AD, which is associated with loss of gray matter volume in neocortex and hippocampus.239,240 Reduced DMN connectivity only occurs in individuals with elevated baseline Aβ-PET indexes, accelerating cortical atrophy.241 Consistent with the findings in humans, aging and AD animal models also show disruptions of functional connectivity in the DMN.242 The salience network (SN) identifies salient stimuli and plays an important role in the coordination of the central executive (CEN) and the DMN, whose functional impairment is related to learning and episodic memory deficits in both amnestic mild cognitive impairment (aMCI) and AD.243 There is an increased Aβ-PET signal within the CEN and the SN in the progression of AD.244,245 A spatial covariance between Aβ aggregates with reduced connectivity and metabolism in the CEN and SN has also been found in AD.246,247
Discovery and development of Aβ-based biomarkers
Based on the amyloid cascade hypothesis, Aβ measurement has been considered as a valuable indicator to assist the diagnosis of AD. In clinical settings, Aβ peptides are most frequently measured in the cerebrospinal fluid (CSF) or through brain imaging of Aβ fibrils with positron emission tomography (PET).248 CSF analysis offers a quantitative result of the net effect of Aβ peptides, while.249,250 There are four tracers used to detected levels of amyloid in the human brain, including 11C-Pittsburgh compound B (11C-PiB),251 AmyvidTM (flobetapir F18),252 NeuraceqTM (florbetaben F18)253 and VizamylTM (flutemetamol F18).254 In practice, reduced concentrations of Aβ42 in CSF and increased retention of Aβ tracers in the brain have been considered as early biomarkers of AD.255,256,257,258 Both biomarkers have been demonstrated to have high diagnostic and prognostic value as they start changing decades before the onset of dementia symptoms.259,260,261,262,263,264,265,266 However, CSF- and PET-based measures are not suitable for large-scale screening due to their invasiveness, high cost and low accessibility. Considering the greater availability of blood sampling, blood-based biomarkers become the primary goal in screening for and diagnosing AD in the population and many studies now focus on examining the role of peripheral Aβ and APP in AD development.267,268,269 One such study found that plasma concentrations of soluble β-secretase cleaved n-terminal APP (sAPPβ) were significantly reduced in AD patients compared with age-matched cognitively healthy individuals or patients with behavioral variant frontotemporal dementia (bvFTD), indicating the potential role of sAPPβ as a promising new biomarker of AD.270 In addition, there is increasing evidence to support that plasma Aβ acts as an endophenotype of AD, which simultaneously changes with Aβ status in the brain.271,272,273 The blood levels of APP669-711/Aβ42 and Aβ40/Aβ42 ratios, as well as peripheral Aβ-bound extracellular vesicles (EVs), have been shown to predict brain Aβ burden.274,275 Our group has also identified that circulating Aβ could pass the blood brain barrier (BBB) and enter the brain, contributing to the development of AD.276 In contrast, Aβ peptides in the CNS can also move into the circulatory system, where the peptides are phagocytosed by the monocytes or neutrophils, directly degraded by the enzymes, or further transported to the peripheral organs or tissues for degradation or excretion.28,277 Recently, the development of single molecular assay (Simoa), an ultra-sensitive immunoassay technology, allows the measurement of Aβ40 and Aβ42 levels at sub-femtomolar concentration. The availability of reliable and sensitive detection of Aβ peptides in blood makes a promise for early diagnosis and better prognosis of AD.
The progression of Anti-Aβ therapy
To date, five drugs have been approved for the treatment of AD. Four of these medications are classified as cholinesterase inhibitors (CIs), including tacrine, donepezil, rivastigmine, and galantamine. Most of them are approved to treat Alzheimer’s type in the mild-to-moderate stages, except for donepezil which is administered to patients with severe or late-stage AD. Tacrine has been discontinued in the US due to severe liver toxicity. Unlike these four medications, memantine is an N-methyl-D-aspartate (NMDA) receptor antagonist, which exerts its neuronal protective effects by inhibiting glutamate activity. However, these drugs can only help alleviate the symptoms instead of modifying the disease. Thus, development of effective disease-modifying therapies for AD is urgent and necessary.
According to ALZFORUM (March 2023, www.alzforum.org), 298 AD therapies have been under clinical trials. 76 of them target the Aβ peptide or its aggregates, including small molecules (Table 1) and immunotherapies (Table 2), which can be classified into four categories: (1) to reduce Aβ generation;278,279,280,281,282 (2) to enhance the degradation and clearance of Aβ and its aggregates;283,284,285 (3) to neutralize soluble Aβ monomers or its toxicity;286,287,288,289,290,291,292,293,294 (4) to directly inhibit Aβ aggregation.295,296,297,298,299 So far, two antibody-based drugs aducanumab and lecanemab have been approved by the FDA and 38 of them have been discontinued due to ineffectiveness or toxic side effects.
BACE1 inhibitors
In 1999, BACE1 was identified as an enzyme required for Aβ production.300,301,302,303 Since then, inhibiting BACE1 activity has been pursued as a key method of halting the amyloid cascade and the development of effective BACE1 inhibitors has become a focus of many drug trials. LY2886721 was the first BACE inhibitor to reach Phase 2 clinical trials.304 Compared to the previous compound, it has better brain penetrance. In 2012, Eli Lilly announced that the application of LY2886721 produced the expected results in Phase 1 studies with reduced CSF levels of Aβ40 and Aβ42 as well as increased sAPPα levels (P3-359, Alzheimer’s Association International Conference, 2012). However, it was halted in the Phase 2 study due to abnormal liver biochemistry values in four participants. Its toxicity was considered to be an off-target effect of the compound, which was not related to BACE1 inhibition (The 11th International Conference on Alzheimer’s & Parkinson’s Disease, 2013). Besides LY2886721, many other candidates have also reached late stages of clinical trials, including atabecestat (Phase 2/3),305 elenbecestat (Phase 3),306 lanabecestat (Phase 2/3)307,308 and umibecestat (Phase 2/3).309 However, all of them have failed to receive final approval to reach the market. Several obstacles have been found in the development of effective BACE1 inhibitors. BACE1 possesses structural similarities with many other aspartyl proteases, such as BACE2, pepsin, renin, cathepsin D and cathepsin E, a significant challenge to achieve the selectivity in BACE1 inhibition without affecting other proteases that cause off-target side effects.310 In addition, the size of the BACE1 active site is relatively large, including catalytic aspartic acid residues, flap, and 10 S loop.311 Since all the developed BACE1 inhibitors are small molecules, it may be difficult to occupy this large active site to efficiently block BACE1 activity. Low penetrance of blood-brain barrier (BBB) is also another concern.312
γ-secretase inhibitors/modulators
γ-secretase inhibitors (GSIs) have been widely investigated as potential therapeutic approaches for AD due to their ability to inhibit Aβ production. However, the existing GSIs act too generally, which causes serious side effects through inhibiting the processing of other proteins, such as Notch, a transmembrane receptor involved in regulating cell-fate decisions.15,313 Thus, researchers have tried to develop a much more specific γ-secretase inhibitor, which only disrupts the production of Aβ but not others. Avagacestat is a recently developed arylsulfonamide γ-secretase inhibitor with high selectivity for APP over Notch, which successfully reduces CSF Aβ levels in the animal models without any Notch-related toxicity.314 Avagacestat was considered as a promising AD treatment with the ability to selectively inhibit the APP processing without affecting the Notch pathway. However, it was terminated in Phase 2 trials due to gastrointestinal and dermatological side effects.315 These failures popularized the development of γ-secretase modulators (GSMs) as an alternative approach. GSMs aim to regulate but not totally block the enzyme’s activity. A recent study found that treatment with one potential candidate, SGSM-36, which successfully reduced the level of toxic Aβ42 peptides, without changing the proteolytic processing of Notch or α- and β-secretase processing of APP.316 EVP-0962 is another GSM that was shown to reduce Aβ42 levels and increase Aβ38 levels without affecting Notch signaling in vitro. It also improved the memory deficits in AD model mice.317 Unfortunately, all of them have been discontinued in the clinical trials.
Active and passive immunotherapy
Immunotherapy has been considered as one of the most promising strategies aimed at the modification of AD development. This approach involves designing synthetic peptides or monoclonal antibodies (mAbs) to decrease brain Aβ load and slow the disease progression. The first AD vaccine tested in a clinical study was AN1792, a synthetic full-length Aβ42 peptide.318 Although the vaccine showed some therapeutic effects, including slowed cognitive decline, the clinical trials were terminated due to the occurrence of aseptic meningoencephalitis in 6% of the participants.319,320,321 A possible explanation for this side effect is the induction of T helper 2 (Th2) cell responses by the excipients applied to produce C-terminus region of Aβ peptides.321 Accordingly, the subsequent vaccines do not include this region of Aβ peptides. Vanutide cridificar (ACC-001) is a conjugate of multiple short Aβ fragments to avoid the safety concerns associated with AN1792.322 Preclinical data showed that vanutide cridificar induced the generation of N-terminal anti-Aβ antibodies and successfully improved cognitive impairment in AD animal models. However, all clinical trials using vanutide cridificar were also discontinued following a serious adverse event.323 Another example is Lu AF20513, which is a mixed peptide containing three repeats of the first 12 amino acids of Aβ peptide interspersed with tetanus toxin sequences. The peptide was designed to activate a B cell response to produce polyclonal antibodies against Aβ. While Lu AF20513 was shown to successfully remove brain amyloid deposits in the initial preclinical study, clinical trials were terminated due to a lack of efficacy.324 Currently, four vaccines are under the clinical trials, including ALZ-101 (Phase 1), ACI-24 (Phase 2), ABvac 40 (Phase 2) and UB311 (Phase 3). ALZ-101 is a vaccine specific to soluble Aβ oligomers rather than Aβ monomers or fibrils.325 It is undergoing a Phase 1B study. ACI-24 is a liposome vaccine based on the Aβ1-15 sequences. It is designed to generate antibodies specifically against the β-sheet folding of Aβ. In the preclinical studies, ACI-24 was shown to generate high titers of anti-Aβ IgG1 and IgG2b antibodies and improve novel object recognition in AD mice.324 Its Phase 2 trials have been started, in which ACI-24 becomes the first anti-Aβ vaccine to be evaluated for treating AD in Down’s syndrome patients. Another vaccine called ABvac40 targets the C-terminus of Aβ peptides and is also currently being evaluated in Phase 2 clinical studies.326 UB-311 consists of the Aβ1-14 peptides in combination with a Th-cell epitope, which was designed to specifically stimulate Th2 cells regulatory immune responses over Th1-mediated autoimmune responses. UB-311 was shown to neutralize Aβ toxicity and enhance plaque clearance in preclinical studies.327 In the Phase 2 studies, UB-311 also showed its safety and generated Aβ antibodies in 96% of the patients with mild AD (14th International Conference on Alzheimer’s and Parkinson’s Disease, 2019). In 2020, it was announced that UB-311 would begin a Phase 3 clinical testing, in which two double-blind, placebo-controlled studies will be conducted. However, the data related to this clinical trial have not been released. In May 2022, UB-311 was granted fast-track designation by the FDA for Alzheimer’s treatment.
Passive immunotherapy prevents some issues of the active immunization by using monoclonal antibodies (mAbs) directly targeting different forms of Aβ peptides, including monomers, oligomers and fibrils to inhibit the formation of toxic aggregates.328,329,330,331 The Fc domain of mAbs binds to the Fc-γ receptors on the microglia, leading to the phagocytosis of the Aβ-mAb complex.332 In addition, the Aβ-mAb complex induces the complement-dependent cytotoxicity, resulting in the lysis of the target cells. In the blood, the mAbs interact with Aβ to reduce Aβ concentration, resulting in a concentration gradient that stimulates the efflux of Aβ from the brain.333 Bapineuzumab is the first antibody to be tested in clinical trials. It is a humanized version of the mouse anti-Aβ monoclonal 3D6 antibody specifically targeting the N-terminal region of Aβ (residues 1–5). Humanized antibodies are generated by modifying protein sequences from non-human species to increase their similarity to natural antibody variants produced in humans, which reduce the immunogenicity of the antibodies, enhance human effector functions, and increase the serum half-life of the antibodies in humans.330 In the preclinical studies, 3D6 binds to monomeric, oligomeric and fibril forms of Aβ, leading to the reduced levels of Aβ and improved cognitive deficits in AD model mice.334 However, the Phase 3 clinical trials revealed that bapineuzumab could not improve clinical outcomes in mild to moderate AD patients.335 There are also other candidates under the Phase 3 trials, including gantenerumab, crenezumab, donanemab, solaneuzumab and lecanemab (BAN2401). Gantenerumab is a human mAb designed to bind with a conformational epitope on Aβ aggregates. It reduces the plaques by stimulating the microglia-mediated phagocytosis. The antibody was found to be safe and well tolerated during the Phase 1 clinical trials, except that transient amyloid-related imaging abnormalities (ARIA) appeared in some patients given a high dosage.336 The initial results of phase 2 studies suggested gantenerumab may have no efficacy in the enrolled cohort. However, subsequent post-hoc analyses showed a slight benefit in patients with fast disease progression. It was also tested in a Phase 2/3 study called the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) aimed at preventing dementia in 210 people who were in the progression to Alzheimer’s disease due to an inherited autosomal-dominant mutation in APP, PSEN1, or PSEN2.337 Gantenerumab treatment significantly reduced the amyloid loads and normalized CSF Aβ42 levels.338 However, cognitive data revealed that gantenerumab did not reach its therapeutic point. In addition, two Phase 3 trials were conducted in prodromal or mild AD patients with amyloid deposition. Just several months ago (Nov 14, 2022), Roche and Genentech announced that the outcome of the Phase 3 trials were disappointing, in which the drugs failed to slow cognitive impairment. A new version of ganterumab, called trontinemab is currently under Phase 1 trial, which contains a Fab fragment for better penetration to the BBB. Compared with unmodified ganterumab, 50 folds more trontinemab entered the brain and bound to Aβ plaques. Similar to gantenerumab, crenezumab also recognizes multiple forms of Aβ aggregates. It has high affinity with the oligomeric and fibril species and amyloid plaques.339,340 Crenezumab is being tested in both prevention and treatment paradigms.341,342,343 Unfortunately, most of the initial trials including the prevention trial failed to achieve their primary endpoints, and crenezumab is now discontinued. Donanemab is a humanized IgG1 monoclonal antibody targeting the existing amyloid plaques and clearing them from the brain.344 Early results from the Phase 1 and 2 clinical studies offered some compelling evidence that donanemab could slow down the amyloid and tau burden. As a result, donanemab has been granted the Breakthrough Therapy designation by the FDA and two Phase 3 trials, including those for prevention and treatment ones, are currently underway. In early of this month (May 3, 2023), Eli Lilly announced partial results of the Phase 3 study showing that donanemab significantly slowed cognitive and functional decline by 35% in patients with early symptomatic AD. In addition, 47% of the participants with donanemab for 1 year showed no clinical progression compared with 29% participants on placebo. The drug achieved its best effect in patients with moderate levels of tau proteins. However, its side effects of bleeding and seizures caused by ARIA also raise big concerns. Solaneuzumab is the humanized version of the murine m266 IgG1 mAbs that target the central region of Aβ. It has more affinity to Aβ monomers than the toxic aggregates. Although solanezumab was well tolerated in the participants, it was not able to show the significant therapeutic benefits to AD patients.345,346 The failure may be due to the too low concentrations of the antibody reaching to the brain.
Aducanumab is the first FDA-approved therapy for Alzheimer’s.47,48 It is a human IgG1 mAb against a conformational epitope found on the N-terminus of Aβ (residues 3–6), and thus specifically targeting aggregates rather than monomers. It has been shown to reduce plaques in imaging studies.347 However, in 2019, Biogen and Eisai announced they would not start an anticipated Phase 3 secondary prevention program and would terminate all ongoing trials as aducanumab treatment was predicted to miss its primary endpoint based on the interim analysis (Mar 2019 news, www.eisai.com). Later, Biogen announced that the interim futility analysis was wrong and the highest dose of aducanumab treatment significantly improved cognitive deficit in the participants (Oct 2019 news, investors.biogen.com). In June 2021, aducanumab was approved by the FDA for medical use.47,48 However, it is considered controversial due to the lack of sufficient evidence to support its efficacy.348,349 As required by the FDA, a Phase 4 confirmatory trial called ENVISION was planned in May 2022. The study will recruit 1500 patients with early AD including at least 18% of participants from black and Hispanic communities in the US (Jan 2022 news, investors.biogen.com).
Lecanemab (BAN2401) is the humanized IgG1 version of the mouse mAb158, which specifically binds to large, soluble Aβ protofibrils. The antibody has been proved to be safe without serious adverse events in the Phase 1 trials.350 In the Phase 2 trial, it had been identified to successfully reduce brain amyloid and improved cognitive decline in the highest-dose group (twice-monthly 10 mg/kg).351 A Phase 3 study called Clarity AD was initiated in March 2019 to determine the therapeutic efficacy of lecanemab on 1795 people with mild cognitive impairment (MCI) or early Alzheimer’s disease. The results were just published and showed that patients with lecanemab treatment had lower brain amyloid levels and reduced cognitive and functional decline as measured by the Clinical Dementia Rating-Sum of Boxes (CDR-SB), which quantifies symptom severity across a range of cognitive and function domains, by 27% compared with placebo.50 The positive results made lecanemab become another FDA-approved treatment of Alzheimer patients with mild cognitive impairment. However, routine MRI scans showed around 21% of individuals on lecanemab experienced side effects such as ARIA, compared with just over 9% in placebo-treated controls.352 ARIA may further cause brain atrophy showing as the increased size of the ventricle. In Feb 2020, it was announced that a large lecanemab study called AHEAD3-45 would run from July 2020 to October 2027 to measure the preventive effect of lecanemab treatment on amyloid and tau tangle formation.353
The stumbling block of anti-Aβ therapy
Disturbed physiological functions of soluble Aβ
Aβ peptides exist in both the brain and blood throughout an individual’s life.354 Although the aggregates have been considered to be toxic, soluble Aβ at physiological levels have been identified to have biological functions, including enhancement of long-term potentiation (LTP),355,356,357,358 stimulation of neuronal differentiation,359 improvement of the brain’s ability to recover from injuries,360,361,362,363 inhibition of oxidative stress,364 antimicrobial activity365 and tumor suppression366,367 (Fig. 4). These physiological functions must be taken into consideration when strategies are developed to lower Aβ levels in AD. Ideally, such strategies should have more precise targeting of conformations, which are fibrils protofibrils or oligomers, and maintain normal physiological level of Aβ monomers.

The physiological functions of soluble Aβ. Soluble Aβ at physiological levels has been identified to have some important functions, including induction of long-term potentiation (LTP), stimulation of neuronal differentiation, improvement of brain recover from injuries, inhibition of oxidative stress, antimicrobial activity and tumor suppression
Modulation of synaptic function
Although Aβ aggregates, especially the soluble oligomeric species impair synaptic plasticity by inhibition of LTP and induction of LTD, growing evidence indicates that a normal level of Aβ peptides may play a key role in the maintenance of synaptic function and cognition.368,369 It has been shown that the KLVFF (16~20 amino acid sequence) of Aβ peptides has a protective effect against excitotoxicity, which prevents neuronal death.370 In addition, both synthetic and endogenous Aβ42 monomers in nanomolar concentrations stimulated the activity of cyclic adenosine monophosphate (cAMP) responsive element-binding protein (CREB) and brain-derived neurotrophic factor (BDNF), which possessed key roles in the regulation of gene expressions related to neuronal functions and survival in normal brains.371,372 In contrast, removal of endogenous Aβ by injection of anti-Aβ antibodies or genetic manipulation greatly decreased LTP and impaired memory, which could be rescued by the addition of human Aβ42.357,373,374,375,376 Together, the possible role of Aβ peptide in the modulation of synaptic function as well as learning and memory has been suggested. Aβ monomers stimulated astrocytes to increase the clearance of synaptic glutamate and therefore protect neurons from glutamate excitotoxicity.377,378 Aβ can also be released into the synaptic cleft, where it acts on presynaptic neurons to induce the release of neurotransmitters (e.g. acetylcholine) or directly activates α7-nicotinic acetylcholine receptors (α7-nAChRs) to enhance long-term potentiation (LTP).355,356,357,358 In the CNS, the nicotinic acetylcholine receptors (nAChRs) are expressed in both neurons and non-neuronal cells.379,380 As ligand-gated ion channels, nAChRs opened in response to the depolarization of the membrane, allowing Na+, K+ and Ca2+ to enter the cells.381,382 Among the isofroms, the α7-nAChRs had the highest Ca2+ permeability.381 The mechanism behind Aβ-induced α7-nAChR activation could be due to the disruption of intracellular signal transduction to stimulate the calcium influx.383 α7-nAChRs are involved in a variety of biological functions, including neurotransmitter release, synaptic plasticity and neurogenesis.384,385 In AD brain, nAChRs have been detected in Aβ42-positive neurons and their reduction is associated with disease progression.386 Furthermore, there was an increase of Aβ/nAChR-like complexes in carriers of APOE ε4, a strong risk factor for LOAD.387 In fact, Aβ might interact with specific subtypes of nAChRs with different structures to mediate its physiological effects or toxicity to cholinergic neurons. Under physiological conditions, low level of Aβ particularly interacted with the α7 isoform via the nitric oxide/cGMP/protein kinase G pathway to activate the channels.388,389 Thus, α7-nAChR KO mice at 12-month-old showed Aβ elevation as a compensatory response of α7-nAChRs and exhibited AD-like pathologies.390
Inhibition of APP changed the expressions of post-synaptic proteins such as GluA1subunit of AMPA receptors, suggesting the involvement of APP in synaptic formation.391 An obvious reduction of LTP was found in cultured hippocampal neurons with knockdown of APP expression.392 Similarly, conditional KO of PSEN1 and PSEN2 to inhibit Aβ production also led to impaired synaptic plasticity and cognitive deficits in animal models.393 In contrast, application of nanomolar synthetic Aβ successfully enhanced the cognitive and memory performance of the mice.357 However, the nanomolar concentrations of Aβ used in the study deviate too far from the physiological level of Aβ in picomolar concentrations. To address this concern, other studies injected picomolar concentrations of Aβ peptides into the mice, which also significantly enhanced synaptic plasticity and memory formation.394 These findings suggest that physiological levels of Aβ monomers are crucial to maintain a normal synaptic function while only Aβ aggregates have the inhibitory and toxic effects.
Promotion of injury recovery
Evidence from patients and animal models also shows rapidly increased Aβ expressions after being injured are beneficial,360,361,362,363 indicating the role of Aβ in stimulating the brain to recover from traumatic and ischemic injuries. There is an elevation of Aβ peptides during traumatic brain injury (TBI), indicating that Aβ may belong to the pathological cascade of TBI or be an agent for improving recovery.395,396 To answer this question, Aβ40 peptides were intracerebroventricularly injected into TBI-impacted BACE1–/– mice, which significantly improved motor memory deficits in these injured mice, suggesting the protective effect of Aβ.362 In contrast, reduction of endogenous Aβ levels by using γ-secretase inhibitor DAPT or deleting the enzyme BACE1 attenuated the functional recovery in mice with spinal cord injury (SCI).396 Aside from TBI, Aβ may also have a protective role against other types of brain injury such as cerebral ischemia, which blocks the blood flow in brain. It has been demonstrated that overexpression of human APP (hAPP695) leads to an obvious lower infarct volume in the cortex of mice suffering from cerebral ischemia.363 Experimental autoimmune encephalomyelitis (EAE) is a T cell-mediated autoimmune disease with inflammation in brain. Aβ treatment was found to effectively inhibit the production of proinflammatory T helper cells (TH1 and TH17) and the related cytokines including IL-6, IFN-γ and IL-17, which improved motor paralysis in EAE animal models. In constrast, genetic deletion of APP significantly aggravated the severity of the disease, suggesting the protective role of Aβ against autoimmune inflammation in CNS.397
Anti-microbial activity
Recently, Aβ’s role as an anti-microbial peptide has been demonstrated. Animal models with the expression of human Aβ showed stronger resistance to bacterial and viral infections.365 Moreover, brain tissues from AD patients show higher anti-microbial activity than samples from age-matched non-AD individuals, which was correlated with Aβ levels in brain.398 It is hypothesized that the anti-microbial activity of Aβ is associated with its capacity to bind with microorganisms and form a net to trap the infectious agents.399 This idea fits with the findings that HSV1 and Borrelia DNA have been found in plaque cores of AD brains.400,401 Aβ peptides are able to interact and entrap various bacterial strains and viruses, such as HSV1 and HSV6, block their entry into the host cells to replicate.402,403,404 Interestly, Aβ42 cannot prevent the replication of non-enveloped human adenovirus, suggesting that it probably interacts with viral coat proteins.404 Aβ stimulated the aggregation of viral particles, which facilitated leukocyte-mediated uptake of viruses.405 In addition, the damaged host cells released nucleic acids containing Aβ aggregates, which were immunogenic and elicited the secretion of type I interferons (IFNs) by adjacent microglia to accomplish the antiviral response.406 The produced interferon-γ (IFN-γ) further facilitated Aβ generation to form a positive feedback loop.407 Similar to anti-viral activity, Aβ peptides also bound to fungal cells and stimulated the phagocytosis of microglia.408 Thus, familial AD mutations accelerated the clearance of C. albicans from brains in mice.408 Together, the underlying mechanisms of Aβ peptides exerting their anti-microbial activity including interation with membranes and disruption of membrane integrity; stimulation of phagocytosis by inducing cytokines or altering microorganisms’ conformation.
Suppression of tumor growth
In addition, recent studies show that AD patients have significantly lower incidences of several types of cancers, including skin cancer, lung cancer, breast cancer and bladder cancer.366,367 Aβ has been demonstrated to inhibit tumor cell growth. In vitro, application of media containing Aβ successfully inhibits the proliferation of cells, including human glioblastoma, human breast adenocarcinoma, and mouse melanoma cells.409 In vivo, injection of Aβ into mice transplanted with human glioblastoma and lung adenocarcinoma suppresses the tumor growth.410 In transgenic mice with the expression of human Aβ, the growth rates of implanted glioma tumor masses are inhibited by 40–50% compared to tumor masses in age-matched wild-type mice.411
A hypothesis has been proposed that Aβ may promote apoptosis, which contributes to its anti-tumor effects. Aβ42 peptides enhanced the transcription of p53, which is responsible for controlling cell apoptosis.412,413 In addition, Aβ42 induced oxidative stress and decreased the expression of X-linked inhibitor of apoptosis (XIAP), which directly inhibited key proteases of the apoptosis pathway including caspase 3, 7 and 9.414,415 Bcl-2, another key anti-apoptotic protein, was also shown to be blocked by Aβ42 peptides.416 In contrast, Aβ42 stimulated the expression of Bax, which induced cell apoptosis and was commonly observed in many cancers.416,417
Inhibition of oxidative stress
A large amount of studies have shown the anti-oxidant properties of Aβ peptides.418,419,420 Both Aβ40 and Aβ42 in physiological concentrations prevented lipoprotein oxidation in CSF and plasma.364,421 In addition, the increased generation of Aβ by cells from Alzheimer’s patients with mutant PSEN1 was accompanied by a reduction of ROS levels.422 Conversely, application of Aβ to primary hippocampal neurons from PSEN1 mutant knock-in mice significantly increased superoxide production.423 Physiological amounts (picomolar concentrations) of Aβ peptides could function as anti-oxidants by inhibiting redox metals, such as Cu, Fe and Zn to bind with ligands in redox cycling.364 The absence of Aβ in neurons may inhibit adequate chelation of metal ions and appropriate removal of O2-, resulting in an increased rather than a reduced oxidative stress.424 Thus, the physiological anti-oxidant activity of Aβ peptides should be taken into account when designing therapeutic drugs to lower Aβ levels.
Stimulation of neurogenesis
Adult neurogenesis in humans was first reported in 1998, in which bromodeoxyuridine (BrdU)-positive cells were found in the post-mortem brain tissue of cancer patients.425 Adult brains contain resident neural stem/progenitor cells (NSPCs), which have multipotency and show great potential for self-renewal.426,427 Adult neurogenesis in AD brains was also widely investigated. Compared with brain tissues from non-demented individuals, AD brains had increased expressions of DCX, PSA-NCAM, TOAD-64/Ulip/CRMP (TUC-4) and NeuroD, indicating the enhanced neurogenesis.428 However, some contradictory results have also been reported. It has been demonstrated that the expression of microtubule-associated protein (MAP) isoforms MAP2a, a marker of the mature neuron, was dramatically decreased in the dentate gyrus of human AD brains, indicating a reduction of neuronal maturation in the hippocampus.429 Another study also found a reduced number of DCX- and Sox2-positive cells in the AD hippocampus as compared with non-demented controls.430 Furthermore, a study including 45 Alzheimer’s patients between 52 and 97 years of age identified that the number of DCX-positive cells declined with the neuropathological progression.431 Growing evidence has shown the effects of Aβ on neurogenic process using NSPCs.359,432 Both Aβ40 and Aβ42 peptides have been identified to induce the proliferation and differentiation of neural progenitor cells (NPCs).359,432 Aβ40 mainly drived differentiation of NPCs into neurons, differing from Aβ42, which increased glia markers in NPCs.359 It has been identified that Aβ peptides stimulate neurogenesis in the subventricular zone (SVZ) through interacting with the p75 neurotrophin receptors in adult mice.433
Maintenance of BBB integrity
The blood-brain barrier (BBB) contributes to a stable brain microenvironment and normal neuronal function. Although neurotoxic Aβ aggregates play a key pathological role in the damage of the BBB, a low level of Aβ peptides may act as a seal to maintain the integrity of the BBB.434 This hypothesis is supported by the role of Aβ as a metal chelating antioxidant to maintain structural integrity under stress conditions.435 The ability of binding with copper ion or extracellular matrix molecules allows Aβ with its small size to be an excellent candidate molecule, which could form a “scab” in the brain. Thus, a rapid generation and deposition of Aβ in stroke and after head trauma, which could benefit to maintain the BBB integrity and inhibit the leakage of serum components into the brain, leading to neuroinflammation.436
Insufficient specificity
γ-secretase has dozens of substrates. Previous clinical trials of γ-secretase inhibitors have failed, in large part due to the toxicity induced by lack of substrate-specific inhibition. Particularly notable is toxicity resulting from inhibition of Notch-1 cleavage, which disrupts essential signaling from this receptor.15,313 Thus, we should discover compounds that act as substrate-selective γ-secretase inhibitors, which block the cleavage of C99, the immediate precursor of Aβ, while allowing Notch cleavage to proceed unimpeded. Recently, a study showed that verteporfin only bound with the APP transmembrane domain rather than the transmembrane domain of the Notch-1 receptor, indicating its inhibitory effect is in a C99-specific manner.437 Our study also showed that PSEN1S169del (a deletion mutation in PSEN1 gene exon 6) has distinct effects on APP processing and Notch1 cleavage.39 This AD pathogenic mutation altered APP processing and Aβ generation without affecting Notch-1 cleavage and Notch signaling in vitro and in vivo. The results indicate that serine169 in PS1 could be a critical site as a potential target for the development of novel γ-secretase modulators without affecting Notch-1 cleavage to treat AD.
A lack of selectivity is also a significant barrier to the therapeutic application of BACE1 inhibitors in AD. For instance, BACE2 is a close homolog of BACE1 but plays a neuroprotective role by inhibiting the amyloidogenic pathway of APP processing7,8,10 and reducing potassium channel Kv2.1-induced neuronal apoptosis.438 Thus, a non-selective BACE1 inhibitor also inhibits BACE2’s protective functions, leading to off target side effects. Although the aspartyl protease family (e.g. BACE2, pepsin, renin, cathepsin D and cathepsin E) has conserved catalytic aspartic acid residues, the subsites in the active sites may be unique.439 Targeting these subsites to develop BACE1 inhibitors may increase their specificity. Aβ-targeting antibodies also show off-target effects. A recent study identified that antibodies with Fc fragment reduced Aβ burden but also induced the engulgment of neuronal synapses by activating complement receptor 3 (CR3) or Fcγ receptor IIB (FcγRIIB), which exacerbates cognitive impairment in AD mice.440
Lack of accurate animal models
AD can be classified into a genetic and sporadic form of the disease.441 More than 99% of AD cases occur at an age >60 years in a sporadic manner, potentiated by various risk factors related to lifestyle.442 Less than 1% of all AD cases are early-onset with symptoms developed at an age of 50 s and earlier, and caused by gene mutations in APP, PSEN1 or PSEN2.7,38,39 In order to study Alzheimer’s pathogenesis and therapeutic strategies, better animal models to recapitulate the natural process of the disease are required.443,444 Many transgenic mouse models have been developed and commonly used, including the mice containing mutations in the APP (e.g. Tg2576,445 APP SweDI,446 APP23,447 J20448 and TgCRND8449 mice), PSEN1 (e.g. PS1A246E,450 PS1M146L451), PSEN2 (PS2N141I452,453 mice) or combinations (e.g. APP23xPS1-R278I,454 APP/PS1,455 APPSwe/PSEN1dE9,456,457 APP23/PS45 (APPSwe/PS1G384A),119,458,459 5xFAD (APP SwFILon, PSEN1 M146L, L286V)460 and ARTE10461 mice). Although the human tau gene MAPT mutations per se only cause frontotemporal dementia (FTD) rather than AD,462 tau mediates Aβ toxicity to promote the pathological process of AD.92,137 The interaction between Aβ and tau is under investigation by the generation of transgenic mouse models expressing human tau and APP, including APP/PS1/rTg21221,463 3xTg-AD (APP Swedish, MAPT P301L and PSEN1 M146V)464 and PLB1-triple465 mice. To avoid the “random integration” problem occurring in the transgenic mice, knock-in mice are generated in place to precisely target a specific locus. AD knock-in/out mice have been employed, including APP knock-in/out,466,467 APPNL-F knock-in,468 APPNL-G-F knock-in468 and APPNL-G-F/MAPT double knock-in469,470 mice. However, such mouse models only mimic the familial AD with an early onset of the disease. The late-onset sporadic AD is induced by a combination of genetic (e.g. Apolipoprotein E4 and TREM-2),101,102 lifestyle and environmental factors.471,472,473 Unfortunately, the current animal models are unable to exactly reflect this complexity, such as aging, which is the major risk factor of sporadic AD. The immune system has long been implicated as an important factor in Alzheimer’s development.474 However, murine immune system is notably different from humans.475 Furthermore, the extensive neuronal loss in AD patients has not been replicated in the murine models.476 Thus, a lack of accurate disease models leads to a translational gap between animal research and the clinical setting. Design and exploration of patient-based research models will be required, which will be further discussed in Section “Perspective and Future Direction”.
Late application
PET imaging allows us to visualize Aβ fibrils in patients, which accumulate in an Alzheimer’s brain as early as 15 years before the onset of symptoms.477 A change in CSF Aβ levels can be detected even up to 25 years before a patient begins to show symptoms.478 Thus, the current application of Aβ therapies may be too late for symptomatic patients, whose therapeutic window has already closed. Compared with curing the disease, prevention by reducing the risk of Alzheimer’s development is believed to be more practical. Prevention trials stand a chance to prevent or slow the progression of cognitive decline and dementia in AD. In 2012, DIAN-TU launched the first prevention trial focusing on two drugs: gantenerumab (against Aβ aggregates) and solanezumab (against soluble Aβ monomers).337 The data showed that gantenerumab had a positive impact on the reduction of cortical amyloid, leading to its further study by an exploratory open-label extension (OLE).338 Crenezumab is the first immunotherapy to be evaluated in the Alzheimer’s Prevention Initiative.343 The participants in this trial were carriers of the autosomal-dominant gene mutation (e.g. PSEN1 E280A) but did not meet the criteria for mild cognitive impairment at the time of enrollment.341 Although crenezumab did not significantly improve cognitive impairment in the participants, it showed some favorable effects (Alzheimer’s Association International Conference, 2022). Discovery of new biomarkers to discriminate the very early stage of sporadic AD is essential for the success of AD prevention.
Perspective and future direction
Although the failed trials have fueled debate on the amyloid hypothesis and raised concerns as to if efforts have been properly directed, it has provided valuable lessons to learn from and information that may improve our understanding of Alzheimer’s pathogenesis and drug development. The following are some principle and practical approaches we believe could be beneficial for future Aβ-targeted drug development and therapy.
Combination therapy and mechanism-based therapy
Some current therapeutic approaches, such as BACE inhibitors and γ-secretase inhibitors/modulators, aim to target Aβ production, which is the early stage of the amyloid cascade.304,305,306 Although these inhibitors have been identified to slow down the plaque formation in patients, they were unable to clear the existing Aβ plaques and ameliorate toxic events already initiated by these Aβ aggregates. Accordingly, combination therapy should be considered for the clinical phase of the disease, which is already the standard of care for many diseases, including rheumatoid arthritis and HIV/AIDS.479,480 Growing evidence indicates that Aβ accumulation stimulates tau phosphorylation and fibrillary tangle formation, leading to the process of neurodegeneration.112,113,114 Thus, additional application of tau-phosphorylating kinase inhibitors or compounds that inhibit tau aggregation and/or promote aggregate disassembly should be beneficial. APP and Aβ can be imported into mitochondria, where they can interact with mitochondrial components, impair ATP production, and increase oxidative damage.481,482 Antioxidants such as lipoic acid,483 vitamin E,484,485 vitamin C486 and β-carotene487 may also be the promising combination approaches for AD. In addition, Aβ’s role in the modulation of synapse function has attracted great attention. The neurotoxic soluble Aβ oligomers have been identified to affect synaptic plasticity and synaptic transmission in various AD animal models.488 Targeting synapse loss and dysfunction may be an effective AD treatment strategy.489 Once the pathological cascade has begun, combination therapy targeting multiple AD pathologies will be more effective than a single therapy, which only addresses one abnormal factor.
Growing evidence shows that elevation of brain Aβ levels in AD could be the consequence of upstream problems including neurovascular dysfunction, disturbed glucose homeostasis, failed control of cell cycle and inflammation.490,491,492 Autophagy, a part of the lysosomal system, is crucial for clearance of toxic accumulated proteins and damage organelles. The autophagic process consists of several steps including sequestration, elongation, maturation, fusion and degradation, aiming to deliver unwanted proteins, organelles and cellular debris to the lysosome for degration. It starts with the formation of phagophore, which then elongates and encloses the cargo to form an autophagosome. The autophagosome either directly fuses with the lysosome form an autolysosome or firstly fuses with late endosomes to form amphisomes, which subsequently fuse with lysosomes. Impairment of the autophagy-lysosomal system has been considered as one of the fundamental causes for many neurodegenerative diseases that feature the deposition of toxic amyloid proteins. Growing evidence shows that dysfunction of autophagy is closely linked with Aβ metabolism and accmulation in AD progression. Autophagy is implicated in Aβ metabolism likely via modulation of its production, secretion and clearance. Aβ originates from the cleavage of its precursor protein APP by β-secretase (BACE1) and γ-secretase. It has been identified that ATG5-dependent autophagy regulates APP degradation.493 In addition, the complex of APP and γ-secretases was found in autophagosomes, suggesting the role of authophgic pathway in the generation of Aβ peptides.494 Autophagy is also required for Aβ secretion. ATG7 is an essential molecule for the autophagosome formation. AD model mice with ATG7 KO showed deficient autophagy associated with drastically reduced extracellular Aβ plaques and markedly accumulated intraneuronal Aβ, suggesting that Aβ secretion was compromised due to the impaired autophagy.495,496 In addition, autophagy regulates the clearance of Aβ peptides. The cysteine protease cathepsin B (CatB) is a key lysosomal protease required for degrading autophagic substrates. It has been demonstrated that genetic deletion of CatB significantly increased Aβ42 burden and worsened amyloid deposition in AD mice, whereas overexpression of CatB reduced amyloid plaques.497 Accumulation of immature autophagosome in dystrophic neurites has been observed in the brain of Alzheimer’s patients due to the defective axonal transportation of autophagosomes.498 Thus, autophagy modulation becomes a promising stategy for Alzheimer’s treatment.499,500 Rapamycin is a commonly used autophagy activator, which inhibits the mTOR pathway by binding with immunophilin FK506-binding protein (FKBP12).501 Recent studies identified that 3xTg-AD mice had enhanced mTOR activity in the hippocampus and neocortex, two areas known to have high concentrations of Aβ plaques.502 Treatment with rapamycin significantly stimulated autophagy associated with markedly reduced both intracellular Aβ and extracellular amyloid deposition in brains as well as improved cognitive deficits in AD mice.503,504
Mechanism-based therapies to target these pathological processes will have optimal benefit when initiated in the asymptomatic stage. Traditional Chinese medicine (TCM) has been established in the Chinese health care system for thousands of years. Most TCM treatment are derived from natural products with multi-target, multi-pathway capacity and mild adverse events. It has preventive and therapeutic effects on many chronic diseases such as cancer, allergy, diabetes and infections by the regulation of cell growth and differentiation, reduction of inflammation, or increase of carbohydrate utilization.505,506,507,508 TCM treatment such as morroniside, rutin, resveratrol, triptolide and berberine have already shown their beneficial effects for AD509,510,511,512,513,514,515,516,517,518,519,520,521,522,523,524,525,526,527 (Table 3).
Patient-based research models
Three-dimensional brain organoids derived from human pluripotent stem cells (hPSCs) have shown significant advantages in modeling neurological disorders including autism, microcephaly and Parkinson’s disease.528,529,530 Three methods have been established to recapitulate Alzheimer’s phenotype in brain organoids: application of Aftin-5 (an Aβ42 agonist) to induce Aβ42 production in brain organoids;531 generation of brain organoids from induced pluripotent stem cells (iPSCs) of familial AD patients;532,533 and creation of differentiated sporadic Alzheimer’s brain organoids by converting APOE3 to APOE4 in patient-derived iPSCs.534 Unlike cell models, AD brain organoids are capable of generating the blood-brain barrier (BBB) as well as connections with other organs.535 This enables them to potentially function as a superior approach in the understanding Alzheimer’s pathogenesis, as well as a better tool for exploration of Alzheimer’s modification. In addition, transplantation of brain organoids may be a novel way to recover neuronal function and neural network after neuronal death during AD.536 However, some limitations still exist and will need to be improved upon. So far, brain organoids can only be cultured within six months, otherwise volume shrinkage and cellular apoptosis occur as neither the oxygen nor nutrients will be able to reach the innermost organoid regions. This limitation leads to the concern that brain organoids are unable to grow “old” enough to mimic the aging human brain. To address this issue, obtaining brain organoids with a vascular system becomes a critical issue.537,538
Identification of early Alzheimer’s biomarkers
Biomarkers that can identify patients at very early stages of AD will greatly benefit the development of disease-modifying therapies.539 In addition to the typical pathologies (e.g. Aβ and tau), other molecules associated with inflammation, synaptic plasticity, may also serve as the accurate and specific biomarkers for early diagnosing AD.540,541,542 Progranulin is a growth factor expressed in neurons and microglia, which modulates neuroinflammatory to reduce microgliosis and astrogliosis.543 It has been observed that the CSF level of progranulin elevates as early as ten years before the presentation of symptoms in patients with familial or sporadic AD.544 Neurogranin is expressed in the cortex and hippocampus, the brain areas most affected by AD.545 As a synaptic marker, it is involved in the modulation of synaptic strength and plasticity.546 Several studies have revealed an elevation of CSF neurogranin in AD and MCI individuals compared to healthy controls.547,548 The CSF neurogranin levels correlated with the brain amyloid load in patients with preclinical AD. It can also successfully predict the rates of cognitive decline in both early Alzheimer’s patients and cognitively healthy controls. In contrast, there is a significant reduction of plasma neuronal-derived exosomal neurogranin in AD patients compared with the healthy controls.549 More importantly, the CSF neurogranin increases exclusively in AD patients and has not been observed in other neurodegenerative disorders, such as frontotemporal dementia or Parkinson’s disease.550
MicroRNA (miRNA) are noncoding RNA molecules of 20–25 nucleotides that can manipulate gene expression post-transcriptionally by binding to the 3ʹ-untranslated region (3ʹUTR) of mRNA to block protein translation or accelerating the degradation of target mRNAs. It has been found that miRNAs are involved in Alzheimer’s pathogenesis and are easily detected in body fluids, including CSF, plasma and serum. Therefore, they become an attractive target for developing AD biomarkers.551,552 In addition to body fluids, ocular markers also gain increasing interest. Abundant evidence from animal and clinical studies shows a correlation between ocular pathology and AD development.553,554,555 A recent study also suggests that depressive symptoms in middle-aged individuals correlates with time to onset of cognitive decline, suggesting the role of psychiatric disorders as early markers of Alzheimer’s disease.556
Conclusions
Since Aβ aggregates act as the unique specific pathological hallmark of AD and play a causative role in the disease development, they are believed as a promising target for Alzheimer’s modification. Most Aβ-targeting drug trials have failed as a consequence by lack of sufficient specificity and accurate translational models, loss of Aβ physiological homeostasis, and failure to be administered during the best therapeutic window. Nevertheless, learning from these failures will be beneficial to the design of better therapeutic approaches. Biomarkers are needed for identifying patients with preclinical Alzheimer’s disease so that treatment such as mechanism-based therapy could prevent or slow down the disease. Translational models and tools to mimic the nature of AD more closely are also required to bridge the gap between basic research and the clinical practice. Combination therapy that targets different mechanisms and pathologies would be directed by biomarkers and customized to the individual. We hope that these solutions could pave the way for exploration and development of more refined Aβ-based therapy for AD.
References
Glenner, G. G. & Wong, C. W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys. Res Commun. 120, 885–890 (1984).
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992).
Lemere, C. A. & Masliah, E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat. Rev. Neurol. 6, 108–119 (2010).
Liu, X. et al. Clusterin transduces Alzheimer-risk signals to amyloidogenesis. Signal Transduct. Target Ther. 7, 325 (2022).
Sun, B. L. et al. Critical thinking on amyloid-beta-targeted therapy: challenges and perspectives. Sci. China Life Sci. 64, 926–937 (2021).
Karran, E. & Hardy, J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 76, 185–205 (2014).
Deng, Y. et al. Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur. J. Neurosci. 37, 1962–1969 (2013).
Zhang, S. et al. BACE1 cleavage site selection critical for amyloidogenesis and Alzheimer’s pathogenesis. J. Neurosci. 37, 6915–6925 (2017).
Sun, X. et al. Distinct transcriptional regulation and function of the human BACE2 and BACE1 genes. FASEB J. 19, 739–749 (2005).
Sun, X., He, G. & Song, W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 20, 1369–1376 (2006).
Liu, X., Wang, Z., Wu, Y., Wang, J. & Song, W. BACE2 degradation mediated by the macroautophagy-lysosome pathway. Eur. J. Neurosci. 37, 1970–1977 (2013).
Li, Y. M. et al. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc. Natl Acad. Sci. USA 97, 6138–6143 (2000).
De Strooper, B. et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 (1999).
De Strooper, B. et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 (1998).
Song, W. et al. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc. Natl Acad. Sci. USA 96, 6959–6963 (1999).
Zhang, Z. et al. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat. Cell Biol. 2, 463–465 (2000).
Takasugi, N. et al. The role of presenilin cofactors in the gamma-secretase complex. Nature 422, 438–441 (2003).
Zhang, S., Zhang, M., Cai, F. & Song, W. Biological function of Presenilin and its role in AD pathogenesis. Transl. Neurodegener. 2, 15 (2013).
Bateman, R. J. et al. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861 (2006).
Engelhardt, B. et al. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 132, 317–338 (2016).
Sweeney, M. D., Sagare, A. P. & Zlokovic, B. V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150 (2018).
Yamada, K. et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J. Biol. Chem. 283, 34554–34562 (2008).
Elali, A. & Rivest, S. The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer’s disease. Front Physiol. 4, 45 (2013).
Deane, R. et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med 9, 907–913 (2003).
Zlokovic, B. V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201 (2008).
Mawuenyega, K. G. et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774 (2010).
Hampel, H. et al. The amyloid-beta pathway in Alzheimer’s Disease. Mol. Psychiatry 26, 5481–5503 (2021).
Tarasoff-Conway, J. M. et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470 (2015).
Silverberg, G. D., Mayo, M., Saul, T., Rubenstein, E. & McGuire, D. Alzheimer’s disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol. 2, 506–511 (2003).
Baranello, R. J. et al. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimer Res 12, 32–46 (2015).
Eckman, E. A. et al. Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J. Biol. Chem. 281, 30471–30478 (2006).
Iwata, N. et al. Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550–1552 (2001).
Sims, R., Hill, M. & Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 23, 311–322 (2020).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458 (2013).
Glenner, G. G. & Wong, C. W. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys. Res Commun. 122, 1131–1135 (1984).
Burger, P. C. & Vogel, F. S. The development of the pathologic changes of Alzheimer’s disease and senile dementia in patients with Down’s syndrome. Am. J. Pathol. 73, 457–476 (1973).
Lemere, C. A. et al. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 3, 16–32 (1996).
Goate, A. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706 (1991).
Zhang, S. et al. A presenilin-1 mutation causes Alzheimer disease without affecting Notch signaling. Mol. Psychiatry 25, 603–613 (2020).
Jonsson, T. et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99 (2012).
DeMattos, R. B. et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41, 193–202 (2004).
Ridler, C. Alzheimer Disease: Misfolded diabetes-mellitus peptide seeds amyloid-beta aggregation. Nat. Rev. Neurol. 13, 128 (2017).
Fewlass, D. C. et al. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 18, 1870–1878 (2004).
Ashok, A., Rai, N. K., Tripathi, S. & Bandyopadhyay, S. Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 143, 64–80 (2015).
Brown, B. M. et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing. Mol. Psychiatry 18, 875–881 (2013).
Sevigny, J. et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56 (2016).
Alexander, G. C., Emerson, S. & Kesselheim, A. S. Evaluation of aducanumab for Alzheimer disease: scientific evidence and regulatory review involving efficacy, safety, and futility. JAMA 325, 1717–1718 (2021).
Dunn, B., Stein, P. & Cavazzoni, P. Approval of aducanumab for Alzheimer disease-The FDA’s perspective. JAMA Intern Med 181, 1276–1278 (2021).
Mintun, M. A., Wessels, A. M. & Sims, J. R. Donanemab in early Alzheimer’s disease. Reply. N. Engl. J. Med 385, 667 (2021).
van Dyck, C. H. et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 388, 9–21 (2023).
Lahmy, V. et al. Blockade of Tau hyperphosphorylation and Abeta(1)(-)(4)(2) generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and sigma(1) receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 38, 1706–1723 (2013).
Hampel, H. et al. A precision medicine framework using artificial intelligence for the identification and confirmation of genomic biomarkers of response to an Alzheimer’s disease therapy: Analysis of the blarcamesine (ANAVEX2-73) Phase 2a clinical study. Alzheimers Dement (N. Y.) 6, e12013 (2020).
Zhang, Y. & Song, W. Islet amyloid polypeptide: Another key molecule in Alzheimer’s pathogenesis? Prog. Neurobiol. 153, 100–120 (2017).
Masters, C. L. & Beyreuther, K. Henryk M. Wisniewski and the amyloid theory of Alzheimer’s disease. J. Alzheimers Dis. 3, 83–86 (2001).
Mott, R. T. & Hulette, C. M. Neuropathology of Alzheimer’s disease. Neuroimaging Clin. N. Am. 15, 755–765 (2005). ix.
Walker, L. C. Abeta plaques. Free Neuropathol. 1, 31 (2020).
Serrano-Pozo, A., Frosch, M. P., Masliah, E. & Hyman, B. T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med 1, a006189 (2011).
Iwatsubo, T., Saido, T. C., Mann, D. M., Lee, V. M. & Trojanowski, J. Q. Full-length amyloid-beta (1-42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am. J. Pathol. 149, 1823–1830 (1996).
Duyckaerts, C., Delatour, B. & Potier, M. C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36 (2009).
Liu, F. et al. Focal-type, but not diffuse-type, amyloid beta plaques are correlated with alzheimer’s neuropathology, cognitive dysfunction, and neuroinflammation in the human hippocampus. Neurosci. Bull. 38, 1125–1138 (2022).
Lambert, M. P. et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl Acad. Sci. USA 95, 6448–6453 (1998).
Bode, D. C., Freeley, M., Nield, J., Palma, M. & Viles, J. H. Amyloid-beta oligomers have a profound detergent-like effect on lipid membrane bilayers, imaged by atomic force and electron microscopy. J. Biol. Chem. 294, 7566–7572 (2019).
Yasumoto, T. et al. High molecular weight amyloid beta(1-42) oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 33, 9220–9234 (2019).
Hong, S. et al. Soluble Abeta oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82, 308–319 (2014).
DelBove, C. E. et al. Reciprocal modulation between amyloid precursor protein and synaptic membrane cholesterol revealed by live cell imaging. Neurobiol. Dis. 127, 449–461 (2019).
Sathya, M. et al. Resveratrol intervenes cholesterol- and isoprenoid-mediated amyloidogenic processing of AbetaPP in familial Alzheimer’s disease. J. Alzheimers Dis. 60, S3–S23 (2017).
Xiong, H. et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis. 29, 422–437 (2008).
Kojro, E., Gimpl, G., Lammich, S., Marz, W. & Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. USA 98, 5815–5820 (2001).
Kim, Y., Kim, C., Jang, H. Y. & Mook-Jung, I. Inhibition of cholesterol biosynthesis reduces gamma-secretase activity and amyloid-beta generation. J. Alzheimers Dis. 51, 1057–1068 (2016).
Panchal, M. et al. Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res 51, 598–605 (2010).
Terakawa, M. S. et al. Impact of membrane curvature on amyloid aggregation. Biochim. Biophys. Acta Biomembr. 1860, 1741–1764 (2018).
Matsuzaki, K. Formation of toxic amyloid fibrils by amyloid beta-protein on ganglioside clusters. Int J. Alzheimers Dis. 2011, 956104 (2011).
Henry, S. et al. Interaction of Abeta(1–42) peptide or their variant with model membrane of different composition probed by infrared nanospectroscopy. Nanoscale 10, 936–940 (2018).
Yang, D. S. et al. Cyclodextrin has conflicting actions on autophagy flux in vivo in brains of normal and Alzheimer model mice. Hum. Mol. Genet 26, 843–859 (2017).
DeKosky, S. T. & Scheff, S. W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464 (1990).
Shankar, G. M. et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med 14, 837–842 (2008).
Townsend, M., Shankar, G. M., Mehta, T., Walsh, D. M. & Selkoe, D. J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J. Physiol. 572, 477–492 (2006).
Luscher, C. & Malenka, R. C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 4, a005710 (2012).
Magdesian, M. H. et al. Amyloid-beta binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling. J. Biol. Chem. 283, 9359–9368 (2008).
Li, S. et al. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31, 6627–6638 (2011).
Lu, W. et al. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268 (2009).
Terashima, A., Suh, Y. H. & Isaac, J. T. R. The AMPA receptor subunit GluA1 is required for CA1 hippocampal long-term potentiation but is not essential for synaptic transmission. Neurochem Res 44, 549–561 (2019).
Diering, G. H. & Huganir, R. L. The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329 (2018).
Wenthold, R. J., Petralia, R. S., Blahos, J. II & Niedzielski, A. S. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 16, 1982–1989 (1996).
Hsieh, H. et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843 (2006).
Zhang, Y., Guo, O., Huo, Y., Wang, G. & Man, H. Y. Amyloid-beta Induces AMPA Receptor Ubiquitination and Degradation in Primary Neurons and Human Brains of Alzheimer’s Disease. J. Alzheimers Dis. 62, 1789–1801 (2018).
Moreno, H. et al. Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc. Natl. Acad. Sci. USA 106, 5901–5906 (2009).
Pigino, G. et al. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc. Natl. Acad. Sci. USA 106, 5907–5912 (2009).
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C. & Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 (2004).
Zhou, Q., Homma, K. J. & Poo, M. M. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757 (2004).
Chabrier, M. A., Cheng, D., Castello, N. A., Green, K. N. & LaFerla, F. M. Synergistic effects of amyloid-beta and wild-type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer’s disease. Neurobiol. Dis. 64, 107–117 (2014).
Ittner, L. M. et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397 (2010).
Marcatti, M. et al. Abeta/tau oligomer interplay at human synapses supports shifting therapeutic targets for Alzheimer’s disease. Cell Mol. Life Sci. 79, 222 (2022).
Tai, H. C. et al. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol. Commun. 2, 146 (2014).
Kaniyappan, S., Chandupatla, R. R., Mandelkow, E. M. & Mandelkow, E. Extracellular low-n oligomers of tau cause selective synaptotoxicity without affecting cell viability. Alzheimers Dement 13, 1270–1291 (2017).
Koch, G. et al. Reversal of LTP-like cortical plasticity in Alzheimer’s disease patients with tau-related faster clinical progression. J. Alzheimers Dis. 50, 605–616 (2016).
Zhou, L. et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 8, 15295 (2017).
Liu, C., Song, X., Nisbet, R. & Gotz, J. Co-immunoprecipitation with Tau Isoform-specific Antibodies Reveals Distinct Protein Interactions and Highlights a Putative Role for 2N Tau in Disease. J. Biol. Chem. 291, 8173–8188 (2016).
Moreno, H. et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 325, 30–38 (2016).
Mondragon-Rodriguez, S. et al. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 287, 32040–32053 (2012).
Zhao, X. et al. Caspase-2 cleavage of tau reversibly impairs memory. Nat. Med. 22, 1268–1276 (2016).
Shipton, O. A. et al. Tau protein is required for amyloid beta-induced impairment of hippocampal long-term potentiation. J. Neurosci. 31, 1688–1692 (2011).
Pallo, S. P., DiMaio, J., Cook, A., Nilsson, B. & Johnson, G. V. W. Mechanisms of tau and Abeta-induced excitotoxicity. Brain Res. 1634, 119–131 (2016).
Grundke-Iqbal, I. et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 83, 4913–4917 (1986).
Ihara, Y., Nukina, N., Miura, R. & Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem 99, 1807–1810 (1986).
Goedert, M., Spillantini, M. G., Cairns, N. J. & Crowther, R. A. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168 (1992).
Greenberg, S. G., Davies, P., Schein, J. D. & Binder, L. I. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J. Biol. Chem. 267, 564–569 (1992).
Oddo, S. et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421 (2003).
Busche, M. A. & Hyman, B. T. Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat. Neurosci. 23, 1183–1193 (2020).
Jack, C. R. et al. The bivariate distribution of amyloid-beta and tau: relationship with established neurocognitive clinical syndromes. Brain 142, 3230–3242 (2019).
Hanseeuw, B. J. et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: A longitudinal study. JAMA Neurol. 76, 915–924 (2019).
Ising, C. et al. NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673 (2019).
Zhang, Y., Dong, Z. & Song, W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target Ther. 5, 37 (2020).
He, Z. et al. Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 24, 29–38 (2018).
Pickett, E. K. et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 29, 3592–3604 (2019). e3595.
Plattner, F., Angelo, M. & Giese, K. P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 281, 25457–25465 (2006).
Hernandez, P., Lee, G., Sjoberg, M. & Maccioni, R. B. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25–35): involvement of lipid rafts. J. Alzheimers Dis. 16, 149–156 (2009).
Terwel, D. et al. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 172, 786–798 (2008).
Ly, P. T. et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest 123, 224–235 (2013).
Iijima, K., Gatt, A. & Iijima-Ando, K. Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer’s disease. Hum. Mol. Genet 19, 2947–2957 (2010).
Liu, F., Iqbal, K., Grundke-Iqbal, I. & Gong, C. X. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 530, 209–214 (2002).
Mazanetz, M. P. & Fischer, P. M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Disco. 6, 464–479 (2007).
Medina, M. & Avila, J. Glycogen synthase kinase-3 (GSK-3) inhibitors for the treatment of Alzheimer’s disease. Curr. Pharm. Des. 16, 2790–2798 (2010).
Zheng, W. H., Bastianetto, S., Mennicken, F., Ma, W. & Kar, S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115, 201–211 (2002).
Swatton, J. E. et al. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. Eur. J. Neurosci. 19, 2711–2719 (2004).
Salazar, S. V. & Strittmatter, S. M. Cellular prion protein as a receptor for amyloid-beta oligomers in Alzheimer’s disease. Biochem Biophys. Res Commun. 483, 1143–1147 (2017).
Kostylev, M. A. et al. Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple alzheimer mouse models. J. Biol. Chem. 290, 17415–17438 (2015).
Rezaie, P., Pontikis, C. C., Hudson, L., Cairns, N. J. & Lantos, P. L. Expression of cellular prion protein in the frontal and occipital lobe in Alzheimer’s disease, diffuse Lewy body disease, and in normal brain: an immunohistochemical study. J. Histochem. Cytochem. 53, 929–940 (2005).
Takahashi, R. H. et al. Accumulation of cellular prion protein within dystrophic neurites of amyloid plaques in the Alzheimer’s disease brain. Neuropathology 31, 208–214 (2011).
Velayos, J. L. et al. The cellular prion protein and its role in Alzheimer disease. Prion 3, 110–117 (2009).
Um, J. W. et al. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235 (2012).
Lau, D. H. et al. Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer’s disease. Acta Neuropathol. Commun. 4, 49 (2016).
Larson, M. et al. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857–16871a (2012).
Gamblin, T. C. et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 100, 10032–10037 (2003).
Shafiei, S. S., Guerrero-Munoz, M. J. & Castillo-Carranza, D. L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci. 9, 83 (2017).
Nilson, A. N. et al. Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J. Alzheimers Dis. 55, 1083–1099 (2017).
Bloom, G. S. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508 (2014).
Leroy, K. et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 181, 1928–1940 (2012).
Miyamoto, T. et al. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 12, 41 (2017).
Avila, J. Our working point of view of tau protein. J. Alzheimers Dis. 62, 1277–1285 (2018).
Campion, D., Pottier, C., Nicolas, G., Le Guennec, K. & Rovelet-Lecrux, A. Alzheimer disease: modeling an Abeta-centered biological network. Mol. Psychiatry 21, 861–871 (2016).
Petersen, R. C. et al. Neuropathologic features of amnestic mild cognitive impairment. Arch. Neurol. 63, 665–672 (2006).
Roberson, E. D. et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754 (2007).
Kaufman, A. C. et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 77, 953–971 (2015).
Nygaard, H. B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 83, 369–376 (2018).
Hanseeuw, B. J. et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann. Neurol. 81, 583–596 (2017).
Schultz, A. P. et al. Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J. Neurosci. 37, 4323–4331 (2017).
Albert, M. et al. Predicting progression from normal cognition to mild cognitive impairment for individuals at 5 years. Brain 141, 877–887 (2018).
Kazim, S. F. et al. Neuronal network excitability in alzheimer’s disease: the puzzle of similar versus divergent roles of amyloid beta and tau. eNeuro 8, ENEURO.0418-20.2020 (2021).
Busche, M. A. et al. Tau impairs neural circuits, dominating amyloid-beta effects, in Alzheimer models in vivo. Nat. Neurosci. 22, 57–64 (2019).
Ransohoff, R. M. How neuroinflammation contributes to neurodegeneration. Science 353, 777–783 (2016).
Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 8, 26 (2011).
Combs, C. K., Johnson, D. E., Karlo, J. C., Cannady, S. B. & Landreth, G. E. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 20, 558–567 (2000).
Griffin, W. S., Sheng, J. G., Roberts, G. W. & Mrak, R. E. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J. Neuropathol. Exp. Neurol. 54, 276–281 (1995).
Rich, J. B. et al. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology 45, 51–55 (1995).
Rogers, J., Luber-Narod, J., Styren, S. D. & Civin, W. H. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 9, 339–349 (1988).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Baik, S. H. et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 30, 493–507 (2019). e496.
Pastore, A., Raimondi, F., Rajendran, L. & Temussi, P. A. Why does the Abeta peptide of Alzheimer share structural similarity with antimicrobial peptides? Commun. Biol. 3, 135 (2020).
Arispe, N., Rojas, E. & Pollard, H. B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 90, 567–571 (1993).
Greter, M. & Merad, M. Regulation of microglia development and homeostasis. Glia 61, 121–127 (2013).
Glenn, J. A., Ward, S. A., Stone, C. R., Booth, P. L. & Thomas, W. E. Characterisation of ramified microglial cells: detailed morphology, morphological plasticity and proliferative capability. J. Anat. 180, 109–118 (1992).
Eyo, U. B. & Dailey, M. E. Microglia: key elements in neural development, plasticity, and pathology. J. Neuroimmune Pharm. 8, 494–509 (2013).
Madry, C. & Attwell, D. Receptors, ion channels, and signaling mechanisms underlying microglial dynamics. J. Biol. Chem. 290, 12443–12450 (2015).
Bolmont, T. et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292 (2008).
Baik, S. H., Kang, S., Son, S. M. & Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia 64, 2274–2290 (2016).
Grubman, A. et al. Transcriptional signature in microglia associated with Abeta plaque phagocytosis. Nat. Commun. 12, 3015 (2021).
Johansson, J. U. et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J. Clin. Invest 125, 350–364 (2015).
Griciuc, A. et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643 (2013).
Michelucci, A., Heurtaux, T., Grandbarbe, L., Morga, E. & Heuschling, P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 210, 3–12 (2009).
Hickman, S. E., Allison, E. K. & El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360 (2008).
Lai, A. Y. & McLaurin, J. Clearance of amyloid-beta peptides by microglia and macrophages: the issue of what, when and where. Future Neurol. 7, 165–176 (2012).
Hawkes, C. A. & McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl Acad. Sci. USA 106, 1261–1266 (2009).
Jay, T. R. et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med 212, 287–295 (2015).
Ulrich, J. D. et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 9, 20 (2014).
Tan, Y. J. et al. Higher Peripheral TREM2 mRNA Levels Relate to Cognitive Deficits and Hippocampal Atrophy in Alzheimer’s Disease and Amnestic Mild Cognitive Impairment. J. Alzheimers Dis. 58, 413–423 (2017).
Hu, N. et al. Increased expression of TREM2 in peripheral blood of Alzheimer’s disease patients. J. Alzheimers Dis. 38, 497–501 (2014).
Jay, T. R. et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37, 637–647 (2017).
Farhy-Tselnicker, I. & Allen, N. J. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev. 13, 7 (2018).
Wilhelmsson, U. et al. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl. Acad. Sci. USA 103, 17513–17518 (2006).
Brambilla, R. et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med 202, 145–156 (2005).
Brambilla, R. et al. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol. 182, 2628–2640 (2009).
van Tijn, P. et al. Mutant ubiquitin decreases amyloid beta plaque formation in a transgenic mouse model of Alzheimer’s disease. Neurochem Int 61, 739–748 (2012).
Vehmas, A. K., Kawas, C. H., Stewart, W. F. & Troncoso, J. C. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 24, 321–331 (2003).
Hughes, C. et al. Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun. Biol. 3, 79 (2020).
Yang, J., Wise, L. & Fukuchi, K. I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front Immunol. 11, 724 (2020).
Zhao, J., O’Connor, T. & Vassar, R. The contribution of activated astrocytes to Abeta production: implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 8, 150 (2011).
Allen, N. J. & Lyons, D. A. Glia as architects of central nervous system formation and function. Science 362, 181–185 (2018).
Wegiel, J. et al. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol. Aging 22, 49–61 (2001).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Lian, H. et al. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 36, 577–589 (2016).
Devine, M. J. & Kittler, J. T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80 (2018).
Ashrafi, G., de Juan-Sanz, J., Farrell, R. J. & Ryan, T. A. Molecular tuning of the axonal mitochondrial Ca(2+) uniporter ensures metabolic flexibility of neurotransmission. Neuron 105, 678–687 (2020). e675.
Guo, L., Tian, J. & Du, H. Mitochondrial dysfunction and synaptic transmission failure in Alzheimer’s disease. J. Alzheimers Dis. 57, 1071–1086 (2017).
Ryu, J. C., Zimmer, E. R., Rosa-Neto, P. & Yoon, S. O. Consequences of metabolic disruption in Alzheimer’s disease pathology. Neurotherapeutics 16, 600–610 (2019).
Ashleigh, T., Swerdlow, R. H. & Beal, M. F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement 19, 333–342 (2023).
Wang, X. et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103 (2009).
Park, J. et al. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem 132, 687–702 (2015).
Cho, D. H. et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105 (2009).
Wang, X. et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl Acad. Sci. USA 105, 19318–19323 (2008).
Shields, L. Y. et al. Mitochondrial fission is a critical modulator of mutant APP-induced neural toxicity. J. Biol. Chem. 296, 100469 (2021).
Fox, T. D. Mitochondrial protein synthesis, import, and assembly. Genetics 192, 1203–1234 (2012).
Sorrentino, V. et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552, 187–193 (2017).
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G. & Anandatheerthavarada, H. K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068 (2006).
Cenini, G., Rub, C., Bruderek, M. & Voos, W. Amyloid beta-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 27, 3257–3272 (2016).
Csordas, G., Weaver, D. & Hajnoczky, G. Endoplasmic reticulum-mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 28, 523–540 (2018).
Marchi, S., Patergnani, S. & Pinton, P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys. Acta 1837, 461–469 (2014).
Hedskog, L. et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl Acad. Sci. USA 110, 7916–7921 (2013).
Area-Gomez, E. et al. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 9, 335 (2018).
Schreiner, B., Hedskog, L., Wiehager, B. & Ankarcrona, M. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 43, 369–374 (2015).
Calvo-Rodriguez, M., Hernando-Perez, E., Nunez, L. & Villalobos, C. Amyloid beta oligomers increase ER-mitochondria Ca(2+) cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca(2+) remodeling. Front Cell Neurosci. 13, 22 (2019).
Pera, M. et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 36, 3356–3371 (2017).
Turrens, J. F. & Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 191, 421–427 (1980).
Sugioka, K. et al. Mechanism of O2- generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochim. Biophys. Acta 936, 377–385 (1988).
Balaban, R. S., Nemoto, S. & Finkel, T. Mitochondria, oxidants, and aging. Cell 120, 483–495 (2005).
Chakrabarti, S. et al. Mitochondrial dysfunction during brain aging: role of oxidative stress and modulation by antioxidant supplementation. Aging Dis. 2, 242–256 (2011).
Smith, M. A., Harris, P. L., Sayre, L. M. & Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl Acad. Sci. USA 94, 9866–9868 (1997).
Mao, P. & Reddy, P. H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochim Biophys. Acta 1812, 1359–1370 (2011).
Nakamura, M. et al. Three histidine residues of amyloid-beta peptide control the redox activity of copper and iron. Biochemistry 46, 12737–12743 (2007).
Bousejra-ElGarah, F., Bijani, C., Coppel, Y., Faller, P. & Hureau, C. Iron(II) binding to amyloid-beta, the Alzheimer’s peptide. Inorg. Chem. 50, 9024–9030 (2011).
Leuner, K. et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal 16, 1421–1433 (2012).
Snyder, E. M. et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8, 1051–1058 (2005).
Busche, M. A. et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689 (2008).
Malinow, R. New developments on the role of NMDA receptors in Alzheimer’s disease. Curr. Opin. Neurobiol. 22, 559–563 (2012).
Brito-Moreira, J. et al. Abeta oligomers induce glutamate release from hippocampal neurons. Curr. Alzheimer Res 8, 552–562 (2011).
Kullmann, D. M. & Lamsa, K. P. Long-term synaptic plasticity in hippocampal interneurons. Nat. Rev. Neurosci. 8, 687–699 (2007).
Li, S. et al. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801 (2009).
Talantova, M. et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl Acad. Sci. USA 110, E2518–E2527 (2013).
Shankar, G. M. et al. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875 (2007).
Wei, W. et al. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196 (2010).
Reinders, N. R. et al. Amyloid-beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. USA 113, E6526–E6534 (2016).
Chang, E. H. et al. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc. Natl. Acad. Sci. USA 103, 3410–3415 (2006).
Geula, C. & Mesulam, M. M. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb. Cortex 6, 165–177 (1996).
Kerbler, G. M. et al. Basal forebrain atrophy correlates with amyloid beta burden in Alzheimer’s disease. Neuroimage Clin. 7, 105–113 (2015).
Grothe, M. J., Heinsen, H., Amaro, E. Jr., Grinberg, L. T. & Teipel, S. J. Cognitive Correlates of Basal Forebrain Atrophy and Associated Cortical Hypometabolism in Mild Cognitive Impairment. Cereb. Cortex 26, 2411–2426 (2016).
Chiesa, P. A. et al. Relationship between basal forebrain resting-state functional connectivity and brain amyloid-beta deposition in cognitively intact older adults with subjective memory complaints. Radiology 290, 167–176 (2019).
Beach, T. G., Honer, W. G. & Hughes, L. H. Cholinergic fibre loss associated with diffuse plaques in the non-demented elderly: the preclinical stage of Alzheimer’s disease? Acta Neuropathol. 93, 146–153 (1997).
Lai, M. K. et al. Selective effects of the APOE epsilon4 allele on presynaptic cholinergic markers in the neocortex of Alzheimer’s disease. Neurobiol. Dis. 22, 555–561 (2006).
Chhatwal, J. P. et al. Preferential degradation of cognitive networks differentiates Alzheimer’s disease from ageing. Brain 141, 1486–1500 (2018).
Buckley, R. F. et al. Functional network integrity presages cognitive decline in preclinical Alzheimer disease. Neurology 89, 29–37 (2017).
Hampton, O. L. et al. Resting-state functional connectivity and amyloid burden influence longitudinal cortical thinning in the default mode network in preclinical Alzheimer’s disease. Neuroimage Clin. 28, 102407 (2020).
Morrissey, Z. D. et al. Hippocampal functional connectivity across age in an App knock-in mouse model of Alzheimer’s disease. Front Aging Neurosci. 14, 1085989 (2022).
He, X. et al. Abnormal salience network in normal aging and in amnestic mild cognitive impairment and Alzheimer’s disease. Hum. Brain Mapp. 35, 3446–3464 (2014).
Myers, N. et al. Within-patient correspondence of amyloid-beta and intrinsic network connectivity in Alzheimer’s disease. Brain 137, 2052–2064 (2014).
Grothe, M. J. & Teipel, S. J., Alzheimer’s Disease Neuroimaging, I. Spatial patterns of atrophy, hypometabolism, and amyloid deposition in Alzheimer’s disease correspond to dissociable functional brain networks. Hum. Brain Mapp. 37, 35–53 (2016).
Brier, M. R. et al. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J. Neurosci. 32, 8890–8899 (2012).
Lin, C. et al. The effect of amyloid deposition on longitudinal resting-state functional connectivity in cognitively normal older adults. Alzheimers Res Ther. 12, 7 (2020).
Pannee, J. et al. Reference measurement procedure for CSF amyloid beta (Abeta)(1-42) and the CSF Abeta(1–42) /Abeta(1–40) ratio - a cross-validation study against amyloid PET. J. Neurochem 139, 651–658 (2016).
Krishnadas, N., Villemagne, V. L., Dore, V. & Rowe, C. C. Advances in brain amyloid imaging. Semin Nucl. Med 51, 241–252 (2021).
Hansson, O., Lehmann, S., Otto, M., Zetterberg, H. & Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res Ther. 11, 34 (2019).
Klunk, W. E. et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 55, 306–319 (2004).
Wong, D. F. et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J. Nucl. Med 51, 913–920 (2010).
Rowe, C. C. et al. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 7, 129–135 (2008).
Serdons, K. et al. Synthesis of 18F-labelled 2-(4’-fluorophenyl)-1,3-benzothiazole and evaluation as amyloid imaging agent in comparison with [11C]PIB. Bioorg. Med Chem. Lett. 19, 602–605 (2009).
Olsson, B. et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 15, 673–684 (2016).
Seppala, T. T. et al. CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology 78, 1568–1575 (2012).
Wolk, D. A. et al. Association between in vivo fluorine 18-labeled flutemetamol amyloid positron emission tomography imaging and in vivo cerebral cortical histopathology. Arch. Neurol. 68, 1398–1403 (2011).
Palmqvist, S., Mattsson, N., Hansson, O. & Alzheimer’s Disease Neuroimaging, I. Cerebrospinal fluid analysis detects cerebral amyloid-beta accumulation earlier than positron emission tomography. Brain 139, 1226–1236 (2016).
Jack, C. R. Jr. Advances in Alzheimer’s disease research over the past two decades. Lancet Neurol. 21, 866–869 (2022).
Pike, K. E. et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain 130, 2837–2844 (2007).
Rowe, C. C. et al. Predicting Alzheimer disease with beta-amyloid imaging: results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann. Neurol. 74, 905–913 (2013).
Blennow, K., Mattsson, N., Scholl, M., Hansson, O. & Zetterberg, H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharm. Sci. 36, 297–309 (2015).
Shaw, L. M. et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. 121, 597–609 (2011).
Villemagne, V. L. et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367 (2013).
Toledo, J. B., Shaw, L. M. & Trojanowski, J. Q. Plasma amyloid beta measurements - a desired but elusive Alzheimer’s disease biomarker. Alzheimers Res Ther. 5, 8 (2013).
Jack, C. R. Jr. & Holtzman, D. M. Biomarker modeling of Alzheimer’s disease. Neuron 80, 1347–1358 (2013).
Halle, M. et al. Methods to monitor monocytes-mediated amyloid-beta uptake and phagocytosis in the context of adjuvanted immunotherapies. J. Immunol. Methods 424, 64–79 (2015).
Frenkel, D. et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 4, 2030 (2013).
Cheng, Y., Tian, D. Y. & Wang, Y. J. Peripheral clearance of brain-derived Abeta in Alzheimer’s disease: pathophysiology and therapeutic perspectives. Transl. Neurodegener. 9, 16 (2020).
Perneczky, R. et al. Soluble amyloid precursor protein beta as blood-based biomarker of Alzheimer’s disease. Transl. Psychiatry 3, e227 (2013).
Palmqvist, S. et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol. Med 11, e11170 (2019).
Palmqvist, S. et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease-Related beta-Amyloid Status. JAMA Neurol. 76, 1060–1069 (2019).
Damotte, V. et al. Plasma amyloid beta levels are driven by genetic variants near APOE, BACE1, APP, PSEN2: A genome-wide association study in over 12,000 non-demented participants. Alzheimers Dement 17, 1663–1674 (2021).
Nakamura, A. et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 554, 249–254 (2018).
Yuyama, K. et al. Immuno-digital invasive cleavage assay for analyzing Alzheimer’s amyloid ss-bound extracellular vesicles. Alzheimers Res Ther. 14, 140 (2022).
Bu, X. L. et al. Blood-derived amyloid-beta protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 23, 1948–1956 (2018).
Wang, J., Gu, B. J., Masters, C. L. & Wang, Y. J. A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 13, 612–623 (2017).
Endres, K. et al. Increased CSF APPs-alpha levels in patients with Alzheimer disease treated with acitretin. Neurology 83, 1930–1935 (2014).
Rosenberg, J. B. et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene Ther. Clin. Dev. 29, 24–47 (2018).
Decourt, B. et al. MCLENA-1: A phase II clinical trial for the assessment of safety, tolerability, and efficacy of lenalidomide in patients with mild cognitive impairment due to Alzheimer’s disease. Open Access J. Clin. Trials 12, 1–13 (2020).
Bakker, A., Albert, M. S., Krauss, G., Speck, C. L. & Gallagher, M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 7, 688–698 (2015).
Maccecchini, M. L. et al. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-beta peptide and tau levels: target engagement, tolerability and pharmacokinetics in humans. J. Neurol. Neurosurg. Psychiatry 83, 894–902 (2012).
Cummings, J. L. et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res Ther. 8, 4 (2016).
Ismail, R. et al. The effect of 40-Hz light therapy on amyloid load in patients with prodromal and clinical Alzheimer’s disease. Int J. Alzheimers Dis. 2018, 6852303 (2018).
Baruch, K. et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 6, 7967 (2015).
Haas, L. T. et al. Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing Alzheimer’s mouse phenotypes. Cell Rep. 20, 76–88 (2017).
Spurrier, J. et al. Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci. Transl. Med 14, eabi8593 (2022).
Krafft, G. A., Jerecic, J., Siemers, E. & Cline, E. N. ACU193: An immunotherapeutic poised to test the amyloid beta oligomer hypothesis of Alzheimer’s disease. Front Neurosci. 16, 848215 (2022).
Izzo, N. J. et al. Preclinical and clinical biomarker studies of CT1812: A novel approach to Alzheimer’s disease modification. Alzheimers Dement 17, 1365–1382 (2021).
Craft, S. et al. Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: A randomized clinical trial. JAMA Neurol. 77, 1099–1109 (2020).
Kellar, D. et al. Intranasal insulin reduces white matter hyperintensity progression in association with improvements in cognition and CSF biomarker profiles in mild cognitive impairment and Alzheimer’s disease. J. Prev. Alzheimers Dis. 8, 240–248 (2021).
Kellar, D. et al. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: a randomized trial. Sci. Rep. 12, 1346 (2022).
Wang, H. Y. et al. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol. Aging 55, 99–114 (2017).
Wang, H. Y. et al. PTI-125 reduces biomarkers of Alzheimer’s disease in patients. J. Prev. Alzheimers Dis. 7, 256–264 (2020).
Hey, J. A. et al. Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer’s disease. Clin. Pharmacokinet. 57, 315–333 (2018).
Kutzsche, J. et al. Safety and pharmacokinetics of the orally available antiprionic compound PRI-002: A single and multiple ascending dose phase I study. Alzheimers Dement (N. Y.) 6, e12001 (2020).
de la Torre, R. & Dierssen, M. Therapeutic approaches in the improvement of cognitive performance in Down syndrome: past, present, and future. Prog. Brain Res. 197, 1–14 (2012).
Lannfelt, L. et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786 (2008).
Scheltens, P. et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res. Ther. 10, 107 (2018).
Vassar, R. et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 (1999).
Hussain, I. et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell Neurosci. 14, 419–427 (1999).
Yan, R. et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402, 533–537 (1999).
Sinha, S. et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540 (1999).
May, P. C. et al. The potent BACE1 inhibitor LY2886721 elicits robust central Abeta pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 35, 1199–1210 (2015).
Sperling, R. et al. Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical Alzheimer disease: A truncated randomized phase 2b/3 clinical trial. JAMA Neurol. 78, 293–301 (2021).
Madrasi, K. et al. Systematic in silico analysis of clinically tested drugs for reducing amyloid-beta plaque accumulation in Alzheimer’s disease. Alzheimers Dement 17, 1487–1498 (2021).
Sakamoto, K. et al. BACE1 inhibitor lanabecestat (AZD3293) in a phase 1 study of healthy Japanese subjects: Pharmacokinetics and effects on plasma and cerebrospinal fluid abeta peptides. J. Clin. Pharm. 57, 1460–1471 (2017).
Zimmer, J. A. et al. Lanabecestat: Neuroimaging results in early symptomatic Alzheimer’s disease. Alzheimers Dement (N. Y) 7, e12123 (2021).
Neumann, U. et al. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 10, e9316 (2018).
Al-Tel, T. H. et al. Design, synthesis, and qualitative structure-activity evaluations of novel beta-secretase inhibitors as potential Alzheimer’s drug leads. J. Med Chem. 54, 8373–8385 (2011).
Benjannet, S. et al. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 276, 10879–10887 (2001).
Yuan, J. et al. Structure-based design of beta-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer’s disease. J. Med Chem. 56, 4156–4180 (2013).
Artavanis-Tsakonas, S., Rand, M. D. & Lake, R. J. Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 (1999).
Albright, C. F. et al. Pharmacodynamics of selective inhibition of gamma-secretase by avagacestat. J. Pharm. Exp. Ther. 344, 686–695 (2013).
Coric, V. et al. Targeting prodromal Alzheimer disease with avagacestat: A randomized clinical trial. JAMA Neurol. 72, 1324–1333 (2015).
Raven, F. et al. Soluble gamma-secretase modulators attenuate Alzheimer’s beta-amyloid pathology and induce conformational changes in presenilin 1. EBioMedicine 24, 93–101 (2017).
Rogers, K. et al. Modulation of gamma-secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice. Mol. Neurodegener. 7, 61 (2012).
Fox, N. C. et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 64, 1563–1572 (2005).
Orgogozo, J. M. et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61, 46–54 (2003).
Bayer, A. J. et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 64, 94–101 (2005).
Gilman, S. et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553–1562 (2005).
Ryan, J. M. & Grundman, M. Anti-amyloid-beta immunotherapy in Alzheimer’s disease: ACC-001 clinical trials are ongoing. J. Alzheimers Dis. 17, 243 (2009).
Arai, H., Suzuki, H. & Yoshiyama, T. Vanutide cridificar and the QS-21 adjuvant in Japanese subjects with mild to moderate Alzheimer’s disease: results from two phase 2 studies. Curr. Alzheimer Res 12, 242–254 (2015).
Davtyan, H. et al. Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trial. J. Neurosci. 33, 4923–4934 (2013).
Sandberg, A. et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 107, 15595–15600 (2010).
Lacosta, A. M. et al. Safety, tolerability and immunogenicity of an active anti-Abeta40 vaccine (ABvac40) in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res. Ther. 10, 12 (2018).
Wang, C. Y. et al. Site-specific UBITh amyloid-beta vaccine for immunotherapy of Alzheimer’s disease. Vaccine 25, 3041–3052 (2007).
Petrushina, I. et al. Characterization and preclinical evaluation of the cGMP grade DNA based vaccine, AV-1959D to enter the first-in-human clinical trials. Neurobiol. Dis. 139, 104823 (2020).
Davtyan, H. et al. Testing a MultiTEP-based combination vaccine to reduce Abeta and tau pathology in Tau22/5xFAD bigenic mice. Alzheimers Res Ther. 11, 107 (2019).
Waldmann, H. Human monoclonal antibodies: The benefits of humanization. Methods Mol. Biol. 1904, 1–10 (2019).
van Lengerich, B. et al. A TREM2-activating antibody with a blood-brain barrier transport vehicle enhances microglial metabolism in Alzheimer’s disease models. Nat. Neurosci. 26, 416–429 (2023).
Bard, F. et al. Epitope and isotype specificities of antibodies to beta -amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc. Natl. Acad. Sci. USA 100, 2023–2028 (2003).
DeMattos, R. B. et al. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 98, 8850–8855 (2001).
Bard, F. et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919 (2000).
Salloway, S. et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med 370, 322–333 (2014).
Klein, G. et al. Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther. 11, 101 (2019).
Moulder, K. L. et al. Dominantly Inherited Alzheimer Network: facilitating research and clinical trials. Alzheimers Res. Ther. 5, 48 (2013).
Salloway, S. et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 27, 1187–1196 (2021).
Adolfsson, O. et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J. Neurosci. 32, 9677–9689 (2012).
Meilandt, W. J. et al. Characterization of the selective in vitro and in vivo binding properties of crenezumab to oligomeric Abeta. Alzheimers Res Ther. 11, 97 (2019).
Rios-Romenets, S. et al. Baseline demographic, clinical, and cognitive characteristics of the Alzheimer’s Prevention Initiative (API) Autosomal-Dominant Alzheimer’s Disease Colombia Trial. Alzheimers Dement 16, 1023–1030 (2020).
Ostrowitzki, S. et al. Evaluating the safety and efficacy of crenezumab vs placebo in adults with early Alzheimer disease: Two phase 3 randomized placebo-controlled trials. JAMA Neurol. 79, 1113–1121 (2022).
Tariot, P. N. et al. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement (N. Y.) 4, 150–160 (2018).
Demattos, R. B. et al. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron 76, 908–920 (2012).
Honig, L. S. et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 378, 321–330 (2018).
Schwarz, A. J. et al. Magnetic resonance imaging measures of brain atrophy from the EXPEDITION3 trial in mild Alzheimer’s disease. Alzheimers Dement (N. Y.) 5, 328–337 (2019).
Sevigny, J. et al. Addendum: The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 546, 564 (2017).
Knopman, D. S., Jones, D. T. & Greicius, M. D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement 17, 696–701 (2021).
Mullard, A. FDA approval for Biogen’s aducanumab sparks Alzheimer disease firestorm. Nat. Rev. Drug Disco. 20, 496 (2021).
Logovinsky, V. et al. Safety and tolerability of BAN2401-a clinical study in Alzheimer’s disease with a protofibril selective Abeta antibody. Alzheimers Res Ther. 8, 14 (2016).
Swanson, C. J. et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res Ther. 13, 80 (2021).
Couzin-Frankel, J. & Piller, C. Alzheimer’s drug stirs excitement-and concerns. Science 378, 1030–1031 (2022).
Rafii, M. S. et al. The AHEAD 3–45 Study: Design of a prevention trial for Alzheimer’s disease. Alzheimers Dement, (2022).
Ullah, R., Park, T. J., Huang, X. & Kim, M. O. Abnormal amyloid beta metabolism in systemic abnormalities and Alzheimer’s pathology: Insights and therapeutic approaches from periphery. Ageing Res Rev. 71, 101451 (2021).
Cirrito, J. R. et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922 (2005).
Abramov, E. et al. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 12, 1567–1576 (2009).
Morley, J. E. et al. A physiological role for amyloid-beta protein:enhancement of learning and memory. J. Alzheimers Dis. 19, 441–449 (2010).
Dineley, K. T., Bell, K. A., Bui, D. & Sweatt, J. D. beta -Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J. Biol. Chem. 277, 25056–25061 (2002).
Chen, Y. & Dong, C. Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 16, 386–394 (2009).
Scott, G. et al. Amyloid pathology and axonal injury after brain trauma. Neurology 86, 821–828 (2016).
Bird, S. M. et al. Cerebral amyloid-beta accumulation and deposition following traumatic brain injury-A narrative review and meta-analysis of animal studies. Neurosci. Biobehav Rev. 64, 215–228 (2016).
Mannix, R. C., Zhang, J., Berglass, J., Qui, J. & Whalen, M. J. Beneficial effect of amyloid beta after controlled cortical impact. Brain Inj. 27, 743–748 (2013).
Clarke, J. et al. Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. Eur. J. Neurosci. 26, 1845–1852 (2007).
Kontush, A. et al. Amyloid-beta is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic. Biol. Med 30, 119–128 (2001).
Kumar, D. K. et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med 8, 340ra372 (2016).
Frain, L. et al. Association of cancer and Alzheimer’s disease risk in a national cohort of veterans. Alzheimers Dement 13, 1364–1370 (2017).
Shafi, O. Inverse relationship between Alzheimer’s disease and cancer, and other factors contributing to Alzheimer’s disease: a systematic review. BMC Neurol. 16, 236 (2016).
Lanni, C. et al. Beta-amyloid short- and long-term synaptic entanglement. Pharm. Res. 139, 243–260 (2019).
Cai, W., Li, L., Sang, S., Pan, X. & Zhong, C. Physiological Roles of beta-amyloid in Regulating Synaptic Function: Implications for AD Pathophysiology. Neurosci. Bull. https://doi.org/10.1007/s12264-022-00985-9 (2022).
Giuffrida, M. L. et al. Monomeric ss-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front Cell Neurosci. 9, 297 (2015).
Zimbone, S. et al. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 17, e12684 (2018).
Nagahara, A. H. & Tuszynski, M. H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Disco. 10, 209–219 (2011).
Seabrook, G. R. et al. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38, 349–359 (1999).
Garcia-Osta, A. & Alberini, C. M. Amyloid beta mediates memory formation. Learn Mem. 16, 267–272 (2009).
Puzzo, D. et al. Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 69, 819–830 (2011).
Galanis, C. et al. Amyloid-beta mediates homeostatic synaptic plasticity. J. Neurosci. 41, 5157–5172 (2021).
Kamenetz, F. et al. APP processing and synaptic function. Neuron 37, 925–937 (2003).
Ikegaya, Y. et al. Beta-amyloid enhances glial glutamate uptake activity and attenuates synaptic efficacy. J. Biol. Chem. 277, 32180–32186 (2002).
Sudweeks, S. N. & Yakel, J. L. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J. Physiol. 527, 515–528 (2000).
Papouin, T., Dunphy, J. M., Tolman, M., Dineley, K. T. & Haydon, P. G. Septal cholinergic neuromodulation tunes the astrocyte-dependent gating of hippocampal NMDA receptors to wakefulness. Neuron 94, 840–854.e847 (2017).
Castro, N. G. & Albuquerque, E. X. alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys. J. 68, 516–524 (1995).
Unwin, N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: insights from Torpedo postsynaptic membranes. Q Rev. Biophys. 46, 283–322 (2013).
Dougherty, J. J., Wu, J. & Nichols, R. A. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 23, 6740–6747 (2003).
Letsinger, A. C., Gu, Z. & Yakel, J. L. alpha7 nicotinic acetylcholine receptors in the hippocampal circuit: taming complexity. Trends Neurosci. 45, 145–157 (2022).
Townsend, M. et al. alpha7-nAChR agonist enhances neural plasticity in the hippocampus via a GABAergic circuit. J. Neurophysiol. 116, 2663–2675 (2016).
Nagele, R. G., D’Andrea, M. R., Anderson, W. J. & Wang, H. Y. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110, 199–211 (2002).
Belloy, M. E., Napolioni, V. & Greicius, M. D. A quarter century of APOE and Alzheimer’s disease: Progress to date and the path forward. Neuron 101, 820–838 (2019).
Cecon, E. et al. Quantitative assessment of oligomeric amyloid beta peptide binding to alpha7 nicotinic receptor. Br. J. Pharm. 176, 3475–3488 (2019).
Gulisano, W. et al. Neuromodulatory action of picomolar extracellular Abeta42 oligomers on presynaptic and postsynaptic mechanisms underlying synaptic function and memory. J. Neurosci. 39, 5986–6000 (2019).
Tropea, M. R. et al. Genetic deletion of alpha7 nicotinic acetylcholine receptors induces an age-dependent Alzheimer’s disease-like pathology. Prog. Neurobiol. 206, 102154 (2021).
Martinsson, I. et al. APP depletion alters selective pre- and post-synaptic proteins. Mol. Cell Neurosci. 95, 86–95 (2019).
Young-Pearse, T. L. et al. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 27, 14459–14469 (2007).
Saura, C. A. et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42, 23–36 (2004).
Puzzo, D. et al. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545 (2008).
Chen, X. H., Johnson, V. E., Uryu, K., Trojanowski, J. Q. & Smith, D. H. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 19, 214–223 (2009).
Pajoohesh-Ganji, A. et al. Inhibition of amyloid precursor protein secretases reduces recovery after spinal cord injury. Brain Res. 1560, 73–82 (2014).
Grant, J. L. et al. Reversal of paralysis and reduced inflammation from peripheral administration of beta-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci. Transl. Med 4, 145ra105 (2012).
Soscia, S. J. et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5, e9505 (2010).
Bourgade, K. et al. Protective effect of amyloid-beta peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheimers Dis. 50, 1227–1241 (2016).
Wozniak, M. A., Mee, A. P. & Itzhaki, R. F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 217, 131–138 (2009).
Miklossy, J. Bacterial amyloid and DNA are important constituents of senile plaques: Further evidence of the spirochetal and biofilm nature of senile plaques. J. Alzheimers Dis. 53, 1459–1473 (2016).
Spitzer, P. et al. Amyloidogenic amyloid-beta-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 6, 32228 (2016).
Eimer, W. A. et al. Alzheimer’s disease-associated beta-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99, 56–63.e53 (2018).
Bourgade, K. et al. beta-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 16, 85–98 (2015).
White, M. R. et al. Alzheimer’s associated beta-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One 9, e101364 (2014).
Di Domizio, J. et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc. Natl Acad. Sci. USA 109, 14550–14555 (2012).
Mastrangelo, M. A., Sudol, K. L., Narrow, W. C. & Bowers, W. J. Interferon-gamma differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am. J. Pathol. 175, 2076–2088 (2009).
Wu, Y. et al. Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat. Commun. 10, 58 (2019).
Zhao, H. et al. Bioluminescence imaging reveals inhibition of tumor cell proliferation by Alzheimer’s amyloid beta protein. Cancer Cell Int 9, 15 (2009).
Paris, D. et al. Inhibition of angiogenesis by Abeta peptides. Angiogenesis 7, 75–85 (2004).
Paris, D. et al. Impaired orthotopic glioma growth and vascularization in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 30, 11251–11258 (2010).
Ohyagi, Y. et al. Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 19, 255–257 (2005).
Alves da Costa, C. et al. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. J. Neurosci. 26, 6377–6385 (2006).
Yamamori, H., Tanaka, T., Kudo, T. & Takeda, M. Amyloid-beta down-regulates XIAP expression in human SH-SY5Y neuroblastoma cells. Neuroreport 15, 851–854 (2004).
Chaudhary, A. K. et al. A potential role of X-linked inhibitor of apoptosis protein in mitochondrial membrane permeabilization and its implication in cancer therapy. Drug Disco. Today 21, 38–47 (2016).
Clementi, M. E. et al. Alzheimer’s amyloid beta-peptide (1–42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem Biophys. Res. Commun. 342, 206–213 (2006).
Liu, Z. et al. Direct activation of Bax protein for cancer therapy. Med Res Rev. 36, 313–341 (2016).
Baruch-Suchodolsky, R. & Fischer, B. Abeta40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry 48, 4354–4370 (2009).
Faller, P. Copper and zinc binding to amyloid-beta: coordination, dynamics, aggregation, reactivity and metal-ion transfer. Chembiochem 10, 2837–2845 (2009).
Smith, D. G., Cappai, R. & Barnham, K. J. The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim. Biophys. Acta 1768, 1976–1990 (2007).
Zou, K., Gong, J. S., Yanagisawa, K. & Michikawa, M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 22, 4833–4841 (2002).
Gibson, G. E., Zhang, H., Sheu, K. R. & Park, L. C. Differential alterations in antioxidant capacity in cells from Alzheimer patients. Biochim Biophys. Acta 1502, 319–329 (2000).
Guo, Q. et al. Increased vulnerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid beta-peptide toxicity: central roles of superoxide production and caspase activation. J. Neurochem 72, 1019–1029 (1999).
Wang, L. et al. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 9, 10 (2020).
Eriksson, P. S. et al. Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317 (1998).
Alvarez-Buylla, A. & Garcia-Verdugo, J. M. Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629–634 (2002).
Babu, H., Ramirez-Rodriguez, G., Fabel, K., Bischofberger, J. & Kempermann, G. Synaptic Network Activity Induces Neuronal Differentiation of Adult Hippocampal Precursor Cells through BDNF Signaling. Front Neurosci. 3, 49 (2009).
Jin, K. et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 101, 343–347 (2004).
Li, B. et al. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J. Neuropathol. Exp. Neurol. 67, 78–84 (2008).
Crews, L. et al. Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J. Neurosci. 30, 12252–12262 (2010).
Tobin, M. K. et al. Human hippocampal neurogenesis persists in aged adults and Alzheimer’s disease patients. Cell Stem Cell 24, 974–982 (2019). e973.
Lopez-Toledano, M. A. & Shelanski, M. L. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J. Neurosci. 24, 5439–5444 (2004).
Sotthibundhu, A., Li, Q. X., Thangnipon, W. & Coulson, E. J. Abeta(1–42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol. Aging 30, 1975–1985 (2009).
Atwood, C. S., Bishop, G. M., Perry, G. & Smith, M. A. Amyloid-beta: a vascular sealant that protects against hemorrhage? J. Neurosci. Res. 70, 356 (2002).
Atwood, C. S. et al. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273, 12817–12826 (1998).
Roberts, G. W. et al. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 57, 419–425 (1994).
Castro, M. A. et al. Verteporfin is a substrate-selective gamma-secretase inhibitor that binds the amyloid precursor protein transmembrane domain. J. Biol. Chem. 298, 101792 (2022).
Liu, F. et al. Cleavage of potassium channel Kv2.1 by BACE2 reduces neuronal apoptosis. Mol. Psychiatry 23, 1542–1554 (2018).
Turner, R. T. 3rd, Hong, L., Koelsch, G., Ghosh, A. K. & Tang, J. Structural locations and functional roles of new subsites S5, S6, and S7 in memapsin 2 (beta-secretase). Biochemistry 44, 105–112 (2005).
Sun, X. Y. et al. Fc effector of anti-Abeta antibody induces synapse loss and cognitive deficits in Alzheimer’s disease-like mouse model. Signal Transduct. Target Ther. 8, 30 (2023).
Bekris, L. M., Yu, C. E., Bird, T. D. & Tsuang, D. W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 23, 213–227 (2010).
Daviglus, M. L. et al. National Institutes of Health State-of-the-Science Conference statement: preventing alzheimer disease and cognitive decline. Ann. Intern Med 153, 176–181 (2010).
Chen, Z. Y. & Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 43, 1026–1040 (2022).
Drummond, E. & Wisniewski, T. Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133, 155–175 (2017).
Hsiao, K. et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 (1996).
Davis, J. et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 279, 20296–20306 (2004).
Sturchler-Pierrat, C. et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 94, 13287–13292 (1997).
Mucke, L. et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058 (2000).
Chishti, M. A. et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570 (2001).
Schneider, I. et al. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J. Biol. Chem. 276, 11539–11544 (2001).
Duff, K. et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713 (1996).
Levy-Lahad, E. et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977 (1995).
Rogaev, E. I. et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778 (1995).
Saito, T. et al. Potent amyloidogenicity and pathogenicity of Abeta43. Nat. Neurosci. 14, 1023–1032 (2011).
Radde, R. et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946 (2006).
Jankowsky, J. L. et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17, 157–165 (2001).
Jankowsky, J. L. et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet 13, 159–170 (2004).
Qing, H. et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J. Exp. Med 205, 2781–2789 (2008).
Zhang, S. et al. Upregulation of MIF as a defense mechanism and a biomarker of Alzheimer’s disease. Alzheimers Res. Ther. 11, 54 (2019).
Oakley, H. et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006).
Willuweit, A. et al. Early-onset and robust amyloid pathology in a new homozygous mouse model of Alzheimer’s disease. PLoS One 4, e7931 (2009).
D’Souza, I. et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. USA 96, 5598–5603 (1999).
Jackson, R. J. et al. Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur. J. Neurosci. 44, 3056–3066 (2016).
Stover, K. R., Campbell, M. A., Van Winssen, C. M. & Brown, R. E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res 289, 29–38 (2015).
Platt, B. et al. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS One 6, e27068 (2011).
Serneels, L. et al. Modeling the beta-secretase cleavage site and humanizing amyloid-beta precursor protein in rat and mouse to study Alzheimer’s disease. Mol. Neurodegener. 15, 60 (2020).
Zheng, H. et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81, 525–531 (1995).
Saito, T. et al. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663 (2014).
Saito, T. et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem. 294, 12754–12765 (2019).
Hashimoto, S. et al. Tau binding protein CAPON induces tau aggregation and neurodegeneration. Nat. Commun. 10, 2394 (2019).
Sims-Robinson, C., Kim, B., Rosko, A. & Feldman, E. L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 6, 551–559 (2010).
Justice, N. J. et al. Posttraumatic stress disorder-like induction elevates beta-amyloid levels, which directly activates corticotropin-releasing factor neurons to exacerbate stress responses. J. Neurosci. 35, 2612–2623 (2015).
Migliore, L. & Coppede, F. Gene-environment interactions in Alzheimer disease: the emerging role of epigenetics. Nat. Rev. Neurol. 18, 643–660 (2022).
Jevtic, S., Sengar, A. S., Salter, M. W. & McLaurin, J. The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res Rev. 40, 84–94 (2017).
Mestas, J. & Hughes, C. C. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 (2004).
Franco, R. & Cedazo-Minguez, A. Successful therapies for Alzheimer’s disease: why so many in animal models and none in humans? Front Pharm. 5, 146 (2014).
Benzinger, T. L. et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 110, E4502–E4509 (2013).
McDade, E. et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 91, e1295–e1306 (2018).
Dale, J., Alcorn, N., Capell, H. & Madhok, R. Combination therapy for rheumatoid arthritis: methotrexate and sulfasalazine together or with other DMARDs. Nat. Clin. Pr. Rheumatol. 3, 450–458 (2007). quiz, following 478.
Bartlett, J. A. et al. An updated systematic overview of triple combination therapy in antiretroviral-naive HIV-infected adults. AIDS 20, 2051–2064 (2006).
Spuch, C., Ortolano, S. & Navarro, C. New insights in the amyloid-Beta interaction with mitochondria. J. Aging Res. 2012, 324968 (2012).
Wilkins, H. M. Interactions between amyloid, amyloid precursor protein, and mitochondria. Biochem Soc. Trans. 51, 173–182 (2023).
Fava, A. et al. The effect of lipoic acid therapy on cognitive functioning in patients with Alzheimer’s disease. J. Neurodegener. Dis. 2013, 454253 (2013).
Sano, M. et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 336, 1216–1222 (1997).
Dias-Santagata, D., Fulga, T. A., Duttaroy, A. & Feany, M. B. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Invest 117, 236–245 (2007).
Murakami, K. et al. Vitamin C restores behavioral deficits and amyloid-beta oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 26, 7–18 (2011).
Hira, S. et al. beta-Carotene: A natural compound improves cognitive impairment and oxidative stress in a mouse model of streptozotocin-induced Alzheimer’s disease. Biomolecules 9, 441 (2019).
Klyubin, I. et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 11, 556–561 (2005).
Peng, L., Bestard-Lorigados, I. & Song, W. The synapse as a treatment avenue for Alzheimer’s Disease. Mol. Psychiatry 27, 2940–2949 (2022).
Sagare, A. P., Bell, R. D. & Zlokovic, B. V. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med 2, a011452 (2012).
Drachman, D. A. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimers Dement 10, 372–380 (2014).
Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799 (2015).
Cavieres, V. A. et al. Tetrahydrohyperforin inhibits the proteolytic processing of amyloid precursor protein and enhances its degradation by Atg5-dependent autophagy. PLoS One 10, e0136313 (2015).
Di Meco, A., Curtis, M. E., Lauretti, E. & Pratico, D. Autophagy dysfunction in Alzheimer’s disease: Mechanistic insights and new therapeutic opportunities. Biol. Psychiatry 87, 797–807 (2020).
Nilsson, P. et al. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69 (2013).
Nilsson, P. et al. Autophagy-related protein 7 deficiency in amyloid beta (Abeta) precursor protein transgenic mice decreases Abeta in the multivesicular bodies and induces Abeta accumulation in the Golgi. Am. J. Pathol. 185, 305–313 (2015).
Mueller-Steiner, S. et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron 51, 703–714 (2006).
Nixon, R. A. et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122 (2005).
Luo, R. et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 16, 52–69 (2020).
Deng, Z., Dong, Y., Zhou, X., Lu, J. H. & Yue, Z. Pharmacological modulation of autophagy for Alzheimer’s disease therapy: Opportunities and obstacles. Acta Pharm. Sin. B 12, 1688–1706 (2022).
Hay, N. & Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 (2004).
Juenemann, K. & Reits, E. A. Alternative macroautophagic pathways. Int J. Cell Biol. 2012, 189794 (2012).
Caccamo, A., Majumder, S., Richardson, A., Strong, R. & Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120 (2010).
Majumder, S., Richardson, A., Strong, R. & Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 6, e25416 (2011).
Liu, J., Wang, S., Zhang, Y., Fan, H. T. & Lin, H. S. Traditional Chinese medicine and cancer: History, present situation, and development. Thorac. Cancer 6, 561–569 (2015).
Chen, H. Y., Lin, Y. H., Huang, J. W. & Chen, Y. C. Chinese herbal medicine network and core treatments for allergic skin diseases: Implications from a nationwide database. J. Ethnopharmacol. 168, 260–267 (2015).
Xie, W., Zhao, Y. & Zhang, Y. Traditional chinese medicines in treatment of patients with type 2 diabetes mellitus. Evid. Based Complement Altern. Med 2011, 726723 (2011).
Huang, K. et al. Traditional Chinese Medicine (TCM) in the treatment of COVID-19 and other viral infections: Efficacies and mechanisms. Pharm. Ther. 225, 107843 (2021).
Durairajan, S. S. et al. Berberine ameliorates beta-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol. Aging 33, 2903–2919 (2012).
Wu, Y. et al. Berberine reduces Abeta42 deposition and tau hyperphosphorylation via ameliorating endoplasmic reticulum stress. Front Pharm. 12, 640758 (2021).
Zang, C. et al. Gardenia jasminoides J.Ellis extract GJ-4 alleviated cognitive deficits of APP/PS1 transgenic mice. Phytomedicine 93, 153780 (2021).
Zhang, Z. et al. GJ-4 alleviates Abeta25-35-induced memory dysfunction in mice through protecting the neurovascular unit. Biomed. Pharmacother. 127, 110131 (2020).
Li, F., Zhang, Y., Lu, X., Shi, J. & Gong, Q. Icariin improves the cognitive function of APP/PS1 mice via suppressing endoplasmic reticulum stress. Life Sci. 234, 116739 (2019).
Zhu, T. et al. Long-term icariin treatment ameliorates cognitive deficits via CD4(+) T cell-mediated immuno-inflammatory responses in APP/PS1 mice. Clin. Inter. Aging 14, 817–826 (2019).
Liu, Q. et al. Characterization of a pectin from Lonicera japonica Thunb. and its inhibition effect on Abeta42 aggregation and promotion of neuritogenesis. Int J. Biol. Macromol. 107, 112–120 (2018).
Wang, P. et al. A glucan isolated from flowers of Lonicera japonica Thunb. inhibits aggregation and neurotoxicity of Abeta42. Carbohydr. Polym. 110, 142–147 (2014).
Chen, K. et al. Morroniside prevents H2O2 or Abeta1-42-induced apoptosis via attenuating JNK and p38 MAPK phosphorylation. Eur. J. Pharm. 834, 295–304 (2018).
Ji, Y. J. et al. Crude saponin from platycodon grandiflorum attenuates abeta-induced neurotoxicity via antioxidant, anti-inflammatory and anti-apoptotic signaling pathways. Antioxid. (Basel) 10, 1968 (2021).
Nam, Y. et al. Platycodon grandiflorum root protects against abeta-induced cognitive dysfunction and pathology in female models of Alzheimer’s disease. Antioxid. (Basel) 10, 1968 (2021).
Marambaud, P., Zhao, H. & Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J. Biol. Chem. 280, 37377–37382 (2005).
El-Sayed, N. S. & Bayan, Y. Possible role of resveratrol targeting estradiol and neprilysin pathways in lipopolysaccharide model of Alzheimer disease. Adv. Exp. Med Biol. 822, 107–118 (2015).
Bermejo-Bescos, P., Jimenez-Aliaga, K. L., Benedi, J. & Martin-Aragon, S. A diet containing rutin ameliorates brain intracellular redox homeostasis in a mouse model of Alzheimer’s disease. Int J. Mol. Sci. 24, 4863 (2023).
Pan, R. Y. et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-beta clearance. Sci. Adv. 5, eaau6328 (2019).
Sun, X. Y. et al. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J. Neuroinflammation 18, 131 (2021).
Ding, B. et al. Tanshinone IIA attenuates neuroinflammation via inhibiting RAGE/NF-kappaB signaling pathway in vivo and in vitro. J. Neuroinflammation 17, 302 (2020).
He, Y. et al. Tanshinone IIA ameliorates cognitive deficits by inhibiting endoplasmic reticulum stress-induced apoptosis in APP/PS1 transgenic mice. Neurochem Int 133, 104610 (2020).
Xu, P. et al. Neuroprotection of triptolide against amyloid-Beta1-42-induced toxicity via the Akt/mTOR/p70S6K-mediated autophagy pathway. Acad. Bras. Cienc. 94, e20210938 (2022).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Chan, W. K., Griffiths, R., Price, D. J. & Mason, J. O. Cerebral organoids as tools to identify the developmental roots of autism. Mol. Autism 11, 58 (2020).
Smits, L. M. et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 5, 5 (2019).
Pavoni, S. et al. Small-molecule induction of Abeta-42 peptide production in human cerebral organoids to model Alzheimer’s disease associated phenotypes. PLoS One 13, e0209150 (2018).
Gonzalez, C. et al. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 23, 2363–2374 (2018).
Raja, W. K. et al. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One 11, e0161969 (2016).
Lin, Y. T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e1147 (2018).
Cazzaniga, A., Fedele, G., Castiglioni, S. & Maier, J. A. The presence of blood-brain barrier modulates the response to magnesium salts in human brain organoids. Int J. Mol. Sci. 23, 5133 (2022).
Dong, X. et al. Human cerebral organoids establish subcortical projections in the mouse brain after transplantation. Mol. Psychiatry 26, 2964–2976 (2021).
Pham, M. T. et al. Generation of human vascularized brain organoids. Neuroreport 29, 588–593 (2018).
Cakir, B. et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175 (2019).
Cummings, J. The role of biomarkers in Alzheimer’s disease drug development. Adv. Exp. Med Biol. 1118, 29–61 (2019).
Park, J. C., Han, S. H. & Mook-Jung, I. Peripheral inflammatory biomarkers in Alzheimer’s disease: a brief review. BMB Rep. 53, 10–19 (2020).
Colom-Cadena, M. et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res Ther. 12, 21 (2020).
Kubis-Kubiak, A., Dyba, A. & Piwowar, A. The interplay between diabetes and alzheimer’s disease-in the hunt for biomarkers. Int J. Mol. Sci. 21, 2744 (2020).
Ahmed, Z. et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 177, 311–324 (2010).
Suarez-Calvet, M. et al. CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM2, neurodegeneration and cognitive decline. EMBO Mol. Med. 10, e9712 (2018).
Guadano-Ferraz, A., Vinuela, A., Oeding, G., Bernal, J. & Rausell, E. RC3/neurogranin is expressed in pyramidal neurons of motor and somatosensory cortex in normal and denervated monkeys. J. Comp. Neurol. 493, 554–570 (2005).
Zhong, L. & Gerges, N. Z. Neurogranin and synaptic plasticity balance. Commun. Integr. Biol. 3, 340–342 (2010).
Portelius, E. et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer’s disease. Brain 138, 3373–3385 (2015).
Kester, M. I. et al. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 72, 1275–1280 (2015).
Goetzl, E. J. et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 30, 4141–4148 (2016).
Wellington, H. et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 86, 829–835 (2016).
Dehghani, R., Rahmani, F. & Rezaei, N. MicroRNA in Alzheimer’s disease revisited: implications for major neuropathological mechanisms. Rev. Neurosci. 29, 161–182 (2018).
Xia, X. et al. Exosomal miRNAs in central nervous system diseases: biomarkers, pathological mediators, protective factors and therapeutic agents. Prog. Neurobiol. 183, 101694 (2019).
Tsai, Y. et al. Ocular changes in TgF344-AD rat model of Alzheimer’s disease. Invest Ophthalmol. Vis. Sci. 55, 523–534 (2014).
Chang, L. Y. et al. Alzheimer’s disease in the human eye. Clinical tests that identify ocular and visual information processing deficit as biomarkers. Alzheimers Dement 10, 251–261 (2014).
Frost, S. et al. Retinal vascular biomarkers for early detection and monitoring of Alzheimer’s disease. Transl. Psychiatry 3, e233 (2013).
Chan, C. K. et al. Depressive symptoms and CSF Alzheimer’s disease biomarkers in relation to clinical symptom onset of mild cognitive impairment. Alzheimers Dement (Amst.) 12, e12106 (2020).
Acknowledgements
We thank Brian J. Song’s editing and comments. This work was supported by the National Natural Science Foundation of China (82201576), Beijing Hospitals Authority Youth Programme (QML20210804) and Beijng Medical Research 2021-8 (YZ), and the National Natural Science Foundation of China (82150710557, 82230043 and 82293642) to WS. WS was the Canada Research Chair in Alzhaimer’s Disease.
Author information
Authors and Affiliations
Contributions
Y.Z. and W.S. conceived and designed this project. Y.Z. wrote the draft of the manuscript. Y.Z., Q.C., R.L. and K.S. did the literature search and review. Y.Z., K.S. and W.S. revised the manuscript, and Y.Z. and W.S. supervised the project. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Chen, H., Li, R. et al. Amyloid β-based therapy for Alzheimer’s disease: challenges, successes and future. Sig Transduct Target Ther 8, 248 (2023). https://doi.org/10.1038/s41392-023-01484-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01484-7
- Springer Nature Limited
This article is cited by
-
Photobiomodulation in experimental models of Alzheimer’s disease: state-of-the-art and translational perspectives
Alzheimer's Research & Therapy (2024)
-
Gut microbiota-host lipid crosstalk in Alzheimer’s disease: implications for disease progression and therapeutics
Molecular Neurodegeneration (2024)
-
Updates in Alzheimer's disease: from basic research to diagnosis and therapies
Translational Neurodegeneration (2024)
-
Alpha-lipoic acid alleviates cognitive deficits in transgenic APP23/PS45 mice through a mitophagy-mediated increase in ADAM10 α-secretase cleavage of APP
Alzheimer's Research & Therapy (2024)
-
Amyloid β oligomer induces cerebral vasculopathy via pericyte-mediated endothelial dysfunction
Alzheimer's Research & Therapy (2024)