
Abstract
Chromosomal instability (CIN) is a hallmark of cancer and is associated with tumor cell malignancy. CIN triggers a chain reaction in cells leading to chromosomal abnormalities, including deviations from the normal chromosome number or structural changes in chromosomes. CIN arises from errors in DNA replication and chromosome segregation during cell division, leading to the formation of cells with abnormal number and/or structure of chromosomes. Errors in DNA replication result from abnormal replication licensing as well as replication stress, such as double-strand breaks and stalled replication forks; meanwhile, errors in chromosome segregation stem from defects in chromosome segregation machinery, including centrosome amplification, erroneous microtubule–kinetochore attachments, spindle assembly checkpoint, or defective sister chromatids cohesion. In normal cells, CIN is deleterious and is associated with DNA damage, proteotoxic stress, metabolic alteration, cell cycle arrest, and senescence. Paradoxically, despite these negative consequences, CIN is one of the hallmarks of cancer found in over 90% of solid tumors and in blood cancers. Furthermore, CIN could endow tumors with enhanced adaptation capabilities due to increased intratumor heterogeneity, thereby facilitating adaptive resistance to therapies; however, excessive CIN could induce tumor cells death, leading to the “just-right” model for CIN in tumors. Elucidating the complex nature of CIN is crucial for understanding the dynamics of tumorigenesis and for developing effective anti-tumor treatments. This review provides an overview of causes and consequences of CIN, as well as the paradox of CIN, a phenomenon that continues to perplex researchers. Finally, this review explores the potential of CIN-based anti-tumor therapy.
Similar content being viewed by others


Introduction
Cancer is a widespread and devastating disease, which according to the World Health Organization claimed 10 million lives in 2020.1 Cancer is closely associated with mutations and aberrant expressions of a series of oncogenes and tumor suppressor genes. For many years, studies have focused on identifying genes that influence tumorigenesis, such as oncogenes and tumor suppressor genes.2 However, it has become increasingly clear that the development and progression of tumors do not rely exclusively on the alteration of a single gene.3 Chromosomal instability (CIN), a phenomenon characterized by chromosomal alterations, is observed in over 90% of solid tumors and many blood cancers.4,5,6 These alterations can result in large-scale changes, rearrangements, or disruptions to cellular genetic information, affecting the expression of numerous genes.2,3,6,7
The maintenance of genomic stability is a fundamental requirement for the normal functioning of cells.8,9,10,11 Under normal conditions, cells have developed a series of checkpoints and mechanisms to stringently control the passage of intact and correct genetic information, serving as safeguards that help cells maintain genomic stability and prevent harmful alterations.8,9,10,11 Thus, CIN, characterized by chromosomal abnormalities, presents a significant challenge to normal cells, often leading to decreased fitness and cell death.3,12 Interestingly, in simpler organisms such as bacteria and viruses, while excessive genomic instability is also harmful, a certain increase in genomic instability can be beneficial, as it could increase the heterogeneity of the population, thereby promoting the survival and proliferation of cells with specific genetic aberrations that provide a growth advantage in a stressful environment.13 This complex situation reveals that while a moderate level of CIN can be beneficial, extremely high levels result in genetic catastrophe and cell death, highlighting the importance of maintaining a balance.13 This delicate balance is not exclusive to simpler organisms but extends to more complex systems, including mammalian cells.14,15,16,17,18,19,20 Interestingly, a similar paradoxical observation emerges when studying tumor cells, where the role of CIN in tumorigenesis exhibits complexity.21,22 Analogous to a double-edged sword, CIN in tumor cells exhibits both tumorigenic and tumor-suppressing effects.21,22,23,24,25,26 On one hand, CIN can promote tumor progression by increasing heterogeneity, thus playing significant roles in tumor development and influencing treatment outcomes,3,27,28 while on the other hand, excessive CIN can lead to growth arrest and even cell death.29 The precise roles of CIN in tumors remain active areas of research. Elucidating the complex interplay between CIN and tumor progression, as well as treatment response, will not only provide a more comprehensive understanding of a major aspect of tumor development and progression, but also important new perspectives for the development of more effective anti-tumor therapies.
CIN manifests in two distinct forms: numerical CIN and structural CIN (Fig. 1).30 Numerical CIN arises from errors in chromosome segregation due to the defects in the mechanisms that guarantee proper sister chromatid segregation, including mitotic checkpoint, centrosome amplification, and abnormalities in microtubules during cell division, and is characterized by the gain or loss of the entire chromosomes.31 While numerical CIN does not change the nucleotide sequences within the chromosomes, it alters the copy number of chromosomes, thereby changing the genetic landscape.32 In contrast, structural CIN can lead to the gain or loss of chromosomal fragments, which are pieces of chromosomes that have broken off, leading to the alteration in nucleotide sequences of large segments of chromosomes.32 This type of CIN is driven by the amplification or deletion of chromosome segments, the formation of extrachromosomal structures, and complex rearrangements of large nucleotide sequences.31 Its origins are linked to the mechanisms involved in repairing double-strand breaks (DSBs), managing replication stress, and regulating non-allelic homologous recombination.31 Interestingly, structural and numerical CIN often coexist in the majority of tumor cells, thereby creating a complex interplay.33,34,35

Types of chromosomal instability. CIN is classified into numerical CIN and structural CIN. Numerical CIN corresponds to the gain or loss of whole chromosomes (aneuploidy) or gain of extra set of chromosomes (polyploidy), while structural CIN refers to the gain or loss of chromosome segments due to deletion, amplification, inversion, and translocation
Although the study of CIN has a long history in tumor research, recent advancements in next-generation sequencing technology and a deeper understanding of tumor biology have brought CIN back into the spotlight. From a broader biological perspective, genome diversity is a fundamental aspect of evolution and speciation.36 It provides the raw material upon which natural selection acts, driving the evolution of new species. In the context of tumors, CIN-induced tumor evolution is crucial for creating this genome diversity. The constant reshuffling of the genome creates a vast pool of genetic variants within the tumor population, known as heterogeneity.25 This CIN-induced increased heterogeneity is believed to endow tumors with enhanced evolutionary capabilities due to increased intratumor heterogeneity, facilitating acquisition of malignant phenotypes and adaptive resistance to therapies.23,24,25,26,37,38,39 Moreover, accumulating research has revealed other consequences and associations with CIN, such as its links to metastasis and tumor immune regulation.30 However, despite these advancements, our understanding of CIN remains incomplete. The complex nature of CIN, its causes and consequences, as well as the paradox of CIN, necessitate a systematic review. This is particularly important, given the potential of CIN as a therapeutic target. Revealing the complex nature of CIN is crucial for understanding one of the major causes of tumor progression, as well as for developing more effective anti-tumor treatments. Therefore, in this review, we aim to provide a comprehensive overview of CIN, exploring its research history, causes, paradoxical nature, and multifaceted influence on tumor biology, as well as discussing the potential and progress of CIN-based anti-tumor therapy.
Milestones in CIN research
The study of CIN has evolved over a century, marked by pivotal milestones (Fig. 2). The journey began with Theodor Boveri who, in 1902, performed the first systematic analysis of the effects of aneuploidy on cell and organismal physiology in sea urchins. Boveri observed that embryos resulting from eggs fertilized by two sperms exhibited developmental defects and died, concluding that chromosome abnormality leads to defect in development and lethality, marking the first hypothesis that connects between chromosomal abnormality and disease. His subsequent work in 1914, “Concerning the Origin of Malignant Tumors,” linked chromosomal abnormality to cancer, marking the first hypothesis that connected chromosomal abnormality to cancer.40,41 Sixteen years later, Barbara McClintock introduced the terms ‘laggards’ or ‘lagging chromosomes’ to signify chromatin lagging between daughter nuclear masses during anaphase, providing a deeper understanding of chromosomal behavior during cell division.42 The field of clinical cytogenetics was initiated in 1956 when Tjio and Levan discovered that humans have 46 chromosomes. This discovery not only corrected the previously held belief of 48 chromosomes, but also paved the way for the study of chromosomal abnormalities in humans.43 In 1994, Rieder and colleagues performed their classic experiment using laser ablation, which revealed the role of unattached kinetochores in extending the duration of mitosis, providing crucial insights into the mechanisms of mitotic checkpoint control.44

Timeline of key milestones in the CIN research. Green represents milestones in basic CIN research, pink represents milestones in CIN clinical translation, and blue represents milestones in CIN meta-analysis and databases
The relation between CIN and diseases was first revealed in 1959, when two significant discoveries were made. Peter Nowell identified the Philadelphia chromosome, a consequence of an abnormal rearrangement between human chromosome 9 and chromosome 22 that could be found in approximately 90% of chronic myeloid leukemia patients, providing the initial evidence of structural chromosome aberration as a malignant factor.45,46 In the same year, Lejeune et al. discovered that an additional copy of chromosome 21, a condition now known as trisomy 21, caused Down syndrome.47 Lejeune et al. continued his research and identified another chromosomal disorder known as cri du chat (cry of the cat) syndrome, a condition that arises when a segment of chromosome 5 is missing.48 These discoveries underscored the detrimental effects associated with chromosome aberrations and emphasized the importance of chromosomal stability.45,47 In 1997, Lengauer et al. quantified CIN in human cancer cell lines, proposing its universality across cancers;49,50 while Angelika Amon’s works from 1999 until 2010 elucidated the molecular aspects of checkpoint crucial in CIN.51,52 These works provided valuable insights regarding the widespread nature of CIN and its molecular mechanisms in cancer, thereby underscoring the significance of CIN in cancer biology. This eventually leads to the recognition of CIN as one of the hallmarks of cancer in 2011.49 Meanwhile, in 2018, Bakhoum et al. provided compelling evidence that CIN could drive tumor metastasis, significantly advancing our understanding of the role of CIN in tumor progression.53
The beginning of the 21st century marked another crucial milestone in CIN research. In 2000, Max Dobles et al. developed the first CIN mice model, enabling in vivo studies of CIN.54 In the same year, Felix Mitelman launched The Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer, which provides a valuable resource for CIN research, marking the opening of the era of meta-analysis in CIN studies.55 This was continued with the initiation of Pan-Cancer Analysis of Whole Genomes (PCAWG) by the International Cancer Genome Consortium (ICGC) in 2014.31,33,56,57 Concurrently, the TRAcking Cancer Evolution through therapy/Rx (TRACERx) clinical study was launched under the guidance of Charles Swanton in 2014. These initiatives marked a significant shift towards large-scale, collaborative efforts in understanding CIN, and paved the way for the development of therapeutic interventions targeting CIN.6,58,59,60,61,62 The discovery of Taxol in 1966 marked a significant milestone in anti-tumor treatment, setting the stage for a novel anti-tumor therapeutic strategy. It was the first drug to successfully demonstrate the potential of targeting mitosis, laying the groundwork for treatments based on CIN.63 Then, half a century later in 2017, this strategy was further realized with the first clinical trial for drugs targeting CIN, starting with the phase I trial of CFI-402257. Although this drug has not yet received full approval, it was granted Fast Track Designation by the FDA in 2023.64,65,66,67 To date, two anti-tumor therapies targeting CIN has been approved, while more than 50 are in clinical trial Phase I/II, with the most recent one initiated in October 2023 for VLS-1488.68 These developments have significantly advanced the translation of CIN research into potential therapeutic interventions. To date, research into CIN continues to progress, with each new discovery providing further insight into this complex field and opening up new avenues for potential cancer treatments.
Causes of CIN
CIN is characterized by changes in chromosome structure and number during cell division.30,32,69 There are several key indicators of CIN, including lagging chromosomes, chromosome bridges, micronuclei, aneuploidy, and polyploidy.70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86 As will be discussed below, aberrant spindle assembly checkpoint (SAC) activity, impaired sister chromatid segregation, aberrant centrosome number, and microtubule-kinetochore attachment error could lead to chromosome missegregation.87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112 This could in turn increase the formation of lagging chromosomes, which are chromosome that moves to the poles of the cell during cell division slower than other chromosomes, and chromosome bridges, which are structures formed when part of sister chromatids intertwines and fails to completely segregate.113,114 Lagging chromosomes and chromosome bridges subsequently could lead to the formation of micronucleus, a small, extra-nuclear body that contains chromosomal fragments or whole chromosome that are not incorporated into the main nucleus.71,115,116,117 Furthermore, chromosome missegregation, along with replication stress, sister chromatid defect, and abnormal centrosome number, could also lead to numerical CIN, as they could promote the occurrence of aneuploidy, a condition where a cell has an abnormal number of individual chromosomes, as well as polyploidy, a condition in where a cell has multiple sets of chromosomes.34,113,114,118,119 These indicators reflect the level of CIN, and are commonly used to assess and study CIN in a cell population (Fig. 3).

Indicators of CIN. Example of indicators commonly used to assess CIN, including lagging chromosomes, chromosome bridges, micronuclei, aneuploidy, and polyploidy
The causes of CIN are multifaceted and can be attributed to a variety of factors (Fig. 4). At its core, CIN is often the result of errors in DNA replication, which can lead to the formation of cells with incomplete or excess genetic material; as well as errors in chromosome segregation during cell division, which can lead to the formation of cells with an abnormal number of chromosomes.120,121 Errors in DNA replication can arise from the abnormal replication licensing as well as replication stress, such as DSBs and stalled replication forks; while errors in chromosome segregation can arise from defects in chromosome segregation machinery, including issues with centrosome amplification, erroneous microtubule-kinetochore attachments, and defects affecting the mitotic checkpoint or impaired sister chromatid segregation. Furthermore, some events induced by CIN could further trigger instability of chromosomes. For instance, chromothripsis is an event of incorporation of chromosome fragments originated from micronuclear chromosome into nuclear chromosome, causing rearrangement of nuclear chromosome.78,115,122,123,124,125,126,127,128,129,130,131,132,133 Thus, while chromothripsis itself is a consequence of CIN, it could also be a cause of subsequent CIN.

Causes of CIN. a Replication stress leads to stalling and collapse of replication forks, which results in DSBs. b Sister chromatid defect allows premature separation of sister chromatids before full alignment, leading to chromosome missegregation. c Aberrant centrosome number such as monopolar spindle and multipolar spindle could lead to aneuploidy. d Microtubule kinetochore attachment error causes failure to form bi-orientation, where each kinetochore is attached to microtubules from only one spindle pole, leading to chromosome missegregation. e Aberrant SAC could lead to aneuploidy, as weakened SAC causes premature chromatid separation, while hyperactivated SAC results in a lagging chromosome. f Extra set of chromosomes as seen in polyploidy could arise from cytokinesis failure, mitotic slippage, or endoreduplication
Replication stress and defective DNA repair
Replication stress is a condition that occurs when the DNA replication machinery is disrupted during the S phase of the cell cycle, leading to a stalled replication fork.134,135 Replication stress can be driven by oncogenes, low nucleotide concentrations, and DNA sequences or structures that are difficult to replicate.136 In response to replication stress, cells activate the DNA damage response (DDR), a cellular response that requires a network of repair proteins. This network includes key proteins such as ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR), which are kinases that help the stabilization of the stalled replication fork, preventing it from collapsing.137,138 As has been observed in various precancerous and cancerous lesions, failure in resolving the stalled replication forks can cause DSBs, and subsequently, rearrangements of parts of chromosomes (translocations) or deletions, thus contributing to structural CIN.34,137,138,139,140,141,142,143,144
In addition to the DDR pathway, other DNA repair-related factors, such as Fanconi anemia (FA) proteins are also important for preventing CIN.145,146,147 The FA pathway is important for repairing interstrand cross-links, which are toxic lesions that prevent DNA strand separation, block replication, and hinder transcription, thereby playing a critical role in responding to replication stress and maintaining chromosome stability.148,149,150 Defects in the FA pathway, specifically in the Fanconi anemia complementation group D2 (FANCD2), can lead to increased translocations and abnormal chromatin structures, contributing to structural CIN.42,147 Moreover, replication stress can also lead to lagging chromosomes and micronuclei.34,118,151,152 Together, these studies demonstrated that replication stress and defects in DNA repair systems contribute to the generation of structural and numerical CIN.
Impaired sister chromatid segregation
The separation of the chromosomes at anaphase requires the loss of sister cohesion in a timely manner. This is facilitated by the cohesin complex, a multi-protein complex composed of four core subunits: either stromal antigen 1 (STAG1) or stromal antigen 2 (STAG2), structural maintenance of chromosomes 1A (SMC1A), structural maintenance of chromosomes 3 (SMC3), and RAD21 cohesin complex component (RAD21).153,154,155,156,157,158 During prophase, the bulk of the cohesin complex, which consists of a ring-shaped structure formed by SMC1A, SMC3, RAD21, and STAG1 in the chromosome arm is removed.159,160 This process involves several proteins, including the WAPL cohesin release factor (WAPL), PDS5 cohesin-associated factor (PDS5), and polo-like kinase 1 (PLK1). These proteins assist in opening the cohesin ring, facilitating its removal from the chromosome arms.161 At anaphase, when separase is activated, the cohesin complex consisting of SMC1A, SMC3, RAD21, and STAG2 in the centromere was cleaved at the RAD21 subunit, leading to the opening of the cohesin ring at the centromere and the final separation of sister chromatids.153,159
Genetic alterations of any of the cohesin subunits, including mutations and inactivation, have been associated with CIN in various human tumors, as this dysregulation in turn leads to aberrant chromatid cohesion and allows premature separation of sister chromatids before full alignment, leading to chromosome bridge, lagging chromosome, and micronuclei.153 Defects in cohesin subunits, such as STAG1 or SMC1A, cause premature separation of chromosome arms and increased aneuploidy.87,88 Moreover, WAPL overexpression induces premature separation of chromosome arms, thereby increasing the rate of chromosome bridge and micronuclei;89 while PDS5 defect disrupts the regulation of cohesin ring removal from chromosome arms, leading to increased DSBs through an as-yet-unknown mechanism.90
Meanwhile, mutations in STAG2 as well as RAD21, a core component of cohesin complex at the centromere crucial for holding the centromeres of sister chromatids together from the time of DNA replication in S phase until their segregation in mitosis, also cause premature separation of sister chromatids and subsequently increase aneuploidy.91,92,93 Interestingly, mutation in STAG2 could also disrupt the interaction between cohesin and the replication machinery, thereby triggering DSBs and subsequently translocation by increasing stalling and collapse of replication forks.94,95 Furthermore, mutation in RAD21, a core component of the cohesin complex that plays a crucial role in holding the centromeres of sister chromatids together from the time of DNA replication in S phase to their segregation in mitosis, induces premature separation of sister chromatids, and increased aneuploid.92 Together, these studies demonstrated that sister chromatid defect contributes to the generation of structural and numerical CIN.
Aberrant centrosome number
CIN can also arise from aberrant centrosome amplification and separation, both of which are critical processes in cell division. Failure in centrosome amplification, for example due to the defects in specific motor proteins such as kinesin family member 2A (KIF2A), kinesin-like protein at 10A (KLP10A), and kinesin-like protein at 67A (KLP67A), or defects in centrosome proteins such as γ-tubulin, gamma complex component 2 (GCP2), and gamma complex component 3 (GCP3), can cause formation of monopolar spindle due to poor centriole separation.162 Monopolar spindle in turn leads to improper sister chromatid separation, as there is only one pole for them to move towards.96 Eventually, all chromosomes end up in a single daughter cell when the cell completely divides, resulting in the formation of polyploid cell.163
Improper timing of centrosome separation prior to cell division, both delayed and accelerated centrosome separation, would also lead to the formation of monopolar spindle.164,165 Loss of ubiquitin-specific peptidase 44 (USP44), a deubiquitinase that localizes at the centrosome, results in incomplete centrosome separation as well as increased monopolar spindle, lagging chromosome, and chromosome bridge. Moreover, USP44 knockout mice are prone to increase in numerical CIN as observed by elevated levels of aneuploidy.166 Defective centrosome separation can also occur in cells with overexpressed kinesin family member 11 (KIF11), a motor protein that drives centrosome separation. KIF11 overexpression can disrupt the normal timing and coordination of centrosome separation, by causing centrosomes to separate too quickly before the completion of centrosome duplication. In such cases, the spindle poles would be formed by imperfectly duplicated centrosomes, leading to the formation of monopolar spindle, and eventually, polyploidy.97
Meanwhile, the mitogen-activated protein kinase (MAPK) pathway, known for its role in cell proliferation, differentiation, and survival, has been implicated in the induction of CIN through centrosome amplification, as constitutive activation of MAPK through rat sarcoma virus (ras)-overexpression resulted in the increase of centrosome amplification, leading to multipolar spindle.167 Furthermore, overexpression of polo-like kinase 4 (PLK4), a master regulator of centrosome amplification, can result in overduplication of centrosome.98 This consequently lead to the formation of cell with multipolar spindles. When anaphase occurs in these cells, the chromosomes are separated abnormally, resulting in aneuploid daughter cells.98 Together, defects in centrosome duplication and separation contribute to the formation of monopolar and multipolar spindles, which are frequently observed in human tumors and are associated with CIN.168,169,170,171,172,173
Microtubule kinetochore attachment error
The process of spindle microtubules binding to kinetochores is asynchronous and stochastic, occurring at different times and in a random manner. This randomness and lack of synchronization can sometimes lead to erroneous attachments, such as merotelic attachments where a single kinetochore binds to microtubules anchored at both spindle poles, and could lead to chromosome missegregation.113,174,175,176,177 Despite the erroneous nature of merotelic attachments, cells often manage to segregate these chromosomes correctly during anaphase. This is due to the cell mechanism that corrects these erroneous kinetochore-microtubule (K-MT) attachments by converting them into bi-oriented attachments, where each kinetochore is attached to microtubules from only one spindle pole.178,179,180
The efficient correction of merotelic attachments requires the dynamic turnover of K-MT interactions. The optimal stability of K-MT attachments, which is neither too loose nor too hyperstable, is crucial for this process. A decrease in this turnover rate could result in persistent merotelic attachments and increased chromosome segregation errors, such as lagging chromosomes.99,100 For instance, some tumors with CIN are characterized by hyperstable K-MT interactions, a state that is more stable compared to chromosomally euploid cells, leading to failure in correcting merotelic attachments and increased CIN; while reducing the K-MT attachment stability from this hyperstable state can restore normal chromosome segregation in cells with CIN.99,100,181 However, while previous studies have shown that loss of STAG2, a cohesin subunit that has been reported to have roles beyond sister chromatids cohesion, results in hyperstabilized K-MT attachments, the exact molecular mechanisms are not fully understood.182,183 To fully characterize the contribution of microtubule-kinetochore attachment errors in CIN, a more detailed study of the complex and dynamic process of spindle microtubules binding to kinetochores is required.
SAC defects
The primary goal of a cell undergoing mitosis is to segregate the replicated chromosomes into two new daughter cells. This is achieved through the attachment of chromosomes to microtubules of the mitotic spindle apparatus.184 Chromosomes attach to the ends of microtubules at kinetochores, which are specialized protein structures that bind to the chromatin centromere.184 Normally, each chromosome has two kinetochores, and it is essential for mitotic cells to form bi-orientation.184 This state is achieved when each sister kinetochore binds microtubules oriented toward opposite spindle poles.
A checkpoint mechanism known as the SAC delays the separation of the sister chromatids at anaphase until every kinetochore has correctly attached to spindle microtubules and all sister chromatids have aligned at the metaphase equatorial plate.185,186,187,188 Thus, the SAC is a safeguard for guaranteeing chromosome bi-orientation on the mitotic spindle by monitoring the proper kinetochore attachment as well as chromosome alignment. As long as improperly attached or unaligned chromosomes remain, SAC halts cells in mitosis and prevents their progress into the final phases of cell division.185,186,187,188 Components of the SAC, including mitotic arrest deficient 2 (MAD2),107,189,190,191,192 budding uninhibited by benzimidazoles 1 (BUB1),193,194,195,196 budding uninhibited by benzimidazoles 1 beta (BUBR1),110,195,197 and budding uninhibited by benzimidazoles 3 (BUB3),193,194,198,199,200,201,202 migrate to unattached kinetochores and form mitotic checkpoint complex (MCC) along with cell division cycle 20 (CDC20). MCC is a key effector of SAC that inhibits the activation of the CDC20-bound anaphase-promoting complex/cyclosome (APC/CCdc20), an E3 ubiquitin ligase that targets cyclin B and securin for degradation by the proteasome.185,186,187,203,204,205,206,207,208,209,210,211 Once sister chromatids have properly attached and aligned, the SAC is inactivated, allowing the MCC to dissociate, freeing CDC20 to activate the APC/C.212 The activation of the APC/CCDC20 triggers securin and cyclin B degradation.203,207,209,213 Securin destruction frees separase, an enzyme that cleaves and inactivates the cohesin complex, allowing sister chromatid separation and the onset of anaphase.203 Meanwhile, cyclin B degradation inactivates cyclin-dependent kinase 1 (Cdk1), allowing the cells to proceed to mitotic exit and complete cell division.185,186,214,215
In eukaryotic cells, SAC plays a crucial role in genomic integrity and its abnormality leads to chromosome segregation errors.10,101 Defects in SAC result in the failure of proper monitoring and controlling the timing of sister chromatid segregation.10,101,102 This in turn leads to increased chromosomal abnormalities, such as chromosome bridge and lagging chromosome, and eventually, errors in equal distribution of genetic material to daughter cells.103,104,105 Moreover, as described above, lagging chromosome, as well as chromosome bridge, could reassemble and form micronucleus, a nucleus-like structure consisting of a bilayer membrane covering a piece of extrachromosomal DNA.175
Cells lacking MAD2, an SAC component, can proliferate in vitro and in vivo but with increased levels of CIN.103,104 Moreover, weakening the checkpoint in mice by partially reducing the expression of various SAC genes including MAD1, MAD2, BUB1, BUBR1, and BUB3, results in premature separation of sister chromatids, chromosome missegregation, and subsequently, CIN.106,107,108,109,110,111,112 In addition to the SAC components, the CDK pathway also plays a significant role in CIN. Gao et al. reported that CUE domain containing 2 (CUEDC2) is phosphorylated by CDK1 during mitosis. This phosphorylated CUEDC2 promotes spindle checkpoint inactivation by promoting MCC dissociation from the APC/C, leading to premature inactivation of SAC and increased CIN.216 Furthermore, chromosome missegregation can be caused by mutations that weaken the SAC, which subsequently results in premature anaphase onset.105 However, mutations in SAC genes are rarely found in human tumors, suggesting that while SAC mutation is one of contributors to CIN in tumor cells, aberrant transcriptional, post-translational modification, and epigenetic regulations might also contribute to SAC defects.121,217,218,219
Interestingly, while weakened SAC can cause CIN, overactivity of the checkpoint induced by, for example, overexpression of SAC gene such as MAD2, or knockdown of genes involved in SAC silencing pathways such as p31/comet or TRIP3, can also induce CIN.220,221,222,223,224 Similar to SAC defect, SAC hyperactivation could lead to the increase of chromosome bridge, lagging chromosome, and micronucleus.220,221,222,223,224,225 However, in contrast to weakened SAC which accelerates mitotic progression and tumor cells proliferation, SAC hyperactivation delays the onset of anaphase and prolongs mitotic arrest.220,221,222,223,224,225 Furthermore, unlike chromosome missegregation induced by SAC defect, which stems from the premature anaphase progression before the erroneous chromosomes-microtubules attachments are corrected, the mechanism of SAC hyperactivation-induced chromosome missegregation is not entirely understood. One possible explanation is that persistent SAC signaling could lead to cohesion fatigue, where the cohesin complexes that hold sister chromatids together become exhausted over time, resulting in aberrant sister chromatid segregation.226,227 Together, while the SAC plays a vital role in ensuring accurate chromosome segregation during mitosis, both its defect and hyperactivation can paradoxically lead to CIN.
Polyploidy-related cell cycle dysregulation
Polyploidy is a condition where a cell has multiple sets of chromosomes and could be both a consequence as well as a cause of CIN.228,229,230,231,232 Polyploidy can occur due to various reasons, including cytokinesis failure, mitotic slippage, endoreduplication, or cell fusion.233 Cytokinesis failure occurs when daughter cells fail to separate after accomplishing telophase.74,234,235,236 This can happen due to various reasons, such as problems with the contractile ring that separates the two daughter cells or the presence of chromosome bridges that physically prevent the cells from separating.234 When cytokinesis failure occurs, the two daughter cells remain connected and form a binucleated cell with twice of the normal number of chromosomes.234 Furthermore, polyploidy can be caused by mitotic slippage, which is a process of premature mitotic exit. This can occur when the SAC activity is weakened, leading to the misinterpretation that all chromosomes are correctly attached to the spindle and the failure to inhibit the activation of the APC/CCDC20.108,237,238 This failure then promotes the premature degradation of cyclin B1, which in turn leads to a decrease in Cdk1 activity, and, as a consequence, promotes the onset of anaphase and premature exit from mitosis without proper chromosome segregation.108,237,238
Endoreduplication is a process in which cells undergo multiple rounds of DNA replication without mitosis, resulting in cells with multiple copies of their genome.239 Endoreduplication can occur due to various reasons, such as problems with the cell cycle machinery.239 One key cell cycle machinery associated with this endoreduplication is a defect in the pre-replication complex (pre-RC).240 The pre-RC, which includes the origin recognition complex, cell division cycle 6, chromatin licensing and DNA replication factor 1 (CDT1), and minichromosome maintenance complex 2-7, assembles at replication origins during G1 phase to license DNA replication at S phase.240 This complex then dissociates from the replication origins after DNA replication is started to prevent another round of DNA replication before the cell completes cytokinesis, thereby guaranteeing the proper number of chromosomes being passed to daughter cells.240 Hence, dysregulation in pre-RC could lead to whole-genome doubling (WGD), a form of polyploidy.228,241,242,243,244,245,246
Cell fusion is a process in which two or more cells fuse together to form a single cell with multiple nuclei, also known as a synkaryote.239,247 This can occur due to various reasons, such as exposure to certain viruses or chemicals.239,247 Following fusion, the parental chromosomes mix and redistribute to the fused cells, thereby producing polyploid fused cells.239,247 Therefore, cell fusion can also result in polyploid cells and contribute to CIN.
The increase in polyploidy, therefore, signifies an increase in CIN, underlining the critical role these cellular processes play in causing CIN. However, as mentioned above, besides as a consequence of CIN, polyploidy is also an important cause of CIN. The extra set of chromosomes in a cell can lead to errors in chromosome segregation during subsequent cell division, forming aneuploid cells with abnormal chromosome numbers.74,248,249,250,251 For instance, a study using tetraploid cells demonstrated that these cells, which contain twice the number of chromosome sets along with two extra centrosomes, can lead to the formation of multipolar spindles. This, in turn, results in the formation of aneuploid cells.252 On the other hand, cells with CIN can also become polyploid due to errors during cell cycle dysregulation as mentioned above.253 Together, this highlights the complex interplay between polyploidy and CIN.
CIN paradox
Maintaining genomic stability is essential for the normal functioning of the cells, and for ensuring the accurate transmission of genetic information to progeny, thereby preserving the continuity of the species.8,9,10,11 Normal cells have intrinsic potentials for maintaining their genome integrity through various mechanisms, such as the ability to repair their damaged DNA, as well as for preventing the passage of damaged, unrepairable DNA to their progenies by triggering apoptosis.254 Given the critical role of genomic stability, any deviation from this state can have deleterious consequences. One such challenge is CIN, which can be detrimental when present in normal cells, as it could decrease cellular fitness.3,12 As observed by Theodor Boveri over a century ago, chromosomal abnormalities are typically intolerable in normal cells, often culminating in cell death.41 However, it is essential to note that CIN is not universally detrimental. There are examples, notably in simpler organisms like bacteria, viruses, and fungi where elevated genomic instability can confer advantages in stressful environments.13
For instance, clinical isolates of the yeast Candida albicans that are resistant to the anti-fungal drug fluconazole carry extra copies of chromosome 5, where genes encoding the drug target, lanosterol 14-α-demethylase (ERG11), and a main regulator of drug efflux pumps, transcription activator of CDR genes 1 (TAC1), are located.255 This indicates that CIN could provide drug resistance through the increased expression of these genes.256 Furthermore, in other yeasts, Saccharomyces cerevisiae and Candida glabrata, increased CIN-induced aneuploidy results in phenotypic advantages by promoting their resistance to fluconazole.257,258,259 Similarly, in bacteria and viruses, elevated genomic instability benefits the population in stressful environments by promoting the survival and proliferation of cells with specific genetic aberrations that confer a growth advantage.13 This suggests a nuanced perspective on CIN, highlighting its potential benefits under specific circumstances. However, it is important to emphasize that the intensity of CIN cannot exceed certain thresholds. Indeed, in bacteria and viruses, cells with drastic instability never become dominant in a population, as their excessive instability levels exceed the cellular threshold, leading to genetic catastrophe and cell death.13 This observation suggests that while moderate CIN can be beneficial, excessive CIN may lead to genetic catastrophe and is lethal. Therefore, understanding the balance between beneficial and detrimental effects of CIN is crucial to comprehend its role, not only in simpler organisms but also in more complex systems like mammalian cells.14,15,16,17,18,19,20
With the advancement of our knowledge regarding tumor biology, a similar observation emerges, where CIN resembles a double-edged sword (Table 1). On one hand, CIN can promote tumorigenesis. As exemplified in yeast and mammalian cells above, CIN could contribute to clonal evolution, providing selective advantages under stressful conditions encountered by tumor cells.257,260,261 This clonal evolution, driven by CIN, can be a key factor in promoting tumorigenesis, as it not only helps tumor cells to survive in harsh environments but can also foster the evolution of the clones with the most tumorigenic phenotypes, that is, clones that have new karyotype that brings them growth advantage and the ability to outcompete others.260 This is also supported by studies using animal models. For example, mice carrying heterozygous deletions of SAC genes, such as MAD1, MAD2, and BUB1B, exhibit increased CIN and develop spontaneous tumors.106,107,108 Similar evidence comes from human patients with mosaic variegated aneuploidy syndrome (MVA), which is characterized by increased CIN and a predisposition to childhood cancer.109,110,262,263 These results suggest that tumor cells may exploit CIN to harness the potential of clonal evolution for optimal adaptation. However, on the other hand, CIN has also been reported to have anti-proliferative effects,264,265,266 and can induce cell death,267,268 senescence,269,270,271 as well as anti-tumor immune response.270,272,273 Clinical observations further complicate the picture, with high CIN signatures in various tumors associated with improved prognosis.274,275,276,277,278,279 These observations have led to the establishment of the “just-right” hypothesis, proposing a moderate level of CIN that benefits tumorigenesis and tumor progression. However, while the concept of how populations with genetic instability evolve over time has been observed in a study based on mathematical modeling,280,281 experimental evidence to support the “just-right” hypothesis for the relation between CIN level and tumor cell fate determination, as well as the molecular mechanisms underlying it are still lacking. One possible explanation for this, at least in part, is the use of different models to elucidate the role of CIN in cancers, emphasizing the need for further research. In addition, it is noteworthy that the specific context of aneuploidy induced by CIN can also influence tumor cell fate. While tumor cells may be able to tolerate the effects of additional chromosomes, excessive loss of chromosomes that contain essential genes for cell survival is detrimental. Therefore, the balance between gain and loss of chromosomes is crucial for tumor cell viability.282,283
Together, the role of CIN in tumorigenesis is complex and paradoxical (Fig. 5), influenced by factors such as the degree of CIN and the specific conditions faced by tumor cells. The intricate balance between the harmful and advantageous effects of CIN underscores the complexity of CIN paradox, emphasizes the need for further research. In the following section, we will discuss further the consequences of CIN and how its seemingly negative effects can, under certain circumstances, confer advantages to tumor cells.

CIN paradox according to the “just-right” model. A moderate, “just-right” level” for CIN could induce a tumor-promoting phenotype by increasing tumor cells adaptability, while excessive CIN is deleterious to tumor cells due to excessive chromosome gain or loss leading to cell death
The multifaceted impacts of CIN on tumor biology
CIN exerts a multifaceted influence on tumor biology, playing significant roles in both tumorigenesis and tumor progression. However, the relationship between CIN and tumorigenesis is complex and resulting in CIN paradox, as mentioned in the previous section. In this section, we will discuss about the diverse consequences of CIN, starting with those closely related to tumorigenesis. These include DNA damage, proteotoxic stress, and metabolic alteration, which potentially have both beneficial and deleterious effects. We will then explore the generally deleterious effects of CIN on cell cycle arrest and senescence. Lastly, we will discuss the beneficial effects of CIN on metastasis, tumor immune regulation, and drug resistance (Fig. 6, Fig. 7, and Table 2).

Consequences of CIN. a Chromothripsis, also known as “chromosome shattering” is induced from rupture of micronuclei, followed by fragmentation of micronuclear DNA and its massive rearrangements. b Protein stoichiometry imbalance caused by changes in the copy number of chromosomes, leading to proteotoxic stress. c Other cellular functions altered by CIN, including metabolic alteration, cell cycle arrest, and senescence

Impact of CIN on drug resistance. CIN confer resistance to anti-tumor drug treatment or immune response through increased intratumor heterogeneity
DNA damage
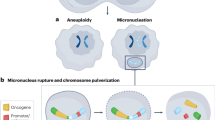
CIN can lead to errors in chromosome segregation during cell division, resulting in the formation of lagging chromosomes and chromosome bridges. These lagging chromosome and chromosomal fragments from chromosome bridges often partition into micronuclei, whose nuclear envelope lacks several non-core membrane proteins such as lamin B, lamin B receptor (LBR), and nucleoporins, making them relatively fragile and prone to rupture.116,129,284,285,286 This rupture exposes the DNA content within the micronuclei to the cytoplasm, leading to chromothripsis, also known as “chromosome shattering”, a process of fragmentation of micronuclear chromosome followed by their massive rearrangements into the main nucleus. Hence, while itself a consequence of CIN as it originates from the micronuclear chromosome, chromothripsis could, at the same time, induce the succeeding CIN, as it could lead to the formation of a karyotype with complex chromosomal rearrangements.78,115,122,123,124,125,126,127,128,129,130,131,132,133
Chromothripsis has been associated with various tumor types and can lead to both numerical and structural CIN, altering gene expression patterns and driving changes in cellular behavior.122,287,288,289 Chromothripsis can contribute to tumorigenesis in multiple ways, either beneficial or deleterious. For instance, while DNA damage can be a cause of CIN as discussed in DSBs from replication stress, it is also a significant consequence of CIN, as chromothripsis could induce extensive DNA damage and destabilize tumor cell growth, leading to apoptosis.122 Interestingly, it can also create a tumor-promoting environment under certain circumstances, including rearrangement that results in karyotype with a better survival advantage. For example, reparation of shattered chromosomal fragments can lead to the incorporation of genetic material from the shattered fragments into double minute chromosomes, which are small, circular chromosomes without centrosomes distributed asymmetrically to daughter cells during cell division.290 Double minutes (also known as extrachromosomal DNA) were reported to contain high copy numbers of MYC proto-oncogene (MYC). Previously, Martins et al. reported that high MYC is an early event selected in many tumors with CIN, thereby providing a selective growth advantage to the tumor cells.291,292,293 Incorrect repair can also lead to a complete loss of gene function, such as the loss of key tumor suppressor gene mothers against decapentaplegic homolog 4 (SMAD4),294 as well as generation of novel oncogenic proteins by chromosome fusion. For example, a novel fusion oncoprotein which could promote AKT signaling activity, ubiquitin specific peptidase 9 X-linked-ES cell expressed Ras (USP9X-ERAS), is formed by chromothripsis involving the US9PX and ERAS genes in colon cancer cells.295
Collectively, chromothripsis is the consequence and at the same time, the cause of further CIN. The cycle of CIN leading to DNA damage, which in turn exacerbates CIN, forms a complex interaction that plays a crucial role in tumor development and progression. Moreover, recent studies suggest that CIN is associated with changes in both chromatin accessibility and transcription resulting from micronuclei formation.296,297 This complex, bidirectional relationship between chromothripsis and CIN, underscores the intricate dynamics of genomic instability in cancer.
Proteotoxic stress
Proteins in cells are often composed of more than one subunit. Multi-subunit protein complexes require balanced stoichiometry to function properly.268,298,299,300,301,302,303 This is achieved by regulating the ratio of protein subunits, and by degrading excessive, unassembled protein subunits.300,304 Unassembled protein subunits must be bound by chaperones to remain in solution until they are degraded by the ubiquitin-proteasome degradation pathway, thus, excessive production of subunits can overwhelm the protein quality control systems, impairing the stoichiometry and homeostasis of proteins in multi-subunit complexes.300,304 Furthermore, the increased demand for degradation puts the cell under proteotoxic stress, a form of cellular stress, and impairs cellular proliferation.268 This is often observed in aneuploid cells, where excessive protein subunits encoded by the altered chromosome could lead to imbalanced protein stoichiometry.305,306,307,308
In contrast, polyploid cells, which contain multiple sets of chromosomes, are likely to suffer less from the effects of genetic alteration and stoichiometry imbalance.230 When an essential gene is altered in polyploid cells, they still have more copies of functional genes compared to aneuploid cells. This redundancy provides a buffer against genetic alterations that might otherwise be detrimental. Kuznetsova et al. revealed that tetraploid cells proliferate almost as efficiently as diploid cells, and exhibit only some detrimental phenotypes observed in aneuploid cells.230 One reason for this could be that the multiple sets of chromosomes in polyploid cells help maintain a balance in protein stoichiometry by keeping the ratio of subunits forming a protein complex constant;230 while in aneuploid cells, gain or loss of certain chromosomes disrupts this ratio, eventually causes the dysfunction of protein complexes that require a specific stoichiometry of their subunits.
Imbalance protein stoichiometry could impair specific cellular functions associated with the affected protein complexes. For example, the gain of chromosome 6, which carries the gene encoding β-tubulin, causes lethality in yeast due to excessive β-tubulin production. However, this lethality can be rescued by additional gain of chromosome 13, which carries the α-tubulin gene, thus restoring the stoichiometry of α/β-tubulin dimers.309 Furthermore, imbalanced protein stoichiometry due to aneuploidy can also lead to the misfolding of proteins, which can accumulate and form toxic aggregates.268,298,299,300,301,310,311
Paradoxically, aneuploidy could also be beneficial for tumor cells. Aneuploidy could alter protein stoichiometry at the level of the interactome, which is the complete set of molecular interactions within a cell or organism including protein-protein, protein-DNA, and other types of molecular interactions essential for cellular function.312 A study of aneuploid patient tumor samples indicated that MET proto-oncogene, receptor tyrosine kinase (MET) amplification conferred resistance to epidermal growth factor receptor (EGFR) inhibitors, erlotinib and gefitinib.313 Typically, MET does not directly activate kinases downstream of the EGFR due to its low binding affinities with them. However, excessive MET expression can bypass the effect of EGFR inhibitors by directly interacting with these kinases and activating them, thereby counteracting the anti-tumorigenic effect of EGFR inhibitors.313 This finding illustrates that the aberrant protein interactions, which arise from the excessive proteins due to stoichiometry imbalance, could serve as a mechanism through which aneuploidy reshapes the interactome, thereby promoting tumorigenesis in tumor cells.
Metabolic alteration
Metabolism, an essential biological process involving a series of chemical reactions that convert food into energy and building blocks for the cells, is controlled by a finely-tuned coordination of complex metabolic networks, which depends on the precise balance of enzymes and regulators. Introduction of additional gene copies could disrupt this delicate balance, leading to metabolic alterations. Aneuploid cells, whether in yeast, animal models, or human cells, exhibit altered cellular metabolism.265,299,314,315 For instance, amplification of chromosome 4 in yeast results in the increase of amino acid levels, except the levels of aspartate and isoleucine, and various tricarboxylic acid cycle intermediates, leading to defects in cell growth.316 In MEFs, trisomy in either chromosome 1, 3, 16, or 19 leads to alterations in glutamine metabolism, and subsequently proliferation defects.265 Similarly, extra copy of chromosome 3 or 5 can impair human cell proliferation through downregulation in proteins involved in carbohydrate metabolism264 Therefore, CIN can lead to alterations in cellular metabolism, which can have detrimental effects on cells.
However, metabolic alteration, also known as metabolic reprogramming, is a hallmark of cancer that can provide several advantages for tumor cells.317,318,319 Metabolic alteration in tumor cells was first observed by Otto Warburg, who noted alterations in the glucose metabolism of tumor cells.320 Since then, alterations in other metabolic pathways, such as amino acids and lipid, along with their importance in tumor biology, have also been found.321,322 Normal cells produce energy from glucose effectively in the presence of oxygen by coupling glycolysis with oxidative phosphorylation.320 In contrast, one of the most well-known tumor cells metabolic reprogramming is the Warburg effect, a phenomenon where tumor cells increase their glucose uptake and glycolysis rate, and prefer glycolysis followed by fermentation, or aerobic glycolysis, instead of glycolysis followed by oxidative phosphorylation even when oxygen is sufficient.320,323 This allows tumor cells to cope with the fluctuating oxygen levels often found within tumor tissues.324 Moreover, aerobic glycolysis could also meet the increased demands of rapid cell proliferation for essential building blocks such as nucleotides, amino acids, and lipids.320,325,326,327 Furthermore, it could fulfill the demand of highly proliferating tumor cells for cellular reductants such as nicotinamide adenine dinucleotide phosphate (NADPH), which are crucial for lipid biosynthesis, drug resistance, and for scavenging reactive oxygen species (ROS) generated by high proliferation.320,323,328
CIN can induce the Warburg effect, triggering metabolic changes that promote tumorigenesis. This is evidenced by a correlation between karyotypic heterogeneity, which serves as an indicator of CIN, and increased consumption of glucose and glutamine, as well as increased production of lactate and glutamate.22,299,329,330 However, the association between other metabolic alterations, such as amino acids metabolic alteration and lipid metabolic alteration, and CIN, except the glutamine and glutamate metabolisms,329 has not been reported and still needs further investigation. In addition to the alterations in nutrient-related metabolism, CIN also impacts the cellular redox state. Cells with CIN exhibit changes in mitochondrial numbers and activity, typically resulting in increased ROS.331 While high levels of ROS can lead to oxidative stress and potential cell death, moderate levels of ROS can promote metastasis.332 Therefore, while CIN initially seems detrimental, it can actually benefit tumor cells by providing them with metabolic advantages. This highlights the complex interplay between CIN, metabolic reprogramming, and tumorigenesis.
Cell cycle arrest
CIN has been reported to cause cell cycle arrest.333,334,335,336 Live-cell imaging of human cells with chromosome missegregation demonstrated that missegregation induces cell cycle arrest in a p53-dependent manner.333,334,335,337 Known as the “guardian of the genome”, p53 plays crucial role in controlling cell cycle progression.338,339,340,341 The tumor suppressive function of p53 is closely related with response to CIN and is critical for determining the fate of cells experiencing CIN.333,334,335,342,343,344,345,346
p53 could suppress the propagation of structural CIN following chromosomal missegregation by inducing cell cycle arrest and apoptosis, thereby limiting the proliferation of aneuploid cells.270,334,347,348 p53 inactivation in tumor cells with CIN results in defects in inducing cell cycle arrest and apoptosis, and eventually, in increased tumor heterogeneity.104,349,350,351 This tendency is consistent with the findings in clinical non-small cell lung tumors, whereas p53-mutant tumors display more complex karyotypes than their wild-type counterparts.352,353 Furthermore, apoptosis observed in CIN mice model, presumably triggered by increased CIN, was rescued upon depletion of p53.348,354 In line with these, in a CIN model using SAC-deficient mice, reducing p53 level leads to increased aneuploidy and T-cell lymphoma proliferation, and at the same time, decreased survival.104,348,349 The RAS pathway, a critical signaling pathway in cells, is involved in cellular signal transduction, leading to cell growth and division. Overexpression of Harvey rat sarcoma virus (H-RAS) can induce CIN. However, the activation of RAS and the subsequent induction of CIN can be halted by the activation of p53, resulting in reduced transforming potential in mice model.355 This study further highlights the importance of p53 in monitoring and preventing CIN propagation to progenies.
In addition to p53, the stress kinase p38, which is a proline-directed serine/threonine kinases of the MAPK family, also plays a role in controlling the proliferation of aneuploid cells. p38 is activated in response to various stress stimuli, including CIN induction, and can induce cell cycle arrest by several mechanisms, including the upregulation of CDK inhibitors, growth arrest and DNA damage inducible alpha (GADD45α), and cyclin D, as well as the downregulation of CDC25.333,356,357,358 Moreover, p38 can work side-by-side with p53 to limit the progression of cells with CIN. Upon missegregation events, p38 increases the degradation of MDM2, a negative regulator of p53, through phosphorylation. This leads to the stabilization of p53 protein and apoptosis induction, thereby preventing the proliferation of cells with CIN.333,359 In addition to chromosomal-related events, the p38/p53 axis can also respond to metabolic stress induced by ROS formation as a consequence of CIN.333,334,358,360 These findings highlight the complex interplay between stress pathways, CIN, and cellular responses, underscoring the crucial roles of p53 and p38 in limiting the progression of cells with CIN.
Senescence
Cells with DNA damage and chromosome missegregation often became senescent and acquired the senescence-associated secretory phenotype (SASP). CIN enhanced by the treatment with SAC inhibitor could result in senescence, as indicated by the increase of senescence markers, such as p53, p21, p16, and senescence-associated β-galactosidase activity.271,361,362,363,364 The DDR pathway, activated in response to CIN-induced DNA damage plays a crucial role in this process.139 DDR can lead to the upregulation of p53, which in turn activates the expression of p21.137,365,366 This subsequently induces cell cycle arrest as a temporary response to allow DNA repair, or senescence as a permanent state of cell cycle arrest when the DNA damage is too severe to be repaired.367 Furthermore, CIN could also enhance the level of another senescence marker, p16, with a mechanism which is still unclear.
The consequence of CIN-induced senescent is complex. From a cell-autonomous perspective, senescence is a mechanism of tumor suppression in which aneuploid cells that undergo senescence will stop dividing and unlikely to undergo cellular transformation.362,363 In addition, senescent cells could be further cleared through autophagic cell death.368 However, CIN-induced senescence also triggers SASP-like gene expression signature, which might contribute to tumorigenesis.270,369 The unique secretome from SASP contains biologically active factors that are released into the microenvironment.370,371 These factors, including chemokines, cytokines, growth factors, and immune regulators, can induce a positive feedback loop and cause chronic inflammation, which can have dual effects on tumorigenesis.270 On one hand, it can act as a defense mechanism against tumors by promoting an anti-tumor immune response. On the other hand, it can contribute to tumorigenesis, as the secretome could activate key transcription factors involved in tumorigenesis, such as RAS.372 Furthermore, CIN-induced senescent cells can also increase the migration and invasion capacities of the neighboring tumor cells through SASP, thereby contributing to tumor progression.369 Therefore, the effect of CIN-induced senescence and its SASP-like gene expression is complex, resulting in both tumor-suppressive and pro-tumorigenic outcomes.
Metastatic capacity
As supported by pre-clinical and clinical data, changes in chromosome copy number could also influence cell motility, matrix degradation, epithelial-mesenchymal transition (EMT), and other processes necessary for metastatic behavior.373,374,375,376 For instance, tumor cells harboring an extra copy of chromosome 5 displayed increased metastatic capacity, as it leads to the silencing of epithelial cell-adhesion genes and thereby activates EMT.377 Meanwhile, the loss of chromosome 16q has been associated with downregulation of E-cadherin (CDH1).378 Interestingly, Gao et al. demonstrated that CIN is also associated with mesenchymal-epithelial-transition (MET), a reverse version of EMT required for extravasation and colonization in different tissues in the process of distant metastasis.378,379 Loss of chromosome 10p results in the loss of the zinc finger E-box-binding homeobox 1 (ZEB1) gene, thus promoting MET, and subsequently metastasis378 Furthermore, CIN could drive metastasis by activating Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway, and through the establishment of local immunosuppressive microenvironment.380,381 Moreover, CIN can also indirectly induce EMT. For instance, the loss of chromosome 8p results in the downregulation of 8p-localized genes, such as N-acylsphingosine amidohydrolase 1 (ASAH1), farnesyl-diphosphate farnesyltransferase 1 (FDFT1), leptin receptor overlapping transcript-like 1 (LEPROTL1), epoxide hydrolase 2 (EPHX2), and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like (BNIP3L), thereby altering the mevalonate and fatty acid metabolic pathways. Disruption of these lipid metabolic pathways in turn increases the activities of small GTPases, such as Ras homolog family member (RHO), Ras-related C3 botulinum toxin substrate (RAC), and rat sarcoma (RAS), and subsequently promotes invasion and metastasis.378,382,383 Meanwhile, in the clinical setting, longitudinal studies such as TRACERx, which track the progression of cancer from primary disease to metastasis and recurrence, have reported that elevated CIN correlates with an increase in metastasis and worse survival outcomes, and subsequently poor prognosis.384,385,386,387,388,389 These results demonstrate the positive correlation between CIN and metastasis, as well as poor survival and outcomes. However, it should be noted that a recent study has also reported the anti-metastasis function CIN. For example, changes in chromosome copy number, for example gaining extra copy of chromosome 13 or chromosome 18, could suppress metastasis.377 The underlying mechanisms of how these specific chromosomal changes suppress metastasis remain unclear, highlighting the complex role of CIN in tumor metastatic capacity.
Tumor immune regulation
CIN has dual activity in immune response, as it is capable of inducing either anti-tumor or pro-tumor immune response. In xenograft models, tumor cells with increased aneuploidy and polyploidy tend to form tumors in immunocompromised mice. However, these tumors either fail to grow or grow more slowly in immunocompetent mice, suggesting the anti-tumor role of CIN-induced immune response.390 This could be attributed to genomic alterations that produce neoantigens, which are recognized by the immune cells, and thus activate the adaptive immune response.391 Consequently, CIN can activate anti-tumor immune responses, thereby subjecting tumor cells to the selection pressure imposed by the immune system, which subsequently eliminates them.391
However, CIN could also contribute to pro-tumor immune response. As a contributor to genomic instability, CIN increases intratumor heterogeneity,37 allowing the generation of different tumor cells with variations in antigen presentation, thereby reducing their visibility to the adaptive immune system, which subsequently leads to immune evasion.30 Furthermore, micronucleus, as a product of missegregation of chromosomes, could also trigger CIN-related immune regulation.53,392,393,394,395,396 Exposure of chromosomal double-stranded DNA (dsDNA) in the micronuclei to the cytoplasm activates the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING)-dependent immune response, a type of innate immune response originally discovered as a sensor of viral dsDNA.53,397,398,399,400 Typically, recognition of dsDNA by cGAS activates STING, which could in turn activate anti-tumor immune response through type 1 interferon (IFN) and canonical nuclear factor kappa B (NF-κB) signaling.53,392,401,402 Interestingly, in the context of CIN, STING promotes EMT and the expression of inflammatory genes, enhancing cell migratory capacity, and subsequently promoting metastasis, by activating non-canonical NF-κB signaling.53,392,403 This process indicates that CIN manipulates the innate immune system to promote tumor cells immune evasion.53 Furthermore, this example clearly demonstrates that CIN could alter normal cell function to favor tumor progression. Moreover, the activation of SASP through CIN-induced senescence could also shift the immune response towards pro-tumor.372 Together, these observations suggest that CIN, through increased heterogeneity or by activation of pathways in favor of pro-tumor immune response, can promote tumor cells immune evasion.
Drug resistance
Anti-tumor drugs encompass a wide range, each with its own complex mechanisms. For instance, alkylating agents cause DNA crosslinks that disrupt DNA replication, eventually inhibiting tumor cell division,13,317,401,402 while anti-metabolites can cause improper DNA synthesis by mimicking endogenous molecules.19,127 Irrespective of their mechanism, resistance to anti-tumor drugs has become a significant hurdle in anti-tumor treatment.27,404 While the molecular mechanism of tumor cells drug resistance are complex and have not been totally elucidated, CIN, as a fuel of tumor evolution that causes intratumor heterogeneity, has been assumed as one of the major reason for drug resistance.230,336,405,406 This increase in heterogeneity can be viewed as a survival strategy employed by tumor cells to adapt to unpredictable environments. This strategy, known as biological bet-hedging or “not putting all your eggs in one basket”, allows tumor cells to diversify their phenotypes, spreading the risk and increasing the likelihood of some cells to survive under selective pressures, such as those imposed by anti-tumor drug treatments.23,407,408
Moreover, Ippolito et al. previously reported the link between CIN and drug resistance through the upregulation of ATP binding cassette subfamily G member 2 (ABCG2), a drug efflux pump, due to the amplification of its upstream regulator MAPK13 in topotecan-resistant tumor cells generated from treatment using CIN-inducing drug nocodazole.17 Together, these show that CIN fuels genomic diversity, upon which selection works, leading to the development of drug resistance.
Although the result from the aforementioned study suggests that the generation of intratumor heterogeneity through CIN can shield tumor cells from the selective pressure caused by anti-tumor drugs, the role of CIN in drug response is nevertheless a complex relationship, as CIN could potentially be induced to excessive level, leading to cell death and enhancing the effectiveness of anti-tumor drug treatments. The following section will discuss how the induction of CIN can be leveraged to enhance the efficacy of anti-tumor drugs. A summary of the different types of anti-tumor drugs and their interactions with CIN is provided in Table 3.
CIN-based potential anti-tumor therapy
Previous studies indicated that an excessive level of CIN beyond a certain threshold could potentially induce tumor cell death. Thus, enhancing the CIN level has been proposed as a promising approach to target tumor cells, and strategies to exacerbate CIN for anti-tumor therapy have been explored.66,358,406,409,410,411,412,413,414,415,416,417,418,419,420,421,422 For example, taxol could increase the number and severity of chromosome segregation errors in tumor cells, while cells with excessive CIN were more sensitive to low doses of taxol.423 Indeed, combining taxol and monopolar spindle 1 (MPS1) inhibitor could reduce xenograft growth more effectively than either compound alone.411 Furthermore, combining SAC inhibitor and other non-taxol-based compounds that induce CIN can synergistically reduce tumor growth. For example, combining a SAC inhibitor with microtubule-destabilizing drug SKI606 results in the selective killing of tumor cells exhibiting a CIN phenotype.410 Moreover, combining a p38α inhibitor, which interferes with DNA damage response, with taxane-based chemotherapy increased the efficiency of killing breast tumor cells compared to taxanes alone by boosting CIN.358 While they have not yet been used in clinical settings, there are also other compounds that can induce CIN. These include inhibitors of the SAC proteins MAD2 or BUBR1, which can induce tumor cell death. Moreover, a compound that induces CIN by targeting the highly expressed in cancer 1/NIMA-related kinase 2 (Hec1/Nek2)-related mitotic pathway also demonstrates promising results.424,425,426
While enhancing CIN could potentially be a powerful method to eradicate tumors, the feasibility of such therapies will depend on many factors including CIN status and the capacity of the tumor cells to tolerate CIN.267,427,428,429,430,431,432,433,434,435,436,437 Moreover, it is important to consider that untransformed cells will also be affected by the CIN-inducing agents and thus will suffer from low to moderate CIN rates. This may predispose these cells to become tumorigenic, leading to therapy-induced tumorigenesis subsequently.
To overcome this problem, efforts have been made to develop strategies that more selectively target cells displaying CIN phenotype, either by exploiting specific vulnerabilities of tumor cells with CIN or identifying new weaknesses incurred in tumor cells with new karyotype. For example, a study by Marquis et al., which aims to identify synthetic lethal gene in tumor cells with CIN, has discovered that targeting kinesin family member 18A (KIF18A) is particularly detrimental to aneuploid tumor cells.438 This sensitivity arises from alterations in spindle geometry and microtubule dynamics specific in tumor cell with CIN, which, upon KIF18A knockdown, leads to excessive CIN and reduces tumor cells viability. This suggests that KIF18A could be a promising synthetic lethal candidate for future drug development efforts targeting tumor cells with CIN. Furthermore, Hong et al. found that IL-6/STAT3 signaling axis downstream of cGAS-STING enables the survival of tumor cells with CIN, and blockade of IL-6 signaling by tocilizumab, a clinically used drug that targets the IL-6 receptor (IL-6R), can impair their growth specifically.403 Moreover, aneuploid cells have also been found to contain higher levels of ceramide, and further increasing the levels of ceramide through treatment with N-[2-hydroxy-1-(4-morpholinylmethyl)-2-phenylethyl]-decanamide monohydrochloride (DL-PDMP), an antagonist of UDP-glucose ceramide glucosyltransferase, is significantly more toxic to aneuploid cells compared with diploid cells.439
Another strategy utilizes new weakness that results from gaining specific chromosome. For instance, a gene encoded on chromosome 1, known as uridine-cytidine kinase 2 (UCK2), is required to activate certain pro-drugs, such as RX-3117 and 3-deazauridine. A recent study revealed that cells with an extra copy of chromosome 1 express higher level of UCK2 and are more sensitive to those drugs compared to diploid cells with just two copies,440 suggesting that introducing specific aneuploidies that can exert anti-tumor function, for example using CRISPR-based tools, might also be a potential CIN-based anti-tumor therapeutic strategy.441,442,443,444,445,446,447 This strategy is still not yet translated to clinical trial, nevertheless, several compounds targeting different pathways to increase CIN are now in phase I-III clinical trials,448,449,450 representing promising progress for future research and development in CIN-based anti-tumor therapy (Table 4).
Conclusion
Great progress has been made during this last decade in understanding how chromosome segregation is regulated and what defects might contribute to chromosome missegregation in tumor cells. While the exact source of CIN in tumor cells is complex and diverse, it is clear that a variety of mechanisms can lead to this phenotype. Tumor cells presumably employ different routes to achieve the same CIN phenotype. Therefore, more research to further characterize the mechanisms contributing to CIN is necessary. This will be crucial in laying the foundation for developing strategies that can modulate CIN, with the aim of inhibiting tumor cells ability to adapt to environmental challenges, as well as preventing tumor drug resistance.
From a clinical perspective, although the relative contribution of various mechanisms of CIN has been described in cultured tumor cell lines, whether these models also recapitulate the types of CIN found in primary and metastatic tumors, as well as the epigenetic causes and consequences of CIN, remains to be determined. Enhancing CIN has been proposed as a promising approach to target aneuploid tumor cells. From the perspective of developing new therapeutic strategies, it is crucial to determine whether cells undergoing CIN share common characteristics. It is also important to understand if CIN triggers a common stress response and whether CIN cells need to develop specific adaptations to adapt to their altered genomes. A more comprehensive understanding of these effects and the resulting cellular responses is crucial for the successful exploitation of CIN for therapeutic strategies. Furthermore, while several SAC inhibitors that increase CIN show promising potential for reducing tumor growth and have already entered clinical trial phase, the success of such therapies depends on factors such as CIN status and the capacity of the tumor cells to tolerate CIN.267,427,428,429,430,431,432,433,434,435,436,437 Despite these challenges, recent advances suggest promising possibilities for a new, personalized, CIN-specific approach to anti-tumor therapy.358,406,410,411,423,424,425,426,438,451,452,453,454,455
Moreover, considering CIN as the fuel for genomic diversity, it is important to acknowledge the challenges posed by tumor evolution. This suggests that future research could focus on characterizing the common phenotypes that emerge from various CIN routes. These strategies might not directly target the mechanisms of CIN; instead, they could focus on the phenotypes selected during the stages of tumor initiation and progression. Adopting this approach could potentially lead to more effective treatments that are less susceptible to being undermined by tumor adaptation and evolution. Furthermore, it could provide valuable biomarkers for early detection and prognosis, thereby opening up new opportunities for preventive measures.293
Furthermore, while targeting tumor cells with CIN holds promise, it is important to consider the risks. Increasing CIN in untransformed cells could inadvertently lead to therapy-induced tumor due to their potential to become tumorigenic under low to moderate CIN rates. Therefore, future research should aim to selectively target cells with a CIN phenotype, thereby reducing the risk of unintentional transformation of normal cells.
In conclusion, our understanding of CIN and its role in tumorigenesis has greatly improved. Although many questions remain unanswered, and further research is needed to fully understand the mechanisms underlying CIN-related tumorigenesis as well as its potential as a therapeutic target, the study of CIN and its effects on tumor cells have nevertheless laid a promising foundation. These insights could potentially guide the development of new strategies for diagnosing and treating cancer.
References
Wild, C., Weiderpass E., & Stewart B. W. (eds) World Cancer Report Cancer Research for Cancer Prevention (International Agency Research on Cancer, 2020).
Tijhuis, A. E., Johnson, S. C. & McClelland, S. E. The emerging links between chromosomal instability (CIN), metastasis, inflammation and tumour immunity. Mol. Cytogenet. 12, 1–21 (2019).
Vasudevan, A. et al. Aneuploidy as a promoter and suppressor of malignant growth. Nat. Rev. Cancer 21, 89–103 (2021).
Taylor, A. M. et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 33, 676–689.e673 (2018).
Weaver, B. A. A. & Cleveland, D. W. Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 18, 658–667 (2006).
Drews, R. M. et al. A pan-cancer compendium of chromosomal instability. Nature 606, 976–983 (2022).
Zhang, C.-Z. & Pellman, D. Cancer genomic rearrangements and copy number alterations from errors in cell division. Annu. Rev. Cancer Biol. 6, 245–268 (2022).
Matthews, H. K., Bertoli, C. & de Bruin, R. A. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 23, 74–88 (2022).
Sarkar, S. et al. Mitotic checkpoint defects: en route to cancer and drug resistance. Chromosom. Res. 29, 131–144 (2021).
Maiato, H. & Silva, S. Double-checking chromosome segregation. J. Cell Biol. 222, e202301106 (2023).
Khodjakov, A. & Rieder, C. L. The nature of cell-cycle checkpoints: facts and fallacies. J. Biol. 8, 1–5 (2009).
Chunduri, N. K. & Storchova, Z. The diverse consequences of aneuploidy. Nat. Cell Biol. 21, 54–62 (2019).
Cahill, D. P., Kinzler, K. W., Vogelstein, B. & Lengauer, C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 9, M57–M60 (1999).
Lezmi, E. & Benvenisty, N. The tumorigenic potential of human pluripotent stem cells. Stem Cells Transl. Med. 11, 791–796 (2022).
Na, J. et al. Aneuploidy in pluripotent stem cells and implications for cancerous transformation. Protein Cell 5, 569–579 (2014).
Barbaric, I. et al. Time-lapse analysis of human embryonic stem cells reveals multiple bottlenecks restricting colony formation and their relief upon culture adaptation. Stem Cell Rep. 3, 142–155 (2014).
Ippolito, M. R. et al. Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev. Cell 56, 2440–2454.e2446 (2021).
Lukow, D. A. et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 56, 2427–2439 (2021).
Rutledge, S. D. et al. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci. Rep. 6, 22828 (2016).
Ben-David, U. et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 5, 4825 (2014).
Holland, A. J. & Cleveland, D. W. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep. 13, 501–514 (2012).
Sheltzer, J. M. & Amon, A. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet. 27, 446–453 (2011).
Lukow, D. A. & Sheltzer, J. M. Chromosomal instability and aneuploidy as causes of cancer drug resistance. Trends Cancer 8, 43–53 (2022).
Watkins, T. B. et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 587, 126–132 (2020).
Andrade, J. R., Gallagher, A. D., Maharaj, J. & McClelland, S. E. Disentangling the roles of aneuploidy, chromosomal instability and tumour heterogeneity in developing resistance to cancer therapies. Chromosom. Res. 31, 28 (2023).
Vitale, I., Shema, E., Loi, S. & Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 27, 212–224 (2021).
Vasan, N., Baselga, J. & Hyman, D. M. A view on drug resistance in cancer. Nature 575, 299–309 (2019).
van Dijk, E. et al. Chromosomal copy number heterogeneity predicts survival rates across cancers. Nat. Commun. 12, 3188 (2021).
Sansregret, L., Vanhaesebroeck, B. & Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 15, 139–150 (2018).
Bakhoum, S. F. & Cantley, L. C. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 174, 1347–1360 (2018).
Li, Y. et al. Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 (2020).
Geigl, J. B., Obenauf, A. C., Schwarzbraun, T. & Speicher, M. R. Defining ‘chromosomal instability'. Trends Genet. 24, 64–69 (2008).
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Burrell, R. A. et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 (2013).
Mitelman, F., Johansson, B. & Mertens, F. (eds) Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (National Cancer Institute, 2024); http://mitelmandatabase.isb-cgc.org/.
Graphodatsky, A. S., Trifonov, V. A. & Stanyon, R. The genome diversity and karyotype evolution of mammals. Mol. Cytogenet. 4, 1–16 (2011).
Bakhoum, S. F. & Landau, D. A. Chromosomal instability as a driver of tumor heterogeneity and evolution. Cold Spring Harb. Perspect. Med. 7, 2029611 (2017).
Bronder, D. & Bakhoum, S. F. A CIN ful way to overcome addiction: how chromosomal instability enables cancer to overcome its oncogene addiction. EMBO Mol. Med. 12, e12017 (2020).
Salgueiro, L. et al. Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol. Med. 12, e10941 (2020).
Hansford, S. & Huntsman, D. G. Boveri at 100: Theodor Boveri and genetic predisposition to cancer. J. Pathol. 234, 142–145 (2014).
Holland, A. J. & Cleveland, D. W. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10, 478–487 (2009).
Fernandez-Casanas, M. & Chan, K. L. The unresolved problem of DNA bridging. Genes 9, 623 (2018).
Gartler, S. M. The chromosome number in humans: a brief history. Nat. Rev. Genet. 7, 655–660 (2006).
Rieder, C. L., Schultz, A., Cole, R. & Sluder, G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J. Cell Biol. 127, 1301–1310 (1994).
Nowell, P. C. Discovery of the Philadelphia chromosome: a personal perspective. J. Clin. Investig. 117, 2033–2035 (2007).
Dobrovic, A., Peters, G. & Ford, J. Molecular analysis of the Philadelphia chromosome. Chromosoma 100, 479–486 (1991).
Megarbane, A. et al. The 50th anniversary of the discovery of trisomy 21: the past, present, and future of research and treatment of Down syndrome. Genet. Med. 11, 611–616 (2009).
Lejeune, J. Trois cas de délétion partielle du bras court d’un chromosome 5. Comp. Rendus Acad. Sci. 257, 3098–3102 (1963).
Lengauer, C., Kinzler, K. W. & Vogelstein, B. Genetic instability in colorectal cancers. Nature 386, 623–627 (1997).
Lengauer, C., Kinzler, K. W. & Vogelstein, B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998).
Amon, A. The spindle checkpoint. Curr. Opin. Genet. Dev. 9, 69–75 (1999).
D’Amours, D. & Amon, A. At the interface between signaling and executing anaphase—Cdc14 and the FEAR network. Genes Dev. 18, 2581–2595 (2004).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Dobles, M. et al. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell 101, 635–645 (2000).
Denomy, C. et al. Banding together: a systematic comparison of the cancer genome atlas and the Mitelman databases. Cancer Res. 79, 5181–5190 (2019).
Weinstein,J. N. et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Dentro, S. C. et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 184, 2239–2254 e2239 (2021).
Jamal-Hanjani, M. et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol. 12, e1001906 (2014).
Abbosh, C. et al. Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA. Nature 616, 553–562 (2023).
Bailey, C. et al. Tracking cancer evolution through the disease course. Cancer Discov. 11, 916–932 (2021).
Al Bakir, M. et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature 616, 534–542 (2023).
McGranahan, N. et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259–1271.e1211 (2017).
Wani, M. C. & Horwitz, S. B. Nature as a remarkable chemist: a personal story of the discovery and development of Taxol. Anticancer Drugs 25, 482–487 (2014).
Mason, J. M. et al. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. 114, 3127–3132 (2017).
Thu, K. L. et al. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc. Natl. Acad. Sci. 115, E1570–E1577 (2018).
Chan, C. Y.-K. et al. CFI-402257, a TTK inhibitor, effectively suppresses hepatocellular carcinoma. Proc. Natl Acad. Sci. 119, e2119514119 (2022).
Ma, J., Chan, J. J., Toh, C. H. & Yap, Y. S. Emerging systemic therapy options beyond CDK4/6 inhibitors for hormone receptor-positive HER2-negative advanced breast cancer. NPJ Breast Cancer 9, 74 (2023).
Bakhoum, S. F. Targeting the undruggable. Science 380, 47 (2023).
McGranahan, N. et al. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 13, 528–538 (2012).
Bakhoum, S. F. et al. The mitotic origin of chromosomal instability. Curr. Biol. 24, R148–R149 (2014).
He, B. et al. Chromosomes missegregated into micronuclei contribute to chromosomal instability by missegregating at the next division. Oncotarget 10, 2660 (2019).
Zasadil, L. M., Britigan, E. M. C. & Weaver, B. A. 2n or not 2n: aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin. Cell Dev. Biol. 24, 370–379 (2013).
Nicholson, J. M. et al. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. elife 4, e05068 (2015).
Fujiwara, T. et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047 (2005).
Passerini, V. et al. The presence of extra chromosomes leads to genomic instability. Nat. Commun. 7, 10754 (2016).
Ricke, R. M., van Ree, J. H. & van Deursen, J. M. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 24, 457–466 (2008).
Gantchev, J. et al. Tools used to assay genomic instability in cancers and cancer meiomitosis. J. Cell Commun. Signal. 1–19 (2021).
Krupina, K., Goginashvili, A. & Cleveland, D. W. Causes and consequences of micronuclei. Curr. Opin. Cell Biol. 70, 91–99 (2021).
Bolhaqueiro, A. C. et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 51, 824–834 (2019).
Hoevenaar, W. H. M. et al. Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 11, 1501 (2020).
Klaasen, S. J. et al. Nuclear chromosome locations dictate segregation error frequencies. Nature 607, 604–609 (2022).
Thompson, S. L. & Compton, D. A. Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 180, 665–672 (2008).
Fenech, M. et al. Micronuclei as biomarkers of DNA damage, aneuploidy, inducers of chromosomal hypermutation and as sources of pro-inflammatory DNA in humans. Mutat. Res. Rev. Mutat. Res. 786, 108342 (2020).
Gomes, A. M. et al. Micronuclei from misaligned chromosomes that satisfy the spindle assembly checkpoint in cancer cells. Curr. Biol. 32, 4240–4254. e4245 (2022).
van Toorn, M., Gooch, A., Boerner, S. & Kiyomitsu, T. NuMA deficiency causes micronuclei via checkpoint-insensitive k-fiber minus-end detachment from mitotic spindle poles. Curr. Biol. 33, 572–580.e572 (2023).
Piemonte, K. M. et al. Disruption of CDK7 signaling leads to catastrophic chromosomal instability coupled with a loss of condensin-mediated chromatin compaction. J. Biol. Chem. 299, 104834 (2023).
Hodges, C. A. et al. SMC1beta-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat. Genet. 37, 1351–1355 (2005).
Nasmyth, K. Segregating sister genomes: the molecular biology of chromosome separation. Science 297, 559–565 (2002).
Ohbayashi, T. et al. Unscheduled overexpression of human WAPL promotes chromosomal instability. Biochem. Biophys. Res. Commun. 356, 699–704 (2007).
Carvajal-Maldonado, D. et al. Perturbing cohesin dynamics drives MRE11 nuclease-dependent replication fork slowing. Nucleic Acids Res. 47, 1294–1310 (2019).
Leylek, T. R., Jeusset, L. M., Lichtensztejn, Z. & McManus, K. J. Reduced expression of genes regulating cohesion induces chromosome instability that may promote cancer and impact patient outcomes. Sci. Rep. 10, 592 (2020).
Deardorff, M. A. et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 90, 1014–1027 (2012).
Solomon, D. A. et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333, 1039–1043 (2011).
van der Lelij, P. et al. STAG1 vulnerabilities for exploiting cohesin synthetic lethality in STAG2-deficient cancers. Life Sci. Alliance 3, e202000725 (2020).
El Beaino, M. et al. Loss of Stag2 cooperates with EWS-FLI1 to transform murine mesenchymal stem cells. BMC Cancer 20, 1–14 (2020).
Nam, H.-J. & van Deursen, J. M. Cyclin B2 and p53 control proper timing of centrosome separation. Nat. Cell Biol. 16, 535–546 (2014).
Prigent, C. & Uzbekov, R. Duplication and segregation of centrosomes during cell division. Cells 11, 2445 (2022).
Coelho, P. A. et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 5, 150209 (2015).
Bakhoum, S. F., Thompson, S. L., Manning, A. L. & Compton, D. A. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol. 11, 27–35 (2009).
Bakhoum, S. F., Genovese, G. & Compton, D. A. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr. Biol. 19, 1937–1942 (2009).
Rieder, C. L. & Maiato, H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 7, 637–651 (2004).
Jallepalli, P. V. & Lengauer, C. Chromosome segregation and cancer: cutting through the mystery. Nat. Rev. Cancer. 1, 109–117 (2001).
Burds, A. A., Lutum, A. S. & Sorger, P. K. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc. Natl. Acad. Sci. 102, 11296–11301 (2005).
Foijer, F. et al. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc. Natl. Acad. Sci. 111, 13427–13432 (2014).
Simon, J. E., Bakker, B. & Foijer, F. CINcere modelling: what have mouse models for chromosome instability taught us? Recent Results Cancer Res. 200, 39–60 (2015).
Iwanaga, Y. et al. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 67, 160–166 (2007).
Michel, L. S. et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 409, 355–359 (2001).
Dai, W. et al. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 64, 440–445 (2004).
Suijkerbuijk, S. J. et al. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res. 70, 4891–4900 (2010).
Hanks, S. et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 36, 1159–1161 (2004).
Prinz, F. et al. Functional and structural characterization of Bub3·BubR1 interactions required for spindle assembly checkpoint signaling in human cells. J. Biol. Chem. 291, 11252–11267 (2016).
Carvalhal, S. et al. Biallelic BUB1 mutations cause microcephaly, developmental delay, and variable effects on cohesion and chromosome segregation. Sci. Adv. 8, eabk0114 (2022).
Cimini, D. et al. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 153, 517–527 (2001).
Cimini, D., Cameron, L. A. & Salmon, E. D. Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Curr. Biol. 14, 2149–2155 (2004).
Crasta, K. et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 (2012).
Hatch, E. M., Fischer, A. H., Deerinck, T. J. & Hetzer, M. W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60 (2013).
Orr, B. et al. An anaphase surveillance mechanism prevents micronuclei formation from frequent chromosome segregation errors. Cell Rep. 37, 109783 (2021).
Wilhelm, T. et al. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 10, 3585 (2019).
Godinho, S. A., Kwon, M. & Pellman, D. Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 28, 85–98 (2009).
Gordon, D. J., Resio, B. & Pellman, D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 13, 189–203 (2012).
Schvartzman, J.-M., Sotillo, R. & Benezra, R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat. Rev. Cancer. 10, 102–115 (2010).
Rode, A. et al. Chromothripsis in cancer cells: an update. Int. J. Cancer 138, 2322–2333 (2016).
Ly, P. et al. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 19, 68–75 (2017).
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011).
Leibowitz, M. L., Zhang, C.-Z. & Pellman, D. Chromothripsis: a new mechanism for rapid karyotype evolution. Annu. Rev. Genet. 49, 183–211 (2015).
Zhang, C.-Z. et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015).
Shoshani, O. et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141 (2021).
Umbreit, N. T. et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 368, eaba0712 (2020).
Joo, Y. K. et al. ATR promotes clearance of damaged DNA and damaged cells by rupturing micronuclei. Mol. Cell. 83, 3642–3658. e3644 (2023).
Ly, P. et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 51, 705–715 (2019).
Li, Y. et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508, 98–102 (2014).
Sanders, A. D. et al. Single-cell analysis of structural variations and complex rearrangements with tri-channel processing. Nat. Biotechnol. 38, 343–354 (2020).
Li, J. S. Z. et al. Chromosomal fragile site breakage by EBV-encoded EBNA1 at clustered repeats. Nature 1–6 (2023).
Gaillard, H., García-Muse, T. & Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer. 15, 276–289 (2015).
Sahgal, P. et al. Replicative stress in gastroesophageal cancer is associated with chromosomal instability and sensitivity to DNA damage response inhibitors. iScience 26, 108169 (2023).
Shaikh, N. et al. Replication stress generates distinctive landscapes of DNA copy number alterations and chromosome scale losses. Genome Biol. 23, 1–24 (2022).
Huang, R.-X. & Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 5, 60 (2020).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Gorgoulis, V. G. et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 (2005).
Bartkova, J. et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005).
Garribba, L. et al. Short-term molecular consequences of chromosome mis-segregation for genome stability. Nat. Commun. 14, 1353 (2023).
Bakhoum, S. F. et al. Mitotic DNA damage response: at the crossroads of structural and numerical cancer chromosome instabilities. Trends Cancer 3, 225–234 (2017).
Jeggo, P. A., Pearl, L. H. & Carr, A. M. DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer 16, 35–42 (2016).
Li, J. et al. The RPA–RNF20–SNF2H cascade promotes proper chromosome segregation and homologous recombination repair. Proc. Natl. Acad. Sci. 120, e2303479120 (2023).
Chan, K. L., North, P. S. & Hickson, I. D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409 (2007).
Chan, K. L., Palmai-Pallag, T., Ying, S. & Hickson, I. D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760 (2009).
Naim, V. & Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 11, 761–768 (2009).
Howlett, N. G. et al. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 14, 693–701 (2005).
Constantinou, A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 121, 21–36 (2012).
Madireddy, A. et al. FANCD2 facilitates replication through common fragile sites. Mol. Cell. 64, 388–404 (2016).
Boteva, L. et al. Common fragile sites are characterized by faulty condensin loading after replication stress. Cell Rep. 32, 108177 (2020).
Böhly, N. et al. Increased replication origin firing links replication stress to whole chromosomal instability in human cancer. Cell Rep. 41, 111836 (2022).
Di Nardo, M., Pallotta, M. M. & Musio, A. The multifaceted roles of cohesin in cancer. J. Exp. Clin. Cancer Res. 41, 1–11 (2022).
Sun, Y. et al. RAD21 is the core subunit of the cohesin complex involved in directing genome organization. Genome Biol. 24, 1–27 (2023).
Haering, C. H., Löwe, J., Hochwagen, A. & Nasmyth, K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell. 9, 773–788 (2002).
Gruber, S., Haering, C. H. & Nasmyth, K. Chromosomal cohesin forms a ring. Cell 112, 765–777 (2003).
Cuadrado, A. & Losada, A. Specialized functions of cohesins STAG1 and STAG2 in 3D genome architecture. Curr. Opin. Genet. Dev. 61, 9–16 (2020).
Shi, Z., Gao, H., Bai, X.-C. & Yu, H. Cryo-EM structure of the human cohesin-NIPBL-DNA complex. Science 368, 1454–1459 (2020).
Watanabe, Y. Sister chromatid cohesion along arms and at centromeres. Trends Genet. 21, 405–412 (2005).
Gerton, J. Chromosome cohesion: a cycle of holding together and falling apart. PLoS Biol. 3, e94 (2005).
Shintomi, K. & Hirano, T. Releasing cohesin from chromosome arms in early mitosis: opposing actions of Wapl–Pds5 and Sgo1. Genes Dev. 23, 2224–2236 (2009).
Tillement, V. et al. Spindle assembly defects leading to the formation of a monopolar mitotic apparatus. Biol. Cell. 101, 1–11 (2009).
Yoshizawa, K., Yaguchi, K. & Uehara, R. Uncoupling of DNA replication and centrosome duplication cycles is a primary cause of haploid instability in mammalian somatic cells. Front. Cell Dev. Biol. 30, 721 (2020).
van Ree, J. H. et al. Pten regulates spindle pole movement through Dlg1-mediated recruitment of Eg5 to centrosomes. Nat. Cell Biol. 18, 814–821 (2016).
Silkworth, W. T. et al. Timing of centrosome separation is important for accurate chromosome segregation. Mol. Biol. Cell 23, 401–411 (2012).
Zhang, Y. et al. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J. Clin. Investig. 122, 4362–4374 (2012).
Saavedra, H. I., Fukasawa, K., Conn, C. W. & Stambrook, P. J. MAPK mediates RAS-induced chromosome instability. J. Biol. Chem. 274, 38083–38090 (1999).
Chan, J. Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 7, 1122–1144 (2011).
Nigg, E. A. & Holland, A. J. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat. Rev. Mol. Cell Biol. 19, 297–312 (2018).
Ganem, N. J., Godinho, S. A. & Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 (2009).
Silkworth, W. T., Nardi, I. K., Scholl, L. M. & Cimini, D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PloS One 4, e6564 (2009).
Cosper, P. F. et al. HPV16 E6 induces chromosomal instability due to polar chromosomes caused by E6AP-dependent degradation of the mitotic kinesin CENP-E. Proc. Natl. Acad. Sci. 120, e2216700120 (2023).
Bühler, M. et al. GPER1 links estrogens to centrosome amplification and chromosomal instability in human colon cells. Life Sci. Alliance 6, e202201499 (2023).
Gregan, J. et al. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 21, 374–381 (2011).
Thompson, S. L. & Compton, D. A. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl Acad. Sci. 108, 17974–17978 (2011).
Cimini, D. Twenty years of merotelic kinetochore attachments: a historical perspective. Chromosom. Res. 31, 18 (2023).
Kops, G. J. & Gassmann, R. Crowning the kinetochore: the fibrous corona in chromosome segregation. Trends Cell Biol. 30, 653–667 (2020).
Song, X. et al. Dynamic crotonylation of EB1 by TIP60 ensures accurate spindle positioning in mitosis. Nat. Chem. Biol. 17, 1314–1323 (2021).
Cheeseman, I. M. & Desai, A. Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 9, 33–46 (2008).
Navarro, A. P. & Cheeseman, I. M. Kinetochore assembly throughout the cell cycle. Semin. Cell. Dev. Biol. 117, 62–74 (2021).
Ertych, N. et al. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat. Cell Biol. 16, 779–791 (2014).
Kabeche, L. & Compton, D. A. Checkpoint-independent stabilization of kinetochore-microtubule attachments by Mad2 in human cells. Curr. Biol. 22, 638–644 (2012).
Kleyman, M., Kabeche, L. & Compton, D. A. STAG2 promotes error correction in mitosis by regulating kinetochore-microtubule attachments. J. Cell Sci. 127, 4225–4233 (2014).
Chan, G. K., Liu, S.-T. & Yen, T. J. Kinetochore structure and function. Trends Cell Biol. 15, 589–598 (2005).
Musacchio, A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 25, R1002–R1018 (2015).
Musacchio, A. & Salmon, E. D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 (2007).
McAinsh, A. D. & Kops, G. J. Principles and dynamics of spindle assembly checkpoint signalling. Nat. Rev. Mol. Cell Biol. 24, 543–559 (2023).
London, N. & Biggins, S. Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell Biol. 15, 736–747 (2014).
Liu, S. et al. Mad2 promotes Cyclin B2 recruitment to the kinetochore for guiding accurate mitotic checkpoint. EMBO Rep. 23, e54171 (2022).
Waters, J. C., Chen, R. H., Murray, A. W. & Salmon, E. D. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141, 1181–1191 (1998).
Chen, R. H., Waters, J. C., Salmon, E. D. & Murray, A. W. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 274, 242–246 (1996).
Li, Y. & Benezra, R. Identification of a human mitotic checkpoint gene: hsMAD2. Science 274, 246–248 (1996).
Brady, D. M. & Hardwick, K. G. Complex formation between Mad1p, Bub1p and Bub3p is crucial for spindle checkpoint function. Curr. Biol. 10, 675–678 (2000).
Basu, J. et al. Localization of the Drosophila checkpoint control protein Bub3 to the kinetochore requires Bub1 but not Zw10 or Rod. Chromosoma 107, 376–385 (1998).
Jablonski, S. A. et al. The hBUB1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma 107, 386–396 (1998).
Taylor, S. S. & McKeon, F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell 89, 727–735 (1997).
Chen, R.-H. BubR1 is essential for kinetochore localization of other spindle checkpoint proteins and its phosphorylation requires Mad1. J Cell Biol. 158, 487–496 (2002).
Lopes, C. S. et al. The Drosophila Bub3 protein is required for the mitotic checkpoint and for normal accumulation of cyclins during G2 and early stages of mitosis. J. Cell Sci. 118, 187–198 (2005).
Babu, J. R. et al. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 160, 341–353 (2003).
Sudakin, V., Chan, G. K. & Yen, T. J. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936 (2001).
Kalitsis, P., Earle, E., Fowler, K. J. & Choo, K. H. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14, 2277–2282 (2000).
Taylor, S. S., Ha, E. & McKeon, F. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J. Cell Biol. 142, 1–11 (1998).
Zhou, Z., He, M., Shah, A. A. & Wan, Y. Insights into APC/C: from cellular function to diseases and therapeutics. Cell Div. 11, 9 (2016).
Izawa, D. & Pines, J. The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 517, 631–634 (2015).
Chen, O. J. et al. Germline missense variants in CDC20 result in aberrant mitotic progression and familial cancer. Cancer Res. 82, 3499–3515 (2022).
Tsang, M. J. & Cheeseman, I. M. Alternative CDC20 translational isoforms tune mitotic arrest duration. Nature 617, 154–161 (2023).
Alfieri, C. et al. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 536, 431–436 (2016).
Lara-Gonzalez, P. et al. A tripartite mechanism catalyzes Mad2-Cdc20 assembly at unattached kinetochores. Science 371, 64–67 (2021).
Piano, V. et al. CDC20 assists its catalytic incorporation in the mitotic checkpoint complex. Science 371, 67–71 (2021).
Fischer, E. S. et al. Juxtaposition of Bub1 and Cdc20 on phosphorylated Mad1 during catalytic mitotic checkpoint complex assembly. Nat. Commun. 13, 6381 (2022).
Sivakumar, S. & Gorbsky, G. J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 16, 82–94 (2015).
Lara-Gonzalez, P., Pines, J. & Desai, A. Spindle assembly checkpoint activation and silencing at kinetochores. Semin. Cell. Dev. Biol. 117, 86–98 (2021).
Kapanidou, M., Curtis, N. L. & Bolanos-Garcia, V. M. Cdc20: at the crossroads between chromosome segregation and mitotic exit. Trends Biochem. Sci. 42, 193–205 (2017).
Vagnarelli, P. Back to the new beginning: mitotic exit in space and time. Semin. Cell. Dev. Biol. 117, 140–148 (2021).
Moreno-Andrés, D., Holl, K. & Antonin, W. The second half of mitosis and its implications in cancer biology. Semin. Cancer. Biol. 88, 1–17 (2022).
Gao, Y.-F. et al. Cdk1-phosphorylated CUEDC2 promotes spindle checkpoint inactivation and chromosomal instability. Nat. Cell Biol. 13, 924–933 (2011).
Cosper, P. F., Copeland, S. E., Tucker, J. B. & Weaver, B. A. Chromosome missegregation as a modulator of radiation sensitivity. Semin. Radiat. Oncol. 32, 54–63 (2022).
He, Z. et al. JMJD5 (Jumonji Domain-containing 5) associates with spindle microtubules and is required for proper mitosis. J. Biol. Chem. 291, 4684–4697 (2016).
Yatskevich, S. et al. Molecular mechanisms of APC/C release from spindle assembly checkpoint inhibition by APC/C SUMOylation. Cell Rep. 34, 108929 (2021).
Rowald, K. et al. Negative selection and chromosome instability induced by Mad2 overexpression delay breast cancer but facilitate oncogene-independent outgrowth. Cell Rep. 15, 2679–2691 (2016).
Sotillo, R. et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11, 9–23 (2007).
Choi, E., Zhang, X., Xing, C. & Yu, H. Mitotic checkpoint regulators control insulin signaling and metabolic homeostasis. Cell Rep. 166, 567–581 (2016).
Ma, H. T. & Poon, R. Y. C. TRIP13 regulates both the activation and inactivation of the spindle-assembly checkpoint. Cell Rep. 14, 1086–1099 (2016).
Kim, D. H. et al. TRIP13 and APC15 drive mitotic exit by turnover of interphase-and unattached kinetochore-produced MCC. Nat. Commun. 9, 4354 (2018).
Gong, Y. et al. Loss of RanGAP1 drives chromosome instability and rapid tumorigenesis of osteosarcoma. Dev. Cell 58, 192–210. e111 (2023).
Sapkota, H., Wasiak, E., Daum, J. R. & Gorbsky, G. J. Multiple determinants and consequences of cohesion fatigue in mammalian cells. Mol. Biol. Cell 29, 1811–1824 (2018).
Gorbsky, G. J. Cohesion fatigue. Curr. Biol. 23, R986–R988 (2013).
Storchova, Z. & Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 5, 45–54 (2004).
Davoli, T. & de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 27, 585–610 (2011).
Kuznetsova, A. Y. et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 14, 2810–2820 (2015).
Wangsa, D. et al. Near-tetraploid cancer cells show chromosome instability triggered by replication stress and exhibit enhanced invasiveness. FASEB J. 32, 3502 (2018).
Selmecki, A. M. et al. Polyploidy can drive rapid adaptation in yeast. Nature 519, 349–352 (2015).
Zhang, J. et al. Human cell polyploidization: the good and the evil. Semin. Cancer. Biol. 81, 54–63 (2022).
Steigemann, P. et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136, 473–484 (2009).
Ganem, N. J. et al. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 158, 833–848 (2014).
Darp, R., Vittoria, M. A., Ganem, N. J. & Ceol, C. J. Oncogenic BRAF induces whole-genome doubling through suppression of cytokinesis. Nat. Commun. 13, 4109 (2022).
Ghelli Luserna di Rorà, A., Martinelli, G. & Simonetti, G. The balance between mitotic death and mitotic slippage in acute leukemia: a new therapeutic window? J. Hematol. Oncol. 12, 1–16 (2019).
Lok, T. M. et al. Mitotic slippage is determined by p31comet and the weakening of the spindle-assembly checkpoint. Oncogene 39, 2819–2834 (2020).
Sanz-Gómez, N., González-Álvarez, M., De Las Rivas, J. & de Cárcer, G. Whole-genome doubling as a source of cancer: how, when, where, and why? Front. Cell Dev. Biol. 11, 1209136 (2023).
Coulombe, P. et al. The ORC ubiquitin ligase OBI1 promotes DNA replication origin firing. Nat. Commun. 10, 2426 (2019).
Fujita, M. Cdt1 revisited: complex and tight regulation during the cell cycle and consequences of deregulation in mammalian cells. Cell Div. 1, 1–9 (2006).
Vittoria, M. A., Quinton, R. J. & Ganem, N. J. Whole-genome doubling in tissues and tumors. Trends Genet. 39, 954–867 (2023).
Lau, T. Y. & Poon, R. Y. Whole-genome duplication and genome instability in cancer cells: double the trouble. Int. J. Mol. Sci. 24, 3733 (2023).
Prasad, K. et al. Whole-genome duplication shapes the aneuploidy landscape of human cancers. Cancer Res. 82, 1736–1752 (2022).
Bielski, C. M. et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195 (2018).
Gemble, S. et al. Genetic instability from a single S phase after whole-genome duplication. Nature 604, 146–151 (2022).
Zhang, H. et al. Cell Fusion-related proteins and signaling pathways, and their roles in the development and progression of cancer. Front. Cell Dev. Biol. 9, 809668 (2022).
Davoli, T. & de Lange, T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 21, 765–776 (2012).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med 376, 2109–2121 (2017).
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Lambuta, R. A. et al. Whole-genome doubling drives oncogenic loss of chromatin segregation. Nature 615, 925–933 (2023).
Storchova, Z. & Kuffer, C. The consequences of tetraploidy and aneuploidy. J Cell Sci 121, 3859–3866 (2008).
Potapova, T. & Gorbsky, G. J. The consequences of chromosome segregation errors in mitosis and meiosis. Biology 6, 12 (2017).
Santaguida, S. & Amon, A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol 16, 473–485 (2015).
Selmecki, A., Forche, A. & Berman, J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313, 367–370 (2006).
Selmecki, A. et al. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 68, 624–641 (2008).
Pavelka, N. et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468, 321–325 (2010).
Rancati, G. et al. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135, 879–893 (2008).
Chen, G., Bradford, W. D., Seidel, C. W. & Li, R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482, 246–250 (2012).
Rancati, G. & Pavelka, N. Karyotypic changes as drivers and catalyzers of cellular evolvability: a perspective from non-pathogenic yeasts. Semin. Cell Dev. Biol. 24, 332–338 (2013).
Ben-David, U. & Amon, A. Context is everything: aneuploidy in cancer. Nat. Rev. Genet. 21, 44–62 (2020).
Snape, K. et al. Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nat. Genet. 43, 527–529 (2011).
Garcia-Castillo, H., Vasquez-Velasquez, A. I., Rivera, H. & Barros-Nunez, P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: delineation of clinical subtypes. Am. J. Med. Genet. A 146A, 1687–1695 (2008).
Stingele, S. et al. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 8, 608 (2012).
Williams, B. R. et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322, 703–709 (2008).
Torres, E. M. et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924 (2007).
López-García, C. et al. BCL9L dysfunction impairs Caspase-2 expression permitting aneuploidy tolerance in colorectal cancer. Cancer Cell 31, 79–93 (2017).
Ohashi, A. et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat. Commun. 6, 7668 (2015).
Meena, J. K. et al. Telomerase abrogates aneuploidy-induced telomere replication stress, senescence and cell depletion. EMBO J. 36, 2922–2924 (2017).
Santaguida, S. et al. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 41, 638–651.e635 (2017).
Andriani, G. A. et al. Whole chromosome instability induces senescence and promotes SASP. Sci. Rep. 6, 35218 (2016).
Vigano, C. et al. Quantitative proteomic and phosphoproteomic comparison of human colon cancer DLD-1 cells differing in ploidy and chromosome stability. Mol. Biol. Cell 29, 1031–1047 (2018).
Krivega, M. et al. Genotoxic stress in constitutive trisomies induces autophagy and the innate immune response via the cGAS-STING pathway. Commun. Biol. 4, 831 (2021).
Birkbak, N. J. et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 71, 3447–3452 (2011).
Zaki, B. I. et al. Chromosomal instability portends superior response of rectal adenocarcinoma to chemoradiation therapy. Cancer 120, 1733–1742 (2014).
Andor, N. et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 22, 105–113 (2016).
Bakhoum, S. F. et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat. Commun. 6, 5990 (2015).
Jamal-Hanjani, M. et al. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann. Oncol. 26, 1340–1346 (2015).
Roylance, R. et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol. Biomark. Prev. 20, 2183–2194 (2011).
Laughney, A. M., Elizalde, S., Genovese, G. & Bakhoum, S. F. Dynamics of tumor heterogeneity derived from clonal karyotypic evolution. Cell Rep. 12, 809–820 (2015).
Komarova, N. L. & Wodarz, D. The optimal rate of chromosome loss for the inactivation of tumor suppressor genes in cancer. Proc. Natl Acad. Sci. 101, 7017–7021 (2004).
Chunduri, N. K., Barthel, K. & Storchova, Z. Consequences of chromosome loss: why do cells need each chromosome twice? Cells 11, 1530 (2022).
Chunduri, N. K. et al. Systems approaches identify the consequences of monosomy in somatic human cells. Nat. Commun. 12, 5576 (2021).
Karoutas, A. & Akhtar, A. Functional mechanisms and abnormalities of the nuclear lamina. Nat. Cell Biol. 23, 116–126 (2021).
Liu, S. et al. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561, 551–555 (2018).
Mammel, A. E. & Hatch, E. M. Genome instability from nuclear catastrophe and DNA damage. Semin. Cell. Dev. Biol. 123, 131–139 (2022).
Cortés-Ciriano, I. et al. Comprehensive analysis of chromothripsis in 2658 human cancers using whole-genome sequencing. Nat. Genet. 52, 331–341 (2020).
Trivedi, P. et al. Mitotic tethering enables inheritance of shattered micronuclear chromosomes. Nature 618, 1049–1056 (2023).
Lin, Y.-F. et al. Mitotic clustering of pulverized chromosomes from micronuclei. Nature 618, 1041–1048 (2023).
Yi, E., Chamorro González, R., Henssen, A. G. & Verhaak, R. G. Extrachromosomal DNA amplifications in cancer. Nat. Rev. Genet. 23, 760–771 (2022).
Yamamoto, K. et al. Micronuclei‐associated MYC amplification in the form of double minute chromosomes in acute myeloid leukemia. Am. J. Hematol. 88, 717–718 (2013).
Nones, K. et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat. Commun. 5, 5224 (2014).
Martins, F. C. et al. Clonal somatic copy number altered driver events inform drug sensitivity in high-grade serous ovarian cancer. Nat. Commun. 13, 6360 (2022).
Notta, F. et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538, 378–382 (2016).
Kloosterman, W. P. et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 77, 3814–3822 (2017).
Agustinus, A. S. et al. Epigenetic dysregulation from chromosomal transit in micronuclei. Nature 619, 176–183 (2023).
Papathanasiou, S. et al. Heritable transcriptional defects from aberrations of nuclear architecture. Nature 619, 184–192 (2023).
Donnelly, N. & Storchová, Z. Aneuploidy and proteotoxic stress in cancer. Mol. Cell Oncol. 2, e976491 (2015).
Zhu, J., Tsai, H.-J., Gordon, M. R. & Li, R. Cellular stress associated with aneuploidy. Dev. Cell 44, 420–431 (2018).
Oromendia, A. B. & Amon, A. Aneuploidy: implications for protein homeostasis and disease. Dis. Model Mech. 7, 15–20 (2014).
Oromendia, A. B., Dodgson, S. E. & Amon, A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 26, 2696–2708 (2012).
Schukken, K. M. & Sheltzer, J. M. Extensive protein dosage compensation in aneuploid human cancers. Genome Res. 32, 1254–1270 (2022).
Cheng, P. et al. Proteogenomic analysis of cancer aneuploidy and normal tissues reveals divergent modes of gene regulation across cellular pathways. Elife 11, e75227 (2022).
Ishikawa, K. Multilayered regulation of proteome stoichiometry. Curr. Genet. 67, 883–890 (2021).
Taggart, J. C. et al. Keeping the proportions of protein complex components in check. Cell Syst. 10, 125–132 (2020).
McShane, E. et al. Kinetic analysis of protein stability reveals age-dependent degradation. Cell 167, 803–815 e821 (2016).
Torres, E. M. Consequences of gaining an extra chromosome. Chromosom. Res. 31, 1–19 (2023).
Krivega, M. & Storchova, Z. Consequences of trisomy syndromes–21 and beyond. Trends Genet. 39, 172–174 (2022).
Anders, K. R. et al. A strategy for constructing aneuploid yeast strains by transient nondisjunction of a target chromosome. BMC genet. 10, 1–11 (2009).
Veitia, R. A., Bottani, S. & Birchler, J. A. Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet. 24, 390–397 (2008).
Papp, B., Pál, C. & Hurst, L. D. Dosage sensitivity and the evolution of gene families in yeast. Nature 424, 194–197 (2003).
Bowers, R. R. et al. SWAN pathway-network identification of common aneuploidy-based oncogenic drivers. Nucleic Acids Res. 50, 3673–3692 (2022).
Bean, J. et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl Acad. Sci. 104, 20932–20937 (2007).
Lulli, M. et al. DNA damage response protein CHK2 regulates metabolism in liver cancer. Cancer Res. 81, 2861–2873 (2021).
Li, Y. et al. PDSS2 deficiency induces hepatocarcinogenesis by decreasing mitochondrial respiration and reprogramming glucose metabolism. Cancer Res. 78, 4471–4481 (2018).
Thorburn, R. R. et al. Aneuploid yeast strains exhibit defects in cell growth and passage through START. Mol. Biol. Cell 24, 1274–1289 (2013).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Martínez-Reyes, I. & Chandel, N. S. Cancer metabolism: looking forward. Nat. Rev. Cancer. 21, 669–680 (2021).
Koppenol, W. H., Bounds, P. L. & Dang, C. V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 11, 325–337 (2011).
Siegel, J. J. & Amon, A. New insights into the troubles of aneuploidy. Annu. Rev. Cell Dev. Biol. 28, 189–214 (2012).
Huang, M. et al. Autonomous glucose metabolic reprogramming of tumour cells under hypoxia: opportunities for targeted therapy. J. Exp. Clin. Cancer Res. 39, 1–13 (2020).
Wu, S. et al. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Cancer Res. 78, 4549–4562 (2018).
Li, Z. et al. NeuroD1 promotes tumor cell proliferation and tumorigenesis by directly activating the pentose phosphate pathway in colorectal carcinoma. Oncogene 40, 6736–6747 (2021).
Wang, Y. et al. Yin Yang 1 promotes the Warburg effect and tumorigenesis via glucose transporter GLUT3. Cancer Sci. 109, 2423–2434 (2018).
Ciccarese, F. & Ciminale, V. Escaping death: mitochondrial redox homeostasis in cancer cells. Front. Oncol. 7, 117 (2017).
Sheltzer, J. M. A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res. 73, 6401–6412 (2013).
Santaguida, S., Vasile, E., White, E. & Amon, A. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 29, 2010–2021 (2015).
Joy, J. et al. Proteostasis failure and mitochondrial dysfunction leads to aneuploidy-induced senescence. Dev. Cell 56, 2043–2058. e2047 (2021).
Liao, Z., Chua, D. & Tan, N. S. Reactive oxygen species: a volatile driver of field cancerization and metastasis. Mol. Cancer 18, 1–10 (2019).
Simões-Sousa, S. et al. The p38α stress kinase suppresses aneuploidy tolerance by inhibiting Hif-1α. Cell Rep. 25, 749–760.e746 (2018).
Thompson, S. L. & Compton, D. A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 188, 369–381 (2010).
Marques, J. F. & Kops, G. J. Permission to pass: on the role of p53 as a gatekeeper for aneuploidy. Chromosom. Res. 31, 31 (2023).
Replogle, J. M. et al. Aneuploidy increases resistance to chemotherapeutics by antagonizing cell division. Proc. Natl. Acad. Sci. 117, 30566–30576 (2020).
Murai, K. et al. p53 mutation in normal esophagus promotes multiple stages of carcinogenesis but is constrained by clonal competition. Nat. Commun. 13, 6206 (2022).
Huang, C. et al. Identification of XBP1-u as a novel regulator of the MDM2/p53 axis using an shRNA library. Sci. Adv. 3, e1701383 (2017).
Joerger, A. C. & Fersht, A. R. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404 (2016).
Bieging, K. T., Mello, S. S. & Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370 (2014).
Levine, A. J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20, 471–480 (2020).
Kuffer, C., Kuznetsova, A. Y. & Storchová, Z. Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma 122, 305–318 (2013).
Levine, D. A. The Cancer Genome Atlas Research Network Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013).
Tang, R. et al. Colorectal cancer without high microsatellite instability and chromosomal instability-an alternative genetic pathway to human colorectal cancer. Carcinogenesis 25, 841–846 (2004).
Adell, M. A. Y. et al. Adaptation to spindle assembly checkpoint inhibition through the selection of specific aneuploidies. Genes Dev. 37, 171–190 (2023).
Hinchcliffe, E. H. et al. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat. Cell Biol. 18, 668–675 (2016).
Soto, M. et al. p53 prohibits propagation of chromosome segregation errors that produce structural aneuploidies. Cell Rep. 19, 2423–2431 (2017).
Li, M. et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl. Acad. Sci. 107, 14188–14193 (2010).
Baker, D. J., Jin, F., Jeganathan, K. B. & van Deursen, J. M. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 16, 475–486 (2009).
Foijer, F. et al. Deletion of the MAD2L1 spindle assembly checkpoint gene is tolerated in mouse models of acute T-cell lymphoma and hepatocellular carcinoma. eLife 6 (2017).
Bao, C. et al. Genomic signatures of past and present chromosomal instability in Barrett’s esophagus and early esophageal adenocarcinoma. Nat. Commun. 14, 6203 (2023).
Marei, H. E. et al. p53 signaling in cancer progression and therapy. Cancer Cell Int. 21, 1–15 (2021).
Xu, D. et al. Acetylation halts missense mutant p53 aggregation and rescues tumor suppression in non-small cell lung cancers. iScience 26, 107003 (2023).
Chi, Y. H. et al. Spindle assembly checkpoint and p53 deficiencies cooperate for tumorigenesis in mice. Int. J. Cancer 124, 1483–1489 (2009).
Woo, R. A. & Poon, R. Y. Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev. 18, 1317–1330 (2004).
Cannell, I. G. et al. A pleiotropic RNA-binding protein controls distinct cell cycle checkpoints to drive resistance of p53-defective tumors to chemotherapy. Cancer Cell 28, 623–637 (2015).
Dolado, I. & Nebreda, A. R. in Stress-Activated Protein Kinases 1st edn, Vol. 20 (eds Posas, F. & Nebreda, R. A.) Ch. 6 (Spinger Berlin, 2008).
Cánovas, B. et al. Targeting p38α Increases DNA damage, chromosome instability, and the anti-tumoral response to taxanes in breast cancer cells. Cancer Cell 33, 1094–1110.e1098 (2018).
Zhang, T. et al. 125I seed promotes apoptosis in non-small lung cancer cells via the p38 mapk-mdm2-p53 signaling pathway. Front. Oncol. 11, 582511 (2021).
Clemente-Ruiz, M. et al. Gene dosage imbalance contributes to chromosomal instability-induced tumorigenesis. Dev. Cell 36, 290–302 (2016).
Maehara, K., Takahashi, K. & Saitoh, S. CENP-A reduction induces a p53-dependent cellular senescence response to protect cells from executing defective mitoses. Mol. Cell Biol. 30, 2090–2104 (2010).
Collado, M. & Serrano, M. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57 (2010).
Prieur, A. & Peeper, D. S. Cellular senescence in vivo: a barrier to tumorigenesis. Curr. Opin. Cell Biol. 20, 150–155 (2008).
Kandala, S. et al. Chronic chromosome instability induced by Plk1 results in immune suppression in breast cancer. Cell Rep. 42, 113266 (2023).
Bunz, F. et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 (1998).
Uetake, Y. & Sluder, G. Activation of the apoptotic pathway during prolonged prometaphase blocks daughter cell proliferation. Mol. Biol. Cell 29, 2632–2643 (2018).
Brady, C. A. & Attardi, L. D. p53 at a glance. J. Cell Sci. 123, 2527–2532 (2010).
Nassour, J. et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663 (2019).
He, Q. et al. Chromosomal instability-induced senescence potentiates cell non-autonomous tumourigenic effects. Oncogenesis 7, 62 (2018).
Freund, A., Orjalo, A. V., Desprez, P. Y. & Campisi, J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246 (2010).
Pawlikowski, J. S., Adams, P. D. & Nelson, D. M. Senescence at a glance. J. Cell Sci. 126, 4061–4067 (2013).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Wörmann, S. M. et al. APOBEC3A drives deaminase domain-independent chromosomal instability to promote pancreatic cancer metastasis. Nat. Cancer 2, 1338–1356 (2021).
Dongre, A. et al. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 77, 3982–3989 (2017).
Gerstberger, S., Jiang, Q. & Ganesh, K. Metastasis. Cell 186, 1564–1579 (2023).
Shoshani, O. et al. Transient genomic instability drives tumorigenesis through accelerated clonal evolution. Genes Dev. 35, 1093–1108 (2021).
Vasudevan, A. et al. Single-chromosomal gains can function as metastasis suppressors and promoters in colon cancer. Dev. Cell 52, 413–428. e416 (2020).
Gao, C. et al. Chromosome instability drives phenotypic switching to metastasis. Proc. Natl. Acad. Sci. 113, 14793–14798 (2016).
Bakir, B., Chiarella, A. M., Pitarresi, J. R. & Rustgi, A. K. EMT, MET, plasticity, and tumor metastasis. Trends Cell Biol. 30, 764–776 (2020).
Li, J. et al. Non-cell-autonomous cancer progression from chromosomal instability. Nature 620, 1080–1088 (2023).
Barrio, L. et al. Chromosomal instability-induced cell invasion through caspase-driven DNA damage. Curr. Biol. 33, 4446–4457. e4445 (2023).
Cai, Y. et al. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell 29, 751–766 (2016).
Lu, C. et al. Hypoxia-activated neuropeptide Y/Y5 receptor/RhoA pathway triggers chromosomal instability and bone metastasis in Ewing sarcoma. Nat. Commun. 13, 2323 (2022).
Nguyen, B. et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 185, 563–575. e511 (2022).
Lengel, H. B. et al. Genomic mapping of metastatic organotropism in lung adenocarcinoma. Cancer Cell 41, 970–985.e973 (2023).
Skakodub, A. et al. Genomic analysis and clinical correlations of non-small cell lung cancer brain metastasis. Nat. Commun. 14, 4980 (2023).
Turajlic, S. et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell 173, 581–594.e512 (2018).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019).
Pradat, Y. et al. Integrative pan-cancer genomic and transcriptomic analyses of refractory metastatic cancer. Cancer Discov. 13, 1116–1143 (2023).
Senovilla, L. et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 337, 1678–1684 (2012).
Kuang, X. & Li, J. Chromosome instability and aneuploidy as context-dependent activators or inhibitors of antitumor immunity. Front. Immunol. 13, 895961 (2022).
Yan, H., Lu, W. & Wang, F. The cGAS-STING pathway: a therapeutic target in chromosomally unstable cancers. Signal Transduct. Target. Ther. 8, 45 (2023).
Kwon, J. & Bakhoum, S. F. The cytosolic DNA-sensing cGAS–STING pathway in cancer. Cancer Discov. 10, 26–39 (2020).
Samson, N. & Ablasser, A. The cGAS–STING pathway and cancer. Nat. Cancer. 3, 1452–1463 (2022).
Ablasser, A. & Chen, Z. J. cGAS in action: expanding roles in immunity and inflammation. Science 363, eaat8657 (2019).
Huang, Y. et al. DNAJA2 deficiency activates cGAS-STING pathway via the induction of aberrant mitosis and chromosome instability. Nat. Commun. 14, 5246 (2023).
Mackenzie, K. J. et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017).
Harding, S. M. et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017).
Vanpouille-Box, C., Demaria, S., Formenti, S. C. & Galluzzi, L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 34, 361–378 (2018).
Hu, J. et al. STING inhibits the reactivation of dormant metastasis in lung adenocarcinoma. Nature 616, 806–813 (2023).
Boukhaled, G. M., Harding, S. & Brooks, D. G. Opposing roles of type I interferons in cancer immunity. Annu. Rev. Pathol. 16, 167–198 (2021).
Wang, R. W. et al. Aneuploid senescent cells activate NF‐κB to promote their immune clearance by NK cells. EMBO Rep. 22, e52032 (2021).
Hong, C. et al. cGAS–STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 607, 366–373 (2022).
Holohan, C., Van Schaeybroeck, S., Longley, D. B. & Johnston, P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 13, 714–726 (2013).
Lee, A. J. X. et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 71, 1858–1870 (2011).
Cohen-Sharir, Y. et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 590, 486–491 (2021).
Brutovský, B. Scales of cancer evolution: selfish genome or cooperating cells? Cancers 14, 3253 (2022).
Beaumont, H. J. et al. Experimental evolution of bet hedging. Nature 462, 90–93 (2009).
Scribano, C. M. et al. Chromosomal instability sensitizes patient breast tumors to multipolar divisions induced by paclitaxel. Sci. Transl. Med. 13, eabd4811 (2021).
Schukken, K. M. et al. Altering microtubule dynamics is synergistically toxic with spindle assembly checkpoint inhibition. Life Sci. Alliance 3, e201900499 (2020).
Jemaà, M. et al. Characterization of novel MPS1 inhibitors with preclinical anticancer activity. Cell Death Differ. 20, 1532–1545 (2013).
Soria-Bretones, I. et al. The spindle assembly checkpoint is a therapeutic vulnerability of CDK4/6 inhibitor–resistant ER+ breast cancer with mitotic aberrations. Sci. Adv. 8, eabq4293 (2022).
Martino, J. et al. Inhibitors of Rho kinases (ROCK) induce multiple mitotic defects and synthetic lethality in BRCA2-deficient cells. Elife 12, e80254 (2023).
Gallo, D. et al. CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature 604, 749–756 (2022).
Maia, A. R. R. et al. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann. Oncol. 26, 2180–2192 (2015).
Maia, A. R. R. et al. Mps1 inhibitors synergise with low doses of taxanes in promoting tumour cell death by enhancement of errors in cell division. Br. J. Cancer 118, 1586–1595 (2018).
Zasadil, L. M. et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 6, 229ra243 (2014).
Lucken, K. et al. EML4-ALK variant 3 promotes mitotic errors and spindle assembly checkpoint deficiency leading to increased microtubule poison sensitivity. Mol. Cancer Res. 20, 854–866 (2022).
Bai, Z. et al. Perspectives and mechanisms for targeting mitotic catastrophe in cancer treatment. Biochim. Biophys. Acta Rev. Cancer 1878, 188965 (2023).
Normandin, K. et al. Genetic enhancers of partial PLK1 inhibition reveal hypersensitivity to kinetochore perturbations. PLoS Genet. 19, e1010903 (2023).
Martinez, M. J. et al. Inhibition of the serine/threonine kinase BUB1 reverses taxane resistance in prostate cancer. iScience 26, 107681 (2023).
Portelinha, A. et al. Synthetic lethality of drug-induced polyploidy and BCL-2 inhibition in lymphoma. Nat. Commun. 14, 1522 (2023).
Janssen, A., Kops, G. J. P. L. & Medema, R. H. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl Acad. Sci. 106, 19108–19113 (2009).
Kops, G. J. P. L., Foltz, D. R. & Cleveland, D. W. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl Acad. Sci. 101, 8699–8704 (2004).
Wu, G. et al. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 68, 8393–8399 (2008).
Hu, C. M. et al. Novel small molecules disrupting Hec1/Nek2 interaction ablate tumor progression by triggering Nek2 degradation through a death-trap mechanism. Oncogene 34, 1220–1230 (2015).
Dhital, B. & Rodriguez-Bravo, V. Mechanisms of chromosomal instability (CIN) tolerance in aggressive tumors: surviving the genomic chaos. Chromosom. Res. 31, 15 (2023).
Sansregret, L. et al. APC/C dysfunction limits excessive cancer chromosomal instability. Cancer Discov. 7, 218–233 (2017).
Dhital, B. et al. Harnessing transcriptionally driven chromosomal instability adaptation to target therapy-refractory lethal prostate cancer. Cell Rep. Med. 4, 100937 (2023).
Gronroos, E. & Lopez-Garcia, C. Tolerance of chromosomal instability in cancer: mechanisms and therapeutic opportunities. Cancer Res. 78, 6529–6535 (2018).
Perelli, L. et al. Interferon signaling promotes tolerance to chromosomal instability during metastatic evolution in renal cancer. Nat. Cancer. 4, 984–100 (2023).
Clarke, M. N. et al. Adaptation to high rates of chromosomal instability and aneuploidy through multiple pathways in budding yeast. EMBO J. 42, e111500 (2023).
Haase, M. A. et al. DASH/Dam1 complex mutants stabilize ploidy in histone‐humanized yeast by weakening kinetochore‐microtubule attachments. EMBO J. 42, e112600 (2023).
Du, M. et al. Nondiploid cancer cells: Stress, tolerance and therapeutic inspirations. Biochim. Biophys. Acta Rev. Cancer 1877, 188794 (2022).
Orr, B. et al. Adaptive resistance to an inhibitor of chromosomal instability in human cancer cells. Cell Rep. 17, 1755–1763 (2016).
Su, X. A. et al. RAD21 is a driver of chromosome 8 gain in Ewing sarcoma to mitigate replication stress. Genes Dev. 35, 556–572 (2021).
Schiavoni, F. et al. Aneuploidy tolerance caused by BRG1 loss allows chromosome gains and recovery of fitness. Nat. Commun. 13, 1731 (2022).
Marquis, C. et al. Chromosomally unstable tumor cells specifically require KIF18A for proliferation. Nat. Commun. 12, 1213 (2021).
Tang, Y.-C. et al. Aneuploid cell survival relies upon sphingolipid homeostasis. Cancer Res. 77, 5272–5286 (2017).
Girish, V. et al. Oncogene-like addiction to aneuploidy in human cancers. Science 381, eadg4521 (2023).
Mays, J. C. et al. KaryoTap enables aneuploidy detection in thousands of single human cells. Preprint at http://www.biorxiv.org/content/10.1101/2023.09.08.555746v1 (2023).
Angrisani, A. & Fachinetti, D. The KaryoCreate technology generates specific aneuploid karyotypes on demand. Cell Rep. Meth. 3, 100514 (2023).
Truong, M. A., Cané-Gasull, P. & Lens, S. Modeling specific aneuploidies: from karyotype manipulations to biological insights. Chromosom. Res. 31, 1–27 (2023).
Truong, M. A. et al. A kinesin‐based approach for inducing chromosome‐specific mis‐segregation in human cells. EMBO J. 15, e111559 (2023).
Lakhani, A. A., Thompson, S. L. & Sheltzer, J. M. Aneuploidy in human cancer: new tools and perspectives. Trends Genet. 39, P968–P980 (2023).
Tovini, L. et al. Targeted assembly of ectopic kinetochores to induce chromosome‐specific segmental aneuploidies. EMBO J. 42, e111587 (2023).
Shih, J. et al. Cancer aneuploidies are shaped primarily by effects on tumour fitness. Nature 619, 793–800 (2023).
Dominguez-Brauer, C. et al. Targeting mitosis in cancer: emerging strategies. Mol. Cell. 60, 524–536 (2015).
Veitch, Z. W. et al. Safety and tolerability of CFI-400945, a first-in-class, selective PLK4 inhibitor in advanced solid tumours: a phase 1 dose-escalation trial. Br. J. Cancer 121, 318–324 (2019).
Lorusso, P. et al. First-in-human study of the monopolar spindle 1 (Mps1) kinase inhibitor BAY 1161909 in combination with paclitaxel in subjects with advanced malignancies. Ann. Oncol. 29, VIII138 (2018).
Cheng, A. et al. A mitotic NADPH upsurge promotes chromosome segregation and tumour progression in aneuploid cancer cells. Nat. Metab. 5, 1141–11158 (2023).
Quinton, R. J. et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 590, 492–497 (2021).
Herman, J. A. et al. Hyper-active RAS/MAPK introduces cancer-specific mitotic vulnerabilities. Proc. Natl Acad. Sci. 119, e2208255119 (2022).
Martín, A. et al. Mitochondrial RNA methyltransferase TRMT61B is a new, potential biomarker and therapeutic target for highly aneuploid cancers. Cell Death Differ. 30, 37–53 (2023).
Andor, N., Altrock, P. M., Jain, N. & Gomes, A. P. Tipping cancer cells over the edge: the context-dependent cost of high ploidy. Cancer Res. 82, 741–748 (2022).
Li, M. et al. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J. Cell Biol. 185, 983–994 (2009).
Rao, C. V. et al. Colonic tumorigenesis in BubR1+/-ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc. Natl Acad. Sci. 102, 4365–4370 (2005).
Caldwell, C. M., Green, R. A. & Kaplan, K. B. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol. 178, 1109–1120 (2007).
Meraldi, P., Honda, R. & Nigg, E. A. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 21, 483–492 (2002).
Anand, S., Penrhyn-Lowe, S. & Venkitaraman, A. R. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 3, 51–62 (2003).
Zhang, D. et al. Cre-loxP-controlled periodic Aurora-A overexpression induces mitotic abnormalities and hyperplasia in mammary glands of mouse models. Oncogene 23, 8720–8730 (2004).
Zhang, D. et al. Aurora A overexpression induces cellular senescence in mammary gland hyperplastic tumors developed in p53-deficient mice. Oncogene 27, 4305–4314 (2008).
Jeganathan, K. et al. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J. Cell Biol. 179, 255–267 (2007).
Wang, Q. et al. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood 103, 1278–1285 (2004).
Baker, D. J. et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat. Cell Biol. 15, 96–102 (2013).
Weaver, B. A. et al. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25–36 (2007).
Diaz-Rodriguez, E., Sotillo, R., Schvartzman, J. M. & Benezra, R. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc. Natl Acad. Sci. 105, 16719–16724 (2008).
Wang, Z., Yu, R. & Melmed, S. Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol. Endocrinol. 15, 1870–1879 (2001).
Chesnokova, V. et al. Pituitary hypoplasia in Pttg-/- mice is protective for Rb+/- pituitary tumorigenesis. Mol. Endocrinol. 19, 2371–2379 (2005).
Aguirre-Portoles, C. et al. Tpx2 controls spindle integrity, genome stability, and tumor development. Cancer Res. 72, 1518–1528 (2012).
van Ree, J. H., Jeganathan, K. B., Malureanu, L. & van Deursen, J. M. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J. Cell Biol. 188, 83–100 (2010).
Fujita, T. et al. Overexpression of UbcH10 alternates the cell cycle profile and accelerate the tumor proliferation in colon cancer. BMC Cancer 9, 1–10 (2009).
Garcia-Higuera, I. et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 10, 802–811 (2008).
Remeseiro, S. et al. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 31, 2076–2089 (2012).
Abbud, R. A. et al. Early multipotential pituitary focal hyperplasia in the alpha-subunit of glycoprotein hormone-driven pituitary tumor-transforming gene transgenic mice. Mol. Endocrinol. 19, 1383–1391 (2005).
Donangelo, I. et al. Pituitary tumor transforming gene overexpression facilitates pituitary tumor development. Endocrinology 147, 4781–4791 (2006).
de Cárcer, G. et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 9, 3012 (2018).
Dharanipragada, P. et al. Blocking genomic instability prevents acquired resistance to MAPK inhibitor therapy in melanoma. Cancer Discov. 13, 880–909 (2023).
Zerbib, J. et al. Human aneuploid cells depend on the RAF/MEK/ERK pathway for overcoming increased DNA damage. Preprint at http://www.biorxiv.org/content/10.1101/2023.01.27.525822v1 (2023).
Zhou, W. et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 23, 48–62 (2013).
Gu, C. et al. CHEK1 and circCHEK1_246aa evoke chromosomal instability and induce bone lesion formation in multiple myeloma. Mol. Cancer 20, 84 (2021).
Tang, X. et al. BUB1B and circBUB1B_544aa aggravate multiple myeloma malignancy through evoking chromosomal instability. Signal Transduct. Target. Ther. 6, 361 (2021).
Davoli, T., Uno, H., Wooten, E. C. & Elledge, S. J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355, eaaf8399 (2017).
Manfredi, M. G. et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 17, 7614–7624 (2011).
Dees, E. C. et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin. Cancer Res. 18, 4775–4784 (2012).
Matulonis, U. A. et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol. Oncol. 127, 63–69 (2012).
Dickson, M. A. et al. Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. Ann. Oncol. 27, 1855–1860 (2016).
Beltran, H. et al. A Phase II trial of the aurora kinase A inhibitor Alisertib for patients with castration-resistant and neuroendocrine prostate cancer: efficacy and biomarkers. Clin. Cancer Res. 25, 43–51 (2019).
Falchook, G. et al. Alisertib in combination with weekly paclitaxel in patients with advanced breast cancer or recurrent ovarian cancer: a randomized clinical trial. JAMA Oncol. 5, e183773 (2019).
Necchi, A. et al. An open-label, single-arm, phase 2 study of the aurora kinase A inhibitor Alisertib in patients with advanced urothelial cancer. Investig. New Drugs 34, 236–242 (2016).
Owonikoko, T. K. et al. Randomized phase II study of paclitaxel plus alisertib versus paclitaxel plus placebo as second-line therapy for SCLC: primary and correlative biomarker analyses. J. Thorac. Oncol. 15, 274–287 (2020).
Graff, J. N. et al. Open-label, multicenter, phase 1 study of alisertib (MLN8237), an aurora A kinase inhibitor, with docetaxel in patients with solid tumors. Cancer 122, 2524–2533 (2016).
Haddad, T. C. et al. Phase I trial to evaluate the addition of alisertib to fulvestrant in women with endocrine-resistant, ER+ metastatic breast cancer. Breast Cancer Res. Treat. 168, 639–647 (2018).
Goff, L. W. et al. Phase I study combining the aurora kinase a inhibitor alisertib with mFOLFOX in gastrointestinal cancer. Investig. New Drugs 37, 315–322 (2019).
DuBois, S. G. et al. Phase II trial of alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma. Clin. Cancer Res. 24, 6142–6149 (2018).
Shah, H. A. et al. Phase I study of aurora a kinase inhibitor alisertib (MLN8237) in combination with selective VEGFR inhibitor pazopanib for therapy of advanced solid tumors. Am. J. Clin. Oncol. 42, 413–420 (2019).
Jing, X. L. & Chen, S. W. Aurora kinase inhibitors: a patent review (2014-2020). Expert Opin. Ther. Pat. 31, 625–644 (2021).
Di Noia, V. et al. Malignant pleural mesothelioma: is tailoring the second-line therapy really “raising the bar? Curr. Treat. Options. Oncol 20, 23 (2019).
Chen, J. A. et al. A phase I dose escalation, dose expansion and pharmacokinetic trial of gemcitabine and alisertib in advanced solid tumors and pancreatic cancer. Cancer Chemother. Pharmacol. 90, 217–228 (2022).
O’Shaughnessy, J. et al. Efficacy and safety of weekly paclitaxel with or without oral alisertib in patients with metastatic breast cancer: a randomized clinical trial. JAMA Netw. Open. 4, e214103 (2021).
Nguyen, T. T., Silva, F. N. & Golemis, E. A. Aurora kinases as therapeutic targets in head and neck cancer. Cancer J. 28, 387–400 (2022).
Venkadakrishnan, V. B. et al. Significance of RB loss in unlocking phenotypic plasticity in advanced cancers. Mol. Cancer Res. 21, 497–510 (2023).
Bain, N. T., Wang, Y. & Arulananda, S. Minimal residual disease in EGFR-mutant non-small-cell lung cancer. Front. Oncol. 12, 1002714 (2022).
Li, S. et al. Emerging targeted therapies in advanced non-small-cell lung cancer. Cancers 15, 2899 (2023).
Farnsworth, D. A., Chen, Y. T., de Rappard Yuswack, G. & Lockwood, W. W. Emerging molecular dependencies of mutant EGFR-Driven non-small cell lung cancer. Cells 10, 3553 (2021).
Kelly, K. R. et al. Phase I study of MLN8237-investigational Aurora A kinase inhibitor-in relapsed/refractory multiple myeloma, non-Hodgkin lymphoma and chronic lymphocytic leukemia. Investig. New Drugs 32, 489–499 (2014).
Barr, P. M. et al. Phase II intergroup trial of alisertib in relapsed and refractory peripheral T-Cell lymphoma and transformed mycosis fungoides: SWOG 1108. J. Clin. Oncol. 33, 2399–2404 (2015).
Zhang, Z. et al. Modulation of oxidative phosphorylation augments antineoplastic activity of mitotic aurora kinase inhibition. Cell Death Dis. 12, 893 (2021).
Friedberg, J. W. et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J. Clin. Oncol. 32, 44–50 (2014).
Kelly, K. R. et al. Phase I study of the investigational aurora a kinase inhibitor alisertib plus rituximab or rituximab/vincristine in relapsed/refractory aggressive B-cell lymphoma. Clin. Cancer Res. 24, 6150–6159 (2018).
Rosenthal, A. et al. A phase Ib study of the combination of the aurora kinase inhibitor alisertib (MLN8237) and bortezomib in relapsed multiple myeloma. Br. J. Haematol. 174, 323–325 (2016).
Brunner, A. M. et al. Alisertib plus induction chemotherapy in previously untreated patients with high-risk, acute myeloid leukaemia: a single-arm, phase 2 trial. Lancet Haematol. 7, e122–e133 (2020).
Mosse, Y. P. et al. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a children’s oncology group phase I consortium study. Clin. Cancer Res. 18, 6058–6064 (2012).
Mosse, Y. P. et al. A phase II study of alisertib in children with recurrent/refractory solid tumors or leukemia: children’s oncology group phase I and pilot consortium (ADVL0921). Clin. Cancer Res. 25, 3229–3238 (2019).
Zhang, C. & Li, H. Molecular targeted therapies for pediatric atypical teratoid/rhabdoid tumors. Pediatr. Investig. 6, 111–122 (2022).
Rechberger, J. S., Nesvick, C. L. & Daniels, D. J. Atypical teratoid rhabdoid tumor (ATRT): disease mechanisms and potential drug targets. Expert. Opin. Ther. Targets 26, 187–192 (2022).
Fletcher, G. C. et al. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol. Cancer Ther. 10, 126–137 (2011).
Diamond, J. R. et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of ENMD-2076, a novel angiogenic and Aurora kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 17, 849–860 (2011).
Matulonis, U. A. et al. ENMD-2076, an oral inhibitor of angiogenic and proliferation kinases, has activity in recurrent, platinum resistant ovarian cancer. Eur. J. Cancer 49, 121–131 (2013).
Lheureux, S. et al. A clinical and molecular phase II trial of oral ENMD-2076 in ovarian clear cell carcinoma (OCCC): a study of the princess margaret phase II consortium. Clin. Cancer Res. 24, 6168–6174 (2018).
Veitch, Z. et al. A phase II study of ENMD-2076 in advanced soft tissue sarcoma (STS). Sci. Rep. 9, 7390 (2019).
Diamond, J. R. et al. A phase II clinical trial of the Aurora and angiogenic kinase inhibitor ENMD-2076 for previously treated, advanced, or metastatic triple-negative breast cancer. Breast Cancer Res. 20, 82 (2018).
Abou-Alfa, G. K. et al. Phase II multicenter, open-label study of oral ENMD-2076 for the treatment of patients with advanced fibrolamellar carcinoma. Oncol. 25, e1837–e1845 (2020).
Yee, K. W. et al. A phase I trial of the aurora kinase inhibitor, ENMD-2076, in patients with relapsed or refractory acute myeloid leukemia or chronic myelomonocytic leukemia. Investig. New Drugs 34, 614–624 (2016).
Lloyd, M. R., Spring, L. M., Bardia, A. & Wander, S. A. Mechanisms of resistance to CDK4/6 blockade in advanced hormone receptor-positive, HER2-negative breast cancer and emerging therapeutic opportunities. Clin. Cancer Res. 28, 821–830 (2022).
Chu, Q. S. et al. Aurora kinase A inhibitor, LY3295668 erbumine: a phase 1 monotherapy safety study in patients with locally advanced or metastatic solid tumors. Investig. New Drugs 39, 1001–1010 (2021).
Lum, C. & Alamgeer, M. Technological and therapeutic advances in advanced small cell lung cancer. Cancers 11, 1570 (2019).
Moreno, L. et al. Accelerating drug development for neuroblastoma: summary of the second neuroblastoma drug development strategy forum from innovative therapies for children with cancer and international society of paediatric oncology europe neuroblastoma. Eur. J. Cancer 136, 52–68 (2020).
Dees, E. C. et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 67, 945–954 (2011).
Macarulla, T. et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol. Cancer Ther. 9, 2844–2852 (2010).
Shimomura, T. et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol. Cancer Ther. 9, 157–166 (2010).
Amin, M. et al. A phase I study of MK-5108, an oral aurora a kinase inhibitor, administered both as monotherapy and in combination with docetaxel, in patients with advanced or refractory solid tumors. Investig. New Drugs 34, 84–95 (2016).
Miura, A. et al. TAS-119, a novel selective Aurora A and TRK inhibitor, exhibits antitumor efficacy in preclinical models with deregulated activation of the Myc, beta-Catenin, and TRK pathways. Investig. New Drugs 39, 724–735 (2021).
Robbrecht, D. G. J. et al. A first-in-human phase 1 and pharmacological study of TAS-119, a novel selective Aurora A kinase inhibitor in patients with advanced solid tumours. Br. J. Cancer 124, 391–398 (2021).
Sootome, H. et al. Aurora A inhibitor TAS-119 enhances antitumor efficacy of taxanes in vitro and in vivo: preclinical studies as guidance for clinical development and trial design. Mol. Cancer Ther. 19, 1981–1991 (2020).
Shiotsu, Y. et al. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood 114, 1607–1617 (2009).
Cheung, C. H. et al. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011 - 2013). Expert Opin. Ther. Pat. 24, 1021–1038 (2014).
Yang, J. et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood 110, 2034–2040 (2007).
Schwartz, G. K. et al. Phase I study of barasertib (AZD1152), a selective inhibitor of Aurora B kinase, in patients with advanced solid tumors. Investig. New Drugs 31, 370–380 (2013).
Boss, D. S. et al. Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B kinase, in patients with solid malignant tumors. Ann. Oncol. 22, 431–437 (2011).
Lowenberg, B. et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood 118, 6030–6036 (2011).
Tsuboi, K. et al. A Phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leuk. Res. 35, 1384–1389 (2011).
Kantarjian, H. M. et al. Stage I of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer 119, 2611–2619 (2013).
Dennis, M. et al. Phase I study of the Aurora B kinase inhibitor barasertib (AZD1152) to assess the pharmacokinetics, metabolism and excretion in patients with acute myeloid leukemia. Cancer Chemother. Pharmacol. 70, 461–469 (2012).
Kantarjian, H. M. et al. Phase I study assessing the safety and tolerability of barasertib (AZD1152) with low-dose cytosine arabinoside in elderly patients with AML. Clin. Lymphoma Myeloma Leuk. 13, 559–567 (2013).
Collins, G. P. et al. A phase II trial of AZD1152 in relapsed/refractory diffuse large B-cell lymphoma. Br. J. Haematol. 170, 886–890 (2015).
Dittrich, C. et al. A phase 1 dose escalation study of BI 831266, an inhibitor of Aurora kinase B, in patients with advanced solid tumors. Investig. New Drugs 33, 409–422 (2015).
Mross, K. et al. A phase I study of BI 811283, an Aurora B kinase inhibitor, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 78, 405–417 (2016).
Dohner, H. et al. A phase I trial investigating the Aurora B kinase inhibitor BI 811283 in combination with cytarabine in patients with acute myeloid leukaemia. Br. J. Haematol. 185, 583–587 (2019).
Zhou, Y. et al. CS2164, a novel multi-target inhibitor against tumor angiogenesis, mitosis and chronic inflammation with anti-tumor potency. Cancer Sci. 108, 469–477 (2017).
Sun, Y. et al. Phase I dose-escalation study of chiauranib, a novel angiogenic, mitotic, and chronic inflammation inhibitor, in patients with advanced solid tumors. J. Hematol. Oncol. 12, 9 (2019).
Cao, J., Chow, L. & Dow, S. Strategies to overcome myeloid cell induced immune suppression in the tumor microenvironment. Front. Oncol. 13, 1116016 (2023).
Gao, T. et al. Inhibition of extranodal NK/T-cell lymphoma by chiauranib through an AIF-dependent pathway and its synergy with L-asparaginase. Cell Death Dis. 14, 316 (2023).
Lv, P. et al. Pathogenesis and therapeutic strategy in platinum resistance lung cancer. Biochim. Biophys. Acta Rev. Cancer 1876, 188577 (2021).
Howard, S. et al. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J. Med. Chem. 52, 379–388 (2009).
Dent, S. F. et al. NCIC CTG IND.181: phase I study of AT9283 given as a weekly 24 hour infusion in advanced malignancies. Investig. New Drugs 31, 1522–1529 (2013).
Foran, J. et al. A phase I and pharmacodynamic study of AT9283, a small-molecule inhibitor of aurora kinases in patients with relapsed/refractory leukemia or myelofibrosis. Clin. Lymphoma Myeloma Leuk. 14, 223–230 (2014).
Moreno, L. et al. A phase I trial of AT9283 (a selective inhibitor of aurora kinases) in children and adolescents with solid tumors: a Cancer Research UK study. Clin. Cancer Res. 21, 267–273 (2015).
Peter, B. et al. Drug-induced inhibition of phosphorylation of STAT5 overrides drug resistance in neoplastic mast cells. Leukemia 32, 1016–1022 (2018).
Mita, M. et al. A phase l study of three different dosing schedules of the oral aurora kinase inhibitor MSC1992371A in patients with solid tumors. Target Oncol. 9, 215–224 (2014).
Raymond, E. et al. A phase I schedule dependency study of the aurora kinase inhibitor MSC1992371A in combination with gemcitabine in patients with solid tumors. Investig. New Drugs 32, 94–103 (2014).
Graux, C. et al. A phase I dose-escalation study of MSC1992371A, an oral inhibitor of aurora and other kinases, in advanced hematologic malignancies. Leuk. Res. 37, 1100–1106 (2013).
Kim, J. T. et al. The discovery of aurora kinase inhibitor by multi-docking-based virtual screening. Int. J. Mol. Sci. 15, 20403–20412 (2014).
Jani, J. P. et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol. Cancer Ther. 9, 883–894 (2010).
Schoffski, P. et al. Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. Eur. J. Cancer 47, 2256–2264 (2011).
Farrell, P. et al. Biological characterization of TAK-901, an investigational, novel, multitargeted Aurora B kinase inhibitor. Mol. Cancer Ther. 12, 460–470 (2013).
Carpinelli, P. et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 6, 3158–3168 (2007).
Meulenbeld, H. J. et al. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int. 111, 44–52 (2013).
Adams, N. D. et al. Discovery of GSK1070916, a potent and selective inhibitor of Aurora B/C kinase. J. Med. Chem. 53, 3973–4001 (2010).
Glaser, K. B. et al. Preclinical characterization of ABT-348, a kinase inhibitor targeting the aurora, vascular endothelial growth factor receptor/platelet-derived growth factor receptor, and Src kinase families. J. Pharmacol. Exp. Ther. 343, 617–627 (2012).
Maitland, M. L. et al. Clinical pharmacodynamic/exposure characterisation of the multikinase inhibitor ilorasertib (ABT-348) in a phase 1 dose-escalation trial. Br. J. Cancer 118, 1042–1050 (2018).
Garcia-Manero, G. et al. Phase 1 dose escalation trial of ilorasertib, a dual Aurora/VEGF receptor kinase inhibitor, in patients with hematologic malignancies. Investig. New Drugs 33, 870–880 (2015).
Payton, M. et al. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 70, 9846–9854 (2010).
Carducci, M. et al. A phase 1, first-in-human study of AMG 900, an orally administered pan-Aurora kinase inhibitor, in adult patients with advanced solid tumors. Investig. New Drugs 36, 1060–1071 (2018).
Kantarjian, H. M. et al. A phase 1 study of AMG 900, an orally administered pan-aurora kinase inhibitor, in adult patients with acute myeloid leukemia. Am. J. Hematol. 92, 660–667 (2017).
Sini, P. et al. Pharmacological Profile of BI 847325, an Orally Bioavailable, ATP-Competitive Inhibitor of MEK and Aurora Kinases. Mol. Cancer Ther. 15, 2388–2398 (2016).
Schoffski, P. et al. A phase I study of two dosing schedules of oral BI 847325 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 77, 99–108 (2016).
Oslob, J. D. et al. Discovery of a potent and selective aurora kinase inhibitor. Bioorg. Med. Chem. Lett. 18, 4880–4884 (2008).
Harrington, E. A. et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 10, 262–267 (2004).
Traynor, A. M. et al. Phase I dose escalation study of MK-0457, a novel Aurora kinase inhibitor, in adult patients with advanced solid tumors. Cancer Chemother. Pharmacol. 67, 305–314 (2011).
Giles, F. J. et al. MK-0457, an Aurora kinase and BCR-ABL inhibitor, is active in patients with BCR-ABL T315I leukemia. Leukemia 27, 113–117 (2013).
Wood, K. W. et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl Acad. Sci. 107, 5839–5844 (2010).
Chung, V. et al. First-time-in-human study of GSK923295, a novel antimitotic inhibitor of centromere-associated protein E (CENP-E), in patients with refractory cancer. Cancer Chemother. Pharmacol. 69, 733–741 (2012).
Blagden, S. P. et al. A phase I trial of ispinesib, a kinesin spindle protein inhibitor, with docetaxel in patients with advanced solid tumours. Br. J. Cancer 98, 894–899 (2008).
Garcia-Saez, I. & Skoufias, D. A. Eg5 targeting agents: From new anti-mitotic based inhibitor discovery to cancer therapy and resistance. Biochem. Pharmacol. 184, 114364 (2021).
Beer, T. M. et al. Southwest Oncology Group phase II study of ispinesib in androgen-independent prostate cancer previously treated with taxanes. Clin. Genitourin. Cancer 6, 103–109 (2008).
Tang, P. A. et al. Phase II study of ispinesib in recurrent or metastatic squamous cell carcinoma of the head and neck. Investig. New Drugs 26, 257–264 (2008).
Lee, C. W. et al. A phase II study of ispinesib (SB-715992) in patients with metastatic or recurrent malignant melanoma: a National Cancer Institute of Canada Clinical Trials Group trial. Investig. New Drugs 26, 249–255 (2008).
Knox, J. J. et al. A phase II and pharmacokinetic study of SB-715992, in patients with metastatic hepatocellular carcinoma: a study of the National Cancer Institute of Canada Clinical Trials Group (NCIC CTG IND.168). Investig. New Drugs 26, 265–272 (2008).
Lee, R. T. et al. A University of Chicago consortium phase II trial of SB-715992 in advanced renal cell cancer. Clin. Genitourin. Cancer 6, 21–24 (2008).
Gomez, H. L. et al. Phase I dose-escalation and pharmacokinetic study of ispinesib, a kinesin spindle protein inhibitor, administered on days 1 and 15 of a 28-day schedule in patients with no prior treatment for advanced breast cancer. Anticancer Drugs 23, 335–341 (2012).
Souid, A. K. et al. A pediatric phase I trial and pharmacokinetic study of ispinesib: a Children’s Oncology Group phase I consortium study. Pediatr. Blood Cancer 55, 1323–1328 (2010).
Holen, K. D. et al. A first in human study of SB-743921, a kinesin spindle protein inhibitor, to determine pharmacokinetics, biologic effects and establish a recommended phase II dose. Cancer Chemother. Pharmacol. 67, 447–454 (2011).
O’Connor, O. A. et al. The addition of granulocyte-colony stimulating factor shifts the dose limiting toxicity and markedly increases the maximum tolerated dose and activity of the kinesin spindle protein inhibitor SB-743921 in patients with relapsed or refractory lymphoma: results of an international, multicenter phase I/II study. Leuk. Lymphoma 56, 2585–2591 (2015).
LoRusso, P. M. et al. First-in-human phase 1 study of filanesib (ARRY-520), a kinesin spindle protein inhibitor, in patients with advanced solid tumors. Investig. New Drugs 33, 440–449 (2015).
Khoury, H. J. et al. A phase 1 dose-escalation study of ARRY-520, a kinesin spindle protein inhibitor, in patients with advanced myeloid leukemias. Cancer 118, 3556–3564 (2012).
Shah, J. J. et al. A Phase 1 and 2 study of Filanesib alone and in combination with low-dose dexamethasone in relapsed/refractory multiple myeloma. Cancer 123, 4617–4630 (2017).
Chari, A. et al. A phase 1 dose-escalation study of filanesib plus bortezomib and dexamethasone in patients with recurrent/refractory multiple myeloma. Cancer 122, 3327–3335 (2016).
Lee, H. C. et al. A phase 1 study of filanesib, carfilzomib, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Blood Cancer J. 9, 80 (2019).
Ocio, E. M. et al. Filanesib in combination with pomalidomide and dexamethasone in refractory MM patients: safety and efficacy, and association with alpha 1-acid glycoprotein (AAG) levels. Phase Ib/II Pomdefil clinical trial conducted by the Spanish MM group. Br. J. Haematol. 192, 522–530 (2021).
Tabernero, J. et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 3, 406–417 (2013).
Ye, X. S. et al. A novel Eg5 Inhibitor (LY2523355) causes mitotic arrest and apoptosis in cancer cells and shows potent antitumor activity in xenograft tumor models. Mol. Cancer Ther. 14, 2463–2472 (2015).
Infante, J. R. et al. Two phase 1 dose-escalation studies exploring multiple regimens of litronesib (LY2523355), an Eg5 inhibitor, in patients with advanced cancer. Cancer Chemother. Pharmacol. 79, 315–326 (2017).
Wakui, H. et al. A phase 1 and dose-finding study of LY2523355 (litronesib), an Eg5 inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 74, 15–23 (2014).
Masanas, M. et al. The oral KIF11 inhibitor 4SC-205 exhibits antitumor activity and potentiates standard and targeted therapies in primary and metastatic neuroblastoma models. Clin. Transl. Med. 11, e533 (2021).
Esaki, T. et al. Phase I study to assess the safety, tolerability and pharmacokinetics of AZD4877 in Japanese patients with solid tumors. Arch. Drug Inf. 4, 23–31 (2011).
Infante, J. R. et al. A phase I study to assess the safety, tolerability, and pharmacokinetics of AZD4877, an intravenous Eg5 inhibitor in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 69, 165–172 (2012).
Jones, R. et al. Phase II study to assess the efficacy, safety and tolerability of the mitotic spindle kinesin inhibitor AZD4877 in patients with recurrent advanced urothelial cancer. Investig. New Drugs 31, 1001–1007 (2013).
Kantarjian, H. M. et al. Phase I/II multicenter study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of AZD4877 in patients with refractory acute myeloid leukemia. Investig. New Drugs 30, 1107–1115 (2012).
Holen, K. et al. A phase I trial of MK-0731, a kinesin spindle protein (KSP) inhibitor, in patients with solid tumors. Investig. New Drugs 30, 1088–1095 (2012).
Zheng, L. et al. Tyrosine threonine kinase inhibition eliminates lung cancers by augmenting apoptosis and polyploidy. Mol. Cancer Ther. 18, 1775–1786 (2019).
Novais, P., Silva, P. M. A., Amorim, I. & Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics 13, 1011 (2021).
Wengner, A. M. et al. Novel Mps1 Kinase Inhibitors with Potent Antitumor Activity. Mol. Cancer Ther. 15, 583–592 (2016).
Schoffski, P. et al. First-in-man, first-in-class phase I study with the monopolar spindle 1 kinase inhibitor S81694 administered intravenously in adult patients with advanced, metastatic solid tumours. Eur. J. Cancer 169, 135–145 (2022).
Woodward, H. L. et al. Introduction of a methyl group curbs metabolism of Pyrido[3,4- d]pyrimidine Monopolar Spindle 1 (MPS1) inhibitors and enables the discovery of the phase 1 clinical candidate N(2)-(2-Ethoxy-4-(4-methyl-4 H-1,2,4-triazol-3-yl)phenyl)-6-methyl- N(8)-neopentylpyrido[3,4- d]pyrimidine-2,8-diamine (BOS172722). J. Med. Chem. 61, 8226–8240 (2018).
Anderhub, S. J. et al. High proliferation rate and a compromised spindle assembly checkpoint confers sensitivity to the MPS1 inhibitor BOS172722 in triple-negative breast cancers. Mol. Cancer Ther. 18, 1696–1707 (2019).
Batalini, F. et al. Phase 1b clinical trial with alpelisib plus olaparib for patients with advanced triple-negative breast cancer. Clin. Cancer Res. 28, 1493–1499 (2022).
Fennell, D. A. et al. Rucaparib in patients with BAP1-deficient or BRCA1-deficient mesothelioma (MiST1): an open-label, single-arm, phase 2a clinical trial. Lancet Respir. Med. 9, 593–600 (2021).
Lenart, P. et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 17, 304–315 (2007).
Sebastian, M. et al. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. J. Thorac. Oncol. 5, 1060–1067 (2010).
Ellis, P. M. et al. A phase I open-label dose-escalation study of intravenous BI 2536 together with pemetrexed in previously treated patients with non-small-cell lung cancer. Clin. Lung Cancer 14, 19–27 (2013).
Mross, K. et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naive patients with unresectable exocrine adenocarcinoma of the pancreas - a study within the central european society anticancer drug research (CESAR) collaborative network. Br. J. Cancer 107, 280–286 (2012).
Muller-Tidow, C. et al. A randomized, open-label, phase I/II trial to investigate the maximum tolerated dose of the Polo-like kinase inhibitor BI 2536 in elderly patients with refractory/relapsed acute myeloid leukaemia. Br. J. Haematol. 163, 214–222 (2013).
Vose, J. M. et al. The Plk1 inhibitor BI 2536 in patients with refractory or relapsed non-Hodgkin lymphoma: a phase I, open-label, single dose-escalation study. Leuk. Lymphoma 54, 708–713 (2013).
Schoffski, P. et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 48, 179–186 (2012).
Nokihara, H. et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with advanced solid tumors. Investig. New Drugs 34, 66–74 (2016).
Lin, C. C. et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br. J. Cancer 110, 2434–2440 (2014).
de Braud, F. et al. A phase I, dose-escalation study of volasertib combined with nintedanib in advanced solid tumors. Ann. Oncol. 26, 2341–2346 (2015).
Machiels, J. P. et al. A phase I study of volasertib combined with afatinib, in advanced solid tumors. Cancer Chemother. Pharmacol. 76, 843–851 (2015).
Awada, A. et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, plus platinum agents in solid tumors: safety, pharmacokinetics and activity. Investig. New Drugs 33, 611–620 (2015).
Kobayashi, Y. et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with acute myeloid leukemia. Cancer Sci. 106, 1590–1595 (2015).
Stadler, W. M. et al. An open-label, single-arm, phase 2 trial of the Polo-like kinase inhibitor volasertib (BI 6727) in patients with locally advanced or metastatic urothelial cancer. Cancer 120, 976–982 (2014).
Ellis, P. M. et al. A randomized, open-label phase II trial of volasertib as monotherapy and in combination with standard-dose pemetrexed compared with pemetrexed monotherapy in second-line treatment for non-small-cell lung cancer. Clin. Lung Cancer 16, 457–465 (2015).
Dohner, H. et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood 124, 1426–1433 (2014).
Tontsch-Grunt, U. et al. Synergistic activity of BET inhibitor BI 894999 with PLK inhibitor volasertib in AML in vitro and in vivo. Cancer Lett. 421, 112–120 (2018).
Kadia, T. M., Ravandi, F., Cortes, J. & Kantarjian, H. New drugs in acute myeloid leukemia. Ann. Oncol. 27, 770–778 (2016).
Ma, W. W. et al. Phase I study of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin. Cancer Res. 18, 2048–2055 (2012).
Navada, S. C. et al. Rigosertib in combination with azacitidine in patients with myelodysplastic syndromes or acute myeloid leukemia: Results of a phase 1 study. Leuk. Res. 94, 106369 (2020).
Tartaglia, G., Cao, Q., Padron, Z. M. & South, A. P. Impaired wound healing, fibrosis, and cancer: the paradigm of recessive dystrophic epidermolysis bullosa. Int. J. Mol. Sci. 22, 5104 (2021).
Veluswamy, R. et al. KRAS G12C-mutant non-small cell lung cancer: biology, developmental therapeutics, and molecular testing. J. Mol. Diagn. 23, 507–520 (2021).
Garcia-Manero, G. et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol. 17, 496–508 (2016).
O’Neil, B. H. et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann. Oncol. 26, 1923–1929 (2015).
Navada, S. C. et al. A phase 1/2 study of rigosertib in patients with myelodysplastic syndromes (MDS) and MDS progressed to acute myeloid leukemia. Leuk. Res. 64, 10–16 (2018).
Gilmartin, A. G. et al. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. 69, 6969–6977 (2009).
Olmos, D. et al. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin. Cancer Res. 17, 3420–3430 (2011).
Nie, Z. et al. Discovery of TAK-960: an orally available small molecule inhibitor of polo-like kinase 1 (PLK1). Bioorg. Med. Chem. Lett. 23, 3662–3666 (2013).
Hikichi, Y. et al. TAK-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol. Cancer Ther. 11, 700–709 (2012).
Valsasina, B. et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol. Cancer Ther. 11, 1006–1016 (2012).
Weiss, G. J. et al. Phase I dose escalation study of NMS-1286937, an orally available Polo-Like Kinase 1 inhibitor, in patients with advanced or metastatic solid tumors. Investig. New Drugs 36, 85–95 (2018).
Zeidan, A. M. et al. A phase Ib study of onvansertib, a novel oral PLK1 inhibitor, in combination therapy for patients with relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 26, 6132–6140 (2020).
Garcia, I. A., Garro, C., Fernandez, E. & Soria, G. Therapeutic opportunities for PLK1 inhibitors: Spotlight on BRCA1-deficiency and triple negative breast cancers. Mutat. Res. 821, 111693 (2020).
Patterson, J. C. et al. Plk1 inhibitors and abiraterone synergistically disrupt mitosis and kill cancer cells of disparate origin independently of androgen receptor signaling. Cancer Res. 83, 219–238 (2023).
Wang, D. et al. A novel PLK1 inhibitor onvansertib effectively sensitizes MYC-driven medulloblastoma to radiotherapy. Neuro. Oncol. 24, 414–426 (2022).
El Dika, I. et al. An open-label, multicenter, phase I, dose escalation study with phase ii expansion cohort to determine the safety, pharmacokinetics, and preliminary antitumor activity of intravenous TKM-080301 in subjects with advanced hepatocellular carcinoma. Oncologist 24, 747–e218 (2019).
Zhou, L. Y. et al. Current RNA-based therapeutics in clinical trials. Curr. Gene Ther. 19, 172–196 (2019).
Uchino, K., Ochiya, T. & Takeshita, F. RNAi therapeutics and applications of microRNAs in cancer treatment. Jpn. J. Clin. Oncol. 43, 596–607 (2013).
Yim, H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anticancer Drugs 24, 999–1006 (2013).
Kim, H. J., Kim, A., Miyata, K. & Kataoka, K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv Rev 104, 61–77 (2016).
Ramos Perez, J. & Montalban-Bravo, G. Emerging drugs for the treatment of chronic myelomonocytic leukemia. Expert Opin. Emerg. Drugs 25, 515–529 (2020).
Kawakami, M. et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc. Natl Acad. Sci. 115, 1913–1918 (2018).
Fu, S. et al. Multicenter Phase II Trial of the WEE1 inhibitor adavosertib in refractory solid tumors harboring CCNE1 amplification. J. Clin. Oncol. 41, 1725–1734 (2023).
Vakili-Samiani, S. et al. Targeting Wee1 kinase as a therapeutic approach in hematological malignancies. DNA Repair 107, 103203 (2021).
Elbaek, C. R., Petrosius, V. & Sorensen, C. S. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat. Res. 819-820, 111694 (2020).
Chandrasekaran, A. & Elias, K. M. Synthetic lethality in ovarian vancer. Mol. Cancer Ther. 20, 2117–2128 (2021).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (32070715, 32270778, 82173029, and 82372655); and the Natural Science Foundation of Chongqing (CSTB2022NSCQ-MSX0612 and CSTB2022NSCQ-MSX0611). Our intention is to summarize the state of art. However, due to space limitations, we would like to apologize to authors whose works are not cited here. Their contributions should not be considered less important than those that are cited.
Author information
Authors and Affiliations
Contributions
R.H., S.W., V.K. designed the review; R.H. wrote original draft preparation; R.H. and S.H. drafted original figures preparation; R.H., S.H., and S.N. drafted original tables preparation; R.H., S.W., V.K. revised the manuscript. Corresponding authors S.W. and V.K. provided financial support and supervision. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hosea, R., Hillary, S., Naqvi, S. et al. The two sides of chromosomal instability: drivers and brakes in cancer. Sig Transduct Target Ther 9, 75 (2024). https://doi.org/10.1038/s41392-024-01767-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01767-7
- Springer Nature Limited
This article is cited by
-
Spatial multi-omics: deciphering technological landscape of integration of multi-omics and its applications
Journal of Hematology & Oncology (2024)