
Abstract
Receptor tyrosine kinases (RTKs), a category of transmembrane receptors, have gained significant clinical attention in oncology due to their central role in cancer pathogenesis. Genetic alterations, including mutations, amplifications, and overexpression of certain RTKs, are critical in creating environments conducive to tumor development. Following their discovery, extensive research has revealed how RTK dysregulation contributes to oncogenesis, with many cancer subtypes showing dependency on aberrant RTK signaling for their proliferation, survival and progression. These findings paved the way for targeted therapies that aim to inhibit crucial biological pathways in cancer. As a result, RTKs have emerged as primary targets in anticancer therapeutic development. Over the past two decades, this has led to the synthesis and clinical validation of numerous small molecule tyrosine kinase inhibitors (TKIs), now effectively utilized in treating various cancer types. In this manuscript we aim to provide a comprehensive understanding of the RTKs in the context of cancer. We explored the various alterations and overexpression of specific receptors across different malignancies, with special attention dedicated to the examination of current RTK inhibitors, highlighting their role as potential targeted therapies. By integrating the latest research findings and clinical evidence, we seek to elucidate the pivotal role of RTKs in cancer biology and the therapeutic efficacy of RTK inhibition with promising treatment outcomes.
Similar content being viewed by others

Introduction
Beginning in the early 1950s, notable progress was achieved in the field of cellular biology through the discovery of receptor tyrosine kinases (RTKs). Although identified as the receptors for insulin and epidermal growth factor (EGF), RTKs subsequently became the primary focus for understanding cellular signaling systems.1,2 During this time, nerve growth factor and EGF were discovered and found to have significant impacts on the development of neurons and the proliferation of cells, both in living organisms and in laboratory settings.
By the 1960s, extensive research on insulin had deepened understanding of the interactions of its receptor. Scientists performed thorough examinations of insulin’s interaction with its receptor on cells or solubilized receptor preparations utilizing radiolabeled insulin. These findings confirmed the ligand-binding properties and introduced the notion of negative interaction in insulin binding. The understanding of this concept was further intensified during the 1970s. The researchers mapped the precise locations on the surfaces of cells where EGF binds and made a connection between the phosphorylation of proteins on tyrosine residues and the signaling within cells, as well as the potential processes that may lead to the development of cancer.3 During this decade, key features of receptors were identified, such as ligand-dependent down-regulation and desensitization via internalization and degradation, observed in both the insulin receptor and EGFR.4 By the early 1980s, it was well-recognized that certain receptors function as ligand-activated protein tyrosine kinases. These discoveries highlighted the role of RTKs in regulating cellular development, vital physiological functions, and cancer development, significantly enhancing our understanding of cellular mechanisms.5,6
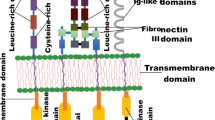
The RTK family encompasses a diverse array of cell surface receptors that respond to growth factors, hormones, and cytokines, mediating a wide range of fundamental cellular and metabolic signaling pathways.7 The common denominators of this receptor family consist of the conserved structural domains, namely, the extracellular ligand-binding domain, the transmembrane helix, and the intracellular tyrosine kinase domain. The extracellular domain of RTKs is a dynamic region that governs ligand binding, receptor activation, and subsequent signaling cascades, making it a key determinant of RTK function and cellular responses.8 Ligand specificity and binding affinity are crucial properties in influencing downstream signaling events.9 Specifically, it consists of distinct structural elements, such as immunoglobulin-like domains, fibronectin type III-like repeats, EGF-like domains, and cysteine-rich regions, which contribute to the classification of RTKs into different families based on their structural extracellular characteristics.10 As such, the number, combination, and arrangement of these domains vary significantly among different RTK families, conferring unique ligand-binding capabilities and regulatory properties to each receptor.11 First, the immunoglobulin-like domains (Ig-like) typically exhibit a sandwich-like structure composed of two β-sheets stabilized by a disulfide bond.12 Named based on their structural similarity to immunoglobulin molecules, they play a crucial role in ligand biding and dimerization. Next, the cysteine-rich domains specific to some classes of RTKs, define loop-rich compact structures that improve the conformational stability of the domain, at the same time influencing the ligand specificity and binding affinity. The fibronectin type III (FN3) repeat is typically comprised of about 90 amino acids and adopts a compact domain structure known for its β-sandwich configuration, also influencing the specific interaction capabilities of FN3-containing RTKs. Lastly, EGF-like repeats are another significant structural motif found in a variety RTKs.13 Named after their identification in the EGF, these repeats play an important role in ligand binding and receptor activation, influencing the signaling pathways in the context of RTKs. The intracellular helix within the kinase domain of receptor tyrosine kinases is a notable structural element. It forms an α-helical structure from a sequence of amino acids and contributes to the overall function and regulation of the kinase.14 Positioned within the kinase domain, this helix aids in maintaining the enzyme’s conformation and is involved in adenosine triphosphate (ATP) binding. Its interactions with other parts of the kinase domain, such as the activation loop, are part of the mechanism controlling the kinase’s activity.15 When the kinases are activated, the intracellular helix often undergoes a shift in position, aligning the required residues allowing catalytic activity. This helix also influences substrate access to the active site and might have a role in interactions with regulatory molecules. The intracellular domain of RTKs is the cornerstone in cellular signal transduction. At the heart of this domain lies the tyrosine kinase domain, an enzymatic center that catalyzes the phosphorylation of specific tyrosine residues on target proteins via ATP.10 The intracellular domain also encompasses regulatory regions, such as the juxta-membrane domains, which can inhibit kinase activity in the absence of a ligand, and C-terminal tails that often contain multiple tyrosine residues.16 These residues, upon phosphorylation, serve as docking sites for adaptor and effector proteins, crucial for signal propagation. The process begins with ligand binding to the RTK’s extracellular domain, triggering receptor dimerization and subsequent autophosphorylation. This autophosphorylation of specific tyrosine residues within the intracellular domain creates binding sites for proteins with Src homology 2 (SH2) or phosphotyrosine binding (PTB) domains.17
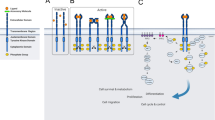
RTKs are grouped into 20 families, based on their amino acid sequence similarities and structural characteristics in their extracellular domains, leading to members within a family binding to similar or same ligands.18 Fig. 1 provides a visual representation of the different domains found in RTKs, highlighting the structural variations that contribute to their diverse roles in cellular signaling.

Structure of the 20 Receptor Tyrosine Kinase Classes. The RTKs structure differs from one receptor to another, with several similarities and differences mostly at the extracellular and intracytoplasmic domains as depicted from left to right in all 20 RTKs classes. Images created with BioRender.com
The activation of RTKs is a multifaceted process, influenced by a delicate balance between external ligand availability and intrinsic receptor conformational dynamics. At the molecular level, RTK activation is not a uniform event but rather a confluence of diverse regulatory mechanisms that reflect the complex biological systems they modulate. The process initiates with the extracellular domain of RTKs, which, upon binding to specific ligands such as growth factors, undergoes structural alterations, a prerequisite for the trans-autophosphorylation of tyrosine residues within the intracellular kinase domains (Fig. 2).

Activation and Intracellular Signaling Mechanisms of Receptor Tyrosine Kinase. Depicted from left to right, the Ligands are binding to the monomer RTKs and trigger the dimerization with a cross-phosphorylation of the protein kinase domains. Further secondary transphosphorylation of the tyrosine kinase domains, juxtamembrane and c-terminal regions occur in the RTKs, which will further create the proper conditions for the recruitment of the intracellular substrates that will further lead to the activation of the key proteins in the downstream signaling pathways. Images created with BioRender.com
As previously described, the specificity of ligand binding is crucially determined by the cumulative properties of the extracellular domain, ensuring that the signal is initiated only in response to the appropriate extracellular cues (i.e., ligand).19 Next, receptor dimerization consists of the pairing of RTK molecules because of ligand binding, facilitating the cross-phosphorylation of tyrosine residues in their intracellular domains.20 Such phosphorylation is essential for activating the kinase function of the receptors, not only by activating the RTKs per se but also by generating binding sites for various intracellular signaling proteins.21 Trans-autophosphorylation, where the kinase domains of the dimerized receptors become strategically aligned, facilitates each kinase domain to phosphorylate specific tyrosine residues on its partner in the dimer.22 This trans-autophosphorylation is a key event, as it activates the kinase domains, marking a transition from a dormant state to an active one. Subsequently, the phosphorylated tyrosines on the RTKs transform into critical docking sites for various intracellular signaling proteins. These proteins, often equipped with SH2 or PTB domains, have a high affinity for the phosphorylated tyrosines.23,24 Their binding to these activated sites on the RTKs is not just a mechanical linkage; it’s the initiation of a complex network of downstream signaling pathways.
Classification of RTK protein families
RTK subfamilies and their members, along with the corresponding discovered ligand, emphasizing on the specific functions and roles of the receptors in cell development, growth and proliferation, metabolism modulation, cell cycle, epithelial to mesenchymal transition and many other physiological and physiopathological involvement are highlighted in Table 1.
Physiological roles of RTKs
Key signaling mechanisms of RTKs
Following the recruitment of signaling proteins to the phosphorylated and activated RTKs, a series of intricate signal transduction cascades is initiated, each targeting specific cellular functions. Prominently, the Mitogen-Activated Protein Kinase (MAPK) pathway is activated, playing a central role in regulating gene expression and orchestrating cellular processes like proliferation and differentiation. Concurrently, the Phosphoinositide 3-Kinase/Protein Kinase B (PI3K/Akt) pathway is mobilized, which is crucial for controlling various aspects of cell survival, growth, and metabolism.25 Another key pathway activated is the Phospholipase C gamma (PLCγ) pathway, influential in modulating calcium signaling and cytoskeletal reorganizations.26
Termination of RTK signaling
To maintain cellular homeostasis and prevent overactivation of these pathways, a set of regulatory mechanisms came into play. These include dephosphorylation of the RTKs by phosphatases, internalization and degradation of the receptors, and feedback inhibition from downstream signaling components.27,28 This regulatory phase is essential for ensuring that the signaling is transient and contextually appropriate, providing a fail-safe against uncontrolled or prolonged activation that could lead to pathological conditions. However, in the context of cancer, dysregulation of these regulatory pathways is common. Altered plasma membrane domains and endocytic trafficking in tumor cells lead to aberrant RTK clustering and signaling29 (Fig. 3).

Termination of RTKs signaling—endocytosis of signaling and Endocytic Trafficking. The red arrows in the figure illustrate variations in the endocytic rate and pathway selection, which are determinant factors for the modulation of RTK surface expression and downstream signaling, potentially driving oncogenic processes. Images created with BioRender.com
Role in cellular growth and proliferation
RTKs are instrumental in regulating cellular growth and proliferation. Upon activation by ligand binding, they initiate a cascade of intracellular signaling, predominantly through the Ras/MAPK pathway, leading to the transcription of genes that drive cell cycle progression. This signaling mechanism is crucial for the controlled growth of cells, ensuring that proliferation occurs in response to appropriate external stimuli. Dysregulation of this process, often seen in the overactivation of RTKs, is a hallmark of various cancers, underlining the critical role of RTK signaling in maintaining normal cell growth and division.30,31,32,33,34,35
RTKs in cellular differentiation and development
RTKs are vital in guiding cellular differentiation and development. They are key players in embryonic development, influencing cell fate decisions and tissue formation. For instance, Fibroblast Growth Factor Receptors (FGFRs) have a well-established role in limb development, while Epidermal Growth Factor Receptors (EGFRs) are crucial in neural development. Through binding with specific ligands, RTKs activate signaling pathways that lead to the differentiation of cells into specialized types, crucial for the proper formation and function of diverse tissues and organs.36
RTKs in metabolism regulation
The metabolic profiles of tumor cells are different from normal cells; thus, tumor cells tend to rewire the metabolism to support tumor progression due to the high metabolic demands.37 Resistance to therapy can occur also due to the metabolic adaptations suggesting that the cellular metabolism can be crucial in tumorigenesis.38 RTKs activation can modulate different metabolic pathways and RTKs driven metabolic reprogramming could lead to different metabolic vulnerabilities that could be targeted, for example, the lactate production can fuel the TCA cycle generating energy in FGFR aberrant cancer cells, serine synthesis can be used for nucleotide biosynthesis and redox homeostasis of EGFR constitutively activated tumors. Jin et al., showed that the RTK involvement in metabolism can induce metabolic reprogramming and provide distinct metabolic vulnerabilities that can be exploited.39 The interplay between RTKs and other metabolic pathways underscores their importance in maintaining metabolic balance within the body.
RTKs in cell survival and apoptosis
The balance between cell survival and programmed cell death (apoptosis) is tightly regulated by RTK signaling. By activating pathways like PI3K/Akt, RTKs promote cell survival and inhibit apoptotic pathways. This protective role is essential for normal cellular function and response to stress. However, aberrant activation of these pathways can lead to uncontrolled cell survival, contributing to the development of cancer. RTKs, therefore, play a dual role in maintaining cellular health, promoting survival under normal conditions, and facilitating apoptosis when cells are damaged or no longer needed. The pathways and kinetics of RTK endocytic trafficking, molecular mechanisms underlying sorting processes, and examples of deviations from the standard trafficking itinerary in the RTK family are discussed in the literature.40 Additionally, overexpression of RTK proteins or functional alterations caused by mutations in the corresponding genes or abnormal stimulation by autocrine growth factor loops contribute to constitutive RTK signaling, resulting in alterations in the physiological activities of cells.41
The cystine-rich domains of variable length are commonly found in RTKs, and multiple RTKs contain Ig domains, with the ectodomain of certain families consisting solely of Ig domains. The role of alternative splicing of RTKs in tumor progression and response to therapies, with a special focus on major RTKs that control proliferation, survival, and angiogenesis, has been discussed in the literature.
RTK dysregulation and cancer connections
The regulation of protein tyrosine phosphatase (PTP) activity plays a significant role in modulating RTK signaling, by acting concurrently.27 The ligand-induced inhibition of PTPs, which conventionally serve to dephosphorylate and deactivate RTKs, results in the prolonged activation of the receptor, thereby amplifying downstream signaling pathways. Moreover, certain RTKs exhibit ligand-independent activation, primarily driven by genetic mutations or overexpression.19 This constitutive activation can lead to aberrant signaling pathways, often implicated in pathological conditions such as oncogenesis.42
Cell communication with the microenvironment involves different paths and membrane receptors that are triggered by different ligands and modulate important pathways. Key biological processes are regulated by ligand-receptor binding, such as cell proliferation, differentiation, migration, cell death mechanisms and others. Tumor cells grow faster than normal cells and some stimuli can be the excess of growth factors in the microenvironment, increased number of receptors for the ligands, or there may be mutations and rearrangements in the chromosomes resulting into different protein structure.43,44 All RTKs consist of an extracellular region with the ligand-binding domain that is linked to the intracellular protein kinase through a transmembrane domain.45,46 RTKs are involved in multiple biological pathways such as differentiation, migration, survival, or apoptosis, thus any abnormality in the RTKs may induce downstream changes and dysregulate biological processes. RTKs can be dysregulated via five main mechanisms: overexpression, TK (tyrosine kinase) domain duplication, autocrine and paracrine activation, genomic rearrangements, and gain/loss of function mutations 17 (Fig. 4).

RTKs dysregulation mechanisms. The dysregulation of RTKs may occur by a gain-of -function mutation, an amplification, chromosomal rearrangements, TK domain duplication or by an autocrine or paracrine activation (left to right). The dysregulations are generating abnormal activation of the RTKs which will be translated into enhanced proliferation, differentiation or angiogenesis, same as into a dysregulated cell cycle and metabolism. Images created with BioRender.com
Due to their importance in cell growth signaling, RTKs play a crucial role in the development and progression of various malignancies. Their significance stems from their ability to trigger the intracellular signaling cascades and influence the key cellular processes such as migration, differentiation, proliferation, and survival. In cancer, many tumor cells exhibit oncogenic addiction to RTKs, and their survival is supported by the RTKs activation.47,48,49 The addiction to oncogenes has been described in 2002, highlighting the fact that tumor cells tend to sustain their survival depending on specific oncoproteins, overcoming the genetic lesions that occur in tumor cells due to their highly proliferative state.50 Some mutations in the RTKs are considered “Driver mutations” and promote a fast cell growth and sustain survival, together with the gene amplification or chromosomal rearrangements.51,52 The RTK activity is well controlled in normal cells, however, the receptors undergo structural changes leading to their overactivation due to a series of factors such as mutations, overexpression, or autocrine/paracrine stimulation. The structural changes or their increased density om the cell membrane increases their affinity to the ligands and overstimulates the downstream signaling.53,54,55
Pathological signaling outcomes arise when RTKs undergo abnormal activation. The oncogenic activation through different mechanisms generates an abnormal and overstimulated signaling via the receptor, increasing the proliferation and survival of tumor cells. RTKs abnormal activation is one of the cancer characteristics which makes RTKs potent targets for therapeutic intervention with specific inhibitors.54 RTK inhibitors can modulate different immunosuppressive cell such as tumor-associated macrophages, regulatory T cells and myeloid-derived suppressor cells that are localized in the tumor microenvironment, thus the immunosuppressive cells can become useful in the combat against tumor cells.56,57
EGFRs, VEGFRs, PDGFRs, FGFRs, ROR1, ROR2, and other RTKs accumulate a series of modifications which trigger their activation and lead to a metabolic reorganization in tumor cells thus increasing their tumorigenicity.58,59 RTKs overexpression was spotted in solid and hematological malignancies, contributing to the enhanced cell proliferation, differentiation, migration, and cell death regulation.60
Further discussions will point out the involvement of RTK subclasses in biological processes and cancer development, summarizing their role in different malignancies, the mechanism of action and the mutations that may occur in the genes encoding RTK proteins and highlighting their oncologic roles.
Deregulation of EGFR in cancer
EGFRs, noted for their high affinity for epidermal growth factors, play a crucial role in cell proliferation and survival by activating Ras/MAPK, PI3K/Akt, and JAK/STAT pathways. Their modulation of critical biological processes makes them potential targets for cancer therapy.61,62
Discovered in 1960s, EGF was described as pro differentiation and growth stimulatory protein when binding to its receptor.63 The purified EGFR had 170 kDa and when binding to the ligand induces receptor clustering. Molecular cloning of the EGFR revealed the similarity with v-erbB oncogene, furthermore, three related members of the receptor family were discovered ErbB2, ErbB3, and Erb4.64,65,66
EGFR binds to EGF and Tumor Necrosis Factor-alpha ligands and control cell growth, differentiation and proliferation. The EGFR family consists of four members: ErbB-1–HER1/epidermal growth factor receptor; ErbB-2–HER2; ErbB-3–HER3, and ErbB-4–HER4.67,68 EGFR mutations and upregulation drive cancer progression, highlighting EGFR as a promising therapeutic target.69
The ErbB family (EGFR, ErbB-3, and HER2) drives cancer proliferation and survival by activating Ras/MAPK, PI3K/Akt, and JAK/STAT pathways. Notably, HER2, prominent in breast cancer (BC), represents an important target for targeted therapies. Unlike other family members, HER2 lacks a known ligand, but is known that its dimerization activates the receptor and triggers the downstream pathways. HER2 overexpression in BC makes it a good target for therapies with Herceptin and other molecules.70
ErbB3, or HER3, is another member of the ErbB receptor family. While it has a lesser kinase activity, it forms heterodimers with other ErbB members, particularly HER2, to activate signaling pathways. ErbB3 is involved in activating the PI3K/Akt pathway, playing a significant role in cancer development and progression. Its role in drug resistance and cancer progression has made it a target of interest in oncological research.
ErbB4, or HER4, is involved in various developmental and physiological processes. Like other ErbB receptors, it activates the Ras/MAPK, PI3K/Akt, and JAK/STAT pathways. ErbB4’s role extends beyond oncology into neurological development, making it a subject of interest in both cancer therapy and neurobiology.
The EGFR family of receptor tyrosine kinases, consisting of four members (ErbB-1/HER1, ErbB-2/HER2, ErbB-3/HER3, and ErbB-4/HER4), plays crucial roles in cell growth, differentiation, and tumor migration regulation.71 The first discovered ErbB receptor is EGFR for which was first described the relationship between overexpression and cancer development.72 Alterations in ErbB family members were found to be correlated with the progression of numerous cancers such as ovarian, esophageal, laryngeal, breast, lung, prostate cancer, and melanoma.73,74,75,76,77,78,79
EGFR, an early oncogene, is a key target in clinical oncology, frequently activated by mutations or overexpression across human cancers, notably in pancreatic adenocarcinoma with poor prognosis, and lung and colon cancers with detected mutations.80,81,82,83,84,85 Table 2 summarizes the information regarding mutations and their role in different diseases.
In non-small cell lung cancer (NSCLC), T790M is a very common point mutation was detected in 60% of patients with EGFR TKI resistance.86,87 C797S mutations were described as responsible for acquired resistance to third generation EGFR-TKI and was found in 40% of patients with mutant NSCLC with T790M mutation. Several activation mutations in EGFR gene are well known at the diagnostic (Exon 19 deletion, L858R, L861Q, S781I, G719A, G719C, G796D, L718Q, L844V, and T790M).87,88
EGFR mutations are frequently discovered in NSCLC, according to Fu et al. the four generations of EGFR-TKIs are efficient if different mutation status of NSCLC cells.89 Tumors with single mutation (Ex19del/L858R; T790M and C797S) can be targeted by all four generations of EGFR-TKIs; Double mutant cells are sensitive to all EGFR-TKIs except 2nd generation while cell with triple mutant status are sensitive only to the 4th generation of EGFR-TKIs.
According to Liu et al., EGFR gene mutations frequency was 2.8% for all tumor samples and 2.4% for all samples collected from patients including 32 types of tumors.90 The most common tumors that had EGFR mutations were glioblastoma multiforme (GBM) with 26.8%, lung adenocarcinoma (LUAD) with 14.4%, diffuse large B-cell lymphoma with 8.3% and skin cutaneous melanoma (SKCM) with 6.5%; on the other hand, patients with uveal melanoma, thyroid carcinoma, kidney chromophobe cell carcinoma or thymoma showed almost undetectable EGFR mutations.90 The 289aa in the Furin-like domain of EGFR was the most frequently mutated position, detected in 27 samples—A289D, A289N, A289I, A289T, A289V, and A289Rfs*9. These mutations were almost exclusively present in GBM samples, and none of these mutations are yet known to be potential targets.90 Mutations in the GF_recep-IV domain were detected in GMB and esophageal squamous cell carcinoma (ESCC) (G598V and G598E), both mutations being related to ligand-receptor binding disfunctions and are treated as oncogenic mutations. In LUAD most mutations were detected in the Pkinase_Tyr domain, in positions 858aa (L858R) and 747-750aa (E746_A750del, L747_E749del and L747_T751del).90
EGFR mutations are divided into seven levels, depending on the clinical targeted therapy implication, with mutations included in level 1 and R1 indicated as targetable, according to Food and Drug Administration.91,92 All level 1 mutations are detected in NSCLC – 28 mutations in LUAD and 2 in lung squamous cell carcinoma and concentrated in exons 19-21 - L858R, L861Q, G719A, S768I, L833F, E796_A750del, L747_E749del, E709_T710delinsD, L747_T751del, and T751_E758del.90
Lung cancer, head and neck cancer and esophagus carcinoma have several commonalities in terms of EGFR with increased expression of EGFR, high frequency of EGFR amplification and low indel mutations. Also, targeted therapy in the case of these three cancers shows promising efficacy. The correlation between EGFR abnormalities and treatment benefits underlines the importance of molecular profiling and the detection of biomarkers for a better selection of treatment.90
Details regarding the EGFR receptors, the genes that encode their proteins and the disease in which the EGFR receptors are involved, with the mutations that are likely oncogenic or not are presented in Table 2.
Deregulation of FGFRs in cancer
Fibroblast growth factor receptors (FGFRs) are integral to developmental processes, with a specific binding affinity to fibroblast growth factors. They activate the Ras/MAPK, PI3K/Akt, and PLCγ pathways, affecting a range of cellular activities including cell division, growth, migration, and angiogenesis. Mutations or dysregulations in FGFRs are associated with various developmental disorders and cancers.43,93
The activation of the signaling pathway of FGFRs is mainly triggered by the binging of fibroblast growth factors (FGFs) and the subsequent dimerization of the receptors, which leads to intracellular kinase trans autophosphorylation.94 Moreover, FGFRs can be triggered in a manner that does not require a specific ligand, such as when the FGFRs gene fuses with other genes that are constantly expressed due to chromosome translocation.95 In a comprehensive analysis using NGS across a diverse range of tumor samples, FGFR mutations were found to be a common occurrence in cancers characterized by FGFR gene abnormalities. For instance, FGFR1 amplifications were notably prevalent in breast and lung cancer, while FGFR2 mutations were frequently in endometrial and gastric cancers (GCs). Also, FGFR mutations, including S249C hotspot mutations, were common in bladder cancer samples.96
FGFR comprise four genes and include seven different receptors that are differentially activated by one of the fibroblast growth factor ligands. Four transmembrane receptors are identified (FGFR1-4) and when binding their ligands, the receptors dimerize and activate downstream pathways that regulate proliferation, survival, angiogenesis and differentiation.96,97,98
Aberrations in FGFR1-4 genes include single-nucleotide variants (SNVs), gene fusions and rearrangements, or copy number amplifications. Due to the increased frequency of alterations in FGFR genes, in solid and hematological malignancies, a molecular diagnostic for accurate detection of these aberrations may indicate which therapy is better.98,99,100,101
SNVs can induce constitutive activation of the FGFRs increasing their affinity to ligands or over activate it. FGFR1 SNVs are rare, with N546K and K656E as the most common mutations identified, and with an unclear consequence, S125L mutation was identified in gallbladder and BC.102,103 The majority of SNVs were identified in FGFR2, which are related to NSCLC, GC and endometrial cancer. The transmembrane mutations Y375C and C382Y, plus the extracellular domain mutations S252W, W290C and P253R are more frequent than the kinase domain mutations N549H/K and K659E, according to Helsten et al.96 The most frequent SNVs in FGFR3 are R248C and S249C in the extracellular domain and G370C and Y373C in the transmembrane domain, with reports in urothelial carcinomas.104,105 Last, but not least, SNVs in FGDR4 are notable in rhabdomyosarcoma, with V550E and N535K contributing to autophosphorylation of the receptor, while Y367C was identified in MDA-MB453 BC cell line.106,107
In FGFR3, K650 and G697 were identified as hotspots for mutations in cancers. The most frequent amino acid changes at K650 were E and M, and N, Q, and T were the least frequent observed. Also, amino acid replacement in N540 position was detected for K, S, D, and H. One frequent mutation specific for FGFR3 was G697C replacement.108
FGFR gene fusions can appear due chromosomal rearrangements or translocations, increasing the receptor dimerization or dysregulating the expression of FGFR. Helsten et al, identified the FGFR2/FGFR3 and TAAC3 fusion as one with high frequency.96 In triple negative breast cancer (TNBC), FGFR2 fusion partners AFF3, CASP7, and CCDC6 aberrantly activate the gene, while in lung cancer other two fusions were detected (FGFR3-TACC3; FGFR2-CIT). Fusion between FGFR3 and TACC3 was also identified in glioblastoma, cervical SC and urothelial carcinoma. Type I FGFR fusions were detected in patients with AML, acute lymphoblastic leukemia (ALL) and peripheral T cell lymphoma (PTCL).109 Therefore, cells harboring these FGFR fusions develop oncogenic properties.
The most frequent genomic alteration of the FGFRs is gene amplification, FGFR1 and FGFR4 having the highest frequencies. FGFR1 amplification is common in HR+ cancers, HER2+ cancers and TNBC, and is associated with poor prognosis.99
Rarely, mutations can occur in residues that are not present in common isoforms, as observed in most cancers. For example, in head and neck squamous cell carcinoma (SCC), a rare mutation, P11362-21 G33R, was detected in an uncommon isoform of FGFR1, while mutations in the common isoforms were not found.110 In the case of FGFR2, mutations in non-common isoforms were identified in various cancers: bladder cancer (P21802-20 M71T), CRC (P21802-20 R88H, R95Q, D221N), lymphoma (P21802-20 M71T), and lung adenocarcinoma (p.R496T).106,111,112 FGFR3 presented a unique mutation in an isoform different from the common ones, specifically P22607-4 P688S in BC.112 For FGFR4, no mutations were detected in isoforms other than the common ones.112
A synthesis of FGFR receptors is presented in Table 3, providing details regarding the genes that encode FGFR proteins, mutations that may be or not be oncogenic and the disease where the FGFR mutations occur. Moreover, different mechanisms of action are presented in the below table, highlighting how diverse the FGFR activity might be in different human oncological diseases.
Deregulation of IR and IGF1R in cancer
Insulin receptors (IR) and insulin-like growth factor 1 receptor (IGF1R) regulate metabolic processes, particularly glucose homeostasis, through modulation of PI3K/Akt and Ras/MAPK pathways, impacting metabolism, cell growth, differentiation, and survival, with implications extending to cancer research.62
Insulin and IGF-1 influence biological mechanisms via the IR and IGF1R. IR and IGFR1 are members of the insulin receptor family, among orphan insulin receptor-related receptor and are responsible for the maintenance of glucose homeostasis, as well as glucose uptake and its conversion into fat, thus modulating the insulin secretion and other metabolic processes.113
IR and IGFR1 play crucial roles in cancer procession and development, overactivation of these receptors is common is cancer cells, with a particular overexpression in dedifferentiated cell, leading to resistance to different anti-tumor therapies.114 Strong evidence suggests the link between type 2 diabetes mellitus, obesity and the development and progression of tumors,115,116,117 thus, even if the IR pathway gained attention for the antidiabetic therapies, nowadays it represents a target for antitumor therapies.
IRs, upon ligand binding, undergo autophosphorylation, activating growth factor receptor-bound protein 2 (GRB2) and the p85 subunit of PI3K. This leads to Akt activation, regulating metabolic enzymes and influencing cell growth, proliferation, and survival, critical processes in tumor development.118
Multiple studies have demonstrated the implication of insulin receptor (IR) pathway in cancer development and progression. Aberrant overactivation of IR pathway is common in cancer cells, mostly in stem-like cells and could be related to drug resistance. Insulin and Insulin-like growth factors I and II bind to IR and IGF-IR, two receptors with high structural similarities that are responsible for glucose metabolism, cell growth and proliferation. As presented in Table 4, in cancer, this pathway is altered and may serve as targets for cancer therapy.119,120,121,122
According to Ullrich et al., due to the high degree of similarity between IR and IGF-IR, hybrid receptors (HRs) can form when an IR alpha-beta hemi receptor combines with an IGF-IR alpha-beta hemi receptor.123 These hybrid receptors are expressed in all tissues along with IR and IGF-IR. The three possible receptors bind the same ligands—insulin, IGF-1, and IGF-2, with different affinities. When ligands bind to the receptors, the receptors become autophosphorylated on their TYR residues and activated intracellular signaling pathways. According to Hers et al., the downstream signaling activates the PI3K and regulates Akt via PDK1, mediating metabolic effects, cell growth, proliferation, and cell survival .118 In adult tissues, IR is responsible for the metabolic functions and IGF-IR are mainly regulators of growth processes.124 Both receptors can overlap in their biological effects in cancer cells, thus latest therapeutical concepts maintain that targeting both IR and IGF-IR would be a better approach than targeting IGF-IR alone.122,125,126,127
In the IGF1R, three cancer associated mutations were described by Craddock and Miller, two of them in the C-terminus (DS1278 and A1347V) and one in the C-terminal lobe of its catalytic domain (M1255I), mutations that disrupt the downstream signaling cascade.128
Table 4 provides a synthetic presentation of Insulin receptors and the oncological diseases in which they are involved, with details regarding the genes that encode the proteins, and different mutations that may act as oncogenic mutations and a description of the various mechanisms that can be disturbed by the mutations in Insulin Receptors.
Deregulation of PDGFRs in cancer
PDGFRs, responding to platelet-derived growth factors, are involved in regulating cell proliferation and migration. They activate pathways like Ras/MAPK, PI3K/Akt, and PLCγ, influencing cell growth, angiogenesis, and wound healing. Dysregulation of PDGFR signaling is implicated in various pathologies, including cancers and fibrotic disorders.129,130 The delicate balance maintained by PDGFR can be disrupted by changes in the receptor or its ligands, or by the crosstalk between the pathways. Dysregulation of PDGFR signaling implies a wide spectrum of disorders, even cancers. In oncological disorders, aberrant PDGFR activation can fuel uncontrolled proliferation and migration. The dysregulation of PDGFR signaling underlies multiple pathological conditions, underscoring the therapeutical potential of the regimens in oncological pathologies and not limited to them.131,132,133
The PDGFR family consists of PDGF alpha, PDGF beta, SCF receptor (Kit), CSF-1 receptor (Fms) and Flt3. PDGF alpha and beta bind homodimers of PDGF-A/B/C and D polypeptides and the heterodimer PDGF-AB. CSF-I bind the IL-34 and CSF-1, while SCF and Flt3 receptors bind one ligand each. All ligands for PDGFR family are dimeric molecules.134
c-KIT, essential for hematopoietic stem cells, melanocytes, and germ cells, activates multiple pathways, including Ras/MAPK, PI3K/Akt, and PLCγ. Its role in cell survival and proliferation, particularly in hematopoietic and melanogenic cells, makes it significant in various cancers. c-KIT mutations are targeted by specific kinase inhibitors in cancer therapy.135,136
PDGFR alpha mutations occur within the autoinhibitory juxta-membrane region (exon 12 mutations) and the kinase domain (exons 14 and 18). V561D mutation occurs in exon 12, while D842V and D842Y in exon 18. All these mutations disrupt the signaling and act as oncogenic mutations in gastrointestinal stromal tumors (GIST).137 Among all the PDGFRA mutations previously discussed, D842V is one of the most widely investigated and clinically significant mutations.138 It results in a gain-of-function in PDGFRA, which enables constitutive kinase activation without the need for ligand binding.139 This ongoing activation stimulates downstream signaling pathways that support cell survival and proliferation, including PI3K/Akt/mTOR and MAPK.140 Moreover, it is linked to a specific subset of GISTs and is present in around 5–6% of these tumors.138 It has been reported that this mutation is susceptible to crenolanib but resistant to certain kinase inhibitors, such as SU11248.139,140 Furthermore, in contrast to other PDGFRA mutations in GISTs, PDGFRA D842V has been connected to certain clinicopathological characteristics.138
V561D mutation occurs in the juxta-membrane domain and is another noteworthy mutation that has oncological implications. The regulatory function of the kinase can be disrupted, resulting in the incorrect activation of the enzyme and consequently the initiation of signaling pathways that lead to cell growth and survival. In contrast to the D842V mutation, the V561D mutation in PDGFRA may still exhibit sensitivity to specific tyrosine kinase inhibitors (TKIs).140,141 Patients with this mutation may exhibit therapeutic responses, as they could potentially respond well to TKI treatment.142 However, apart from GIST, the occurrence of activating c-KIT and PDGFR mutations in other types of human malignancies is extremely uncommon. Hence patients afflicted with these malignancies, although exhibiting an excessive amount of c-KIT and/or PDGFR, are unlikely to derive any advantages from imatinib-targeted therapy.143
PDGFR Gain-of-function mutations are identified in PDGFRA in several diseases such as Y266C, Ins450C, Del (8,9), V536E, Ins544V, N659X, D842X, and Ins491A in glioblastoma; D842X, N659X, and V561D in GIST. In PDGFRB, gain-of-function mutations were identified in unicentric Castleman disease (N666X) and multiple mutations in non-oncologic diseases.144
In patients with pediatric glioma, D842V, N659K, E229K, C235R, Y288C, and C290R were identified as missense mutations, E7del, E10del2, E10del as deletions and C450ins, A491ins and V544ins as insertions. Some oncogenic mutations can confer resistance to small molecule inhibitors; thus, the therapeutic approaches may need improvement.145
In AML, there are two groups of mutations that activate the FMS-like tyrosine kinase-3 (FLT3) gene. These mutations are known as FLT3-internal tandem duplications (FLT3-ITDs) and FLT3 point mutations in the tyrosine-kinase domain (FLT3-TKD). FLT3-ITDs occur in the juxta-membrane (JM) domain and are present in up to 25% of patients with AML. FLT3-TKD mutations, on the other hand, occur in the tyrosine-kinase domain and are found in up to 10% of AML patients. However, a novel category of activating point mutations (PMs) has been discovered, which are concentrated in a 16-amino acid segment of the FLT3 juxta-membrane domain (FLT3-JM-PMs).146
PDGFR mutations have a significant impact on the protein function. Multiple mutations are detected in the genes that encode PGDFR proteins and most of them seem to be oncogenic and produce imbalance in the normal physiological function of the receptor. The details related to the mechanism of action and the oncological disorders in which PDGFRs are involved are detailed in Table 5.
Deregulation of VEGFRs in cancer
Vascular endothelial growth factor receptors (VEGFR-1, -2, and -3) play crucial roles in angiogenesis and vascular permeability by binding vascular endothelial growth factor (VEGF). Activation of the Ras/MAPK, PI3K/Akt, and PLCγ pathways influences wound healing, angiogenesis and vascular development. Targeting VEGFRs is critical in anti-cancer therapies, especially in inhibiting tumor blood supply due to their involvement in pathological angiogenesis.147
Among its ligands, VEGFR plays critical roles in physiological and pathological angiogenesis, being a key target in cancer. VEGFR family includes receptors which have different roles: VEGFR-1 (Flt-1), VEGFR-2 (KDR/Flk-1) and VEGFR-3 (Flt-4), the first two with roles in angiogenesis and Flt-4 in lymphangiogenesis.148,149 Furthermore, VEGFRs pathways are connected by a crosstalk with other pathways involved in cell survival, cell migration, actin reorganization, focal adhesion and proliferation, thus any structural and functional changes in the receptors may lead to imbalance in many other biological processes.150
Mokhdomi et al. investigated mutational patterns in 10 exons of VEGFR-1, identifying 10 genotypic variations with distinct allelic frequencies, including 8 novel variants and 2 known ones. Notably, analysis of the global SNP database unveiled the rs730882263:C>G mutation in VEGFR-1, resulting in the VEGFR-1 p.Cys1110Ser variant within the catalytic domain. This mutation potentially contributes to colon cancer pathogenesis.151 Moreover, VEGFR-1 rs7993418 polymorphism was associated with hematogenous metastases in GC.152
Two frequent gain-of-function mutations in VEGFR-2, R1051Q and D1052N were related to an increased enzymatic activity of the receptor. R1051Q variant stimulates PI3K/Akt signaling in tumor cells leading to resistance to therapy.153 In melanoma, using SK-MEL-31 cells as a model, R1051Q mutation activated the receptor, stimulating melanoma progression without ligand-binding.154 In the absence of a ligand, VEGFR-2 can form phosphorylated dimers. In this case, conformational switch of extracellular, intracellular, and transmembrane domains of the receptor are of major importance. Engineered transmembrane domain mutations such as E764I-T771I-F778I and N762I-V769I-G770I, have a crucial role in VEGRF-2 dimer stabilization by affecting its phosphorylation status.155 On the other hand, C482R pathogenic mutation leads to an increase in phosphorylation even in the absence of ligands. This mutation is linked to infantile hemangioma.156
VEGFR-2 high expression and single nucleotide polymorphisms rs1870377 A>T and rs7692791 were correlated with GC prognosis and poor survival.157,158 In the case of CRC, VEGFR-2 1192C/T and −604T/C single nucleotide polymorphisms are associated with microvessel density in tumor tissue.159 Other VEGFR-2 alterations might be correlated with Alzheimer’s disease based on a study performed on plasma samples obtained from mild cognitive impairment and Alzheimer’s disease patients.160
VEGFR-3 expression is correlated with tumor progression by means of lymphatic metastasis (in the case of breast, lung, ovarian, renal cell, colorectal, gastric, oral, cervical, prostate, pancreatic cancer and basal cell carcinoma) or angiogenesis (in the case of ovarian, colorectal, gastric, cervical, prostate, pancreatic, melanoma, laryngeal cancer).161 On the other hand, VEGFR-3 missense mutations are associated with different forms of autosomal dominant primary lymphedema,162 for example Milroy disease.163
Table 6 depicts the importance of VEGFR in the development of several malignancies and highlights the genes that encode the proteins and specific mutations that occur within these genes leading to an overstimulated receptor, or an unfunctional protein that creates the proper conditions for malignant cells to develop and proliferate.
Deregulation of HGFRs in cancer
The primary HGFR, c-Met, is involved in cell motility, invasion, and metastasis. Its activation primarily leads to the stimulation of the Ras/MAPK, PI3K/Akt, and STAT pathways. Dysregulation of c-Met is linked to various cancers, making it a target for therapies aimed at inhibiting metastatic spread.164
c-Met, known as hepatocyte growth factor receptor, orchestrates cell motility, invasion, and metastasis by activating signaling pathways such as Ras/MAPK, PI3K/Akt, and JAK/STAT, aberrant activation of which is linked to diverse cancers, notably driving invasive and metastatic growth, rendering c-Met a prominent therapeutic target, with multiple inhibitors devised to counter its oncogenic activity in cancer therapy.165
c-MET and RON have similar biochemical properties and share similar structures. C-MET is recognized by HGF while RON has its specific ligand, the macrophage-stimulating protein. RON distribution is restricted to cells that have epithelial origin and studies have demonstrated that RON expression is required for attenuating the inflammatory response, controlling the macrophages activities during infections.166 RON overexpression was observed in cancers localized in pancreas, bladder, lung, breast, colon, thyroid and skin, its overexpression is correlated with advanced clinical stages, and it seems that RON can modulate cell growth and migration via MAPK/Akt pathways sustaining tumorigenicity.167,168,169,170
Hepatocyte growth factor receptor protein is a single pass tyrosine kinase receptor which is key in embryogenesis and wound healing. Abnormal activation of MET in different cancers correlates with poor prognosis, enhanced angiogenesis and Epithelial to Mesenchymal Transition (EMT).171,172,173,174,175,176
The c-Met pathway is a potential target therapy in cancers.172 The activation or hyperresponsiveness of the HGFR/HGF pathway involves two primary mechanisms: mutations in MET within the extracellular or cytoplasmatic domain, resulting in prolonged biochemical signaling, and ligand-independent activation, characterized by the overexpression of the wild-type protein. The two mechanisms can act individually or concomitantly. Multiple point mutations were identified in the semaphoring, immunoglobulin plexin transcription, juxta-membrane (JM) or tyrosine kinase (TK) domains of MET, as Sattler and Reddy stated in their work.172 N375S mutation was identified in NSCLC, small cell lung cancer (SCLC), mesothelioma, and melanoma177,178,179,180; T992I in NSCLC, SCLC, mesothelioma and BCs and other mutations specifically identified in other cancer subtypes.181,182,183,184
Although there are limitations on the activation of cMET induced by HGF, dysregulated signaling of HGF-cMET has been detected in several malignant neoplasms.185 Abnormal cMET activation is possible through processes that are not dependent on HGF, such as MET mutations, gene amplification, and transcriptional upregulation.172
Jeffers and his group generated several fibroblast cell mutants (NIH 3T3 cells) that were inoculated in mice models, and compared to the wild type, M1258T; Y1248H; D1248H; D1246N; Y1248C; V1238I; V1206L, and M1149T clones induced tumor growth in mice.186 These mutations were identified with high frequency in patients with RCC.187,188,189,190 Furthermore, several in vivo studies have demonstrated that the activation of the HGF–cMET signaling pathway is a critical factor in promoting cancer invasion and metastasis.191,192
Elevated levels of HGF in both tumor tissues and plasma have been observed in patients with various types of cancers, such as invasive breast carcinoma, glioma, and multiple myeloma.193,194,195 The information regarding mutational status of these receptors and the mechanism of action in different malignancies are detailed in Table 7, with specific details regarding the encoding genes and the oncogenic status of the mutations that were detected in each receptor.
Deregulation of MuSK
Muscle-specific Kinase (MuSK) is crucial in neuromuscular junction formation. Its activation stimulates pathways like PI3K/Akt and MAPK, which are essential for the clustering of acetylcholine receptors and development of the postsynaptic membrane. MuSK’s role in muscle function makes it a focus in neuromuscular disorder studies.196,197
MuSK is a RTK that is required for the maintenance and formation of the neuromuscular junction and its ligand is agrin, which triggers the signaling cascade via casein kinase 2 (CK2), Dok-7 and rapsyn.198,199,200,201 The activation of MuSK requires also its coreceptor LRP4 and a stoichiometric ratio of 1:1:1 agrin: LRP4: MuSK complex being essential for its function.202With around 100 kD and a single-pass transmembrane RTK, Musk interacts with agrin and LRP4 to modulate the postsynaptic apparatus.203 Communication from the motoneuron to the muscle is essential for the formation and maintenance of the neuromuscular junction, thus Musk plays a crucial role in the acetylcholine receptor clustering during the development of the neuromuscular junction.204
Because of the role in neuromuscular junction maintenance, Musk is not studied in cancer related subjects, and the literature is focused on its role in autoimmune diseases such as Myasthenia gravis or other neuromuscular disorders.205 Although not related to oncological disorders, there is one mutation in MuSK that might be responsible for myasthenic syndrome, M835V, which induces changes in the receptor structure, therefore disturbing the downstream signaling.206 Other two mutations (c.2062C>T (p.Q688X) non-sense mutation and c.2324T>C (p.F775S) missense mutation) that are not recorded in Human Gene Mutation Database, were identified in a case of Chinese neonatal congenital myasthenic syndrome. The missense mutation was inherited from the mother and was predicted as pathogenic and severe by various bioinformatics programs.207
Deregulation of ALK in cancer
Anaplastic lymphoma kinase (ALK) plays a role in the development of the nervous system and is implicated in various cancers. It activates pathways such as Ras/MAPK, PI3K/Akt, and JAK/STAT, contributing to its oncogenic potential. ALK is a target for cancer therapy, especially in anaplastic large cell lymphoma and NSCLC.208,209,210
In addition to its role in cancer, ALK and its receptor family member Leukocyte tyrosine kinase receptor (LTK) have a key role in the normal physiology of the central nervous system. It was observed that in the absence of LTK and ALK, neuronal migration, neural progenitor populations and cortical layers patterning were disrupted, thus some researchers suggest that the use of ALT/LTK inhibitors in cancer patients should be carefully monitored to avoid brain dysfunctions.211 Moreover, the mammalian RTK ALK was first described as the product of the t(2;5) chromosomal translocation found in non-Hodgkin’s lymphoma.212,213
The physiological roles of LTK while not fully understood, have been partially explained through some in vivo studies. It was observed that mice that had ALK/LTK knockout genes had significant reduction in newborn neurons, suggesting that ALK may play an important role in the generation or survival of these neurons, during neurodevelopment.214,215 The status of LTK and ALK as “orphan” receptors changed with the identification of their ligands, ALKAL1 and ALKAL2.216,217,218
Roll and Reuther evaluated the ALK activating mutations in LTK, using a benign tumor model (pheochromocytoma—adrenal gland benign tumor) in mice.219 This study identified specific ALK mutations like F1147L and R1275Q, and the corresponding LTK mutations F568L and R669Q. Moreover, the F1147L and R1275Q mutations are frequently detected in neuroblastomas.220,221,222 The two mutations are responsible for the constitutive activation of ALK in neuroblastoma.223 ALK/LTK mutations and their role in this specific malignancy are presented in Table 8.
Deregulation of ROS1 in cancer
ROS1 belongs to the human RTKs and is the only member of the ROS1 family. ROS1 was evaluated first in solid tumors in 1987 and it was revealed that in glioblastoma cell line U118MG, ROS1 was altered.224 ROS1 rearrangements were observed in NSCLC, initially in 2007, together with the discovery of ALK rearrangements in NSCLC.225,226,227 Moreover, multiple fusions were detected, most of them in less than 2–3% of these cases. However, fusions with CD74 were detected in almost half of the cases,228 fusions with EZR in not more than 24%,229 with SDC4, TPM3 and SLC34A2 in less than 15%.228,229,230 Such rearrangements were also reported in cholangiocarcinoma, in 8.7% of the samples that were analyzed.231 ROS1 acts like a driver in various cancers, including NSCLC.232,233 It activates signaling pathways like Ras/MAPK, PI3K/Akt, and JAK/STAT, which contribute to its role in oncogenesis. ROS1 has become an important focus in cancer therapy, with targeted inhibitors showing effectiveness against ROS1-driven cancers.234
Point mutations in ROS1 such as D2033N, S1986F, L2000V, G2032K, G2032R and L2086F, have been associated with resistance to therapy. These mutations are linked to a poor overall survival rate, as detailed in Table 9 which highlights the mechanism of action of each detected mutation, even if their oncogenic status is not known yet.235,236,237,238
ROS1expression is undetectable in most normal tissues, expressed at low levels in parathyroid glands, eyes, skeletal muscle, larynx and adrenal glands. However, high expression levels were found in the cerebellum, peripheric nerves, colon, kidney and stomach.239,240,241
Deregulation of RET in cancer
Rearrange during transfection (RET) is the RTK that is interacting with ligands at the cell surface and plays a key role in the development of the central and peripheral nervous system.242 RET is crucial for the development of the enteric nervous system and kidneys. It primarily activates the Ras/MAPK, PI3K/Akt, and PLCγ pathways. Mutations in RET are associated with multiple endocrine neoplasia243 and Hirschsprung’s disease,244 making it a significant focus in developmental biology and genetic disease research.245
RET was discovered as a protooncogene; thus, a large body of research was conducted to evaluate the role and the effects of RET and its mutated forms. It seems that RET may play a key role in parathyroid hyperplasia, thyroid cancer, and lung cancer.246 RET fusions were found in around 20% of papillary thyroid carcinomas and 2% of the NSCLC.245,247,248
RET is a receptor tyrosine kinase embedded in the cell membrane, encoded by the proto-oncogene RET.249 RET has been recognized as playing vital roles in various developmental processes, especially in the formation of the kidney and the enteric nervous system during embryonic development.250,251,252 Changes in RET have been linked to several diseases, including Hirschsprung’s disease and various types of cancer.253,254 Over the past thirty years, numerous alterations in RET have been identified that lead to continuous activation of its kinase activity, a key factor in many cancer subtypes.255,256,257
The phosphorylation of specific tyrosine residues on the cytoplasmic portion of the RET receptor is key to its function. This phosphorylation enables the binding of various adaptor proteins, which are essential for transmitting external signals and activating major downstream signaling pathways. These pathways include PI3K/Akt, RAS/RAF/MEK/ERK, JAK2/STAT3, and PLCγ.258
Specifically, RET phosphorylation at Y687 attracts SHP2 phosphatase, activating the PI3K/AKT pathway to promote cell survival.259 The tyrosine residues Y752 and Y928 serve as crucial sites for binding the Signal Transducer and Activator of Transcription 3 (STAT3), leading to its activation and movement into the nucleus, which is important for the transcription of STAT3 target genes.260 Phosphorylation at RET -Y905 maintains RET in an active state and is vital for attaching to adaptor proteins Grb7/10.261,262 Moreover, RET -Y981 is essential for the activation of Src kinase.263
PLC- γ interacts with phospho- RET at Y1015, subsequently activating the PKC pathway.264 The phosphorylation of RET at Y1062 is critical for recruiting adaptor proteins that trigger the activation of the PI3K/Akt, RAS/RAF/MEK/ERK, and MAPK pathways.265 Lastly, Grb2 binds to phospho-RET at Y1096, facilitating the activation of the RAS/RAF/MEK/ERK pathway, which is important for cell proliferation and differentiation.266,267
M918T mutation in RET was identified in thyroid gland carcinoma, as a gain of function mutation, increasing the substrate binding and conferring drug resistance.268,269 Furthermore, RET A883F mutation, also a gain of function mutation, increased RET kinase activity in solid tumors of thyroid cancer.270,271,272
Using new NGS platforms for analysis, human tumor specimens were used to discover RET mutations in other cancers such as breast carcinoma (RET C634R), endometrial and Merkel-cell carcinomas (RET E511K) and RET V804M in CRC and hepatoma.273 RET C634R is a gain of function mutation, resulting in an autophosphorylation of RET,274 RET 3511K, a gain of function mutation, increased RET and ERK phosphorylation.275 Another gain of function mutation in RET, V804M increased the kinase activity and is considered a gatekeeper due to lack of response to inhibitors like cabozantinib in thyroid cancer.276
Deregulation of ROR in cancer
ROR1 and ROR2, involved in developmental processes and cancer progression, primarily modulate the Wnt signaling and JAK/STAT pathways. Their roles in cell migration and cancer progression, particularly in the context of Wnt signaling, make them potential targets in oncology.277
ROR is a small RTK family, including ROR1 and ROR2 which were first characterized in 1992 form SH-SY5Y neuroblastoma cell line.277,278 The two receptors are highly expressed during embryogenesis and not expressed in adult tissue; however, an increased expression of the ROR receptors is observed in tumor tissues with increased cell proliferation.279,280,281,282 According to Zhao et al., no mutation in ROR1 have been found yet,283 however the overexpression of ROR1 itself is a cause of increased proliferation and cell growth, in malignancies, ischemia and diabetes.284,285,286
In the case of ROR2, no mutation was detected in cancers, however, a study focused on 21 patients with short stature, identified 10 missense, one nonsense and one frameshift mutation. The only mutation that had a potential effect on the downstream Wnt5a-ROR2 pathway was G559S which may disturb the subcellular localization and protein expression.287
Deregulation of Eph family of receptors in cancer
Erythropoietin-producing human hepatocellular Receptors (Ephs), including EphA and EphB, are involved in developmental processes in the nervous system. They primarily activate the Ras/MAPK and JAK/STAT pathways. Their implication in cancer progression and metastasis has made them a focus in cancer research.288
Ephs are a group of receptors that become active when binding to their ephrins; Ephs are divided into two subclasses EphA and EphB.289 The Ephs are activated by a cell-cell interaction with their ligands that are membrane-bound proteins, and the signaling is involved in embryogenic development, cell migration, and segmentation290,291 and play a key role in angiogenesis, stem cell differentiation and cancer progression.292,293 Eph receptors represent the biggest RTK family, with nine EphA receptors (A1-A8 and A10) and five EphB receptors (B1-B4 and B6).294,295,296
Deficiency in autophosphorylation of EphA3 was observed for D678E and R728L mutant forms. The N85S and T116N mutations in the binding domain and D219V and S229Y in sushi domain and M269I in EGF domain showed differences in the ephrin-binding ability, compared to wild type, while other mutants showed no differences. G187R mutant was detected as very important, disrupting the conformation of the binding domain. Also, G228R and W250R mutations in sushi domain had drastically disrupted the binding.297
Several mutations were highlighted in different regions of the EphA3, with various degrees of Loss-of-function, impairing the binding or induce structural alterations T116N, G187R, V206L, S229Y, W250R, M269I, F311L, N379K, T393K, A435S, D446Y, S449F, G518L, T660K, D678E, K761N, R728L, G766E, and T933M.298
Exon 17 of EPHB4 gene was sequenced in lung cancer cell lines and patient samples and several mutations were detected: in the extracellular linker region (A230V), first extracellular fibronectin III repeat (A371V, P381S), in the extracellular juxta-membrane domain (W534*, E536K), in the TK domain (G723S, A742V) and a mutation in the intracellular linker region (P881S). Except A371V, the rest were newly discovered by Ferguson et al.299
Chakraborty et al. detected several structural alterations in EphA3 (A749D, W790C, F152S), EphA7 (L749F), EphB1 (G685C) and in EphB4 (V748A), all mutations inducing changes in NSCLC samples.300 The mutations impact in NSCLC remains unclear and needs further functional analysis for each mutation.
Faoro et al. evaluated one mutation in EphA2 which caused constitutive activation of this receptor and increases invasiveness.301 G391R mutation was detected in H2170 cells and 2 out of 28 SCC patient samples, but not in other subtypes of lung cancer.
The mutational status of these receptors is presented in Table 10 highlighting the disease in which each mutation was detected and the mechanism of action that led to the potential oncogenic activity.
Deregulation of RYK in cancer
RYK, a member of the receptor-like tyrosine kinase family, is involved in Wnt signaling, influencing both β-catenin-dependent (canonical) and β-catenin-independent (noncanonical) pathways. The roles of RYK have been investigated in several model organisms, such as Drosophila, zebrafish, Xenopus, and mouse models.302,303,304,305
The study of RYK is significant for understanding complex developmental processes and its aberrant roles in diseases, including cancer, making it a notable focus in both developmental biology and oncology.306 Its implication in cancer progression, particularly in relation to Wnt signaling, has led to its exploration as a potential therapeutic target.307 Its regulation affects various cellular processes such as cell polarity, cell migration, skeletal development, neurogenesis, and axon guidance.306,308 Ryk targeted deletion in mice results in inhibited growth, abnormalities in the development of the skull and skeleton, and death after birth.305 Experiments conducted outside of a living organism have demonstrated that RYK has the ability to attach to Wnt, Frizzled 8, and Dishevelled proteins in order to initiate β-catenin/TCF-dependent transcription.309,310
Moreover, RYK has a genetic interaction Van Gogh-like 2 (Vangl2) proteins. Vangl2−/−; Ryk−/− mice show typical phenotypes associated with planar cell polarity signaling, including problems with neural tube closure, elongation of the body axis, and craniofacial development.311,312 Also, within the hematopoietic system, RYK has been demonstrated to possess a cell-intrinsic influence over hematopoietic stem cells (HSCs), regulating their proliferation, apoptosis, and their capacity for repopulation.313
In a study led by Seon-Yeong Jeong et al., RYK role in regulating Wnt signaling in bone marrow mesenchymal cells was investigated. Previous research had indicated RYK’s influence on HSC proliferation, quiescence, and apoptosis, but the study proposed a role for RYK in the stromal component of the bone marrow. Notably, downregulating RYK in mesenchymal stromal cells (MSCs) had no impact on their cell-autonomous functions, such as proliferation and differentiation. However, it did affect MSC colony-forming activity, independent of Wnt signaling.314
A CRISPR-Cas9 gene-targeting model confirmed the absence of Wnt response in cell-autonomous MSC functions during differentiation and suggested a Wnt-independent role for RYK in maintaining MSC colony-forming populations. Reducing RYK also attenuated Wnt3a’s stimulatory effect on MSC niche activity, particularly in enhancing HPC self-renewal. Significantly, RYK dose-dependently modulated Wnt signaling in MSCs, influencing its intensity. These findings underscore RYK’s physiological role in regulating Wnt signaling in the bone marrow MSC niche, fine-tuning HPC self-renewal, and contributing to the control of hematopoietic activity in a homeostatic manner.314
RYK, along with other receptors like ROR1/2, contributes to the transmission of signals initiated by Wnt ligands, specifically Wnt5a. The signals deviate from the canonical pathway and have the potential to influence cellular processes such as cell polarity, migration, and axonal growth.315
Moreover, in the presence of RYK, Wnt5a influences axon growth and repulsive signal guidance via Ca2+-dependent signaling. Wnt5a can exhibit either tumor-suppressing or oncogenic properties in various types of cancer.316,317
In a study by Katso et al., assessed the H-RYK overexpression’s predictive value in epithelial ovarian cancer. The study also explored the potential role of H-RYK in angiogenesis, a critical process in tumor progression and metastasis. Despite its impaired catalytic activity, H-RYK was shown to signal through the mitogen-activated protein kinase pathway. Interestingly, H-RYK overexpression did not correlate with the proliferative status of the tumor cells, suggesting its contribution to tumorigenesis might not be through inducing excessive cell proliferation. Instead, it may play a role similar to other kinase-impaired receptors in promoting cell survival, thus contributing to carcinogenesis by protecting cells from apoptosis.318
Deregulation of CCK-PTK7 in cancer
CCK4/PTK7 was first described by Mossie et al., in 1995, and its functions are not completely understood.319 Initially named CCK-4 (colon carcinoma kinase 4), PTK7 is regulating Wnt signaling pathways and controls morphogenesis and patterning modulating cell molarity, migration and wound healing.320,321,322 Later, it was shown that PTK7 expression could be correlated with cancer development and metastasis, while mutations in PTK7 are involved in human neural tube closure defects, scoliosis or inner ear polarity defects.323,324,325,326
PTK7 upregulation was detected in gastric, esophageal, colorectal, lung carcinoma or BC, while a downregulation of PTK7 was related to lung SCC, ovarian cancer and melanomas.327,328,329,330 Other studies underlined PTK7 role as a marker for normal colon stem cells and its potential role as a marker for tumor initiating cells in NSCLC, ovarian cancer or TNBC,331,332 while PTK7 inhibition showed a sustained tumor regression, indicating that some anti PTK7 therapies may have a role in tumor inhibition.333,334 However, no mutations have been reported to be responsible for tumor development.
Deregulation of NGFR in cancer
Nerve growth factor receptor (NGFR), also named P75 neurotrophin receptor or CD271, is a 45 kDa receptor, consisting of a single peptide and has nerve growth factor as a ligand.335,336
Nerve growth factor receptor is involved in signaling for tumor development and progression, with an important role in proliferation when overactivated. Wu et al. showed that NGFR is highly expressed in metastatic lung clones of TNBC cells, the overexpression led to the growth and invasion of tumor cells to distant tissues. Moreover, the study demonstrates that NGFR is highly expressed in TNBC patients compared to non-TNBC patients, and negatively correlated with overall survival of the patients.337,338
TRKA is the most common oncogene in the TRK family, with significant presence in human tumors, over 7%, while TRKB and TRKC are less present. The inhibition of activated TRKA is a reliable approach in tumor inhibition. Several compounds showed inhibitory effect on NTRKs and display side effects, however as Wang et al. highlighted, no reports of selective TRKA inhibitors have been published. Wang research group discovered a TRKA selective inhibitor named 32 h which had an IC50 of 72 nmol/L for TRKA while for TRKB and C was above 1 µmol/L and the antitumor effect was demonstrated by cutting-edge determinations such as RNA-seq which underlined the TRKA inhibition and modulation of Wnt pathways. Furthermore, the pharmacokinetic properties have been tested and generated promising results on xenograft models suggesting that TRKA selective inhibitors can represent a therapeutic approach for NTRK1 fusion positive cancers.339
The TRK family, including TRKA, TRKB, and TRKC, is involved in nerve growth and survival. They activate pathways like Ras/MAPK, PI3K/Akt, and PLCγ, playing roles in both neuronal and non-neuronal tissues. Their involvement in neurodegenerative diseases and cancers makes them significant in neurological research and therapy.340
Deregulation of AXL in cancer
The AXL family of RTKs, also known as TAM family includes three main receptors: TYRO3, AXL, and MER. TAM receptors are overexpressed in different malignancies, such as leukemia, melanoma, gastric, colon, lung and BC, promoting cell survival.341,342,343,344,345 AXL biosynthesis is regulated by key transcription factors (AP1, Sp1/Sp3, YAP/TAZ/TEAD, HIF1α, MZF-1) and Toll-like receptor signaling in dendritic cells and macrophages, which increase AXL mRNA expression. This process is further controlled by a feedback mechanism involving other RTKs.346,347,348 AXL regulates cell proliferation, cell cycle, cell growth, and survival.349,350
In adults, AXL expression is usually low but is abnormally high in several cancers, including breast, chronic lymphocytic leukemia, NSCLC, pancreatic, glioblastoma, melanoma, RCC, colorectal, prostate, and esophageal cancers.345,351 The overexpression of AXL is correlated with various cellular processes as follows: epithelial to mesenchymal transition, angiogenesis, chemotherapy resistance and weak antitumor immune response.345 The regulation of AXL involves various factors, including microRNA miR-34a at the translational level and the proteolytic release of its extracellular domain, cleaved by metalloproteinases ADAM10 and ADAM17.352 In addition to this, HIF1α348 and AP-1347 transcription factors and methylation of CpG islands in the promotor region353 were reported as AXL regulators in some studies.
AXL activation by the GAS6 ligand leads to the stimulation of various downstream signaling cascades such as JAK/STAT3, PI3K/Akt, Grb2/RAS/MEK/ERK1/2, and FAK/Src/NF kappa B.18,354
In a study, Salian-Mehta et al. have identified three missense AXL mutations (p.L50F, p.S202C, and p.Q361P) and one intronic variant (c.586-6C>T) in Kallman syndrome and norm osmic idiopathic hypogonadotropic hypogonadism subjects.355 In another study were described 53 AXL, 36 MER and 25 TYRO3 mutations identified on a cohort of 509 female patients diagnosed with endometrial adenocarcinoma.356,357 In all these cases the main majority were missense mutations. It is important to mention that no mutations have been detected in the KW (I/L)A (I/L)ES (a.a 714–720) motif, which is a conserved domain specific to all RTKs of the TAM family.357
Both AXL and TYRO3 were found to be expressed in various cutaneous melanoma cell lines.358 TYRO3 is particularly activated by tumor secreted protein S (ProS1) resulting in the activation of multiple pathways involved in cancer cells survival such as AKT and ERK.359,360 The overexpression of TYRO3 has been associated with poor survival in the case of colorectal, hepatocellular and BCs.361 Various mutations within the kinase domain of TYRO3 such as M592I, N615K, W708fs*5, A709T, C690R have been linked to to colon,362 lung,363 melanoma,364 brain cancer,364 and acute myeloid leukemia respectively.365 In the cytosolic domain have been remarked the following mutations: R462Q, R514Q and G809D which were associated with melanoma,364 pancreatic366 and colon cancer.362 In the extracellular and transmembrane domains have been determined Q67 and H60Q in melanoma367,368 and E340 in lung cancer.363
MER, also known as RP38, c-Eyk, c-mer, and Tyro12 is considered a proto-oncogene, playing important roles in cell survival, migration and differentiation.369 It was found to be upregulated in leukemia,370 lymphoma,371 colorectal,372 gastric,373 and lung374 cancers. Specific MER variants like P802S were found in melanoma,375 while another range of mutations like p.T690I, p.R20S, p.I518V, p.R466K, p.S118N, p.V870I, p.A282T, p.N498S, p.R293H, p.R865W, p.E823Q variants were identified in multiple myeloma.376 Moreover, MER mutations are linked with around 2% of the cases with severe autosomal recessive retinal dystrophies.377 Over the time have been identified 79 variants including missense (33), nonsense (12), splice defects (12), small deletions (12), small insertion-deletions (2), small duplications (3), exonic (2) and gross (3) deletions.377
Deregulation of TIE in cancer
Tyrosine kinase with immunoglobulin-like and EGF-like domains (TIE), including TIE1 and TIE2 as its principal components, represents a critical group within the receptor tyrosine kinase family. TIE1, specifically, is a transmembrane protein predominantly located in endothelial cells.378,379 These receptors demonstrate a notable degree of amino acid similarity in their cytoplasmic domains, while their extracellular regions are less similar but still share some amino acid identity. In the receptors internal structure, there is a split kinase domain that becomes capable of binding to a variety of proteins following self-phosphorylation. The external part of these receptors is characterized by multiple immunoglobulin (Ig)-like domains, dispersed with several EGF-like cysteine repeats and fibronectin type III domains.380
The TIE transmembrane protein family, highly receptive to angiopoietins, includes TIE1 and TIE2, with each playing distinct roles in vascular biology. Originally perceived as an orphan receptor, Tie2’s classification evolved following the identification of angiopoietin-1 (Ang1, ANGPT1) as its ligand, along with other ligands such as Ang2, Ang4, and mouse Ang3. Tie2, notable for its broad expression in various cell types including those in larger blood vessels, is especially active during tumor-related angiogenesis.381
Advanced structural studies have shown that the extracellular region of Tie2 can form dimers even without ligand binding, facilitated by membrane-proximal fibronectin type III domains. This dimerization process is crucial for its angiogenic functions and its roles in vascular biology, indicating a deeper level of regulatory mechanisms for Tie2 activation and signaling.382
Therapies targeting TIE receptors, including monoclonal antibodies against TIE2 and small molecule inhibitors disrupting the TIE-Angiopoietin signaling, are important in the therapeutic landscape. Agents such as Trebananib used in a phase III cancer trial, demonstrated increased progression-free survival in ovarian cancer, highlighting its potential in advanced stages where TIE-1 overexpression is linked to poorer prognosis.383,384 Also, The VE-PTP inhibitor AKB-9778 has shown effectiveness in reducing edema and improving vision in retinal vascular diseases, supporting the therapeutic potential of TIE2 activation.385
Moreover, Ishibashi et al. have identified a crucial role of TIE-1 in ovarian cancer treatment, specifically regarding cisplatin resistance. Their research clearly separates the functions of TIE-1 from TIE-2, revealing that TIE-1 overexpression correlates with poor prognosis and lower effectiveness of cisplatin in advanced ovarian cancer. This finding suggests the importance of TIE-1 in determining treatment outcomes and highlights its potential as a target for developing new therapeutic strategies in ovarian cancer management.386
In a recent study, Marguier et al. demonstrated the significant role of TIE-2 positive (TIE-2+) monocytic myeloid-derived suppressor cells (M-MDSC) in melanoma. They found that these cells are more immunosuppressive than their TIE-2-negative counterparts, particularly in advanced stages of melanoma. High levels of TIE-2+ M-MDSC were linked to reduced effectiveness of melanoma-specific T-cell responses, with ANGPT2 enhancing the suppressive ability of these cells. The study also indicated that an increased presence of TIE-2+ M-MDSC and ANGPT2 in blood is associated with poor prognosis in melanoma. TIE-2 expression on M-MDSC boosts their suppressive features, including the overexpression of inhibitory proteins like PD-L1 and IL-10. ANGPT2 further enhances these immunosuppressive pathways, pointing to the key role of TIE-2 kinase activation in the suppression of T-cell function.387
Deregulation of DDR
Discoidin domain receptors (DDR), specifically DDR1 (CD167a) and DDR2 (CD167b), have been recently recognized as distinctive constituents of the transmembrane RTK family.388 These receptors exemplify a distinct subclass of RTKs. In contrast to conventional RTKs, which commonly interact with peptide-like growth factors as their ligands, DDR1 and DDR2 exhibit a distinctive activation mechanism. These receptors are uniquely stimulated through their binding to collagen, which is the most abundant protein found in the extracellular matrix. One important attribute of DDR is their significant participation in the synthesis and degradation processes of collagen, thereby emphasizing their unique function within the RTK family.389,390 DDRs contribute not only to the processes of cellular proliferation and differentiation, but also to the dynamics of cell movement, invasion, and attachment.391 Moreover, alterations and atypical expression of DDR1 and DDR2 are associated with the advancement of cancer and an unfavorable prognosis.392,393,394
Similar to other RTKs, DDRs are structured into three distinct regions: the outer binding domain, the spanning transmembrane (TM) section, and the inner kinase domain (KD). Both a discoidin (DS) domain and a DS-analogous domain are needed for outer collagen interaction. DDR-connecting proteins bind to TM extracellular juxta-membrane (JM) phosphorylated tyrosines. DDR-connecting proteins bind to phosphorylated tyrosines located in the extracellular juxta-membrane (JM) portion of the TM.395 The helical shape of the TM segment allows for receptor pairing without relying on collagen. The receptor’s internal segment consists of the JM zone and the tyrosine kinase KD, which are both essential for its enzymatic activity. Src interaction with tyrosines in the DDR’s collagen-engaged activation loop promotes the phosphorylation process. This series of events has the potential to result in the self-phosphorylation of more tyrosine residues within the kinase domain’s juxta-membrane area. As a result, it attracts adaptor molecules that regulate other cellular processes.396,397
DDRs were also associated with the process of EMT, which is influenced by the specific ligand and cell type involved. For example, in prostate cancer cells, the activation of DDR1 triggers the phosphorylation of Pyk2 and MKK7, contributing to the progression of EMT.398,399
The invasion of tumor cells depends on matrix metalloproteinases (MMPs), which destroy the extracellular matrix.400 For example, DDR1 promotes invasion in MDA-MB-231 BC cells by boosting MMP-2 and MMP-9 secretion.401 By increasing MMP-2 levels, DDR1 expression can also cause colon cancer cell invasion.402 Recent studies indicate that suppressing DDR2 can decrease B16BL6 melanoma cell invasion by reducing MMP-2 and MMP-9 expression via the ERK/NF-κB pathway.403 In contrast, B16-F10 murine melanoma cells with lower DDR2 expression did not affect lung metastasis.404
In a study of ESCC, involving mostly male patients (88.3%) with a median age of 68, it was found that high phosphorylated DDR1 (pDDR1) staining, observed in 36.7% of the tumors, is a prognostic indicator. This high pDDR1 staining was significantly associated with shorter recurrence-free and overall survival, highlighting its importance in assessing ESCC progression and prognosis. The study’s findings emphasize the potential role of pDDR1 as a valuable biomarker in understanding and possibly guiding the treatment of ESCC, especially considering the diverse patient characteristics such as age, histological grades, and treatment methods within the cohort.405
Deregulation of LMR
LMR (from lemur tail kinase receptors) are an unusual type of receptors, due to their short extracellular domains and very large intracellular domains, thus their name, include LMR1 encoded by AATK gene; LMR2 encoded by LMTK2 gene and LMR3 encoded by LMTK3 gene.406,407,408 These membrane-anchored receptors are involved in cell signaling including cell differentiation, invasiveness, migration and proliferation.409,410 Even if there are many lemurian tyrosine kinases (LMTKs) studies reported in literature, this family of RTKs is still incompletely characterized. The nomenclature for this kinase family has suffered various modifications along the time: AATYK,46 LMR,411 LMTK.408,412 Some authors consider to not include LMR as a separate family of receptors because of their constitution, being recognized as Ser/Thr RTKs.409
With a precise function still to be defined, LMR1 was highlighted as a potential marker for apoptosis, commonly referred to as AATYK (Apoptosis-associated tyrosine kinase), and its overexpression induces differentiation in neuroblastoma SH-SY5Y cells.407 Alternative splicing in LMTK1 has given two isoforms LMTK1A and LMTK1B, from which LMTK1A is involved in endosomes recycling process.409 Recently, LMTK1 has been reported as a risk factor for Alzheimer’s disease being involved in endosomal localization of amyloid proteins.413 LMTK2 is also associated with neurodegenerative dementias,414 but no evidence regarding LMR2 has been reported yet. No ligands have been identified for this type of RTKs. LMTK2 and 3 are two of the 27 understudied kinases as stated.415
LMTK2 gene expression is affected in the early stages of prostate cancer, this gene could be taken into consideration as a potential biomarker for clinical stratification of prostate cancer patients.416 Moreover, LMTK2 gene rs6465657 SNP was detected in the case of prostate cancer in some studies.416,417 The LMTK3 gene has been identified as overexpressed in bladder,418 breast419, and colorectal420 cancers. The vast majority of LMTK3 gene mutations are missense mutations, and some somatic mutations were correlated with neuroblastoma.421
Deregulation of STYK1
STYK1 (from Serine/Threonine/Tyrosine kinase) also called NOK (Novel Oncogene with Kinase domain) receptor was reported as an upstream regulator of autophagy.422 This receptor that shares homology with PDGFR and FGFR RTKs was reported as tumorigenic and metastatic in nude mice.423 Until nowadays no ligand to STYK1 was reported in literature.424
STYK1 is involved in metastasis and epithelial to mesenchymal transition and its overexpression is associated with poor prognosis in NSCLC patients.425 The ligands for these RTKs family are still unknown.424 Overexpression of STYK1 was also noticed in acute leukemia,426,427 hepatocellular carcinoma,428 ovarian cancer,429 NSCLC430 and castration resistant prostate cancer patients.431 At cellular level, overexpression of this receptor involves cell cycle late mitosis arrest and affects cell division.432
Point mutations Y327F and Y356F at the kinase domain might act as tumorigenesis regulators as described by Chen et al in vitro and in vivo studies.433 Y417F point mutation at the carboxyl tail plays an autoinhibitory role in modulating signaling transductions.433
Deletions in the transmembrane domain regulates the oligomerization function of STYK1 and affects RAS/MAPK signaling.434
This receptor is subcellularly found in two different isoforms: dot pattern (DP) and aggregation pattern (AP), from which AP was abundantly found in tumors.435 In addition, AP isoform was found to be involved in endocytosis signaling pathways due to its high distribution in endosomes and is considered to have an important implication in intracellular trafficking together with EGFR.435
RTK signaling nodes, cross-talking, and complementary pathways
RTKs act as critical nodes in cellular signaling networks, intricately linked with several key signaling pathways and regulators. Among the most prominent of these are PTEN, Akt, and mTORC, each playing a distinct yet interconnected role. PTEN serves as a fundamental negative regulator in this network, primarily by dephosphorylating PIP3, thus attenuating PI3K signaling and consequently modulating Akt activity. Akt, a central player in the PI3K pathway, is important for a range of cellular functions, including cell survival, growth, and proliferation. Upon activation by RTKs, Akt phosphorylates a myriad of substrates, leading to diverse cellular outcomes. One of the critical downstream effects of Akt activation is the stimulation of the mTORC1 and mTORC2 complexes. mTORC1, sensitive to nutrient availability and growth factors, regulates protein synthesis and cell growth, while mTORC2 is involved in cytoskeletal organization and cell survival. This cross-talk, facilitated by RTKs, highlights the sophistication of cellular signaling and the potential impact of dysregulation in these pathways, especially in pathological conditions like cancer.436
Multiple interconnected pathways are modulated by the RTKs which interact with crucial biological signaling pathways which contribute to critical processes such as survival, cell death mechanisms, proliferation, migration, invasion, and resistance to therapy. FGFR, PDGFR and EGFR are key elements in cellular biology. When binding their ligands, the downstream signaling is triggered and the above-mentioned biological processes are modulated.437,438,439
The RTKs interact with multiple signaling cascades on a large scale. One notable interaction is the cross-talk between RTKs and the WNT signaling which along with interactions with other pathways such as NF-κB and TGF-β, controls homeostasis, cellular differentiation, and developmental processes.
Understanding the complex interconnections among RTK signaling pathways, their cross-talk mechanisms, and complementary pathways is essential for clarifying typical cellular processes and the ways in which dysregulation in these networks plays a role in illnesses such as cancer, immune-related disorders, and developmental disorders. This information forms the foundation for focused therapy approaches meant to change these signaling networks in a way that is therapeutically advantageous.
RTK-mediated signal transduction and its implications in cellular function and oncogenesis
The activation of RTKs triggers multiple signaling pathways, including the Rac/MEKK1/MEKK/JNK pathway, Ras/Raf/MEK1/2/ERL/1/2 pathwayPI3K/Akt/NFKB pathway and JAK/STAT pathway.440,441 Multiple molecules were developed to target the downstream effectors of these pathways in order to inhibit tumor cells’ growth and proliferation (Fig. 5).

Outline of RTK families—downstream effectors—RTK inhibitors. The RTK downstream signaling is linked to several key biological pathways such as Rac/MEKK/JNK, Ras/Raf/MEK/ERK, PI3K/Akt/mTOR and NF-kB, JAK/STAT or Rho and calcium signaling, modulating gene expression and cellular metabolism. The biological processes that are influenced by the RTKs status are related to cell survival, migration, differentiation, growth, proliferation, and angiogenesis. Images created with BioRender.com
When binding the EGF, the EGFR is signaling through Ras/Raf/Mek/ERK, JAK/PI3K,STAT, and PKC/NFKB pathways leading to cell proliferation and survival.442 The signaling cascade initiated by the IR is focused on the PI3K/PIP3/Akt pathway, which modulates glucose production via FOXO, lipid and protein synthesis via mTORC1, glycogen synthesis via GSK3B and glucose uptake via TBC1D4.443 PDGFR signaling modulates Ras/MAPK, JNK/SAPK, PLCγ, PTEN, and Akt/PKB pathways promoting cellular rearrangements, stimulating cell growth, and motility.444
VEGFRs are key in the angiogenesis biological pathways and when binding their ligand, the downstream signaling modulates PKC/MEK/ERK1/2, PI3K/Akt, MAPK and Src/FAK pathways, promoting migration, survival, and proliferation.445,446Similarly, FGFR signaling is also responsible for survival, differentiation, and proliferation, via signal modulation of RAS/RAF/MAPK/ERK1/2 and PI3K/Akt pathways.447
CCK signaling respond to stimuli by modulating Ras/Raf/MEK1/2/ERK1/2, Rac/MEKK/JNK, and P38/MAPKAPK/Hsp27 pathways inducing changes into the actin dynamics, stimulates survival and proliferation of cells.448 Survival and apoptosis are also regulated by CD271 (NGFR) via TRAF6/JNK/JUN and RIP2/IRAK/IKK/NFκB pathways.449
HFGR/c-MET signaling is important in maintaining survival and increasing proliferation, due to the downstream signaling via Ras/Raf/MAPK, Paxillin/FAK, STAT3/5, and PI3K/Akt/mTOR pathways.450
EPHR signaling is very complex, with multiple cross-talks in order to sustain proliferation, differentiation, survival and migration by modulating JNK/STAT, ERK, PI3K/Akt, ABL1/Cyclin-D1, and Ras/ERK pathways.340,451 Migration, survival and proliferation are also regulated by the downstream signaling via AXL receptor, through ERK, SRC, P38, and PI3K/Akt pathway, AXL overexpression is correlated with poor overall survival in oncological patients, due to it implication is survival and proliferation.452
Besides its role in the tumor microenvironment of metastasis where is directly involved in the blood vessel permeabilization and inflammation,453 TIE receptors are signaling through PI3K/Akt pathway modulating cell growth and proliferation. Pathway that is regulated also by PTEN in the interchange between PIP2 and PIP3.454
Migration and invasion are key for tumor cells to spread and create metastatic sites, and RTKs are directly involved in these biological processes. When WNT5A binds to RYK receptor initiates the downstream signaling through calcium and through RhoA/ROCK/Akt/ERK/MAPK/P38 inducing an enhanced migration, invasion and inflammation.455
DDR1 and DDR2 have key implications in cell survival, growth and adhesion/migration via the downstream signaling through Ras/Raf/MAPK, PI3K/Akt/NFκB, JAK/STAT and Rho pathways.456 Rac/JNK, PI3K/Akt and Ras/Raf/MEK/ERK1/2 pathways are also regulated by the RET receptor signaling which is responsible for an enhanced proliferation and differentiation in normal and tumor cells.457,458
One of the last remaining orphan receptors, ROS1, are involved in proliferation and survival via signaling through Ras/MEK/ERK, PI3K/Akt and JAK/STAT pathways.230 Unlike ROS1, the ROR1 and ROR2 receptors are no longer classified as orphan receptors. Upon ligand binding, they initiate a signaling cascade through the PI3K/Akt/mTOR and Ras/Raf/MAPK pathways, leading to the inactivation of FoxO and the suppression of e-Cadherin. This series of events modulates crucial cellular processes including survival, proliferation, migration, and tumor development.459,460,461,462
ALK/LTK signaling pathway is responsible for the modulation of cell proliferation, invasion, migration, angiogenesis and apoptosis inhibition, via PI3K/Akt/mTOR, JAK/STAT/VEGF, Ras/Raf/MAPK, and PLCγ/PIP2/IP3 pathways in lymphomas, neuroblastoma and NSCLC.463,464
RTKs are crucial in various biological processes such as proliferation, cell survival, angiogenesis, migration, and metastasis formation, via their signaling through key biological pathways. The Ras/RAF/MAPK pathway leads to a cascade of phosphorylation events, which ultimately activate the mitogen-activated protein kinases (MAPKs) which influence proliferation, differentiation, and survival. ERK, the final effector, translocate to the nucleus and further phosphorylates transcription factors leading to gene expression for key pathways for cell development. Another key pathway that is modulated by RTKs is PI3K/Akt/mTOR, which involves the downstream effectors protein kinase B (Akt) and mammalian target of rapamycin (mTOR), regulating metabolism, cell growth and survival. The JAK/STAT pathway is involved in regulating gene expression and immune responses via STAT proteins. RTKs are important in cell signaling by activating these pathways which control the most important cellular processes (Fig. 5). Dysregulations in RTKs or in the downstream effectors can lead to various diseases, including cancer.17,58,411,465,466
Signaling pathways of MAPK in cancer
MAPK pathway is involved in various biological functions such as gene expression, the regulation of blood glucose levels, cell differentiation, tumor progression, drug resistance and survival.436,467 The key players in this pathway are RAS, RAF, MEK and ERK1/2 proteins. The cascade begins with Ras, a small GTPase and an upstream protein regulator, which activates Raf. This promotes MEK1/2 followed by the activation of ERK1/2 which regulates different transcription factors, modulating gene expression.468 In cancers, this pathway can be activated by cytokine mutations, overexpression of wild/mutant receptors (e.g., EGFR). Moreover, this pathway is important in apoptosis, by phosphorylating apoptosis regulators like Bad, Mcl-1, Bim, caspase-9 or Bcl-2.469,470
Ras proteins, encoded by the HRAS, KRAS, NRAS, and additional genes, with several subtypes, including H-Ras, N-Ras, K-Ras, M-Ras or R-Ras, each mediating different pathways within the cell. Specifically, K-Ras is more actively involved in the MEK/ERK pathway, while H-Ras tends to activate the PI3K/Akt pathway. On the other hand, M-Ras and R-Ras also participate in these pathways but are not as frequently mutated in cancers. Mutations in these Ras proteins are detected with different frequencies across cancer types. K-Ras mutations are notably common in a wide array of cancers, whereas N-Ras mutations are predominant only in some cancer.471,472
The Raf protein family, which includes A-Raf, B-Raf, and Raf-1, serves as upstream activators of the ERK pathway and also influences apoptosis. Mutations in Raf proteins are frequent in cancers, B-Raf mutations are specific for melanoma, colon cancer, ovary cancer or thyroid cancer (and not limited to these types of cancer). The mutated form of B-Raf is known to overactivate MEK/ERK signaling and can lead to the subsequent activation of Raf-1.469,473
MEK1 and MEK2 play a regulatory role in an array of cellular processes, including migration, differentiation, metabolism, proliferation, and apoptosis. Aberrant activation of MEK, particularly through mutation, can decrease the cytokine dependence of hematopoietic stem cells, potentially leading to malignancy.474,475
Lastly, ERK1/2 are MAPK superfamily members that mainly modulate apoptosis and proliferation. Activated ERK can phosphorylate various protein kinases in all cellular compartments and can finally lead to the phosphorylation of different transcription factors such as c-Myc, NFκB, c-Jun or Ets-1.469,476
Mutations in RAS proteins
Mutations in RAS proteins are very common events in tumor development, occurring most frequently at codons 12, 13, and 61. Around 30% of human cancers gain RAS mutations with K-Ras being the most mutated type. In CRC, K-Ras mutations are frequent, particularly in codons 12 and 13, a pattern that is similar to pancreatic cancer. It is hypothesized that K-Ras mutations occur in the early stages of tumor development. In lung cancer, K-Ras mutations are highly prevalent, and may be triggered by epigenetic factors such as chemicals. On the other hand, Ras mutations are rare in hematological malignancies.477,478,479,480,481
Several mutations are detected in RAS proteins.482 H-Ras displays several point mutations G12V, G12S, G12A, G13D, and Q61R, affecting codons 12, 13, and 61.483,484,485 K-Ras has mutations at codon 12 and 13 (G12D, G12S, G12R, G12A, G12V, G12C, and G13D).486,487 Mutations G12D and Q61K are observed in N-Ras and G22V and Q71L in M-Ras.488,489,490
According to Prior et al., five frequent mutations account for 70% of all Ras-mutant proteins (G12D, G12V, G12C, G13D, and Q61R).489,490 K-Ras G12C mutation was identified in multiple CRC and lung cancer samples491,492,493 and K-Ras G12D mutation in pancreatic adenocarcinoma492 and K-Ras G12D mutation in pancreatic adenocarcinoma.494,495,496,497
Mutations in RAF proteins
RAF MAPK protein is phosphorylated following RAS activation. RAF acts as a mediator for MEK1/2 activation which further modulates ERK. Among the isoforms, B-RAF is the most potent activator of MEK, whereas A-RAF and C-RAF (RAF-1) are less responsive to RAS stimulation compared to oncogenic Src. Both A and C isoforms require RAS-GTP on the cell membrane for activation.498,499
As in the case of RAS, RAF mutations are common in cancers like melanoma, CRC, ovary and thyroid malignancies.500,501,502,503 T1796A mutation in B-RAF gene change valine with glutamic acid in position 600 (V600E) and is the most frequent mutation.504,505 Three mutations were identified in C-RAF which induce MAPK cascade by activating C-RAF (G466E, G466V, and G596R).506
Smiech et al., synthesized the information related to RAF mutations.507 BRAF D594 mutations were identified as follows: D594A in CRC, D594E in melanoma and multiple myeloma, D594G and D594N in NSCLC, multiple myeloma and CRC, and D594H in NSCLC.508,509,510,511,512
Three classes of BRAF mutations were identified. Class I—V600E in most of the cancers: V600M in melanoma, skin adenocarcinoma, V600K/R/D in skin cancers. Class II—K601E and G469A in most of the cancers, G469V in lung cancer and lymphoma, G469R in skin cancer and melanoma, G464V in lung cancer and biliary tract cancer, L597Q in CLL, skin cancer and melanoma, K601N in hematological malignancies, L597V in colon cancer and biliary tract cancers, G464E in endometrial carcinoma, and K601T in GCs. Class III—D594N, N581S, G466V, G594G, and D594G in bladder cancer, hematological malignancies, CRC, glioma, pancreatic cancer, lung cancer, melanoma, and head and neck cancers, G466E in BC, skin cancer and melanoma, S467L in skin cancer and melanoma, G469E in oral cancer, skin cancer and melanoma, G466A in lung and skin cancer, G596R in lung and bladder cancer, D594A in liver and colon cancers, D594H in colon and lung cancer, G596D in glioblastoma and F595L in bladder cancers.507,513,514,515,516,517 Moreover, the CRAF mutation P261A was identified in lung cancer cell lines and seem to have oncogenic properties, stimulating ERK pathway.518
In a cohort of CRC patients assessed by NGS, several RAF mutations were identified, including S467L, R603L, G466V and V600M with different allelic frequencies.519 BRAF is mutated in over 60% of melanoma cases, with one single-point mutation accounting for 80% of the BRAF mutations. V600E is activating with 500% more times compared to wild type BRAF.505,520
Mutations in MEK1/2
The RAS/RAF/MEK/ERK pathway is crucial in tumor growth, with genomic alterations in RAS and RAF genes that activate MEK to activate a downstream signaling pathway. As a key protein in this pathway, MEK is a promising target for therapies that aim to modulate the pathway and results. MEK is the intermediary between RAS/RAF and ERK. With seven MEK enzymes identified, MEK1 and MEK2 are involved in the RAS/RAF/MEK/ERK pathway, and the primary role of these proteins involves phosphorylation and activation of ERK propagating the signals from receptors such as RTKs to the nucleus. This process regulates gene expression, important for cell survival, differentiation and proliferation.521,522,523
In colon cancer, resistance to MEK inhibitors, often can be due to the presence of specific mutations such as V211D in MEK1.524 Moreover, the C121S mutations in MEK1 lead to abnormal kinase activity compared to the wild type. These mutations are classified into three categories based on their RAF interactions. First class of MEK1 mutants with RAF dependent (D67N, P124L, P124S, L177V), the second class of MEK1 mutants, RAF regulated (E203K, L177M, C121S, F53L, K57E, Q56P, K57N, ΔE51-Q58, ΔF53-Q58) and the third class of MEK1 mutants RAF independent (ΔL98-I103, ΔI99-K104, ΔE102-I103, ΔI103-K104), where Δ is indicating a deletion of an amino acids sequence or the fact that the sequence is missing.525,526 It was demonstrated that P124L and Q56P mutations in MEK1 confer resistance to MEK and RAF inhibitors in melanomas.527
Mutations in ERK1/2
ERK1/2 is part of the MAPK pathway, playing a crucial role in cell differentiation, survival and migration. Activated by external stimuli, ERK1 and ERK2 are essential in the RAS/RAF/MEK/ERK signaling cascade. Once activated, ERK1 and ERK2 translocate to the nucleus and phosphorylate transcription factors regulating gene expression. Dysregulations in ERK1/2 can lead to cancer progression, either due to mutations in upstream proteins or overexpression of pathway-triggering receptors, and the result is an uncontrolled cell growth.528,529,530,531
ERK1/2 is part of the MAPK pathway, playing a crucial role in cell differentiation, survival and migration. ERK1 and ERK2 are activated by extracellular stimuli that are transferred in the downstream cascade via RAS/RAF/MEK/ERK pathway. Once activated, ERK1 and ERK2 translocate to the nucleus and phosphorylate transcription factors regulating gene expression. Dysregulations in ERK1/2 can lead to cancer progression. Even if the dysregulation is a result of mutant upstream proteins or due to overexpression of the receptors that trigger the pathway, the result is an uncontrolled cell growth.528,529,530,531ERK1/2 mutations can be responsible for the enhanced tumorigenic phenotype of the cells and lead to different cancers.532 ERK2 E322K mutation in present in cervical and head and neck cancer.533,534,535
PAM pathway in cancer
PI3K/Akt/mTOR pathway has a crucial role in cell survival, growth, and cell cycle. Its regulation involves crosstalk with other pathways, and when experiencing abnormalities, the downstream signaling can generate a landscape suitable for malignant cell development. This pathway is frequently activated in cancers and can contribute to resistance to therapy. Dysfunctions in PI3K activity, loss of PTEN or Akt overactivation can lead to cancers and drug resistance.536,537,538,539,540,541
Mutations in PI3K and PTEN
PIK3CA is the gene that encodes the p100alpha catalytic subunit of PI3K. Mutations in this gene lead to over-activation of the PAM pathway and uncontrolled cell growth. The D725N and H1047Y mutations in PIK3CA often co-occur with mutations in RAS/RAF pathway.519
In a Phase I clinical trial (NCT01219699), hotspot mutations E542K and H1047R/L in PIK3CA were identified in a BC patient. PTEN loss was noted in lung metastasis, while the primary tumor showed E542K and G725G mutations. Metastatic sites exhibited PTEN deletions and mutations S339fs and K342_splice.542,543
PTEN is a key tumor suppressor which inhibits cell proliferation and increases sensitivity to apoptosis.544,545 Alterations in PTEN function have been identified in a wide spectrum of tumors, indicating that it may control tumorigenesis.546 PTEN dysfunction leads to a prolonged PI3K/Akt signaling which induces abnormal cell growth and proliferation.547
Mutations in AKT
Three isoforms of Akt are encoded by three genes: AKT1, AKT2, and AKT3. AKT1 is involved in survival and antiapoptotic processes, is a key protein in signaling for tissue growth. Due to its involvement in antiapoptotic signaling, it may promote tumor development. AKT2 signals through the insulin pathway and is involved in glucose transport. While AKT3 is more expressed in brain and neural tissue.548,549,550,551,552
One recurrent mutation in AKT is E17K, present in all three isoforms. The AKT1 E17K mutation is responsible for leukemia development in mice models,553 moreover it can induce oncogenic transformation in normal breast epithelial cells (MCF10A).554,555 This mutation is present in different types of cancer, with different frequencies, such as BC, endometrial cancer, skin and bone cancer, thyroid and colon cancer.556,557,558,559
The L52R mutation in AKT1 was detected in endometrial cancer, and D323G in BC that is ER positive and HER2-negative. In prostate cancer, an AKT2 mutation, L78-Q79ins may be involved in therapeutic sensitivity.560,561 In melanomas, E17K and Q79K mutations in AKT1 increase the resistance to vemurafenib therapy.562
Mutations in mTOR
The conserved serine/threonine kinase, mTOR, has two distinct protein complexes, mTORC1 and mTORC2 with specific roles in cell signaling. mTORC1 is involved in regulating cell growth, metabolism, and protein synthesis. The downstream effectors S6K and 4E-BP1 are phosphorylated to promote cell proliferation. mTORC2 regulates migration, cytoskeletal reorganization, and cell proliferation. In cancer, dysregulations in mTOR pathway are leading to uncontrolled cell growth and tumor progression. Mutations in the upstream regulators or in mTOR can sustain a prolonged activity contributing to oncogenesis.563,564,565
mTOR mutations were identified in T cells (MOLT16 T cell leukemia cell line), endometrium cells (HEC59, JHUEM7 endometrial carcinoma cell lines) and kidney cells (SNU349 RCC cell line). The mutations C1483Y and R2430M were detected in T cell leukemia, R460*, S2215Y and E1799K in endometrial carcinoma cells, with E1799K also detected in kidney cancer cells.566 The mTOR mutation L2209V was identified in a specimen of large cell neuroendocrine carcinoma, and it was showing the ability to transform fibroblasts into tumor-like cells.567 The H419R mTOR mutation was detected in thyroid carcinoma cells and G2359E in melanoma tumors.568
In RCC, the Y1974H mTOR mutation was identified in a metastatic site in one 57-year-old patient.569 According to Ghosh et al., three-point mutations in RCC were identified in mTOR and seem responsible for stimulated cell proliferation (Y1463S, K1452N, A1519T).570 H1968Y and P2213S mTOR mutations were described by Kong et al., as gene alterations in melanoma.571 L2185A mTOR mutation confers resistance to therapy in colorectal and lung cancers.572 In a resistant to treatment thyroid cancer tumor, from a female patient, mutation F2108L was highlighted by Wagle et al., and it appears responsible for the increased resistance to treatment, in conjunction with mutations in other proteins.573
JAK/STAT pathway in cancer
Janus Kinase family includes JAK1, JAK2, JAK3, and TYK2. Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway controls cellular response to signals like growth factors and cytokines. Activated by IFNγ, JAK1, and JAK2 phosphorylate STAT1/STAT3, modulating the inflammation and immunity. JAK1 also phosphorylates STAT5/STAT1, impacting the antigen presentation and antiviral response. STAT3/STAT5 is further involved in mechanisms related to survival, proliferation and angiogenesis.574,575,576,577,578,579,580
JAK1, JAK2 and TYK2 are ubiquitously expressed, while JAK3 is mostly expressed in hematopoietic cells.581,582,583,584,585 TYK2 is implicated in IFN signaling through Toll receptor, mediating the response to LPS.586 JAK1 is essential for IL2, IL4, IL15, IL21, and other interleukins, which are also dependent upon JAK3. JAK1 is also key for IL6 and IL11, leukemia inhibitory factor (LIF) and ciliary neurotrophic factor (CNF), INFs and granulocyte colony stimulating factor (G-CSF). On the other hand, JAK2 is essential for hormone-like cytokines such as prolactin, erythropoietin, IL3, IL5, GM-CSF.587
The STAT proteins family consists of seven members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6.588,589 STAT1 is key for IFNγ signaling and STAT4 for IL12 pathway. Both STAT1 and STAT4 are important factors for Th1 cells polarization,590 STAT1 enhancing cell division.591 STAT6 modulates IL4 and IL13 via Th2 signaling and may inhibit Th1 polarization.590,592 STAT4 can also activate NK cells while STAT5 can promote white blood cells formation, and STAT6 promote B cell proliferation and survival.591 STAT3 is excessively activated in AML, multiple myeloma, and various solid tumors like breast, colon, liver, head and neck, lung, and ovarian cancers, correlating with unfavorable clinical outcomes.593,594,595,596,597,598
STAT5 activation has an important role in tumorigenesis. Mutations in STAT5 are few and almost all of them are detected in hematological cancers. When overactivated, STAT5 can enhance the epithelial to mesenchymal transition, induce anti-apoptotic signals and promote invasion and metastases.599,600,601
Mutations in STAT proteins
STAT3 and STAT5b are members of the STAT family that are often activated in cancers. The activation of STAT3 can result from upstream kinases, lack of negative regulation, somatic mutations or positive feedback loops. The STAT3 gene is frequently mutated in hematopoietic neoplasms like T-cell large granular lymphocytic leukemia (T-LGL), PTCL, diffuse large B cell lymphomas (DLBCL), anaplastic large T-cell lymphoma (ALCL), and chronic NK lymphoproliferative disorders (NKTCL).599,602,603,604,605,606,607
Mutations in the SH2 domain of STAT3 have been identified across several types of lymphomas: Y640F, D661Y, D661I, D661H, G618R, S614R, and N647I in T-LGL; Q743H in PTCL; D661Y, D661H, N647I, G618R, S614R, and N647I in ALCL; Y640F and I498Y in cutaneous T-cell lymphoma (CTCL); and D661Y, D661H, S614R, and A703T in NKTCL.599 Numerous mutations within STAT5b have been observed across various malignancies, including N542H detected in TLGL, NKTCL, and CTCL, Y665F exclusively in TLGL and NKTCL, Q706L in CTCL, T-ALL, and T-PLL. Additionally, unique mutations such as Q743H in PTCL-NOS, T648S in T-ALL, and R659C and Y665H in T-PLL were identified. Notably, all STAT5b mutations were localized within the SH2 domain of the protein.599
Mutations in JAK1 proteins
Arulogun et al., identified V658I missense mutation in JAK1 in a patient with myeloproliferative neoplasm.608 Additional activating mutations of JAK1, including V658F, V658L, and V658I were reported in T cell prolymphocytic leukemia and ALL.609,610
Flex et al. evaluated JAK1 mutations in ALL and identified several mutations across different subtypes. In adult B-ALL mutations such as I62V, K204M, A634N and R724H were identified, while no mutations were detected in childhood B-ALL. Moreover, adult T-ALL patients presented eight mutations: I62V, R360W, S512L, A634D, R724H, R879S, R879C and R879H; childhood T-ALL only presented L653F mutation.611
Furthermore, in both B- and T-ALL, several JAK1 mutations were described in different protein regions: I62V,611 K204M,611 R360W,611 S512L,611 L624_R629>W,609 A634D,611 S646F,609 L653F,611 V658F,609 R724H,611 L783F,611 R879C,611 R879H,611 R879S.611 Additionally, V623A and T468S mutations were identified in AML.612
Mutations in JAK2 proteins
A frequently detected mutation in JAK2 is the V617F missense mutation, commonly found in myeloproliferative neoplasms.613,614according to Arulogun et al., 95% of polycythemia vera, 50% of essential thrombocythemia and primary myelofibrosis harbor this mutation.608
Haan et al. synthesized multiple mutations in Janus Kinases in hematological disorders.615 In megakaryoblastic leukemia, M535I and T875N mutations were identified.616,617,618 Additional JAK2 mutations in polycythemia vera includeF537I,619 K539L,620 F537-K539delinsL,620 H538-K539delinsL,621 H538D + K539L + I546S,622 H538-K539del,622 V536-I546dup,622 V536-I546dup11,621 F537-I546dup10 + F547L,621 I540-E543delinsMK,621 R541-E543delinsK,621 N542-E543del,621 E543-D544del,621 D544-L545del,622 C618R + V617F.623 In B-ALL mutations detected include L611S,624 I682F,609 I682AQG,609 R683G,609 R683S,609 R683T,625 R683K,626 R867Q,609 D873N609 andP933R.609
Mutations in JAK3 proteins
Recurrent mutations in both JAK1 and JAK3 have been detected in several hematological malignancies by sequencing studies. More than 10% of patients diagnosed with T-ALL have at least one mutation in the JAK3 gene.627
Additionally, one JAK3 mutation A572V was identified in acute megakaryoblastic leukemia.628 Among these mutations, several others were detected in the JAK3 protein: P132T,628 Q501H + R657Q,629 A573V,630 M576L,631 A593T631 and V722I617 in acute megakaryoblastic leukemia, while S789P609 was detected in childhood B-ALL.
Mutations in TYK2
Mutations in TKY2 lead to an upregulation of the downstream STAT signaling.632 According to Tomasson et al., G363S mutations were specific for TYK2 proteins in AML.633 Multiple mutations were identified in the TYK2 protein, in nonmalignant diseases such as juvenile idiopathic arthritis, rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis or systematic lupus erythematosus and we will not mention them.634
Therapeutic approaches in targeting RTKs
US Food and Drug Administration (FDA) approved small molecule RTK-inhibitors
RTK-inhibitors are used to treat a variety of malignancies (hematologic, pulmonary, gastrointestinal, endocrinologic and uro-genital cancers) both as first-line therapies such as osimertinib, alectinib or entrectinib and in advanced or metastatic disease such as capmatinib, tepotinib or crizotinib. The FDA-approved inhibitors are either non-selective, targeting multiple RTKs (imatinib and sorafenib), with dual specificity, targeting only 2 RTKs with high specificity (lapatinib and afatinib) or highly selective to only one RTK (gefitinib). Table 11 summarizes the 46 RTK-inhibitors we identified as US FDA-approved by the end of 2023. We specified year of approval; the cancer types the drugs were approved for and the target RTKs. Guidelines and a thorough list of indications are outside the scope of this review; each country and medical system has its own set of guidelines for these cancers.
Limitations of current RTK-inhibitor therapies
A variety of factors have to be taken into consideration when choosing RTK-inhibitors: patient characteristics (age, fitness, social and family history, comorbidities, previous lines of treatment, occupation and working habits, presence of risk factors); the characteristics of the tumor (staging and tumor extension, tumor morphology, tumor genetics/genomics); the tumor microenvironment that both directly (by interactions with the tumoral cells) and indirectly (by influencing non-tumoral cells, such as fibroblasts) affects efficacy of RTK-inhibitors; pharmacokinetics and dynamics of the RTK-inhibitor; the presence of treatment-related toxicities; acquisition of primary or secondary drug resistance; cost-efficacy; drug availability; patients’ and physicians’ preferences. Due to the paucity of comprehensive clinical data, selecting the best RTK-inhibitor monotherapy or combination therapy is another constraining factor. These treatment-related limitations will be discussed in detail in the upcoming sections, along with some strategies to help overcome these challenges.
Drug resistance mechanisms to RTK-inhibitors
Cancer cells may have preexisting genetic mutations or alterations that make them less responsive to the inhibitory effects of RTK-inhibitors. This inherent or natural insensitivity of tumoral cells refers to innate or primary resistance. These mutations may affect the target pathway or activate alternative signaling pathways, allowing the cancer cells to bypass the inhibitory effects of the drug. There are several mutations associated with innate resistance to EGFR-inhibitors, such as the oncogenic RAS mutations. The expression of AXL, for instance, is another example of innate resistance to c-Kit (regorafenib) or ROS1-inhibitors (crizotinib, repotrectinib).635,636 A recent study demonstrated that the cell cycle phase is also a regulator of innate sensitivity to RTK-inhibitors. Overexpression of cyclin-D1, a cell cycle regulator, for instance, induced resistance to osimertinib.637 Other biological factors, such as the localization of the tumor are also a predictor of innate resistance. Even after rectifying for stage and tumor size, right-sided CRCs, for example, have a worse prognosis and respond less to EGFR inhibitors.638,639
Another major limitation of current RTK-inhibitors is that resistance may be also acquired during treatment through various mechanisms, including the development of new mutations, activation of compensatory signaling pathways, or changes in the expression of drug efflux pumps. These changes enable cancer cells to survive and proliferate in the presence of the drug. Understanding the specific molecular mechanisms driving both innate and acquired resistance to RTK inhibitors is crucial for the development of more effective therapies. Ongoing research aims not only to identify crucial resistance mechanisms by multimodal clinical, biochemical, histologic, genetic, epigenetic and genomic approaches but also to identify biomarkers and molecular signatures associated with resistance, which can help guide treatment decisions and the development of novel targeted therapies. It is important to note that resistance mechanisms can vary between different types of cancer and even among individual patients with the same type of tumor. Personalized medicine approaches, such as molecular profiling of tumors, are increasingly used to tailor treatment strategies based on the unique characteristics of each patient’s cancer. Resistance to tyrosine kinase inhibitors has been excellently reviewed by Yang et al.640
Thus, in our review we focused on drug resistance pathways specifically to RTK-inhibitors, with a focus on recent findings and novel mechanisms. Table 12 summarizes tumor types and potential mechanisms underlying therapeutic resistance.
On-target mutations as driver of resistance
We have already covered a number of additional on-target genetic alterations that have been described in previous sections. Studies published in the last 5 years have identified new mutations that promote RTK-inhibitor resistance. For instance, resistance to first-generation EGFR-inhibitors may be associated with EGFR point-mutations (T790M, C797S, L792F, V843I).641 Many of them, such as the C797S cis mutation, may reduce sensibility to newer generation inhibitors, such as osimertinib too.642 FGFR-inhibitors, such as erdafitinib, futibatinib or pemigatinib, may also show decreased efficacy after acquisition of several driver mutations in the FGFR3 gene (N540K, V553L, V553M, V555L, V555M).643 Several FLT3 mutations, such as the N701K, F691L induce resistance to gilteritinib, quizartinib or both.644 The same mechanisms (Y1248H, D1246N) have been observed in the case of c-Met-inhibitors, such as crizotinib or capmatinib.645 Double-hit mutations, such as HER L869R/T862A; L869R/L755S; or L755S/T862A, reduce susceptibility to HER2-inhibitors, as seen with neratinib.646 Chromosomal rearrangements of oncogenes may also cause acquired resistance. For EGFR-inhibitors, most of these oncogenic fusions involve the ALK, BRAF, FGFR, NTRK, RET, and ROS1 genes.647 FGFR2 rearrangements have been shown in pemigatinib-resistant patients.643 In order to allow the best possible drug selection and customize personalized therapy, it will be crucial in the future to identify not only oncogenic alterations but their correlation with drug resistance as well.
Off-target genetic mutations as driver of resistance
Amplification of the MET and HER2 genes or mutations in the BIRC5 or TP53 genes lead to EGFR-inhibitor resistance.641,648,649,650,651,652 Mutations of ALK, EGFR, and KRAS, ALK copy-number gains or amplification of KIT have been associated with resistance to ALK- or c-Met inhibitors, such as crizotinib or capmatinib.645,653,654,655,656 Also, MET amplification and overexpression was correlated to resistance to ALK-, ROS1, and RET-inhibitors.657 BRAF-fusion is another mutation reported as a resistance mechanism to osimertinib.658
Epigenetic changes
Genes involved in DNA methylation, such as HOXB9 have been correlated to EGFR-inhibitor resistance.659 Expression level of epigenetic regulator proteins is also a driving mechanism of RTK-inhibitor resistance (Fig. 6). Gefitinib resistance, for instance, was linked to overexpression of the Vir-like m6A methyltransferase-associated (KIAA1429) protein that regulates methylation processes and synthesis of regulatory RNAs.660 Loss of these epigenetic regulators, such as loss of the transcriptional repressor chromobox homolog 5 (CBX5) leads to resistance to EGFR-inhibitors by overexpression of antiapoptotic molecules.661 Another novel epigenetic change linked to osimertinib resistance is chromatin remodeling with changes in chromatin accessibility.662 Other papers demonstrated that alterations in long-coding RNAs may lead to gefitinib resistance.663 Long non-coding RNAs may also play a significant role in RTK-inhibitor resistance by disturbing intercellular communication, the tumor microenvironment, by induction of further epigenetic modifications or by activation of alternative pathways. The role of these RNAs has yet been described for gefitinib, erlotinib, afatinib, and osimertinib, but future research will most likely expand the list beyond EGFR-inhibitors.664 Same resistance mechanisms to EGFR-inhibitors have been revealed by exosomes, extracellular non-coding, long non-coding, circular and micro-RNAs.665 Mutations in pseudogenes are suggested as possible resistance mechanisms too, such as the expression of DUXAP10, which induces resistance to gefitinib.666 Targeting RTKs linked to epigenetic regulators may greatly improve therapeutic efficacy and regulate resistance mechanisms. Epigenetics is an important area of research nowadays. Further basic and translational research data are needed.

Main resistance mechanisms to RTK inhibitors. The hypoxic tumor cells that express HIF have overstimulated proangiogenic status and promote tumor vascularization and resistance to RTK inhibitors (left). The secreted exosomes containing genetic information (RNAs, long noncoding RNAs) stimulate epithelial to mesenchymal transition disturbing cell adhesion and increasing motility and invasiveness. The stimuli from the hypoxic cells influence other cells behavior by inducing RTKs reorganization, changes in their conformation and protein degradation. Oxidative stress (as an external stimuli) disrupts the mitochondrial metabolic pathways changing the balance within the cells leading to drug resistance. Drug uptake and transport are affected by the changes induced in the carriers and membrane transporters which may disrupt the drug uptake leading to an increased resistance to RTK inhibitors by externalizing the drugs via active mechanisms. The low-right box depicts the resistance to therapy induced by genetic mutations by epigenetic changes that induce mutation in the genes encoding the RTK proteins or other downstream proteins involved in key biological processes such as survival, proliferation or programed cell death. Images created with BioRender.com
Activation of metabolic pathways
In case of first-generation EGFR-inhibitors (gefitinib, erlotinib), there are several pathways that may lead to resistance when down- or upregulated, such as phosphorylation of Src family kinase, overexpression of the hepatocyte growth factor, activation of the AXL, NF-kB, GAS6, ADAM17, NOTCH, P53, Wnt, PI3K/AKT/mTOR, RAS-RAF pathways.655,667,668,669,670,671,672 JAK/STAT and PI3K/AKT-pathway upregulation, and overexpression of interleukin-17A, IGF1R or FGFR1 seem to play a role in development of resistance to afatinib, a second-generation EGFR-inhibitor.652,673,674,675,676 Activation and upregulation of the EGFR-pathway, in contrast, leads to adaptive resistance to ALK- and ROS1-inhibitors.677,678 Recent studies demonstrated the role of upregulation of NADPH oxidase 4 (NOX4) in resistance to gefitinib and osimertinib.679 Upregulation of HER3 was shown to induce resistance to osimertinib.680 Upregulation of BIM or EGFR are linked to ALK-inhibitor resistance.681 Alternations in the IGF-pathway may lead to several intracellular modifications that eventually lead to decreased sensitivity to EGFR-, c-MET-, and HER2-inhibitors.682 Upregulation of immune-checkpoints, such as PD-L1 via the mTOR pathway have been recently associated with development of resistance to VEGFR-inhibitors in RCC.683 Urothelial cancers often show resistance to FGFR-inhibitors. One of the main mechanisms revealed is the EGFR and the PI3K/AKT/mTOR pathway bypass activations.643 In the case of the HER2-inhibitor lapatinib, a calcium-dependent activation of the RelA (NF-kB) pathway has been observed in resistant cells.684 In the case of GIST, the implication of the sphingophospholipid metabolic pathway is suggested to cause resistance to the non-selective inhibitor imatinib.685 The number of specific receptors on the surface of tumor cells also influences sensitivity to RTK-inhibitors. For example, the levels of CXCR4 on the surface of acute myeloid leukemia cells regulate responses to the FLT3-inhibitor quizartinib.686 Interestingly, overexpression of tumor suppressor proteins may also lead to resistance. In the case of gefitinib, upregulation of CircSETD3, a tumor suppressor in hepatocellular carcinoma, leads to resistance in NSCLC.687
Activation of vascular pathways
Hypoxia in the tumor microenvironment leads to expression of several hypoxia-induced transcription factors (HIF) that eventually lead to overexpression of downstream proangiogenic factors, such as VEGF, PDGF, or transforming growth factor α. These molecules eventually lead not only to increased tumor vascularization but also to resistance to kinase inhibitors.688,689 A switch from angiogenetic to non-angiogenic pathways may induce tumor progression and by activating alternative pathways it may also cause resistance to RTK-inhibitors. There are two main non-angiogenic mechanisms: the vascular co-option, where tumor cells use pre-existing vasculature or the vascular mimicry where vessel-like structures are formed inside the tumor. Both have been demonstrated to cause sunitinib resistance, for instance.690,691,692 The exact mechanisms are not well understood yet. More research on other RTK-inhibitors is required to determine the importance of vascular pathways in the emergence of drug resistance.
Cell cycle aberrations
Gene amplifications, especially in the genes encoding cyclin D1, D2, E1, and CDK 4 and 6; and deletions in the CDK inhibitor 2A gene (CDKN2A), are the two main cell-cycle-related changes in cancer causing deregulations in the cell cycle checkpoints.693 These mutations may develop during RTK-inhibitor treatment and lead resistance.647 Resistance to HER2-inhibitors, for instance, may be caused by cell cycle aberrations. The inhibition of CDK4 and 6 was shown to resensitize BC cells to HER2-inhibition.694
Modulation of death pathways
Cellular death pathways refer to the intricate and regulated series of events that lead to the demise of a cell. These pathways are fundamental in maintaining the proper functioning and homeostasis of multicellular organisms but may also be implicated in development of cancer and also resistance to RTK-inhibitors. Oxidative stress induced growth inhibitor 1 (OSGIN1) is a regulator of apoptosis, for example. Its overexpression inhibits apoptosis and was shown to induce resistance to gefitinib.695 BCL2, another antiapoptotic protein is overexpressed in HER2-inhibitor resistant cells.696 Beyond apoptosis regulation, modulation of autophagy may also contribute to resistance. When EGFR is activated, autophagy is inhibited; conversely when EGFR is suppressed, autophagy can be triggered. There is increasing proof linking autophagy to resistance to EGFR-inhibitors. Induction of autophagy may be relevant in development of erlotinib and crizotinib resistance, for instance.697,698 Another paper demonstrated the role of improved autophagy in gefitinib desensitization.699 However, EGFR T790M mutated cell lines showed increased sensitivity to RTK-inhibitor when these were combined with histone acetyltransferases, that activated the autophagy pathway.700 This data suggests that both up- and downregulation of autophagy may play a role in development of resistance to RTK-inhibitors. Autophagy is also involved in resistance to FLT3-inhibitors and sorafenib.701,702 Pyroptosis is a form of programmed cell death characterized by a pro-inflammatory response. Unlike apoptosis, which is a non-inflammatory form of cell death. Inhibition of pyroptosis was linked to gefitinib and EGFR-inhibitor resistance.703
Metabolic reprogramming
Proteomic and transcriptomic data reveal that resistance to TKIs is associated with several metabolic changes: increased oxidative stress responses, hypoxia signatures, disruptions from the mitochondrial metabolic pathways to cytosolic one, such as glycolysis or the pentose phosphate pathway.704,705
Histological transformation
Epithelial-to-mesenchymal transition (EMT) is one of the mechanisms observed in RTK-inhibitor desensitization. During EMT epithelial cells undergo molecular changes that lead to the acquisition of mesenchymal characteristics leading to reduced cell adhesion, increased motility, invasiveness, and stem cell properties. EMT has been demonstrated to be associated to EGFR- (afatinib) and ALK-inhibitor (crizotinib) resistance.673,706 EMT-caused resistance to sunitinib has been demonstrated too.707 Inactivation of the tumor suppressors RB1 or TP53 during RTK-inhibitor therapy may result in entire histological shifts, such as from NSCLC to a small-cell subtype.704
Conformational changes of target RTKs
A novel concept of resistance to highly selective RTK-inhibitors, such as gilteritinib is the conformational changes occurring in the active site of the kinases.708 Further studies are required to identify the role of atomistic details behind resistance to tyrosine kinase inhibitors.
Lysosomal sequestration of the RTK-inhibitor
There are a few articles reporting the role of lysosomal sequestration of inhibitors in development of resistance by a decrease in the concentration of the drug.709,710 A recent paper, however, concluded that these hypotheses cannot be reproduced in vitro, and this mechanism does not actually mediate resistance to imatinib and other RTK-inhibitors.711
Modulation of drug uptake and transport
Modulation of transporter molecules has also been reported as a metabolic resistance mechanism. Overexpression of solute carrier family 12 member 8 (SLC12A8), an ion transporter, has been demonstrated to play a role in development of resistance of EGFR-inhibitors by activating alternative pathways.712 Polymorphisms that affect activity of the ATP-binding cassette (ABC) or solute carrier (SLC) membrane transporters may lead to inefficient drug uptake and RTK-inhibitor resistance.713,714 This mechanism has been demonstrated in the case of sunitinib, for instance.715
Tumor microenvironment
Not only metabolic reprogramming but also reprogramming of the tumor microenvironment mediates resistance to RTK-inhibitors.716 Resistance is induced by the microenvironment by multiple mechanisms: activating alternative pathways, suppressing T-cells, promoting EMT or by polarization of macrophages into inhibitory, suppressor cells.717 These changes have been observed in EGFR-inhibitor (gefitinib, osimertinib) resistance.717 Novel macrophage renewal modes have also been identified, with distinct macrophages contributing to various resistance mechanisms in the same tumor microenvironment.718 Extracellular matrix stiffness is another factor contributing to progression and RTK-inhibitor resistance. Upregulation of adaptor-related protein complex 1 subunit sigma 1 (AP1S1), for instance, leads to matrix stiffness and resistance to erlotinib.719 Chen et al. extensively reviewed the tumor microenvironmental changes following tyrosine kinase inhibitor therapies, a detailed description of these is out of the scope of our paper.720
Toxicities of RTK-inhibitors
Adverse events can be classified as on-target (due to inhibition of the target receptor) or off-target (due to simultaneous inhibition of multiple kinases), a positive correlation existing between the risk of toxicity and the number of RTKs inhibited.721 Thus, a limitation of non-selective RTK-inhibitors is the increased risk for all adverse events. RTK inhibitors, and tyrosine kinase inhibitors, in general, show the highest relative risk (RR = 5.6) of high-grade cardiac toxicity among anti-cancer drugs.722 Selective RTK inhibitors that play a major role in development of cardiac manifestations target HER2 and VEGFR.723 In contrast with anthracyclines, tyrosine kinase inhibitors were thought to induce cumulative dose-independent, late-onset, non-progressive and partially reversible cardiac toxicities. Recent studies, however, invalidate these theories since several TKIs may cause irreversible cardiac dysfunction.724 The mechanism is not well known. HER2-inhibitors, such as lapatinib and afatinib, are mostly associated with heart failure with left ventricular dysfunction and systemic hypertension.725 VEGFR (and PDGFR) inhibitors may cause systemic hypertension, the highest incidence being reported with lenvatinib but also observed with cabozantinib, vandetanib and regorafenib; QTc prolongation (axitinib, regorafenib), pulmonary hypertension (lapatinib, lorlatinib) and arterial thrombosis added to the heart failure or systemic hypertension that is associated to all VEGFR inhibitors. Pazopanib was also reported to induce apical ballooning syndrome and fulminant heart failure.726 EGFR inhibitors may also induce arterial thrombotic events (erlotinib) or QTc prolongation in case of osimertinib.727,728 ALK-inhibitors (crizotinib, ceritinib) and pazopanib have been associated with bradycardia.722,729 Non-selective or multi-kinase inhibitors are linked to all beforehand described cardiac events: heart failure (dasatinib), QTc prolongation (nilotinib), right ventricular dysfunction and pulmonary hypertension (dasatinib, bosutinib, ponatinib), accelerated atherosclerosis (ponatinib, nilotinib), increased risk of myocardial infarction and/or cerebrovascular events (ponatinib, nilotinib, sorafenib) and venous thromboembolism.727,728,730,731 Pleuro-pericardial effusions are frequently observed with dasatinib, imatinib, ponatinib, bosutinib and FLT3 inhibitors.732,733
As a conclusion, cardiac toxicity is a significant concern, as it can limit the use of these drugs, especially in patients with pre-existing heart conditions. All inhibitors may also cause dose-dependent, mild/moderate, reversible diarrhea, constipation, nausea, vomiting, abdominal discomfort, or anorexia. These symptoms are more common in solid tumors compared to the hematologic ones.734 Elevated liver enzymes are common in the first months of treatment. These values usually normalize without any intervention. Highest levels have been observed for nilotinib, bosutinib and ponatinib but also for ALK-inhibitors, such as ceritinib and brigatinib.735 Skin reactions are one of the most common during RTK-inhibitor therapies. They are usually self-limited. However, dose reduction, drug desensitization or cessation due to severe skin lesions have been reported in the case of erlotinib, afatinib and imatinib.736,737 In terms of metabolic and endocrine changes, RTK-inhibitors may cause several electrolytic/glycemic/hormonal imbalances: hyper-/hypo-glycemia (imatinib, sunitinib, nilotinib), hypophosphatemia (imatinib, bosutinib, dasatinib, ponatinib), hypothyroidism (imatinib, sunitinib) or hypogonadism with gynecomastia (sunitinib).738,739,740 Pancreatitis has been reported in a higher incidence in sorafenib-exposed patients.741 Pulmonary toxicity may be a significant limitation of RTK-inhibitors since interstitial lung disease (in case of EGFR- and ALK-inhibitors) or drug-induced pneumonitis (following EGFR- or c-MET inhibitors) may be fatal side effects in some patients.742,743 Renal adverse effects are usually not life-threatening but severe proteinuria (mostly following VEGFR-inhibitors such as sorafenib, sunitinib, pazopanib), albuminuria (sorafenib, sunitinib) or a transient decline in glomerular filtration rate (imatinib, bosutinib) have been reported.744,745 RTK inhibitors used for treatment of chronic myeloid leukemia (due to the simultaneous BCR::ABL-1 inhibitory effects), such as imatinib, bosutinib, dasatinib, nilotinib and ponatinib, may impact blood cell counts, leading to anemia, neutropenia, or thrombocytopenia.746,747 This can increase the risk of cardiovascular complications, bleeding, and infections. Because ketoconazole and other antimicrobial drugs may increase plasma levels of RTK-inhibitors, such as sorafenib, regorafenib, lenvatinib, or cabozantinib, while also causing cytopenias, their use may be problematic patient populations.748 Another drawback of currently available RTK inhibitors is their usage during pregnancy. Certain drugs are classified as teratogenic (imatinib, erlotinib, lapatinib), while others have not been tested in these circumstances and hence cannot be advised for pregnant women.749,750
The application of personalized pharmacogenomics for targeted therapy or drug localization and targeted biodistribution by organ- or tissue-specific nanocarriers may be feasible options in the future to minimize the incidence rate of side effects. Reducing toxicities may also be facilitated by the use of highly selective RTK-inhibitors, as discussed in the paragraphs that follow.
Approaches to overcome limitations
Early detection of relapse/resistance to treatment
Early identification of resistance-driver mutations in circulating tumor DNA (ctDNA) may be an option because switching to another therapy, even before relapse or advancing to a metastatic state may improve outcomes. A recent study demonstrated resistance prediction to ALK-inhibitors in NSCLC patients.751 Another study found that an increase in ctDNA levels at the start or during treatment with EGFR inhibitors was associated with resistance and might be used to predict response to therapy in the future.752 We found no papers on early prediction of resistance for other RTK inhibitor classes, highlighting the need for additional study in this field.
Modulation of pharmacokinetics and biodistribution by nanocarriers
Nanodrug formulations or nanotools as delivery agents, such as liposomal osimertinib, may improve response rates and even overcome resistance because of the increased uptake, targeted distribution, and improvement of therapeutic indexes.753,754 Afatinib-loaded nanoparticles showed higher drug concentrations in the tumor tissue, both in lung- and liver cancer patients.755 Lapatinib nanoformulations in breast cancer showed higher efficacy due to preferential accumulation in the cancer cells.756 Nanoparticles also enable efficient co-delivery of drug combinations, for instance gefitinib in association with cyclosporin A in NSCLC.757 The crizotinib-dasatinib combination showed increased permeability through the blood-brain barrier when delivered using micellar formulations in glioblastoma patients.758 Nanocarriers may also reduce the incidence of adverse events of RTK-inhibitors, for example imatinib showed significantly decreased cardiotoxicity when loaded in nanoparticles.759 There are several other examples of the utility of these novel agents. Smidova et al. excellently reviewed their advantages and current clinical trials on RTK-inhibitor containing nanoparticles.759
Modulation of downstream or parallel signaling pathways
In case of acquired resistance, targeting alternative oncogenic pathways in combination with RTK-inhibition may overcome resistance mechanisms. Targeting the NOTCH pathway with gamma-secretase inhibitors may increase the efficacy of osimertinib in non-small-cell lung cancer.670 Targeting antiapoptotic proteins, such as survivin (BIRC5) in combination with EGFR-inhibitors, may also be a therapeutic opportunity in the future.661 In the case of RCC, hypoxia-mediated changes play an important role in the development of resistance. Thus, cabozantinib in combination with hypoxia-induced transcription factor inhibitors is being investigated to inhibit angiogenic vascular pathway activation. Targeting CD70, a marker of EMT, is tested preclinically in EGFR-inhibitor resistant cells.760 PROTACs degrade proteins of interest using the endogenous cell proteasome degradation system. PROTAC-mediated targeting of fusion proteins that affect RTK-inhibitor sensibility may give a further treatment option for refractory patients.761 Also, in case of autophagy-mediated resistance, Bruton’s tyrosine kinase (BTK) inhibitors seem to efficiently inhibit this resistance mechanism and synergize with FLT3-inhibitors in acute myeloid leukemia.702 Another feasible approach would be to combine ALK-inhibitors with PI3Kβ-inhibitors.762 Overcoming epigenetic modifications may be feasible by combining RTK-inhibitors with histone deacetylases.763 Inhibiting EMT with specific inhibitors in combination with EGFR-inhibitors may also be a feasible option in the future.764 Targeting the cell-cycle with CKD-inhibitors combined with osimertinib is now ongoing too (NCT04545710).
Adoptive cellular and other targeted therapies
RTK-inhibitor resistant patients may benefit from antibody-based or adoptive treatments, such as chimeric antigen receptor (CAR) T-cells. For example, c-MET targeting CAR-Ts demonstrated efficacy regardless of RTK inhibitor sensitivity.765 Antibody-drug conjugates, such as telisotuzumab vedotin which is conjugated with a microtubule inhibitor cytotoxic drug, are other options to target c-Met in combination with EGFR-inhibitors in NSCLC.766 CD70, a marker of epithelial-mesenchymal transformation, can be efficiently targeted by CAR T-cells and overcome resistance to EGFR-targeted therapies.760 PanErbB-targeting CAR T-cells also showed promising results in head and neck cancer.767 Translational researchers should prioritize developing cellular therapeutics for RTK-inhibitor resistant patients, as well as conducting randomized, controlled studies for the agents described above. Combining RTK inhibitors with other non-invasive targeted therapies, such as stereotactic ablative radiotherapy, may improve outcomes, reduce risk of resistance, and even overcome it when present.768
Discovery of novel tyrosine kinases as therapeutical targets
Discovery of both non-receptor and receptor tyrosine kinases that are associated with resistance is of high clinical impact. Combining RTK-inhibitors with non-receptor tyrosine kinase inhibitors is suggested to improve outcomes in FLT3-mutated acute myeloid leukemia in vitro and in vivo.769 But RTK-inhibitor resistance may be also overcome by targeting cytoplasmic tyrosine kinases as well. For instance, protein tyrosine kinase 2 (PTK2) has been shown to be hyperphosphorylated in EGFR-resistant NSCLC patients. Moreover, targeting PTK2 may overcome resistance (https://doi.org/10.1186/s12931-019-1244-2). A powerful example for novel RTK targets is EphA2 that has been demonstrated to serve as an escape mechanism in a number of malignancies, including colorectal, breast, liver, and GCs. Because of the documented EphA2-EGFR crosstalk, ALW-II-41-27 is a novel EphA2 small molecule inhibitor that may be crucial in overcoming EGFR-mediated resistance.770 ALW-II-41-27 is awaiting clinical trials. Another promising target is AXL. There is no FDA approved selective AXL-inhibitor yet. Bemcentinib, a novel AXL-targeting, highly selective agent demonstrated efficacy and a good tolerability in unfit, chemotherapy ineligible AML patients in the phase 2 NCT02488408 trial.771 However, the BERGAMO phase 2 (NCT03824080) trial suggested that monotherapy with bemcentinib offers limited efficacy, a possible combination with hypomethylating agents and venetoclax may be a feasible option in the future.772 Promising results have been achieved in NSCLS as well in a phase 1 trial (NCT02922777) but further investigation is needed to determine efficacy of bemcentinib in this context.773 A description of RTK-inhibitors that are being studied only in preclinical stages is, however, outside the scope of this article. Our goal is to stress the importance of basic and translational research in identifying novel targets for improving outcomes.
Novel-generation RTK-inhibitors
The following paragraphs will address relevant clinical trials assessing RTK-inhibitors that have not yet received US FDA approval, either as monotherapies or, more frequently, in combination with immunotherapies, chemotherapy, or other RTK inhibitors. These novel RTK-inhibitors are summarized below in Table 13.
To attain the lowest possible toxicities and maximum efficacy, novel RTK-inhibitors aim to be highly selective. Nonetheless, there is still ongoing research on novel non-selective or multikinase-targeting drugs, with promising results. Foretinib is one such RTK-inhibitor that has been tested in patients with advanced hepatocellular carcinoma. In the phase 2 NCT00920192 study, these patients achieved a median overall survival of 15.7 months (95% CI 7.9-NR).774 Direct comparisons to other agents are awaiting. Preclinical studies suggest that foretinib may be a feasible option as second-line therapy for capmatinib/tepotinib resistant NSCLC patients.775 Clinical studies investigating this hypothesis are required. Anlotinib, a Chinese FDA-approved multikinase-inhibitor, is another promising example. It has shown superiority over placebo in trials for medullary thyroid cancer (phase 3 ALTER01031; NCT02586350), synovial sarcoma patients (phase 3 APROMISS; NCT03016819), and NSCLC (phase 3 ALTER0303; NCT02388919).776,777,778
Switching to a newer generation selective inhibitor of the same family may resensitize patients and improve outcomes. The most novel RTK inhibitors we identified target EGFR. One of the most promising, not yet FDA-approved third-generation EGFR-inhibitor, with several phase 3 trials demonstrating its efficacy, is lazertinib. Direct comparison with gefitinib in the LASER301 phase 3 trial (NCT04248829) demonstrated significant superiority in terms of efficacy in NCSLC. Median PFS achieved for lazertinib was 20.6 months (95% CI, 17.8–26.1 months) compared to 9.7 months (95% CI, 9.2–11.3 months) in the gefitinib arm (P < 0.001).779 Furmonertinib and almonertinib, another third-generation EGFR-inhibitors, also proved to be superior to gefitinib as first line therapy in the phase 3 FURLONG (NCT03787992) and AENEAS (NCT03849768) trials.780,781 To compare furmonertinib, almonertinib and lazertinib, additional randomized, controlled trials are needed. Befotertinib was shown to be superior to the first-generation EGFR-inhibitor icotinib in metastatic NSCLC in the phase 3 NCT04206072 trial.782 Other EGFR-inhibitors, such as limertinib, abivertinib, rezivertinib and sunvozertinib have been tested in metastatic NSCLC and demonstrated promising efficacy and tolerable safety profile.783,784,785,786,787 However, phase 3 trials are required to further assess efficacy. Epitinib is a selective EGFR-inhibitor that proved to be safe and promising in treating GBM and NSCLC with brain metastasis.788 Tesevatinib has been studied in both GBM and NSCLC with brain metastasis showing a good permeability through the blood-brain barrier. We identified no phase 3 trials at the moment for tesevatinib and epitinib.
Novel FGFR inhibitors, such as lirafugratinib, were described as promising agents, but they were only studied in a preclinical settings.789 Trials are needed to confirm their efficacy in the clinic.
In the case of c-Met inhibitors, switching from crizotinib or capmatinib to the newer generation cabozantinib in NSCLC resensitized patients and led to significantly better outcomes.645
In case of ALK-positive NSCLC, iruplinalkib, a new generation ALK-inhibitor showed in the phase 2 INTELLECT trial (NCT04641754) significant antitumoral activity without high-grade toxicities, even in crizotinib-resistant patients.790
Another novel target that demonstrated promising results is the CSF-1R-inhibitor surufatinib which significantly improved progression-free survival in both pancreatic and extrapancreatic neuroendocrine tumors in the phase 3 SANET-p (NCT02589821) and SANET-ep (NCT02588170) trial.791
Novel generation, selective RET-inhibitors, such as zeteletinib, vepafestinib and TPX-0046 have been reviewed by Clark et al.792,793 We identified no phase 2/3 clinical trials on novel RET-inhibitors.
Several RTK-inhibitors demonstrated, however, no improved outcomes or significant toxicities in clinical trials, hence no further studies have been initiated, such as dovitinib for RCC,794 nintedanib for CRC,795 tandutinib for AML.796
RTK-inhibitor dual combinations
When resistance to monotherapy is identified, dual/triple drug-combination may improve efficacy. Studies indicate that combining RTK-inhibitors of the same class may help overcome resistance. This strategy, known as vertical pathway inhibition, tries to doubly inhibit the same signaling pathway. These possible combinations include a number of RTK-inhibitors that are not yet FDA-approved. There are several ways of vertical pathway inhibition: combining a multikinase-inhibitor, such as anlotinib, with a highly selective RTK-inhibitor, such as osimertinib, resulted in resistance reversal, for example.797 Another approach is targeting an RTK and one of its downstream effectors, such as the RET/mTOR dual inhibition.798
EGFR/c-Met dual inhibition by osimertinib and savolitinib was suggested to overcome c-Met resistance in non-small lung cancer patients increasing median PFS from 5.5 (95% CI, 4.1–7.7) to 11.1 months (95% CI, 4.1–22.1).799 Other clinical trials (NCT04816214 and NCT03940703) are also examining the combination of osimertinib with other c-Met inhibitors, such capmatinib and tepotinib. Tepotinib was also combined with gefitinib for NSCLC and showed superiority compared to standard-of-care chemotherapy.800 In case of lorlatinib (ROS1/ALK-inhibitor) resistance, combination with a pan-HER2-inhibitor (afatinib or dacomitinib) seems to overcome resistance in vitro in NSCLC cells.801 Resistance to ROS-1 inhibitor entrectinib may be overcome by adding a MET-inhibitor.760 Doublets of third- and fourth-generation EGFR-inhibitors are also studied. The SYMPHONY phase 1/2 trial combined osimertinib with BLU-945, for instance, and achieved less toxicities and promising efficacy.802 Drug combinations used in ALK-inhibitor resistant patients have been excellently summarized by Desai et al.803 Other studies, such as the phase 3 ACTIVE (NCT02824458) trial, demonstrated that the novel VEGFR-inhibitor apatinib may enhance the effects of EGFR-targeting gefitinib. The combination showed a superior PFS of 13.7 months (95% CI 11.9–14.1 months) compared to a median PFS of 10.2 months (95% CI 10.1–11.9 months) in the placebo + gefitinib arm (P = 0.02)804 in advanced NSCLC. Tolerable safety profile with promising responses to apatinib have been shown in advanced osteosarcoma patients as well in the phase 2 NCT02711007 trial.805
Dual inhibition by the same agent is also possible nowadays, clinical trials studied ripretinib, for example, a dual, highly selective inhibitor of KIT and PDGFRA. Ripretinib significantly improved median PFS, and overall survival compared to placebo in the phase 3 INVICTUS trial.806 Although ripretinib did not prove superior to currently used RTK-inhibitor for GIST, sunitinib in terms of efficacy, significantly reduced toxicities were reported in the phase 3 INTRIGUE trial.807 A novel phase 3 trial (INSIGHT – NCT05734105) is currently recruiting patients to investigate if efficacy is superior compared to sunitinib in specific genetic subtypes of GIST (NCT05734105). Triple inhibition of VEGFR, PDGFR and FGFR by nintedanib has been studied in NSCLC and CRC. Nintedanib showed promising results in combination with chemotherapy only in ovarian cancer, overall survival did not improve in monotherapy in case of metastatic CRC.795,808
RTK-inhibitors plus chemoimmunotherapy
Pottier et al. reviewed the effects on immune cells and the immune niche of several RTK-inhibitors, such as cabozantinib, sunitinib, axitinib, imatinib, other FGFR- or VEGFR1-inhibitors. These agents induce overexpression of immune checkpoints, induce proliferation and polarization of anti-tumoral macrophages, induce proliferation of inhibitory regulatory T-cells and produce several anti-inflammatory cytokines.809 In light of these, RTK inhibitors are currently being studied in combination with immunotherapies to reduce their immunosuppressive effects and increase efficacy. The non-selective RTK-inhibitor famitinib combined with the novel generation PD-1 checkpoint inhibitor camrelizumab demonstrated high overall response rates (53.7%, 95% CI 37–69%) with a median progression-free survival of 16.6 months (95% CI 8.3-not reached) in advanced NSCLC.810 The same combination showed promising antitumor activity in TNBC as well.811 Camrelizumab was also combined with apatinib in the phase 1 SPACE study for advanced gastric adenocarcinoma. Although the study sample size is small, the study showed favorable outcomes with a median OS of 17.9 months (95% CI 7.8-not reached) without any surgical intervention.812 In case of advanced/metastatic BC, the pan-ErbB inhibitor pyrotinib is being currently studied with trastuzumab, an anti-HER2 monoclonal antibody, and docetaxel as adjuvant-therapy in the phase 3 PHILA (NCT03863223); as neoadjuvant therapy in the PHEBA (NCT03588091) trial or with capecitabine in the phase 3 PHOEBE (NCT03080805) trial.813,814,815
Dual inhibition of RTKs with small molecule inhibitors and mono/bispecific antibodies is an emerging strategy in the clinic. Such a combination is lazertinib combined with amivantamab, an EGFR-MET bispecific antibody, which exhibited outstanding results both with and without chemotherapy in osimertinib-resistant NSCLC patients.816,817
However, not in all cases an RTK-inhibitor-immunotherapy combination proved superior to chemotherapy. Such an example is sitravatinib with nivolumab versus docetaxel for NSCLC in the phase 3 SAPPHIRE (NCT03906071) study.818
There are several other RTK-inhibitor-chemotherapy combinations that are currently investigated in trials. We are awaiting results. Research questions in the field would be which checkpoint inhibitor to combine with RTK-inhibitors, should we combine more immunotherapies (such as nivolumab plus ipilimumab), should we use classical or novel-generation checkpoint-inhibitors, such as tislelizumab or toripalimab.
Conclusion and future perspectives
Current research in the field of genomics, proteomics and other related areas facilitated the discovery of various molecular alterations associated with cancer. A significant portion of these alterations have been discovered in RTKs, which impair cellular signaling and contribute to cancer development. The ability to pharmacologically target RTKs due to their functional characteristics has made them a focal point in oncological research. Therapies targeting RTKs have shown remarkable progress in the treatment of cancer, providing survival benefits even for advanced or metastatic cancers. To develop potent and selective inhibitors, researchers and clinicians must have a thorough understanding of the conformation of RTKs and their physiological roles. This allows them to not only aim for better responses but also limit treatment-related toxicities and prevent relapses. The long-term success of these therapies is often held up by the emergence of drug resistance. Ongoing and future research efforts should be concentrated on understanding the molecular basis of these resistance mechanisms. Moreover, it is necessary to conduct research that specifically aims to understand how RTK-inhibitors interact within the cancer microenvironment. This entails developing innovative therapeutic drugs in innovative drug formulations, such as nanocarriers, that can selectively target various RTKs or their subsequent signaling cascades, hence reducing the probability of toxicities and resistance. Personalized approaches, such as analyzing the genetic characteristics of the tumor and tailoring personalized treatment plans, are essential. The objective of these strategies should be to identify patients who are sensitive or primarily resistant to treatment and choose the optimal RTK-inhibitor (combination).
In addition to tumor dynamics, tumor heterogeneity, and other variables, RTK inhibitors exhibit different potency and selectivity within the same class. In the case of BC with brain metastases, for instance, HER2-inhibitors have been compared in a meta-analysis. While the use of lapatinib or tucatinib favored RTK-inhibitor containing regimens, the use of afatinib showed no significant benefit in terms of progression free survival and overall survival.819 Afatinib, however, showed significantly longer median survival in NSCLC patients compared to erlotinib or gefitinib.820 In RCC, c-Met inhibitors, such as cabozantinib, showed superiority against sunitinib.821 However, if cabozantinib is the best currently available option amongst c-Met inhibitors is not well known, since there are no direct comparisons of c-Met inhibitors. Thus, we emphasize the importance of initiation of randomized, controlled studies directly comparing RTK-inhibitors for different cancer types. However, not only efficacy has to be taken into consideration. Several studies showed that nilotinib outperforms dasatinib and imatinib. However, due to financial considerations, imatinib still remains first-line therapy in chronic myeloid leukemia. Other physicians still choose dasatinib instead of nilotinib as second-line therapy. Thus, cost-efficiency, drug availability and physician preferences may also influence which RTK-inhibitor is chosen.
Regardless of the previously mentioned cofounding factors, more molecules must be developed, evaluated, compared, and approved by the FDA in order improve outcomes, reduce toxicities, overcome resistance and maximize the number of patients who benefit from RTK-inhibitors.
References
Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 237, 1555–1562 (1962).
Levi-Montalcini, R. & Booker, B. Excessive growth of the sympathetic ganglia evoked by a protein isolated from mouse salivary glands. Proc. Natl Acad. Sci. USA 46, 373–384 (1960).
De Meyts, P., Roth, J., Neville, D. M., Gavin, J. R. & Lesniak, M. A. Insulin interactions with its receptors: experimental evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 55, 154–161 (1973).
Carpenter, G. & Cohen, S. 125I-labeled human epidermal growth factor. Binding, internalization, and degradation in human fibroblasts. J. Cell Biol. 71, 159–171 (1976).
Ushiro, H. & Cohen, S. Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J. Biol. Chem. 255, 8363–8365 (1980).
Kasuga, M., Zick, Y., Blithe, D. L., Crettaz, M. & Kahn, C. R. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 298, 667–669 (1982).
De Meyts, P. Receptor tyrosine kinase signal transduction and the molecular basis of signalling specificity. in Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease 51–76 (Springer New York, New York, NY, 2015). https://doi.org/10.1007/978-1-4939-2053-2_4.
Zhao, M., Jung, Y., Jiang, Z. & Svensson, K. J. Regulation of energy metabolism by receptor tyrosine kinase ligands. Front. Physiol. 11, 1–23 (2020).
Vaparanta, K. et al. An extracellular receptor tyrosine kinase motif orchestrating intracellular STAT activation. Nat. Commun. 13, 1–19 (2022).
Gonzalez-Magaldi, M., McCabe, J. M., Cartwright, H. N., Sun, N. & Leahy, D. J. Conserved roles for receptor tyrosine kinase extracellular regions in regulating receptor and pathway activity. Biochem. J. 477, 4207–4220 (2020).
Yao, Z. & Stagljar, I. Multiple functions of protein phosphatases in receptor tyrosine kinase signaling revealed by interactome analysis. Mol. Cell Oncol. 4, 1–3 (2017).
Grassot, J., Gouy, M., Perrière, G. & Mouchiroud, G. Origin and molecular evolution of receptor tyrosine kinases with immunoglobulin-like domains. Mol. Biol. Evol. 23, 1232–1241 (2006).
Stuttfeld, E. & Ballmer‐Hofer, K. Structure and function of VEGF receptors. IUBMB Life 61, 915–922 (2009).
Diwanji, D., Thaker, T. & Jura, N. More than the sum of the parts: toward full‐length receptor tyrosine kinase structures. IUBMB Life 71, 706–720 (2019).
Matsushita, C. et al. Transmembrane helix orientation influences membrane binding of the intracellular juxtamembrane domain in Neu receptor peptides. Proc. Natl Acad. Sci. USA 110, 1646–1651 (2013).
Hedger, G., Sansom, M. S. P. & Koldsø, H. The juxtamembrane regions of human receptor tyrosine kinases exhibit conserved interaction sites with anionic lipids. Sci. Rep 5, 1–10 (2015).
Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010).
Hafizi, S. & Dahlblack, B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev. 17, 295–304 (2006).
Aveic, S. & Tonini, G. P. Resistance to receptor tyrosine kinase inhibitors in solid tumors: can we improve the cancer fighting strategy by blocking autophagy? Cancer Cell Int 16, 1–8 (2016).
Sun, X. et al. Receptor tyrosine kinase phosphorylation pattern–based multidrug combination is an effective approach for personalized cancer treatment. Mol. Cancer Ther. 15, 2508–2520 (2016).
Yao, Z. et al. A global analysis of the receptor tyrosine kinase-protein phosphatase interactome. Mol. Cell 65, 347–360 (2017).
Paul, M. D. & Hristova, K. The RTK interactome: overview and perspective on RTK heterointeractions. Chem. Rev. 119, 5881–5921 (2019).
Li, S. C. et al. High-affinity binding of the Drosophila Numb phosphotyrosine-binding domain to peptides containing a Gly-Pro-(p)Tyr motif. Proc. Natl Acad. Sci. USA 94, 7204–7209 (1997).
Li, S. C. et al. Characterization of the phosphotyrosine-binding domain of the Drosophila Shc protein. J. Biol. Chem. 271, 31855–31862 (1996).
Xu, A. M. & Huang, P. H. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 70, 3857–3860 (2010).
Fiorini, M., Alimandi, M., Fiorentino, L., Sala, G. & Segatto, O. Negative regulation of receptor tyrosine kinase signals. FEBS Lett. 490, 132–141 (2001).
Phillips-Mason, P. J., Craig, S. E. L. & Brady-Kalnay, S. M. Should I stay or should I go? Shedding of RPTPs in cancer cells switches signals from stabilizing cell-cell adhesion to driving cell migration. Cell Adh Migr. 5, 298–305 (2011).
Tang, R., Langdon, W. Y. & Zhang, J. Negative regulation of receptor tyrosine kinases by ubiquitination: key roles of the Cbl family of E3 ubiquitin ligases. Front. Endocrinol 13, (2022).
Casaletto, J. B. & McClatchey, A. I. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 12, 387–400 (2012).
Du, Z. & Lovly, C. M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17, 1–13 (2018).
Butti, R. et al. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol. Cancer 17, 34 (2018).
Ferrara, N., Gerber, H.-P. & LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 9, 669–676 (2003).
Ullah, R., Yin, Q., Snell, A. H. & Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 85, 123–154 (2022).
Truong, T. H. & Carroll, K. S. Redox regulation of protein kinases. Crit. Rev. Biochem. Mol. Biol. 48, 332–356 (2013).
Belov, A. A. & Mohammadi, M. Grb2, a double-edged sword of receptor tyrosine kinase signaling. Sci. Signal 5, 1–7 (2012).
Talukdar, S., Emdad, L., Das, S. K. & Fisher, P. B. EGFR: an essential receptor tyrosine kinase-regulator of cancer stem cells. Adv. Cancer Res. 147, 161–188 (2020).
Wei, Q., Qian, Y., Yu, J. & Wong, C. C. Metabolic rewiring in the promotion of cancer metastasis: mechanisms and therapeutic implications. Oncogene 39, 6139–6156 (2020).
Locasale, J. W. Metabolic rewiring drives resistance to targeted cancer therapy. Mol. Syst. Biol 8 (2012).
Jin, N. et al. Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat Commun. 10 (2019).
Lin, C.-C. et al. Receptor tyrosine kinases regulate signal transduction through a liquid-liquid phase separated state. Mol. Cell 82, 1089–1106 (2022).
Huang, H. Proteolytic cleavage of receptor tyrosine kinases. Biomolecules 11 (2021).
Bache, K. G., Slagsvold, T. & Stenmark, H. Defective downregulation of receptor tyrosine kinases in cancer. EMBO J. 23, 2707–2712 (2004).
Hubbard, S. R. & Miller, W. T. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr. Opin. Cell Biol. 19, 117–123 (2007).
Montor WR, Salas Arose, Melo FHM. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: the current arsenal of inhibitors. Mol Cancer 17 (2018).
McDonell, L. M., Kernohan, K. D., Boycott, K. M. & Sawyer, S. L. Receptor tyrosine kinase mutations in developmental syndromes and cancer: two sides of the same coin. Hum. Mol. Genet. 24, 60–66 (2015).
Robinson, D. R., Wu, Y. M. & Lin, S. F. The protein tyrosine kinase family of the human genome. Oncogene 19, 5548–5557 (2000).
Saraon, P. et al. Receptor tyrosine kinases and cancer: oncogenic mechanisms and therapeutic approaches. Oncogene 40, 4079–4093 (2021).
Sheffels, E. & Kortum, R. L. Breaking oncogene addiction: getting RTK/RAS-mutated cancers off the SOS. J. Med. Chem. 64, 6566–6568 (2021).
Orlando, E. et al. An oncogene addiction phosphorylation signature and its derived scores inform tumor responsiveness to targeted therapies. Cell Mol. Life Sci. 80 (2023).
Weinstein, I. B. Addiction to oncogenes-the Achilles heal of cancer. Science 297, 63–64 (2002).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Medves, S. & Demoulin, J. Tyrosine kinase gene fusions in cancer: translating mechanisms into targeted therapies. J. Cell Mol. Med. 16, 237–248 (2012).
Ebrahimi, N. et al. Receptor tyrosine kinase inhibitors in cancer. Cell Mol. Life Sci. 80, (2023).
Lin, C.-C., Suen, K. M., Lidster, J. & Ladbury, J. E. The emerging role of receptor tyrosine kinase phase separation in cancer. Trends Cell Biol. 34, 371–379 (2023).
Jaradat, S. K., Ayoub, N. M., Al Sharie, A. H. & Aldaod, J. M. Targeting receptor tyrosine kinases as a novel strategy for the treatment of triple-negative breast cancer. Technol. Cancer Res. Treat. 23 (2024).
Labani-Motlagh, A., Ashja-Mahdavi, M. & Loskog, A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front. Immunol. 11 (2020).
Gaspar, N. et al. Lenvatinib with etoposide plus ifosfamide in patients with refractory or relapsed osteosarcoma (ITCC-050): a multicentre, open-label, multicohort, phase 1/2 study. Lancet Oncol. 22, 1312–1321 (2021).
Zhang, N. & Li, Y. Receptor tyrosine kinases: biological functions and anticancer targeted therapy. MedComm 4 (2023).
Wirth, T. A role for RTKs in Hodgkin lymphoma. Blood 105, 3766–3766 (2005).
Gupta, R., Knight, C. L. & Bain, B. J. Receptor tyrosine kinase mutations in myeloid neoplasms. Br. J. Haematol. 117, 489–508 (2002).
Kim, Y. et al. Temporal resolution of autophosphorylation for normal and oncogenic forms of EGFR and differential effects of gefitinib. Biochemistry 51, 5212–5222 (2012).
Ferguson, K. M., Hu, C. & Lemmon, M. A. Insulin and epidermal growth factor receptor family members share parallel activation mechanisms. Protein Sci. 29, 1331–1344 (2020).
Cohen, S. The epidermal growth factor (EGF). Cancer 51, 1787–1791 (1983).
Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharm. Res. 87, 42–59 (2014).
Ceresa, B. P. & Peterson, J. L. Cell and molecular biology of epidermal growth factor receptor. Int. Rev. Cell Mol. Biol. 313, 145–178 (2014).
Ullrich, A. et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309, 418–425 (1984).
Yarden, Y. & Pines, G. The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer 12, 553–563 (2012).
Jorissen, R. N. et al. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell Res. 284, 31–53 (2003).
Rosenkranz, A. A. & Slastnikova, T. A. Epidermal growth factor receptor: key to selective intracellular delivery. Biochemistry (Mosc) 85, 967–1092 (2020).
Arteaga, C. L. & Engelman, J. A. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25, 282–303 (2014).
Arienti, C., Pignatta, S. & Tesei, A. Epidermal growth factor receptor family and its role in gastric cancer. Front. Oncol. 9 (2019).
Thompson, D. M. & Gill, G. N. The EGF receptor: structure, regulation and potential role in malignancy. Cancer Surv. 4, 767–788 (1985).
Reschke, M. et al. HER3 is a determinant for poor prognosis in melanoma. Clin. Cancer Res. 14, 5188–5197 (2008).
Koumakpayi, I. H. et al. Expression and nuclear localization of ErbB3 in prostate cancer. Clin. Cancer Res. 12, 2730–2737 (2006).
Tanner, B. et al. ErbB-3 predicts survival in ovarian cancer. J. Clin. Oncol. 24, 4317–4323 (2006).
Wei, Q. et al. EGFR, HER2 and HER3 expression in esophageal primary tumours and corresponding metastases. Int. J. Oncol. 31, 493–499 (2007).
Wei, Q. et al. EGFR, HER2, and HER3 expression in laryngeal primary tumors and corresponding metastases. Ann. Surg. Oncol. 15, 1193–1201 (2008).
Kapitanović, S. et al. Expression of erbB-3 protein in colorectal adenocarcinoma: correlation with poor survival. J. Cancer Res. Clin. Oncol. 126, 205–211 (2000).
Witton, C. J., Reeves, J. R., Going, J. J., Cooke, T. G. & Bartlett, J. M. S. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J. Pathol. 200, 290–297 (2003).
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Russo, A. et al. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): old successes and future perspectives. Oncotarget 6, 26814–26825 (2015).
Nahm, C. B. et al. Biomarker panel predicts survival after resection in pancreatic ductal adenocarcinoma: a multi-institutional cohort study. Eur. J. Surg. Oncol. 45, 218–224 (2019).
Ueda, S. et al. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas 29, 1–8 (2004).
Wang, Z. ErbB receptors and cancer. Methods Mol. Biol. 1652, 3–35 (2017).
Shah, R. B., Ghosh, D. & Elder, J. T. Epidermal growth factor receptor (ErbB1) expression in prostate cancer progression: correlation with androgen independence. Prostate 66, 1437–1444 (2006).
Yu, H. A. et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR -mutant lung cancers. Clin. Cancer Res. 19, 2240–2247 (2013).
Yamaura, T. et al. Genetic alterations in epidermal growth factor receptor-tyrosine kinase inhibitor-naïve non-small cell lung carcinoma. Oncol. Lett. 19, 4169–4176 (2020).
Zheng, D. et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 8, 49671–49679 (2017).
Fu, K., Xie, F., Wang, F. & Fu, L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. J. Hematol. Oncol. 15 (2022).
Liu, H., Zhang, B. & Sun, Z. Spectrum of EGFR aberrations and potential clinical implications: insights from integrative pan-cancer analysis. Cancer Commun. 40, 43–59 (2020).
Wang, F. et al. Identification of genetic alterations associated with primary resistance to EGFR-TKIs in advanced non-small-cell lung cancer patients with EGFR sensitive mutations. Cancer Commun. 39, 1–15 (2019).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 2017 (2017).
Jebali, A. & Dumaz, N. The role of RICTOR downstream of receptor tyrosine kinase in cancers. Mol. Cancer 17 (2018).
Mikhaylenko, D. S., Alekseev, B. Y., Zaletaev, D. V., Goncharova, R. I. & Nemtsova, M. V. Structural alterations in human fibroblast growth factor receptors in carcinogenesis. Biochemistry 83, 930–943 (2018).
Babina, I. S. & Turner, N. C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 17, 318–332 (2017).
Helsten, T. et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res. 22, 259–267 (2016).
Ferguson, H. R., Smith, M. P. & Francavilla, C. Fibroblast growth factor receptors (FGFRs) and noncanonical partners in cancer signaling. Cells 10, 1–35 (2021).
Krook, M. A. et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 124, 880–892 (2021).
Sobhani, N. et al. Current status of fibroblast growth factor receptor-targeted therapies in breast cancer. Cells 7 (2018).
Imamura, T. Physiological functions and underlying mechanisms of fibroblast growth factor (FGF) family members: recent findings and implications for their pharmacological application. Biol. Pharm. Bull. 37, 1081–1089 (2014).
Qin, A. et al. Detection of known and novel FGFR fusions in non-small cell lung cancer by comprehensive genomic profiling. J. Thorac. Oncol. 14, 54–62 (2019).
André, F. & Cortés, J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res. Treat. 150, 1–8 (2015).
Gallo, L. H., Nelson, K. N., Meyer, A. N. & Donoghue, D. J. Functions of fibroblast growth factor receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 26, 425–449 (2015).
Rosty, C. et al. Clinical and biological characteristics of cervical neoplasias with FGFR3 mutation. Mol. Cancer 4 (2005).
Hernández, S. et al. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J. Clin. Oncol. 24, 3664–3671 (2006).
Ruhe, J. E. et al. Genetic alterations in the tyrosine kinase transcriptome of human cancer cell lines. Cancer Res. 67, 11368–11376 (2007).
Tang, S., Hao, Y., Yuan, Y., Liu, R. & Chen, Q. Role of fibroblast growth factor receptor 4 in cancer. Cancer Sci. 109, 3024–3031 (2018).
Patani, H. et al. Landscape of activating cancer mutations in FGFR kinases and their differential responses to inhibitors in clinical use. Oncotarget 7, 24252–24268 (2016).
Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 16, 105–122 (2019).
Stransky, N. et al. The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160 (2011).
Greenman, C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007).
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Belfiore, A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 13, 671–686 (2007).
Pisani, P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch. Physiol. Biochem 114, 63–70 (2008).
Kim, D.-S. & Scherer, P. E. Obesity, diabetes, and increased cancer progression. Diabetes Metab. J. 45, 799–812 (2021).
Scully, T., Ettela, A., LeRoith, D. & Gallagher, E. J. Obesity, type 2 diabetes, and cancer risk. Front. Oncol. 10 (2020).
Hers, I., Vincent, E. E. & Tavaré, J. M. Akt signalling in health and disease. Cell Signal 23, 1515–1527 (2011).
Drakas, R., Tu, X. & Baserga, R. Control of cell size through phosphorylation of upstream binding factor 1 by nuclear phosphatidylinositol 3-kinase. Proc. Natl Acad. Sci. USA 101, 9272–9276 (2004).
Ullrich, A. et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313, 756–761 (1985).
Ebina, Y. et al. Expression of a functional human insulin receptor from a cloned cDNA in Chinese hamster ovary cells. Proc. Natl Acad. Sci. USA 82, 8014–8018 (1985).
Malaguarnera, R. & Belfiore, A. The insulin receptor: a new target for cancer therapy. Front. Endocrinol. 2 (2011).
Ullrich, A. et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 5, 2503–2512 (1986).
Nakae, J., Kido, Y. & Accili, D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr. Rev. 22, 818–835 (2001).
Reeves, R., Edberg, D. D. & Li, Y. Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol. Cell Biol. 21, 575–594 (2001).
Aiello, A. et al. HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur. J. Cancer 46, 1919–1926 (2010).
Paul, S. et al. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 25, 4271–4283 (2006).
Craddock, B. P. & Miller, W. T. Effects of somatic mutations in the C-terminus of insulin-like growth factor 1 receptor on activity and signaling. J. Signal. Transduct 2012, 1–7 (2012).
Gorgisen, G. et al. Identification of novel mutations of Insulin Receptor Substrate 1 (IRS1) in tumor samples of non-small cell lung cancer (NSCLC): Implications for aberrant insulin signaling in development of cancer. Genet Mol. Biol. 42, 15–25 (2019).
Hamilton, T. G., Klinghoffer, R. A., Corrin, P. D. & Soriano, P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol. Cell Biol. 23, 4013–4025 (2003).
Dell, S., Peters, S., Müther, P., Kociok, N. & Joussen, A. M. The role of PDGF receptor inhibitors and PI3-kinase signaling in the pathogenesis of corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 47, 1928–1937 (2006).
Lokker, N. A. et al. Functional importance of platelet-derived growth factor (PDGF) receptor extracellular immunoglobulin-like domains. Identification of PDGF binding site and neutralizing monoclonal antibodies. J. Biol. Chem. 272, 33037–33044 (1997).
Manzat Saplacan, R. M. et al. The role of PDGFs and PDGFRs in colorectal cancer. Mediators Inflamm. 2017, (2017).
Sheikh, E., Tran, T., Vranic, S., Levy, A. & Bonfil, R. D. Role and significance of c-KIT receptor tyrosine kinase in cancer: A review. Bosn. J. Basic Med Sci. 22, 683–698 (2022).
Heldin, C.-H. & Lennartsson, J. Receptor tyrosine kinases and their ligands. in Encyclopedia of Cell Biology Vol. 3, 8–21 (Elsevier, 2016).
Hirota, S. et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279, 577–580 (1998).
Bahlawane, C. et al. Constitutive activation of oncogenic PDGFRα-mutant proteins occurring in GIST patients induces receptor mislocalisation and alters PDGFRα signalling characteristics. Cell Commun. Signal. 13 (2015).
Ravegnini, G. et al. miRNA expression may have implications for immunotherapy in PDGFRA mutant GISTs. Int. J. Mol. Sci. 23 (2022).
Prenen, H. et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin. Cancer Res. 12, 2622–2627 (2006).
Ip, C. K. M. et al. Neomorphic PDGFRA extracellular domain driver mutations are resistant to PDGFRA-targeted therapies. Nat. Commun. 9, 1–14 (2018).
Heinrich, M. C. et al. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin. Cancer Res. 18, 4375–4384 (2012).
Weisberg, E. et al. Effects of PKC412, Nilotinib, and Imatinib Against GIST-Associated PDGFRA Mutants With Differential Imatinib Sensitivity. Gastroenterology 131, 1734–1742 (2006).
Burger, K. et al. Activating mutations in c-KIT and PDGFRα are exclusively found in gastrointestinal stromal tumors and not in other tumors overexpressing these imatinib mesylate target genes. Cancer Biol. Ther. 4, 1270–1274 (2005).
Guérit, E., Arts, F., Dachy, G., Boulouadnine, B. & Demoulin, J.-B. PDGF receptor mutations in human diseases. Cell Mol. Life Sci. 78, 3867–3881 (2021).
Paugh, B. S. et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 73, 6219–6229 (2013).
Reindl, C. et al. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood 107, 3700–3707 (2006).
Rizzo, A., Pantaleo, M. A., Astolfi, A., Indio, V. & Nannini, M. The identity of PDGFRA D842V-mutant gastrointestinal stromal tumors (GIST). Cancers 13 (2021).
Alitalo, K. & Carmeliet, P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 1, 219–227 (2002).
Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2, 1097–1105 (2011).
Zhu, X., Zhou, W. The emerging regulation of VEGFR-2 in triple-negative breast cancer. Front Endocrinol. 6 (2015).
Mokhdomi, T. A. et al. A novel kinase mutation in VEGFR-1 predisposes its αC-helix/activation loop towards allosteric activation: Atomic insights from protein simulation. Eur. J. Hum. Genet. 24, 1287–1293 (2016).
Scartozzi, M. et al. Role of vascular endothelial growth factor (VEGF) and VEGF-R genotyping in guiding the metastatic process in pT4a resected gastric cancer patients. PLoS One 7, 1–7 (2012).
Grillo, E. et al. A novel variant of VEGFR2 identified by a pan-cancer screening of recurrent somatic mutations in the catalytic domain of tyrosine kinase receptors enhances tumor growth and metastasis. Cancer Lett. 496, 84–92 (2021).
Grillo, E. et al. Expression of activated VEGFR2 by R1051Q mutation alters the energy metabolism of Sk-Mel-31 melanoma cells by increasing glutamine dependence. Cancer Lett. 507, 80–88 (2021).
Sarabipour, S., Ballmer-Hofer, K. & Hristova, K. VEGFR-2 conformational switch in response to ligand binding. Elife 5 (2016).
Boye, E., Jinnin, M. & Olsen, B. R. Infantile hemangioma. J. Craniofac. Surg. 20, 678–684 (2009).
Zhu, X. et al. The VEGFR-2 protein and the VEGFR-2 rs1870377 A>T genetic polymorphism are prognostic factors for gastric cancer. Cancer Biol. Ther. 20, 497–504 (2019).
Maeng, C. H. et al. Effects of single nucleotide polymorphisms on treatment outcomes and toxicity in patients treated with sunitinib. Anticancer Res. 33, 4619–4626 (2013).
Hansen, T. F. et al. Microvessel density and the association with single nucleotide polymorphisms of the vascular endothelial growth factor receptor 2 in patients with colorectal cancer. Virchows Arch. 456, 251–260 (2010).
Cho, S.-J., Park, M. H., Han, C., Yoon, K. & Koh, Y. H. VEGFR2 alteration in Alzheimer’s disease. Sci. Rep. 7 (2017).
Hsu, M.-C., Pan, M.-R. & Hung, W.-C. Two birds, one stone: double hits on tumor growth and lymphangiogenesis by targeting vascular endothelial growth factor receptor 3. Cells 8 (2019).
Karkkainen, M. J. et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 25, 153–159 (2000).
Gordon, K. et al. FLT4/VEGFR3 and Milroy disease: novel mutations, a review of published variants and database update. Hum. Mutat. 34, 23–31 (2013).
Gu, Y. et al. Case report: Unique FLT4 variants associated with differential response to anlotinib in angiosarcoma. Front. Oncol. 12 (2022).
Feng, Y., Yang, Z. & Xu, X. c-Met: a promising therapeutic target in bladder cancer. Cancer Manag. Res. 14, 2379–2388 (2022).
Wang, M.-H., Zhou, Y.-Q. & Chen, Y.-Q. Macrophage-stimulating protein and RON receptor tyrosine kinase: potential regulators of macrophage inflammatory activities. Scand. J. Immunol. 56, 545–553 (2002).
Wang, M.-H., Lee, W., Luo, Y.-L., Weis, M. T. & Yao, H.-P. Altered expression of the RON receptor tyrosine kinase in various epithelial cancers and its contribution to tumourigenic phenotypes in thyroid cancer cells. J. Pathol. 213, 402–411 (2007).
Zhou, Y.-Q., He, C., Chen, Y.-Q., Wang, D. & Wang, M.-H. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene 22, 186–197 (2003).
Cheng, H.-L. et al. Co-expression of RON and MET is a prognostic indicator for patients with transitional-cell carcinoma of the bladder. Br. J. Cancer 92, 1906–1914 (2005).
Maggiora, P. et al. The RON and MET oncogenes are co-expressed in human ovarian carcinomas and cooperate in activating invasiveness. Exp. Cell Res. 288, 382–389 (2003).
Johnson, M. et al. Selective tumorigenesis in non-parenchymal liver epithelial cell lines by hepatocyte growth factor transfection. Cancer Lett. 96, 37–48 (1995).
Sattler, M., Reddy, M. M., Hasina, R., Gangadhar, T. & Salgia, R. The role of the c-Met pathway in lung cancer and the potential for targeted therapy. Ther. Adv. Med. Oncol. 3, 171–184 (2011).
Weidner, K. M. et al. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 384, 173–176 (1996).
Jeffers, M. et al. Hepatocyte growth factor/scatter factor-Met signaling induces proliferation, migration, and morphogenesis of pancreatic oval cells. Cell Growth Differ. 7, 1805–1813 (1996).
Rong, S., Segal, S., Anver, M., Resau, J. H. & Vande Woude, G. F. Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc. Natl Acad. Sci. USA 91, 4731–4735 (1994).
Zarnegar, R. Regulation of HGF and HGFR gene expression. Epithelial-Mesenchymal Interactions in Cancer (eds. Goldberg, I. D., & Rosen, E. M.) vol. 74 33–49 (Experientia Supplementum, 1995).
Shieh, J.-M. et al. Lack of association of C-Met-N375S sequence variant with lung cancer susceptibility and prognosis. Int. J. Med. Sci. 10, 988–994 (2013).
Kong, L. R., Binte Mohamed Salleh, N. A., Tan, T. Z., Kappei, D. & Goh, B. C. P1.02-041 characterization of MET-N375S as an activating mutation in squamous cell carcinoma of the lung. J. Thorac. Oncol. 12, S512–S512 (2017).
Jagadeeswaran, R. et al. Functional analysis of c-Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res. 66, 352–361 (2006).
Xu, T.-X. et al. cMET-N375S germline mutation is associated with poor prognosis of melanoma in Chinese patients. Transl. Cancer Res. 7, 248–256 (2018).
Cortot, A. B. et al. Exon 14 deleted MET receptor as a new biomarker and target in cancers. J. Natl Cancer Inst. 109 (2017).
Tyner, J. W. et al. MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res. 70, 6233–6237 (2010).
Neklason, D. W. et al. Activating mutation in MET oncogene in familial colorectal cancer. BMC Cancer 11 (2011).
Sadiq, A. A. & Salgia, R. MET as a possible target for non–small-cell lung cancer. J. Clin. Oncol. 31, 1089–1096 (2013).
Blumenschein, G. R., Mills, G. B. & Gonzalez-Angulo, A. M. Targeting the hepatocyte growth factor–cMET axis in cancer therapy. J. Clin. Oncol. 30, 3287–3296 (2012).
Jeffers, M. et al. Activating mutations for the Met tyrosine kinase receptor in human cancer. Proc. Natl Acad. Sci. USA 94, 11445–11450 (1997).
Iyer, A. et al. Structure, tissue-specific expression, and transforming activity of the mouse met protooncogene. Cell Growth Differ. 1, 87–95 (1990).
Di Renzo, M. F. et al. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene 6, 1997–2003 (1991).
Kan, M. et al. Hepatocyte growth factor/hepatopoietin A stimulates the growth of rat kidney proximal tubule epithelial cells (RPTE), rat nonparenchymal liver cells, human melanoma cells, mouse keratinocytes and stimulates anchorage-independent growth of SV-40 transformed RPTE. Biochem. Biophys. Res. Commun. 174, 331–337 (1991).
Schmidt, L. et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 16, 68–73 (1997).
Zeng, Z.-S. et al. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 265, 258–269 (2008).
Nakamura, M. et al. Hepatocyte growth factor secreted by ovarian cancer cells stimulates peritoneal implantation via the mesothelial–mesenchymal transition of the peritoneum. Gynecol. Oncol. 139, 345–354 (2015).
Seidel, C. et al. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study Group. Blood 91, 806–812 (1998).
Sheen-Chen, S.-M., Liu, Y.-W., Eng, H.-L. & Chou, F.-F. Serum levels of hepatocyte growth factor in patients with breast cancer. Cancer Epidemiol. Biomark. Prev. 14, 715–717 (2005).
Xie, Q. et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc. Natl Acad. Sci. USA 109, 570–575 (2012).
Cruickshanks, N. et al. Role and therapeutic targeting of the HGF/MET pathway in glioblastoma. Cancers 9 (2017).
Borges, L. S. & Richman, D. P. Muscle-specific kinase myasthenia gravis. Front. Immunol. 11, 1–11 (2020).
Valenzuela, D. M. et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron 15, 573–584 (1995).
DeChiara, T. M. et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85, 501–512 (1996).
Gautam, M. et al. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85, 525–535 (1996).
Till, J. H. et al. Crystal structure of the MuSK tyrosine kinase: insights into receptor autoregulation. Structure 10, 1187–1196 (2002).
Xie, T. et al. Structural insights into the assembly of the agrin/LRP4/MuSK signaling complex. Proc. Natl Acad. Sci. USA 120, 1–8 (2023).
Yang, J. et al. Nestin negatively regulates postsynaptic differentiation of the neuromuscular synapse. Nat. Neurosci. 14, 324–330 (2011).
Koneczny, I., Cossins, J. & Vincent, A. The role of muscle-specific tyrosine kinase (MuSK) and mystery of MuSK myasthenia gravis. J. Anat. 224, 29–35 (2014).
Hoch, W. et al. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 7, 365–368 (2001).
Ben Ammar, A. et al. A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS One 8, (2013).
Spengos, K. et al. Dropped head syndrome as prominent clinical feature in MuSK-positive Myasthenia Gravis with thymus hyperplasia. Neuromuscul. Disord. 18, 175–177 (2008).
Golding, B., Luu, A., Jones, R. & Viloria-Petit, A. M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol Cancer 17 (2018).
Reshetnyak, A. V. et al. Mechanism for the activation of the anaplastic lymphoma kinase receptor. Nature 600, 153–157 (2021).
Omar, N. E. et al. Postmarketing safety of anaplastic lymphoma kinase (ALK) inhibitors: an analysis of the FDA Adverse Event Reporting System (FAERS). ESMO Open 6, 1–7 (2021).
Christova, T., Ho, S. K., Liu, Y., Gill, M. & Attisano, L. LTK and ALK promote neuronal polarity and cortical migration by inhibiting IGF1R activity. EMBO Rep. 24, 1–21 (2023).
Stephenson, S.-A. et al. Anti-tumour effects of antibodies targeting the extracellular cysteine-rich region of the receptor tyrosine kinase EphB4. Oncotarget 6, 7554–7569 (2015).
Ma, E. S. K. Recurrent cytogenetic abnormalities in non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. Methods Mol. Biol. 1541, 279–293 (2017).
Weiss, J. B. et al. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharm. Biochem. Behav. 100, 566–574 (2012).
Bilsland, J. G. et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 33, 685–700 (2008).
Reshetnyak, A. V. et al. Augmentor α and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl Acad. Sci. USA 112, 15862–15867 (2015).
Guan, J. et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 4 (2015).
Mo, E. S., Cheng, Q., Reshetnyak, A. V., Schlessinger, J. & Nicoli, S. Alk and Ltk ligands are essential for iridophore development in zebrafish mediated by the receptor tyrosine kinase Ltk. Proc. Natl Acad. Sci. USA 114, 12027–12032 (2017).
Roll, J. D. & Reuther, G. W. ALK-activating homologous mutations in LTK induce cellular transformation. PLoS One 7 (2012).
Chen, Y. et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971–974 (2008).
George, R. E. et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975–978 (2008).
Wulf, A. M., Moreno, M. M., Paka, C., Rampasekova, A. & Liu, K. J. Defining pathological activities of ALK in neuroblastoma, a neural crest-derived cancer. Int. J. Mol. Sci. 22 (2021).
Mazot, P. et al. The constitutive activity of the ALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene 30, 2017–2025 (2011).
Birchmeier, C., Sharma, S. & Wigler, M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc. Natl Acad. Sci. USA 84, 9270–9274 (1987).
Zhu, Q., Zhan, P., Zhang, X., Lv, T. & Song, Y. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl. Lung Cancer Res. 4, 300–309 (2015).
Cai, W. et al. ROS1 fusions in Chinese patients with non-small-cell lung cancer. Ann. Oncol. 24, 1822–1827 (2013).
Zhang, Y. et al. Disease progression patterns and molecular resistance mechanisms to crizotinib of lung adenocarcinoma harboring ROS1 rearrangements. NPJ Precis. Oncol. 6 (2022).
Rikova, K. et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 (2007).
Takeuchi, K. et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 18, 378–381 (2012).
Davies, K. D. & Doebele, R. C. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. 19, 4040–4045 (2013).
Gu, T.-L. et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One 6, 1–9 (2011).
Yu, Z.-Q. et al. ROS1-positive non-small cell lung cancer (NSCLC): biology, diagnostics, therapeutics and resistance. J. Drug Target 30, 845–857 (2022).
Duma, N., Santana-Davila, R. & Molina, J. R. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 94, 1623–1640 (2019).
El-Deeb, I. M., Yoo, K. H. & Lee, S. H. ROS receptor tyrosine kinase: a new potential target for anticancer drugs. Med Res. Rev. 31, 794–818 (2011).
Lin, J. J. et al. Spectrum of mechanisms of resistance to crizotinib and lorlatinib in ROS1 fusion-positive lung cancer. Clin. Cancer Res. 27, 2899–2909 (2021).
Almquist, D. & Ernani, V. The road less traveled: a guide to metastatic ROS1-rearranged non-small-cell lung cancer. JCO Oncol. Pract. 17, 7–14 (2021).
Gendarme, S., Bylicki, O., Chouaid, C. & Guisier, F. ROS-1 fusions in non-small-cell lung cancer: evidence to date. Curr. Oncol. 29, 641–658 (2022).
D’Angelo, A. et al. Focus on ROS1-positive non-small cell lung cancer (NSCLC): crizotinib, resistance mechanisms and the newer generation of targeted therapies. Cancers 12, (2020).
Blume-Jensen, P. & Hunter, T. Oncogenic kinase signalling. Nature 411, 355–365 (2001).
Rimkunas, V. M. et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin. Cancer Res. 18, 4449–4457 (2012).
Cheng, Y., Sun, Y., Wang, L.-Z., Yu, Y.-C. & Ding, X. Cytoplasmic c-ros oncogene 1 receptor tyrosine kinase expression may be associated with the development of human oral squamous cell carcinoma. Oncol. Lett. 10, 934–940 (2015).
Jóri, B. et al. Acquired G2032R resistance mutation in ROS1 to lorlatinib therapy detected with liquid biopsy. Curr. Oncol. 29, 6628–6634 (2022).
Wirth, L. J. et al. LIBRETTO-531: a phase III study of selpercatinib in multikinase inhibitor-naïve RET-mutant medullary thyroid cancer. Future Oncol. 18, 3143–3150 (2022).
Li, A. Y. et al. RET fusions in solid tumors. Cancer Treat. Rev. 81, 1–11 (2019).
Drilon, A., Hu, Z. I., Lai, G. G. Y. & Tan, D. S. W. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 15, 151–167 (2018).
Mulligan, L. M. GDNF and the RET receptor in cancer: new insights and therapeutic potential. Front. Physiol. 9 (2018).
Liang, J. et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J. Pathol. 244, 215–226 (2018).
Romei, C., Ciampi, R. & Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 12, 192–202 (2016).
Takahashi, M. RET receptor signaling: function in development, metabolic disease, and cancer. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 98, 112–125 (2022).
Chi, X. et al. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev. Cell 17, 199–209 (2009).
Enomoto, H. et al. RET signaling is essential for migration, axonal growth and axon guidance of developing sympathetic neurons. Development 128, 3963–3974 (2001).
Schuchardt, A., D’Agati, V., Larsson-Blomberg, L., Costantini, F. & Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367, 380–383 (1994).
Tomuschat, C. & Puri, P. RET gene is a major risk factor for Hirschsprung’s disease: a meta-analysis. Pediatr. Surg. Int. 31, 701–710 (2015).
Jain, S. et al. Mice expressing a dominant-negative Ret mutation phenocopy human Hirschsprung disease and delineate a direct role of Ret in spermatogenesis. Development 131, 5503–5513 (2004).
Smith, D. P., Houghton, C. & Ponder, B. A. Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene 15, 1213–1217 (1997).
Mulligan, L. M. et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363, 458–460 (1993).
Wagner, S. M., Zhu, S., Nicolescu, A. C. & Mulligan, L. M. Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2. Clinics 67, 77–84 (2012).
Li, Q., Tie, Y., Alu, A., Ma, X. & Shi, H. Targeted therapy for head and neck cancer: signaling pathways and clinical studies. Signal Transduct. Target Ther 8, 1–28 (2023).
Perrinjaquet, M., Vilar, M. & Ibáñez, C. F. Protein-tyrosine phosphatase SHP2 contributes to GDNF neurotrophic activity through direct binding to phospho-Tyr687 in the RET receptor tyrosine kinase. J. Biol. Chem. 285, 31867–31875 (2010).
Schuringa, J. J. et al. MEN2A-RET-induced cellular transformation by activation of STAT3. Oncogene 20, 5350–5358 (2001).
Kawamoto, Y. et al. Identification of RET autophosphorylation sites by mass spectrometry. J. Biol. Chem. 279, 14213–14224 (2004).
Iwashita, T., Asai, N., Murakami, H., Matsuyama, M. & Takahashi, M. Identification of tyrosine residues that are essential for transforming activity of the ret proto-oncogene with MEN2A or MEN2B mutation. Oncogene 12, 481–487 (1996).
Encinas, M., Crowder, R. J., Milbrandt, J. & Johnson, E. M. Tyrosine 981, a novel ret autophosphorylation site, binds c-Src to mediate neuronal survival. J. Biol. Chem. 279, 18262–18269 (2004).
Borrello, M. G. et al. The full oncogenic activity of Ret/ptc2 depends on tyrosine 539, a docking site for phospholipase Cgamma. Mol. Cell Biol. 16, 2151–2163 (1996).
Asai, N., Murakami, H., Iwashita, T. & Takahashi, M. A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with shc adaptor proteins. J. Biol. Chem. 271, 17644–17649 (1996).
Alberti, L. et al. Grb2 binding to the different isoforms of Ret tyrosine kinase. Oncogene 17, 1079–1087 (1998).
Liu, X. et al. Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. J. Biol. Chem. 271, 5309–5312 (1996).
Gujral, T. S., Singh, V. K., Jia, Z. & Mulligan, L. M. Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2B. Cancer Res. 66, 10741–10749 (2006).
Liu, X., Shen, T., Mooers, B. H. M., Hilberg, F. & Wu, J. Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharm. 175, 3504–3515 (2018).
Carlomagno, F. et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 23, 6056–6063 (2004).
Mulligan, L. M. RET revisited: expanding the oncogenic portfolio. Nat. Rev. Cancer 14, 173–186 (2014).
De Falco, V. et al. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J. Clin. Endocrinol. Metab. 98, 811–819 (2013).
Kato, S. et al. RET aberrations in diverse cancers: next-generation sequencing of 4,871 patients. Clin. Cancer Res. 23, 1988–1997 (2017).
Iwashita, T. et al. A two-hit model for development of multiple endocrine neoplasia type 2B by RET mutations. Biochem. Biophys. Res. Commun. 268, 804–808 (2000).
Muzza, M. et al. Four novel RET germline variants in exons 8 and 11 display an oncogenic potential in vitro. Eur. J. Endocrinol. 162, 771–777 (2010).
Meng, S., Wu, H., Wang, J. & Qiu, Q. Systematic analysis of tyrosine kinase inhibitor response to RET gatekeeper mutations in thyroid cancer. Mol. Inf. 35, 495–505 (2016).
Stricker, S., Rauschenberger, V. & Schambony, A. ROR-family receptor tyrosine kinases. Curr. Top. Dev. Biol. 123, 105–142 (2017).
Masiakowski, P. & Carroll, R. D. A novel family of cell surface receptors with tyrosine kinase-like domain. J. Biol. Chem. 267, 26181–26190 (1992).
Daneshmanesh, A. H. et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int. J. Cancer 123, 1190–1195 (2008).
Baskar, S. et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 14, 396–404 (2008).
Cui, B. et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood 128, 2931–2940 (2016).
Choudhury, A. et al. Silencing of ROR1 and FMOD with siRNA results in apoptosis of CLL cells. Br. J. Haematol. 151, 327–335 (2010).
Zhao, Y. et al. Tyrosine kinase ROR1 as a target for anti-cancer therapies. Front. Oncol. 11 (2021).
Sánchez-Solana, B., Laborda, J. & Baladrón, V. Mouse resistin modulates adipogenesis and glucose uptake in 3T3-L1 preadipocytes through the ROR1 receptor. Mol. Endocrinol. 26, 110–127 (2012).
Kurita, Y. et al. A high-fat/high-sucrose diet induces WNT4 expression in mouse pancreatic β-cells. Kurum. Med. J. 65, 55–62 (2019).
Heliste, J. et al. Receptor tyrosine kinase profiling of ischemic heart identifies ROR1 as a potential therapeutic target. BMC Cardiovasc. Disord. 18, (2018).
Gui, B. et al. Heterozygous recurrent mutations inducing dysfunction of ROR2 gene in patients with short stature. Front. Cell Dev. Biol. 9 (2021).
Zisch, A. H. & Pasquale, E. B. The Eph family: a multitude of receptors that mediate cell recognition signals. Cell Tissue Res. 290, 217–226 (1997).
Eph Nomenclature Committee. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Eph Nomenclature Committee. Cell 90, 403–404 (1997).
Davy, A. & Soriano, P. Ephrin signaling in vivo: look both ways. Dev. Dyn. 232, 1–10 (2005).
Rohani, N., Canty, L., Luu, O., Fagotto, F. & Winklbauer, R. EphrinB/EphB signaling controls embryonic germ layer separation by contact-induced cell detachment. PLoS Biol. 9 (2011).
Kuijper, S., Turner, C. J. & Adams, R. H. Regulation of angiogenesis by Eph-ephrin interactions. Trends Cardiovasc. Med. 17, 145–151 (2007).
Genander, M. & Frisén, J. Ephrins and Eph receptors in stem cells and cancer. Curr. Opin. Cell Biol. 22, 611–616 (2010).
Arora, S., Scott, A. M. & Janes, P. W. Eph receptors in cancer. Biomedicines 11 (2023).
Giaginis, C. et al. Clinical significance of ephrin (eph)-A1, -A2, -a4, -a5 and -a7 receptors in pancreatic ductal adenocarcinoma. Pathol. Oncol. Res. 16, 267–276 (2010).
Kandouz, M. The Eph/Ephrin family in cancer metastasis: communication at the service of invasion. Cancer Metastasis Rev. 31, 353–373 (2012).
Lisabeth, E. M., Fernandez, C. & Pasquale, E. B. Cancer somatic mutations disrupt functions of the EphA3 receptor tyrosine kinase through multiple mechanisms. Biochemistry 51, 1464–1475 (2012).
Lisabeth, E. M., Falivelli, G. & Pasquale, E. B. Eph receptor signaling and ephrins. Cold Spring Harb. Perspect. Biol. 5, 1–20 (2013).
Ferguson, B. D. et al. Novel EPHB4 receptor tyrosine kinase mutations and kinomic pathway analysis in lung cancer. Sci. Rep. 5 (2015).
Chakraborty, S., Baruah, R., Mishra, N. & Varma, A. K. In-silico and structure-based assessment to evaluate pathogenicity of missense mutations associated with non-small cell lung cancer identified in the Eph-ephrin class of proteins. Genom. Inform. 21 (2023).
Faoro, L. et al. EphA2 mutation in lung squamous cell carcinoma promotes increased cell survival, cell invasion, focal adhesions, and mammalian target of rapamycin activation. J. Biol. Chem. 285, 18575–18585 (2010).
Zhuang, G. et al. Effects of cancer-associated EPHA3 mutations on lung cancer. J. Natl Cancer Inst. 104, 1182–1197 (2012).
Magnus, J. H., Husby, G. & Kolset, S. O. Presence of glycosaminoglycans in purified AA type amyloid fibrils associated with juvenile rheumatoid arthritis. Ann. Rheum. Dis. 48, 215–219 (1989).
Kim, G.-H., Her, J.-H. & Han, J.-K. Ryk cooperates with Frizzled 7 to promote Wnt11-mediated endocytosis and is essential for Xenopus laevis convergent extension movements. J. Cell Biol. 182, 1073–1082 (2008).
Halford, M. M. et al. Ryk-deficient mice exhibit craniofacial defects associated with perturbed Eph receptor crosstalk. Nat. Genet. 25, 414–418 (2000).
Roy, J. P., Halford, M. M. & Stacker, S. A. The biochemistry, signalling and disease relevance of RYK and other WNT-binding receptor tyrosine kinases. Growth Factors 36, 15–40 (2018).
Green, J., Nusse, R. & van Amerongen, R. The role of Ryk and Ror receptor tyrosine kinases in Wnt signal transduction. Cold Spring Harb. Perspect. Biol. 6, 9175–9175 (2014).
Fradkin, L. G., Dura, J.-M. & Noordermeer, J. N. Ryks: new partners for Wnts in the developing and regenerating nervous system. Trends Neurosci. 33, 84–92 (2010).
Schmitt, A. M. et al. Wnt-Ryk signalling mediates medial-lateral retinotectal topographic mapping. Nature 439, 31–37 (2006).
Lu, W., Yamamoto, V., Ortega, B. & Baltimore, D. Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 119, 97–108 (2004).
Macheda, M. L. et al. The Wnt receptor Ryk plays a role in mammalian planar cell polarity signaling. J. Biol. Chem. 287, 29312–29323 (2012).
Andre, P. et al. The Wnt coreceptor Ryk regulates Wnt/planar cell polarity by modulating the degradation of the core planar cell polarity component Vangl2. J. Biol. Chem. 287, 44518–44525 (2012).
Famili, F. et al. The non-canonical Wnt receptor Ryk regulates hematopoietic stem cell repopulation in part by controlling proliferation and apoptosis. Cell Death Dis. 7, (2016).
Jeong, S.-Y., Lyu, J., Kim, J.-A. & Oh, I.-H. Ryk modulates the niche activity of mesenchymal stromal cells by fine-tuning canonical Wnt signaling. Exp. Mol. Med. 52, 1140–1151 (2020).
Kikuchi, A., Yamamoto, H., Sato, A. & Matsumoto, S. New insights into the mechanism of wnt signaling pathway activation. in International Review of Cell and Molecular Biology (ed Kwang, W. J.) Vol. 291, 21–71 (Academic Press, 2011).
Zhu, N. et al. Challenging role of Wnt5a and its signaling pathway in cancer metastasis (Review). Exp. Ther. Med. 8, 3–8 (2014).
Ford, C. E., Qian Ma, S. S., Quadir, A. & Ward, R. L. The dual role of the novel Wnt receptor tyrosine kinase, ROR2, in human carcinogenesis. Int. J. Cancer 133, 779–787 (2013).
Katso, R. M. et al. Overexpression of H-Ryk in epithelial ovarian cancer: prognostic significance of receptor expression. Clin. Cancer Res. 6, 3271–3281 (2000).
Mossie, K. et al. Colon carcinoma kinase-4 defines a new subclass of the receptor tyrosine kinase family. Oncogene 11, 2179–2184 (1995).
Berger, H., Wodarz, A. & Borchers, A. PTK7 Faces the Wnt in development and disease. Front. Cell Dev. Biol. 5 (2017).
Lee, H. K., Chauhan, S. K., Kay, E. & Dana, R. Flt-1 regulates vascular endothelial cell migration via a protein tyrosine kinase-7-dependent pathway. Blood 117, 5762–5771 (2011).
Lander, R. & Petersen, C. P. Wnt, Ptk7, and FGFRL expression gradients control trunk positional identity in planarian regeneration. Elife 5 (2016).
Dunn, N. R. & Tolwinski, N. S. Ptk7 and Mcc, unfancied components in non-canonical Wnt signaling and cancer. Cancers 8 (2016).
Grimes, D. T. et al. Zebrafish models of idiopathic scoliosis link cerebrospinal fluid flow defects to spine curvature. Science 352, 1341–1344 (2016).
Wang, M. et al. Role of the planar cell polarity gene Protein tyrosine kinase 7 in neural tube defects in humans. Birth Defects Res. A Clin. Mol. Teratol. 103, 1021–1027 (2015).
Lu, X. et al. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 430, 93–98 (2004).
Chen, R. et al. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res. 74, 2892–2902 (2014).
Jin, J., Ryu, H. S., Lee, K. B. & Jang, J.-J. High expression of protein tyrosine kinase 7 significantly associates with invasiveness and poor prognosis in intrahepatic cholangiocarcinoma. PLoS One 9, 1–11 (2014).
Lhoumeau, A.-C. et al. Overexpression of the promigratory and prometastatic PTK7 receptor is associated with an adverse clinical outcome in colorectal cancer. PLoS One, 10 (2015).
Wang, H. et al. PTK7 protein is decreased in epithelial ovarian carcinomas with poor prognosis. Int. J. Clin. Exp. Pathol. 7, 7881–7889 (2014).
Jiang, W. et al. PTK7 expression is associated with lymph node metastasis, ALK and EGFR mutations in lung adenocarcinomas. Histol. Histopathol. 35, 489–495 (2020).
Gärtner, S. et al. PTK 7 Is a transforming gene and prognostic marker for breast cancer and nodal metastasis involvement. PLoS One 9, 1–11 (2014).
Damelin, M. et al. A PTK7-targeted antibody-drug conjugate reduces tumor-initiating cells and induces sustained tumor regressions. Sci. Transl. Med. 9 (2017).
Jung, P. et al. Isolation of human colon stem cells using surface expression of PTK7. Stem Cell Rep. 5, 979–987 (2015).
Herrmann, J. L. et al. Mediation of NGF-stimulated extracellular matrix invasion by the human melanoma low-affinity p75 neurotrophin receptor: melanoma p75 functions independently of trkA. Mol. Biol. Cell 4, 1205–1216 (1993).
Mahendram, S., Subapanditha, M. K., McFarlane, N., Venugopal, C. & Singh, S. K. Flow-cytometric identification and characterization of neural brain tumor-initiating cells for pathophysiological study and biomedical applications. in Neural Surface Antigens 199–211 (Elsevier, 2015). https://doi.org/10.1016/B978-0-12-800781-5.00017-7.
Chao, M. V. et al. Structure and functions of NGF receptors. in Botulinum and Tetanus Neurotoxins 17–24 (Springer US, Boston, MA, 1993). https://doi.org/10.1007/978-1-4757-9542-4_3.
Wu, R., Li, K., Yuan, M. & Luo, K. Q. Nerve growth factor receptor increases the tumor growth and metastatic potential of triple-negative breast cancer cells. Oncogene 40, 2165–2181 (2021).
Wang, G. et al. Rational design and crystallographic analysis of novel isoform-selective TRKA inhibitors for cancer therapy. Acta Pharm. Sin. B 13, 440–443 (2023).
Regua, A. T., Doheny, D., Arrigo, A. & Lo, H.-W. Trk receptor tyrosine kinases in metastasis and cancer therapy. Discov. Med. 28, 195–203 (2019).
Okimoto, R. A. & Bivona, T. G. AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer 6, 27–34 (2015).
Falcone, I. et al. AXL Receptor in breast cancer: molecular involvement and therapeutic limitations. Int. J. Mol. Sci. 21 (2020).
Graham, D. K., DeRyckere, D., Davies, K. D. & Earp, H. S. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 14, 769–785 (2014).
Linger, R. M. A., Keating, A. K., Earp, H. S. & Graham, D. K. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 100, 35–83 (2008).
Gay, C. M., Balaji, K. & Byers, L. A. Giving AXL the axe: targeting AXL in human malignancy. Br. J. Cancer 116, 415–423 (2017).
Mudduluru, G., Vajkoczy, P. & Allgayer, H. Myeloid zinc finger 1 induces migration, invasion, and in vivo metastasis through Axl gene expression in solid cancer. Mol. Cancer Res. 8, 159–169 (2010).
Mudduluru, G., Leupold, J. H., Stroebel, P. & Allgayer, H. PMA up-regulates the transcription of Axl by AP-1 transcription factor binding to TRE sequences via the MAPK cascade in leukaemia cells. Biol. Cell 103, 21–33 (2011).
Rankin, E. B. et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl Acad. Sci. USA 111, 13373–13378 (2014).
Zhang, Y., Earp, H. S. & Liu, P. Beyond growth signaling: apoptotic sensor MERTK activates AKT by a novel mechanism. Mol. Cell Oncol. 6 (2019).
Jiang, Y. et al. MERTK mediated novel site Akt phosphorylation alleviates SAV1 suppression. Nat. Commun. 10 (2019).
Zhu, C., Wei, Y. & Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol. Cancer 18 (2019).
Rankin, E. & Giaccia, A. The receptor tyrosine kinase AXL in cancer progression. Cancers 8 (2016).
Mudduluru, G. & Allgayer, H. The human receptor tyrosine kinase Axl gene—promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. Biosci. Rep. 28, 161–176 (2008).
Zhai, X. et al. Gas6/AXL pathway: immunological landscape and therapeutic potential. Front. Oncol. 13 (2023).
Salian-Mehta, S. et al. Functional consequences of AXL sequence variants in hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 99, 1452–1460 (2014).
Dilara Fatma, A. & Özkan, D. Molecular profiling of TAM tyrosine kinase receptors and ligands in endometrial carcinoma: An in silico-study. Taiwan J. Obstet. Gynecol. 62, 311–324 (2023).
Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, 607–613 (2019).
Sensi, M. et al. Human cutaneous melanomas lacking MITF and melanocyte differentiation antigens express a functional Axl receptor kinase. J. Investig. Dermatol 131, 2448–2457 (2011).
Al Kafri, N. & Hafizi, S. Identification of signalling pathways activated by Tyro3 that promote cell survival, proliferation and invasiveness in human cancer cells. Biochem. Biophys. Rep. 28 (2021).
Al Kafri, N. & Hafizi, S. Tumour-secreted protein S (ProS1) activates a tyro3-Erk signalling axis and protects cancer cells from apoptosis. Cancers 11 (2019).
Smart, S. K., Vasileiadi, E., Wang, X., DeRyckere, D. & Graham, D. K. The emerging role of TYRO3 as a therapeutic target in cancer. Cancers (Basel) 10, 1–27 (2018).
Seshagiri, S. et al. Recurrent R-spondin fusions in colon cancer. Nature 488, 660–664 (2012).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008).
Wagle, N. et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 4, 61–68 (2014).
Loriaux, M. M. et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood 111, 4788–4796 (2008).
Jiao, Y. et al. Whole‐exome sequencing of pancreatic neoplasms with acinar differentiation. J. Pathol. 232, 428–435 (2014).
Easty, D. J. et al. Novel and known protein tyrosine kinases and their abnormal expression in human melanoma. J. Investig. Dermatol 101, 679–684 (1993).
Krauthammer, M. et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44, 1006–1014 (2012).
Shi, C. et al. The proto-oncogene Mer tyrosine kinase is a novel therapeutic target in mantle cell lymphoma. J. Hematol. Oncol. 11 (2018).
Lee-Sherick, A. B. et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 3, 1–18 (2018).
Farnworth-McHugh, S. et al. Potential oncogenic effect of the MERTK-dependent apoptotic-cell clearance pathway in starry-sky B-cell lymphoma. Front. Immunol. 11 (2020).
Ohta, S. et al. The role of MerTK in promoting cell migration is enhanced by the oncogenic Ras/IL‐33 signaling axis. FEBS J. 289, 1950–1967 (2022).
Yi, J. H. et al. MerTK is a novel therapeutic target in gastric cancer. Oncotarget 8, 96656–96667 (2017).
Xie, S. et al. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget 6, 9206–9219 (2015).
Tworkoski, K. A. et al. MERTK controls melanoma cell migration and survival and differentially regulates cell behavior relative to AXL. Pigment Cell Melanoma Res. 26, 527–541 (2013).
Hucthagowder, V. et al. Resequencing analysis of the human candidate ras and receptor tyrosine kinase gene family in multiple myeloma. Blood 116, 301–301 (2010).
Audo, I. et al. MERTK mutation update in inherited retinal diseases. Hum. Mutat. 39, 887–913 (2018).
Eklund, L., Kangas, J. & Saharinen, P. Angiopoietin-tie signalling in the cardiovascular and lymphatic systems. Clin. Sci. 131, 87–103 (2017).
Puri, M. C., Partanen, J., Rossant, J. & Bernstein, A. Interaction of the TEK and TIE receptor tyrosine kinases during cardiovascular development. Development 126, 4569–4580 (1999).
Schnürch, H. & Risau, W. Expression of tie-2, a member of a novel family of receptor tyrosine kinases, in the endothelial cell lineage. Development 119, 957–968 (1993).
Duran, C. L. et al. Targeting Tie2 in the tumor microenvironment: from angiogenesis to dissemination. Cancers. 13 (2021).
Madukwe, J. & Ferguson, K. The mechanism of ligand‐induced activation of the tie family of receptor tyrosine kinases. FASEB J. 33, 809.10–809.10 (2019).
Monk, B. J. et al. Anti-angiopoietin therapy with trebananib for recurrent ovarian cancer (TRINOVA-1): a randomised, multicentre, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 15, 799–808 (2014).
Monk, B. J. et al. Final results of a phase 3 study of trebananib plus weekly paclitaxel in recurrent ovarian cancer (TRINOVA-1): long-term survival, impact of ascites, and progression-free survival-2. Gynecol. Oncol. 143, 27–34 (2016).
Campochiaro, P. A. et al. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology 122, 545–554 (2015).
Ishibashi, M. et al. Tyrosine kinase receptor TIE-1 mediates platinum resistance by promoting nucleotide excision repair in ovarian cancer. Sci. Rep. 8 (2018).
Marguier, A. et al. TIE-2 signaling activation by angiopoietin 2 on myeloid-derived suppressor cells promotes melanoma-specific T-cell inhibition. Front. Immunol. 13 (2022).
Dang, N. et al. CD167 acts as a novel costimulatory receptor in T-cell activation. J. Immunother. 32, 773–784 (2009).
Fridman, R. & Agarwal, G. New concepts on the interactions of discoidin domain receptors with collagen. Biochim. Biophys. Acta Mol. Cell Res. 1866 (2019).
Iwai, L. K., Luczynski, M. T. & Huang, P. H. Discoidin domain receptors: a proteomic portrait. Cell Mol. Life Sci. 71, 3269–3279 (2014).
Henriet, E. et al. Multitasking discoidin domain receptors are involved in several and specific hallmarks of cancer. Cell Adh. Migr. 12, 363–377 (2018).
Ford, C. E. et al. Expression and mutation analysis of the discoidin domain receptors 1 and 2 in non-small cell lung carcinoma. Br. J. Cancer 96, 808–814 (2007).
Hammerman, P. S. et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 1, 78–89 (2011).
Yang, S. et al. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung carcinomas. Oncol. Rep. 24, 311–319 (2010).
Yeung, D. A. et al. Clustering, spatial distribution, and phosphorylation of discoidin domain receptors 1 and 2 in response to soluble collagen I. J. Mol. Biol. 431, 368–390 (2019).
Orgel, J. P. R. O. & Madhurapantula, R. S. A structural prospective for collagen receptors such as DDR and their binding of the collagen fibril. Biochim. Biophys. Acta Mol. Cell Res. 1866 (2019).
Leitinger, B. Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 310, 39–87 (2014).
Majo, S. & Auguste, P. The Yin and Yang of discoidin domain receptors (DDRs): implications in tumor growth and metastasis development. Cancers (Basel) 13, 1–27 (2021).
Bonfil, R. D. et al. Expression and subcellular localization of Discoidin Domain Receptor 1 (DDR1) define prostate cancer aggressiveness. Cancer Cell Int. 21 (2021).
Fu, H.-L. et al. Shedding of discoidin domain receptor 1 by membrane-type matrix metalloproteinases. J. Biol. Chem. 288, 12114–12129 (2013).
Castro-Sanchez, L., Soto-Guzman, A., Guaderrama-Diaz, M., Cortes-Reynosa, P. & Salazar, E. P. Role of DDR1 in the gelatinases secretion induced by native type IV collagen in MDA-MB-231 breast cancer cells. Clin. Exp. Metastasis 28, 463–477 (2011).
Le, C. C. et al. LRP-1 promotes colon cancer cell proliferation in 3D collagen matrices by mediating DDR1 endocytosis. Front. Cell Dev. Biol. 8 (2020).
Poudel, B., Lee, Y.-M. & Kim, D.-K. DDR2 inhibition reduces migration and invasion of murine metastatic melanoma cells by suppressing MMP2/9 expression through ERK/NF-κB pathway. Acta Biochim. Biophys. Sin. 47, 292–298 (2015).
Reger de Moura, C. et al. Discoidin domain receptors: a promising target in melanoma. Pigment. Cell Melanoma Res. 32, 697–707 (2019).
Sugimoto, K. et al. Prognostic impact of phosphorylated discoidin domain receptor-1 in esophageal cancer. J. Surg. Res. 235, 479–486 (2019).
Alexander, S. P. et al. The concise guide to PHARMACOLOGY 2015/16: catalytic receptors. Br. J. Pharm. 172, 5979–6023 (2015).
Raghunath, M. et al. A novel kinase, AATYK induces and promotes neuronal differentiation in a human neuroblastoma (SH-SY5Y) cell line. Brain Res. Mol. Brain Res. 77, 151–162 (2000).
Mórotz, G. M. et al. A revised nomenclature for the lemur family of protein kinases. Commun. Biol. 7 (2024).
Ferrari, E., Naponelli, V. & Bettuzzi, S. Lemur tyrosine kinases and prostate cancer: a literature review. Int. J. Mol. Sci. 22 (2021).
Wendler, F., Purice, T.-M., Simon, T., Stebbing, J. & Giamas, G. The LMTK-family of kinases: emerging important players in cell physiology and pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 1867 (2021).
Manning, G., Whyte, D. B., Martinez, R., Hunter, T. & Sudarsanam, S. The protein kinase complement of the human genome. Science 298, 1912–1934 (2002).
Kawa, S. et al. Azoospermia in mice with targeted disruption of the Brek/Lmtk2 (brain-enriched kinase/lemur tyrosine kinase 2) gene. Proc. Natl Acad. Sci. USA 103, 19344–19349 (2006).
Komaki, K. et al. Lemur tail kinase 1 (LMTK1) regulates the endosomal localization of β-secretase BACE1. J. Biochem. 170, 729–738 (2021).
Bencze, J. et al. Neuropathological characterization of Lemur tyrosine kinase 2 (LMTK2) in Alzheimer’s disease and neocortical Lewy body disease. Sci. Rep. 9 (2019).
Vella, V., Giamas, G. & Ditsiou, A. Diving into the dark kinome: lessons learned from LMTK3. Cancer Gene Ther. 29, 1077–1079 (2022).
Vezelis, A. et al. LMTK2 as potential biomarker for stratification between clinically insignificant and clinically significant prostate cancer. J. Oncol. 2021, 1–6 (2021).
Harries, L. W., Perry, J. R., McCullagh, P. & Crundwell, M. Alterations in LMTK2, MSMB and HNF1B gene expression are associated with the development of prostate cancer. BMC Cancer 10 (2010).
Jiang, T. et al. LMTK3 promotes tumorigenesis in bladder cancer via the ERK/MAPK pathway. FEBS Open Biol. 10, 2107–2121 (2020).
Stebbing, J. et al. LMTK3 confers chemo-resistance in breast cancer. Oncogene 37, 3113–3130 (2018).
Shi, H. et al. Lemur tyrosine kinase-3 is a significant prognostic marker for patients with colorectal cancer. Int. J. Clin. Exp. Pathol. 7, 1101–1107 (2014).
Ditsiou, A. et al. The multifaceted role of lemur tyrosine kinase 3 in health and disease. Open Biol. 11 (2021).
Zhou, C. et al. STYK1 promotes autophagy through enhancing the assembly of autophagy-specific class III phosphatidylinositol 3-kinase complex I. Autophagy 16, 1786–1806 (2020).
Liu, L. et al. A novel protein tyrosine kinase NOK that shares homology with platelet-derived growth factor/fibroblast growth factor receptors induces tumorigenesis and metastasis in nude mice. Cancer Res. 64, 3491–3499 (2004).
Trenker, R. & Jura, N. Receptor tyrosine kinase activation: from the ligand perspective. Curr. Opin. Cell Biol. 63, 174–185 (2020).
Lai, Y. et al. STYK1/NOK Promotes metastasis and epithelial-mesenchymal transition in non-small cell lung cancer by suppressing FoxO1 signaling. Front. Cell Dev. Biol. 9 (2021).
Kondoh, T., Kobayashi, D., Tsuji, N., Kuribayashi, K. & Watanabe, N. Overexpression of serine threonine tyrosine kinase 1/novel oncogene with kinase domain mRNA in patients with acute leukemia. Exp. Hematol. 37, 824–830 (2009).
Wang, Z. et al. STYK1 promotes epithelial-mesenchymal transition and tumor metastasis in human hepatocellular carcinoma through MEK/ERK and PI3K/AKT signaling. Sci Rep. 6, 1–12 (2016).
Jackson, K. A., Oprea, G., Handy, J. & Kimbro, K. S. Aberrant STYK1 expression in ovarian cancer tissues and cell lines. J. Ovar. Res. 2 (2009).
Ma, Z. et al. STYK1 promotes tumor growth and metastasis by reducing SPINT2/HAI-2 expression in non-small cell lung cancer. Cell Death Dis. 10 (2019).
Chung, S. et al. Overexpression of the potential kinase serine/threonine/tyrosine kinase 1 (STYK 1) in castration‐resistant prostate cancer. Cancer Sci. 100, 2109–2114 (2009).
Zeng, S.-L. et al. STYK1/NOK affects cell cycle late mitosis and directly interacts with anaphase-promoting complex activator CDH1. Heliyon 8 (2022).
Chen, Y. et al. Point mutation at single tyrosine residue of novel oncogene NOK abrogates tumorigenesis in nude mice. Cancer Res. 65, 10838–10846 (2005).
Li, Y.-H. et al. The carboxyl terminal tyrosine 417 residue of NOK has an autoinhibitory effect on NOK-mediated signaling transductions. Biochem. Biophys. Res. Commun. 356, 444–449 (2007).
Li, Y.-H. et al. Transmembrane helix of novel oncogene with kinase-domain (NOK) influences its oligomerization and limits the activation of RAS/MAPK signaling. Mol. Cells 27, 39–46 (2009).
Ding, X., Jiang, Q.-B., Li, R., Chen, S. & Zhang, S. NOK/STYK1 has a strong tendency towards forming aggregates and colocalises with epidermal growth factor receptor in endosomes. Biochem. Biophys. Res. Commun. 421, 468–473 (2012).
Braicu, C. et al. A Comprehensive review on MAPK: a promising therapeutic target in cancer. Cancers(Basel) 11, 1–25 (2019).
Guo, X. & Wang, X.-F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 19, 71–88 (2009).
Mahajan, K. & Mahajan, N. P. Cross-talk of tyrosine kinases with the DNA damage signaling pathways. Nucleic Acids Res. 43, 10588–10601 (2015).
Regad, T. Targeting RTK signaling pathways in cancer. Cancers 7, 1758–1784 (2015).
Sudhesh Dev, S., Zainal Abidin, S. A., Farghadani, R., Othman, I. & Naidu, R. Receptor tyrosine kinases and their signaling pathways as therapeutic targets of curcumin in cancer. Front. Pharmacol. 12, 1–26 (2021).
Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 (2000).
Lo, H.-W. & Hung, M.-C. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer 94, 184–188 (2006).
Haeusler, R. A., McGraw, T. E. & Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 19, 31–44 (2018).
Andrae, J., Gallini, R. & Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 22, 1276–1312 (2008).
Mineur, P. et al. Newly identified biologically active and proteolysis-resistant VEGF-A isoform VEGF111 is induced by genotoxic agents. J. Cell Biol. 179, 1261–1273 (2007).
Toledo, R. A. et al. Exome sequencing of plasma DNA portrays the mutation landscape of colorectal cancer and discovers mutated VEGFR2 receptors as modulators of antiangiogenic therapies. Clin. Cancer Res 24, 3550–3559 (2018).
Azoury, S. C., Reddy, S., Shukla, V. & Deng, C.-X. Fibroblast growth factor receptor 2 (FGFR2) mutation related syndromic craniosynostosis. Int. J. Biol. Sci. 13, 1479–1488 (2017).
Williams, J. A. et al. Cholecystokinin activates a variety of intracellular signal transduction mechanisms in rodent pancreatic acinar cells. Pharm. Toxicol. 91, 297–303 (2002).
Vidal, A. & Redmer, T. Decoding the role of CD271 in melanoma. Cancers 12 (2020).
Lam, B. Q., Dai, L. & Qin, Z. The role of HGF/c-MET signaling pathway in lymphoma. J. Hematol. Oncol. 9, 1–8 (2016).
Gucciardo, E., Sugiyama, N. & Lehti, K. Eph- and ephrin-dependent mechanisms in tumor and stem cell dynamics. Cell Mol. Life Sci. 71, 3685–3710 (2014).
Pasquale, E. B. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10, 165–180 (2010).
Saharinen, P., Eklund, L. & Alitalo, K. Therapeutic targeting of the angiopoietin–TIE pathway. Nat. Rev. Drug Discov. 16, 635–661 (2017).
Zhang, X. et al. Potential of tyrosine kinase receptor TIE-1 as novel therapeutic target in high-PI3K-expressing ovarian cancer. Cancers 12 (2020).
Rodriguez-Trillo, A. et al. Non-canonical WNT5A signaling through RYK contributes to aggressive phenotype of the rheumatoid fibroblast-like synoviocytes. Front. Immunol. 11, 1–13 (2020).
Mariadoss, A. V. A. & Wang, C.-Z. Exploring the cellular and molecular mechanism of discoidin domain receptors (DDR1 and DDR2) in bone formation, regeneration, and its associated disease conditions. Int. J. Mol. Sci. 24 (2023).
Goto, K., Kawahara, I., Kuniyasu, H. & Takaki, M. A protein tyrosine kinase receptor, c-RET signaling pathway contributes to the enteric neurogenesis induced by a 5-HT4 receptor agonist at an anastomosis after transection of the gut in rodents. J. Physiol. Sci. 65, 377–383 (2015).
Regua, A. T., Najjar, M. & Lo, H.-W. RET signaling pathway and RET inhibitors in human cancer. Front. Oncol. 12 (2022).
Hasan, M. K., Ghia, E. M., Rassenti, L. Z., Widhopf, G. F. & Kipps, T. J. Wnt5a enhances proliferation of chronic lymphocytic leukemia and ERK1/2 phosphorylation via a ROR1/DOCK2-dependent mechanism. Leukemia 35, 1621–1630 (2021).
Chen, Y. et al. Cirmtuzumab blocks Wnt5a/ROR1 stimulation of NF-κB to repress autocrine STAT3 activation in chronic lymphocytic leukemia. Blood 134, 1084–1094 (2019).
Villarroel, A. et al. Src and Fyn define a new signaling cascade activated by canonical and non-canonical Wnt ligands and required for gene transcription and cell invasion. Cell Mol. Life Sci. 77, 919–935 (2020).
Endo, M., Kamizaki, K. & Minami, Y. The Ror-family receptors in development, tissue regeneration and age-related disease. Front. Cell. Dev. Biol. 10, (2022).
Della Corte, C. M. et al. Role and targeting of anaplastic lymphoma kinase in cancer. Mol. Cancer 17 (2018).
Shiota, M. et al. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 9, 1567–1574 (1994).
Fruman, D. A. et al. The PI3K pathway in human disease. Cell 170, 605–635 (2017).
Yu, H., Pardoll, D. & Jove, R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 (2009).
Chang, F. et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 17, 1263–1293 (2003).
Zhang, Z., Zhou, X., Shen, H., Wang, D. & Wang, Y. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: evidence from an in vitro study. BMC Med. 7 (2009).
McCubrey, J. A. et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 1773, 1263–1284 (2007).
Steelman, L. S. et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 18, 189–218 (2004).
Stirewalt, D. L. et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood 97, 3589–3595 (2001).
Yan, J., Roy, S., Apolloni, A., Lane, A. & Hancock, J. F. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem. 273, 24052–24056 (1998).
Garnett, M. J. & Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 6, 313–319 (2004).
Shelton, J. G. et al. B-raf and insulin synergistically prevent apoptosis and induce cell cycle progression in hematopoietic cells. Cell Cycle 3, 189–196 (2004).
Blalock, W. L. et al. A conditionally-active form of MEK1 results in autocrine tranformation of human and mouse hematopoietic cells. Oncogene 19, 526–536 (2000).
Nakano, H. et al. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc. Natl Acad. Sci. USA 95, 3537–3542 (1998).
Nandan, M. O. & Yang, V. W. Genetic and chemical models of colorectal cancer in mice. Curr. Colorectal Cancer Rep. 6, 51–59 (2010).
Reuter, C. W., Morgan, M. A. & Bergmann, L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood 96, 1655–1669 (2000).
Vaughn, C. P., Zobell, S. D., Furtado, L. V., Baker, C. L. & Samowitz, W. S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 50, 307–312 (2011).
Almoguera, C. et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53, 549–554 (1988).
Bizebard, T. et al. Crystallization and preliminary X-ray diffraction studies of a monoclonal antibody Fab fragment specific for an influenza virus haemagglutinin and of an escape mutant of that haemagglutinin. J. Mol. Biol. 216, 513–514 (1990).
Rajasekharan, S. & Raman, T. Ras and Ras mutations in cancer. Open Life Sci. 8, 609–624 (2013).
Bazan, V. et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann. Oncol. 13, 1438–1446 (2002).
Parsons, B. L., Culp, S. J., Manjanatha, M. G. & Heflich, R. H. Occurrence of H-ras codon 61 CAA to AAA mutation during mouse liver tumor progression. Carcinogenesis 23, 943–948 (2002).
Bos, J. L. ras oncogenes in human cancer: a review. Cancer Res. 49, 4682–4689 (1989).
Edkins, S. et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol. Ther. 5, 928–932 (2006).
Imamura, Y. et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res. 18, 4753–4763 (2012).
Quilliam, L. A. et al. M-Ras/R-Ras3, a transforming ras protein regulated by Sos1, GRF1, and p120 Ras GTPase-activating protein, interacts with the putative Ras effector AF6. J. Biol. Chem. 274, 23850–23857 (1999).
Irahara, N. et al. NRAS mutations are rare in colorectal cancer. Diagn. Mol. Pathol. 19, 157–163 (2010).
Omholt, K. et al. Screening of N-ras codon 61 mutations in paired primary and metastatic cutaneous melanomas: mutations occur early and persist throughout tumor progression. Clin. Cancer Res. 8, 3468–3474 (2002).
Cascetta, P. et al. KRAS in NSCLC: state of the art and future perspectives. Cancers 14 (2022).
Veluswamy, R., Mack, P. C., Houldsworth, J., Elkhouly, E. & Hirsch, F. R. KRAS G12C-mutant non-small cell lung cancer. J. Mol. Diagn. 23, 507–520 (2021).
Qunaj, L., May, M. S., Neugut, A. I. & Herzberg, B. O. Prognostic and therapeutic impact of the KRAS G12C mutation in colorectal cancer. Front Oncol 13 (2023).
Chen, K., Zhang, Y., Qian, L. & Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 14 (2021).
Hunter, J. C. et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl Acad. Sci. USA 111, 8895–8900 (2014).
Hallin, J. et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 (2020).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Nandan, M. O. & Yang, V. W. An update on the biology of RAS/RAF mutations in colorectal cancer. Curr. Colorectal Cancer Rep. 7, 113–120 (2011).
Marais, R., Light, Y., Paterson, H. F., Mason, C. S. & Marshall, C. J. Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J. Biol. Chem. 272, 4378–4383 (1997).
Dobre, E.-G., Nichita, L., Popp, C., Zurac, S. & Neagu, M. Assessment of RAS-RAF-MAPK pathway mutation status in healthy skin, benign nevi, and cutaneous melanomas: pilot study using droplet digital PCR. Int. J. Mol. Sci. 25 (2024).
Mayr, D., Hirschmann, A., Löhrs, U. & Diebold, J. KRAS and BRAF mutations in ovarian tumors: a comprehensive study of invasive carcinomas, borderline tumors and extraovarian implants. Gynecol. Oncol. 103, 883–887 (2006).
Jiang, L., Chu, H. & Zheng, H. B-Raf mutation and papillary thyroid carcinoma patients. Oncol. Lett. 11, 2699–2705 (2016).
Clarke, C. N. & Kopetz, E. S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 6, 660–667 (2015).
Rajagopalan, H. et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 418 (2002).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002).
Garnett, M. J., Rana, S., Paterson, H., Barford, D. & Marais, R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell 20, 963–969 (2005).
Śmiech, M., Leszczyński, P., Kono, H., Wardell, C. & Taniguchi, H. Emerging BRAF mutations in cancer progression and their possible effects on transcriptional networks. Genes 11 (2020).
Walker, B. A. et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 132, 587–597 (2018).
Boyle, E. M. et al. BRAF and DIS3 mutations associate with adverse outcome in a long-term follow-up of patients with multiple myeloma. Clin. Cancer Res. 26, 2422–2432 (2020).
Wu, X. et al. Mutations in BRAF codons 594 and 596 predict good prognosis in melanoma. Oncol. Lett. 14, 3601–3605 (2017).
Cremolini, C. et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann. Oncol. 26, 2092–2097 (2015).
Zheng, G. et al. Clinical detection and categorization of uncommon and concomitant mutations involving BRAF. BMC Cancer 15 (2015).
Heidorn, S. J. et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 (2010).
Schirripa, M. et al. Class 1, 2, and 3 BRAF-mutated metastatic colorectal cancer: a detailed clinical, pathologic, and molecular characterization. Clin. Cancer Res. 25, 3954–3961 (2019).
Park, E. et al. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 575, 545–550 (2019).
Haling, J. R. et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26, 402–413 (2014).
Wan, P. T. C. et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 (2004).
Noeparast, A. et al. CRAF mutations in lung cancer can be oncogenic and predict sensitivity to combined type II RAF and MEK inhibition. Oncogene 38, 5933–5941 (2019).
Isnaldi, E. et al. Clinico-pathological associations and concomitant mutations of the RAS/RAF pathway in metastatic colorectal cancer. J. Transl. Med. 17 (2019).
Marino, K. A., Sutto, L. & Gervasio, F. L. The effect of a widespread cancer-causing mutation on the inactive to active dynamics of the B-Raf kinase. J. Am. Chem. Soc. 137, 5280–5283 (2015).
Salzmann, M. et al. MEK inhibitors for pre-treated, NRAS-mutated metastatic melanoma: a multi-centre, retrospective study. Eur. J. Cancer 166, 24–32 (2022).
Marani, A., Gioacchini, H., Paolinelli, M., Offidani, A. & Campanati, A. Potential drug-drug interactions with mitogen-activated protein kinase (MEK) inhibitors used to treat melanoma. Expert Opin. Drug Metab. Toxicol. 19, 555–567 (2023).
Akinleye, A., Furqan, M., Mukhi, N., Ravella, P. & Liu, D. MEK and the inhibitors: from bench to bedside. J. Hematol. Oncol. 6 (2013).
Gao, Y. et al. V211D mutation in MEK1 causes resistance to MEK inhibitors in colon cancer. Cancer Discov. 9, 1182–1191 (2019).
Gao, Y. et al. Allele-specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov. 8, 648–661 (2018).
Kubota, Y. et al. Qualitative differences in disease-associated MEK mutants reveal molecular signatures and aberrant signaling-crosstalk in cancer. Nat. Commun. 13 (2022).
Emery, C. M. et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl Acad. Sci. USA 106, 20411–20416 (2009).
Sang, D. et al. Ancestral reconstruction reveals mechanisms of ERK regulatory evolution. Elife 8 (2019).
Martin-Vega, A. & Cobb, M. H. Navigating the ERK1/2 MAPK Cascade. Biomolecules 13 (2023).
Balmanno, K. et al. ERK1/2 inhibitors act as monovalent degraders inducing ubiquitylation and proteasome-dependent turnover of ERK2, but not ERK1. Biochem. J. 480, 587–605 (2023).
Wortzel, I. & Seger, R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2, 195–209 (2011).
Smorodinsky-Atias, K., Soudah, N. & Engelberg, D. Mutations that confer drug-resistance, oncogenicity and intrinsic activity on the ERK MAP kinases-current state of the art. Cells 9 (2020).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Ojesina, A. I. et al. Landscape of genomic alterations in cervical carcinomas. Nature 506, 371–375 (2014).
Goetz, E. M., Ghandi, M., Treacy, D. J., Wagle, N. & Garraway, L. A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 74, 7079–7089 (2014).
Yu, L., Wei, J. & Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol. 85, 69–94 (2022).
Vivanco, I. & Sawyers, C. L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 (2002).
Mohan, C. D. et al. Trisubstituted-imidazoles induce apoptosis in human breast cancer cells by targeting the oncogenic PI3K/Akt/mTOR signaling pathway. PLoS One 11 (2016).
Janku, F., Yap, T. A. & Meric-Bernstam, F. Targeting the PI3K pathway in cancer: are we making headway? Nat. Rev. Clin. Oncol. 15, 273–291 (2018).
Manning, B. D. & Cantley, L. C. AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 (2007).
Glaviano, A. et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer 22 (2023).
Juric, D. et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518, 240–244 (2015).
Razavi, P. et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 1, 382–393 (2020).
Masson, G. R. & Williams, R. L. Structural mechanisms of PTEN regulation. Cold Spring Harb. Perspect. Med. 10, 1–13 (2020).
Steck, P. A. et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362 (1997).
Lee, Y.-R., Chen, M. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 19, 547–562 (2018).
Papa, A. & Pandolfi, P. P. The PTEN−PI3K axis in cancer. Biomolecules 9 (2019).
Yang, Z.-Z. et al. Physiological functions of protein kinase B/Akt. Biochem Soc. Trans. 32, 350–354 (2004).
Garofalo, R. S. et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J. Clin. Investig. 112, 197–208 (2003).
Chen, W. S. et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 15, 2203–2208 (2001).
Xie, J. & Weiskirchen, R. What does the “AKT” stand for in the name “AKT Kinase”? some historical comments. Front. Oncol. 10 (2020).
Bao, F. et al. Akt scaffold proteins: the key to controlling specificity of Akt signaling. Am. J. Physiol. Cell Physiol. 321, 429–442 (2021).
Carpten, J. D. et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 (2007).
Lauring, J. et al. Knock in of the AKT1 E17K mutation in human breast epithelial cells does not recapitulate oncogenic PIK3CA mutations. Oncogene 29, 2337–2345 (2010).
Davies, B. R. et al. Tumors with AKT1E17K mutations are rational targets for single agent or combination therapy with akt inhibitors. Mol. Cancer Ther. 14, 2441–2451 (2015).
Rudolph, M. et al. AKT1 (E17K) mutation profiling in breast cancer: prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer 16 (2016).
Alves, C. P. et al. AKT1low quiescent cancer cells promote solid tumor growth. Mol. Cancer Ther. 17, 254–263 (2018).
Guo, G. et al. Oncogenic E17K mutation in the pleckstrin homology domain of AKT1 promotes v-Abl-mediated pre-B-cell transformation and survival of Pim-deficient cells. Oncogene 29, 3845–3853 (2010).
Chen, Y. et al. Effect of AKT1 (p. E17K) hotspot mutation on malignant tumorigenesis and prognosis. Front. Cell Dev. Biol. 8 (2020).
Hyman, D. M. et al. AKT inhibition in solid tumors With AKT1 mutations. J. Clin. Oncol. 35, 2251–2259 (2017).
Shrestha Bhattarai, T. et al. AKT mutant allele-specific activation dictates pharmacologic sensitivities. Nat. Commun. 13, 1–11 (2022).
Shi, H. et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 4, 69–79 (2014).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017).
Zou, Z., Tao, T., Li, H. & Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 10, 1–11 (2020).
Panwar, V. et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 8 (2023).
Grabiner, B. C. et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 4, 554–563 (2014).
Yamaguchi, H. et al. Transforming somatic mutations of mammalian target of rapamycin kinase in human cancer. Cancer Sci. 106, 1687–1692 (2015).
Murugan, A. K., Liu, R. & Xing, M. Identification and characterization of two novel oncogenic mTOR mutations. Oncogene 38, 5211–5226 (2019).
Rodríguez-Moreno, J. F. et al. Exceptional response to temsirolimus in a metastatic clear cell renal cell carcinoma with an early novel MTOR-activating mutation. J. Natl Compr. Cancer Netw. 15, 1310–1315 (2017).
Ghosh, A. P. et al. Point mutations of the mTOR-RHEB pathway in renal cell carcinoma. Oncotarget 6, 17895–17910 (2015).
Kong, Y. et al. Analysis of mTOR gene aberrations in melanoma patients and evaluation of their sensitivity to PI3K–AKT–mTOR pathway inhibitors. Clin. Cancer Res. 22, 1018–1027 (2016).
Wu, T.-J. et al. Identification of a non-gatekeeper hot spot for drug-resistant mutations in mTOR kinase. Cell Rep. 11, 446–459 (2015).
Wagle, N. et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 371, 1426–1433 (2014).
Owen, K. L., Brockwell, N. K. & Parker, B. S. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers 11 (2019).
Nan, J., Wang, Y., Yang, J. & Stark, G. R. IRF9 and unphosphorylated STAT2 cooperate with NF-κB to drive IL6 expression. Proc. Natl Acad. Sci. USA 115, 3906–3911 (2018).
Cheon, H. & Stark, G. R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl Acad. Sci. USA 106, 9373–9378 (2009).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Platanias, L. C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 (2005).
Fuertes, M. B., Woo, S.-R., Burnett, B., Fu, Y.-X. & Gajewski, T. F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 34, 67–73 (2013).
Battistini, A. Interferon regulatory factors in hematopoietic cell differentiation and immune regulation. J. Interferon Cytokine Res. 29, 765–780 (2009).
Ragimbeau, J. et al. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J. 22, 537–547 (2003).
Hofmann, S. R. et al. Jak3-independent trafficking of the common gamma chain receptor subunit: chaperone function of Jaks revisited. Mol. Cell Biol. 24, 5039–5049 (2004).
Tortolani, P. J. et al. Regulation of JAK3 expression and activation in human B cells and B cell malignancies. J. Immunol. 155, 5220–5226 (1995).
Musso, T. et al. Regulation of JAK3 expression in human monocytes: phosphorylation in response to interleukins 2, 4, and 7. J. Exp. Med. 181, 1425–1431 (1995).
Kawamura, M. et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc. Natl Acad. Sci. USA 91, 6374–6378 (1994).
Karaghiosoff, M. et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 4, 471–477 (2003).
Yamaoka, K. et al. The Janus kinases (Jaks). Genome Biol. 5, 1–6 (2004).
Copeland, N. G. et al. Distribution of the mammalian Stat gene family in mouse chromosomes. Genomics 29, 225–228 (1995).
Heim, M. H. The STAT protein family. in Signal Transducers and Activators of Transcription (STATs) 11–26 (Springer Netherlands, Dordrecht, 2003). https://doi.org/10.1007/978-94-017-3000-6_2.
O’Shea, J. J. & Murray, P. J. Cytokine signaling modules in inflammatory responses. Immunity 28, 477–487 (2008).
Schindler, C., Levy, D. E. & Decker, T. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063 (2007).
Zhou, L., Chong, M. M. W. & Littman, D. R. Plasticity of CD4+ T cell lineage differentiation. Immunity 30, 646–655 (2009).
Wang, H., Lafdil, F., Kong, X. & Gao, B. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. Int. J. Biol. Sci. 7, 536–550 (2011).
Bar-Natan, M., Nelson, E. A., Xiang, M. & Frank, D. A. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT 1, 55–64 (2012).
Geiger, J. L., Grandis, J. R. & Bauman, J. E. The STAT3 pathway as a therapeutic target in head and neck cancer: barriers and innovations. Oral. Oncol. 56, 84–92 (2016).
Suh, Y.-A., Jo, S.-Y., Lee, H.-Y. & Lee, C. Inhibition of IL-6/STAT3 axis and targeting Axl and Tyro3 receptor tyrosine kinases by apigenin circumvent taxol resistance in ovarian cancer cells. Int. J. Oncol. 46, 1405–1411 (2015).
Ludwig, H., Nachbaur, D., Fritz, E., Krainer, M. & Huber, H. Interleukin-6 is a prognostic factor in multiple myeloma. Blood 77, 2794–2795 (1991).
Chen, Y. et al. STAT3, a poor survival predicator, is associated with lymph node metastasis from breast cancer. J. Breast Cancer 16, 40–49 (2013).
Shahmarvand, N., Nagy, A., Shahryari, J. & Ohgami, R. S. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 109, 926–933 (2018).
Hassel, J. C., Winnemöller, D., Schartl, M. & Wellbrock, C. STAT5 contributes to antiapoptosis in melanoma. Melanoma Res. 18, 378–385 (2008).
Constantinescu, S. N., Girardot, M. & Pecquet, C. Mining for JAK–STAT mutations in cancer. Trends Biochem. Sci. 33, 122–131 (2008).
Teramo, A. et al. Intrinsic and extrinsic mechanisms contribute to maintain the JAK/STAT pathway aberrantly activated in T-type large granular lymphocyte leukemia. Blood 121, 3843–3854 (2013).
Rajala, H. L. M., Porkka, K., Maciejewski, J. P., Loughran, T. P. & Mustjoki, S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann. Med. 46, 114–122 (2014).
Rajala, H. L. M. et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 121, 4541–4550 (2013).
Crescenzo, R. et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 27, 516–532 (2015).
Ohgami, R. S., Ma, L., Monabati, A., Zehnder, J. L. & Arber, D. A. STAT3 mutations are present in aggressive B-cell lymphomas including a subset of diffuse large B-cell lymphomas with CD30 expression. Haematologica 99, 105–107 (2014).
Ohgami, R. S. et al. STAT3 mutations are frequent in CD30+ T-cell lymphomas and T-cell large granular lymphocytic leukemia. Leukemia 27, 2244–2247 (2013).
Arulogun, S. O. et al. JAK1 somatic mutation in a myeloproliferative neoplasm. Haematologica 102, 324–327 (2017).
Mullighan, C. G. et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl Acad. Sci. USA 106, 9414–9418 (2009).
Jeong, E. G. et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin. Cancer Res. 14, 3716–3721 (2008).
Flex, E. et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 205, 751–758 (2008).
Xiang, Z. et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood 111, 4809–4812 (2008).
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005).
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005).
Haan, C., Behrmann, I. & Haan, S. Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J. Cell Mol. Med. 14, 504–527 (2010).
Mercher, T. et al. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood 108, 2770–2779 (2006).
Malinge, S. et al. Activating mutations in human acute megakaryoblastic leukemia. Blood 112, 4220–4226 (2008).
Zhao, L. et al. A JAK2 interdomain linker relays Epo receptor engagement signals to kinase activation. J. Biol. Chem. 284, 26988–26998 (2009).
Sayyah, J. et al. Z3, a novel Jak2 tyrosine kinase small-molecule inhibitor that suppresses Jak2-mediated pathologic cell growth. Mol. Cancer Ther. 7, 2308–2318 (2008).
Percy, M. J. et al. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica 92, 1607–1614 (2007).
Pietra, D. et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 111, 1686–1689 (2008).
Schnittger, S. et al. Detection of JAK2 exon 12 mutations in 15 patients with JAK2V617F negative polycythemia vera. Haematologica 94, 414–418 (2009).
Zhang, S.-J. et al. The investigation of JAK2 mutation in Chinese myeloproliferative diseases-identification of a novel C616Y point mutation in a PV patient. Int. J. Lab. Hematol. 29, 71–72 (2007).
Funakoshi-Tago, M. et al. The acute lymphoblastic leukemia-associated JAK2 L611S mutant induces tumorigenesis in nude mice. J. Biol. Chem. 284, 12680–12690 (2009).
Gaikwad, A. et al. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br. J. Haematol. 144, 930–932 (2009).
Bercovich, D. et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet 372, 1484–1492 (2008).
Degryse, S. et al. Mutant JAK3 signaling is increased by loss of wild-type JAK3 or by acquisition of secondary JAK3 mutations in T-ALL. Blood 131, 421–425 (2018).
Walters, D. K. et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell 10, 65–75 (2006).
Sato, T. et al. Functional analysis of JAK3 mutations in transient myeloproliferative disorder and acute megakaryoblastic leukaemia accompanying Down syndrome. Br. J. Haematol. 141, 681–688 (2008).
De Vita, S. et al. Loss-of-function JAK3 mutations in TMD and AMKL of Down syndrome. Br. J. Haematol. 137, 337–341 (2007).
Kiyoi, H., Yamaji, S., Kojima, S. & Naoe, T. JAK3 mutations occur in acute megakaryoblastic leukemia both in Down syndrome children and non-Down syndrome adults. Leukemia 21, 574–576 (2007).
Kuravi, S. et al. Functional characterization of NPM1–TYK2 fusion oncogene. NPJ Precis. Oncol. 6 (2022).
Tomasson, M. H. et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 111, 4797–4808 (2008).
Li, Z., Rotival, M., Patin, E., Michel, F. & Pellegrini, S. Two common disease-associated TYK2 variants impact exon splicing and TYK2 dosage. PLoS One 15, 1–20 (2020).
Peters, T. L. et al. Intrinsic resistance to ROS1 inhibition in a patient with CD74-ROS1 mediated by AXL overexpression. Thorac. Cancer 14, 3259–3265 (2023).
Breitenecker, K. et al. Synergism of the receptor tyrosine kinase Axl with ErbB receptors mediates resistance to regorafenib in hepatocellular carcinoma. Front Oncol 13 (2023).
Volta, F. et al. Intrinsic resistance to osimertinib in EGFR mutated NSCLC cell lines induced by alteration in cell-cycle regulators. Target Oncol. 18, 953–964 (2023).
Parseghian, C. M., Napolitano, S., Loree, J. M. & Kopetz, S. Mechanisms of innate and acquired resistance to anti-EGFR therapy: a review of current knowledge with a focus on rechallenge therapies. Clin. Cancer Res. 25, 6899–6908 (2019).
Venook, A. P. et al. Effect of first-line chemotherapy combined with cetuximab or bevacizumab on overall survival in patients with KRAS wild-type advanced or metastatic colorectal cancer: a randomized clinical trial. JAMA 317, 2392–2401 (2017).
Yang, Y., Li, S., Wang, Y., Zhao, Y. & Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal. Transduct. Target. Ther. 7, 1–36 (2022).
Zhu, X. et al. Identification and validation of afatinib potential drug resistance gene BIRC5 in non-small cell lung cancer. Front. Oncol. 11 (2021).
Del Re, M. et al. Implications of KRAS mutations in acquired resistance to treatment in NSCLC. Oncotarget 9, 6630–6643 (2018).
Riedel, R. et al. Resistance to MET inhibition in MET-dependent NSCLC and therapeutic activity after switching from type I to type II MET inhibitors. Eur. J. Cancer 179, 124–135 (2023).
Wang, L.-S. et al. Acquired EML4-ALK fusion and EGFR C797S in cis mutation as resistance mechanisms to osimertinib in a non-small cell lung cancer patient with EGFR L858R/T790M. Anticancer Drugs 34, 1146–1150 (2023).
Fukuda, S. et al. The magnitude of CXCR4 signaling regulates resistance to quizartinib in FLT3/ITD+ cells via RUNX1. Leuk. Res. 124 (2023).
Gomatou, G., Syrigos, N. & Kotteas, E. Osimertinib resistance: molecular mechanisms and emerging treatment options. Cancers 15 (2023).
Xia, W. et al. Resistance to ErbB2 tyrosine kinase inhibitors in breast cancer is mediated by calcium-dependent activation of RelA. Mol. Cancer Ther. 9, 292–299 (2010).
Politi, K., Fan, P.-D., Shen, R., Zakowski, M. & Varmus, H. Erlotinib resistance in mouse models of epidermal growth factor receptor-induced lung adenocarcinoma. Dis. Model Mech. 3, 111–119 (2010).
Katayama, R. et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 4, 120ra17–120ra17 (2012).
Yoshida, T. et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin. Cancer Res. 20, 4059–4074 (2014).
Pang, L.-L. et al. Efficacy and potential resistance mechanisms of afatinib in advanced non-small cell lung cancer patients with EGFR G719X/L861Q/S768I. Cancer 128, 3804–3814 (2022).
Nakamura, T. et al. Mechanisms of acquired resistance to afatinib clarified with liquid biopsy. PLoS One 13 (2018).
Reischmann, N. et al. Overcoming MET-mediated resistance in oncogene-driven NSCLC. iScience 26, 1–20 (2023).
Kong, W.-M., Guo, Y.-J., Ma, J. & Shi, C. BTN2A1-BRAF fusion may be a novel mechanism of resistance to osimertinib in lung adenocarcinoma: a case report. Transl. Cancer Res. 12, 186–193 (2023).
Mazières, J. et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J. Clin. Oncol. 31, 1997–2003 (2013).
Peng, Y. & Tan, J. The relationship between IGF pathway and acquired resistance to tyrosine kinase inhibitors in cancer therapy. Front. Biosci. (Landmark Ed.) 28 (2023).
de Reyniès, A., Boige, V., Milano, G., Faivre, J. & Laurent-Puig, P. KRAS mutation signature in colorectal tumors significantly overlaps with the cetuximab response signature. J. Clin. Oncol. 26, 2228–2230 (2008).
Liu, D., Lu, X., Huang, W. & Zhuang, W. Long non-coding RNAs in non-small cell lung cancer: implications for EGFR-TKI resistance. Front. Genet. 14 (2023).
Nguyen, K.-S. H., Kobayashi, S. & Costa, D. B. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin. Lung Cancer 10, 281–289 (2009).
Lin, X. et al. KIAA1429 promotes tumorigenesis and gefitinib resistance in lung adenocarcinoma by activating the JNK/MAPK pathway in an m6A-dependent manner. Drug Resist. Updat. 66, 1–12 (2023).
Bugide, S., Edwards, Y. J. K., Gupta, R., Green, M. R. & Wajapeyee, N. CBX5 loss drives EGFR inhibitor resistance and results in therapeutically actionable vulnerabilities in lung cancer. Proc Natl Acad. Sci. USA 120 (2023).
de Miguel, F. J. et al. Mammalian SWI/SNF chromatin remodeling complexes promote tyrosine kinase inhibitor resistance in EGFR-mutant lung cancer. Cancer Cell 41, 1516–1534 (2023).
Chen, H. et al. Long noncoding RNA RP11-89K21.1 interacts with miR-146a/b-5p to promote proliferation and gefitinib resistance through regulating RHPN2 and RhoA/ROCK pathway in lung adenocarcinoma. Cancer Biother. Radiopharm. 38, 282–292 (2023).
Cheng, D. et al. Exosomal non-coding RNAs-mediated EGFR-TKIs resistance in NSCLC with EGFR mutation. Med. Oncol. 40 (2023).
Azuma, K. et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 5, 5908–5919 (2014).
Ren, S. et al. The pseudogene DUXAP10 contributes to gefitinib resistance in NSCLC by repressing OAS2 expression. Acta Biochim. Biophys. Sin. 55, 81–90 (2023).
Wen, C. et al. CircSETD3 mediates acquired resistance to gefitinib in non-small lung cancer cells by FXR1/ECT2 pathway. Int. J. Biochem. Cell Biol. 154 (2023).
Zhang, Z. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 44, 852–860 (2012).
Huang, S. et al. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer Res. 71, 7071–7079 (2011).
Takahashi, H. et al. Notch pathway regulates osimertinib drug-tolerant persistence in EGFR-mutated non-small-cell lung cancer. Cancer Sci. 114, 1635–1650 (2023).
Baumgart, A. et al. ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non-small cell lung cancer. Cancer Res. 70, 5368–5378 (2010).
Chen, X. et al. Crizotinib overcomes hepatocyte growth factor-mediated resistance to gefitinib in EGFR-mutant non-small-cell lung cancer cells. Anticancer Drugs 24, 1039–1046 (2013).
van der Wekken, A. J. et al. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit. Rev. Oncol. Hematol. 100, 107–116 (2016).
Kim, H. R. et al. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol. Oncol. 7, 1093–1102 (2013).
Lee, K.-L. et al. Sustaining the activation of EGFR signal by inflammatory cytokine IL17A prompts cell proliferation and EGFR-TKI resistance in lung cancer. Cancers (Basel) 15, 1–18 (2023).
Kim, S. M. et al. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 11, 2254–2264 (2012).
Katayama, Y. et al. Adaptive resistance to lorlatinib via EGFR signaling in ALK-rearranged lung cancer. NPJ Precis. Oncol. 7 (2023).
Kondo, N. et al. MIG6 loss confers resistance to ALK/ROS1 inhibitors in NSCLC through EGFR activation by low-dose EGF. JCI Insight 8 (2023).
Liu, W.-J. et al. Elevated NOX4 promotes tumorigenesis and acquired EGFR-TKIs resistance via enhancing IL-8/PD-L1 signaling in NSCLC. Drug Resist. Updat. 70 (2023).
Romaniello, D. et al. Targeting HER3, a catalytically defective receptor tyrosine kinase, prevents resistance of lung cancer to a third-generation EGFR kinase inhibitor. Cancers 12 (2020).
Sang, J. et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 3, 430–443 (2013).
He, M. et al. Sunitinib increases the cancer stem cells and vasculogenic mimicry formation via modulating the lncRNA-ECVSR/ERβ/Hif2-α signaling. Cancer Lett. 524, 15–28 (2022).
Coco, S. et al. Afatinib resistance in non-small cell lung cancer involves the PI3K/AKT and MAPK/ERK signalling pathways and epithelial-to-mesenchymal transition. Target Oncol. 10, 393–404 (2015).
Hammers, H. J. et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: evidence from a xenograft study. Mol. Cancer Ther. 9, 1525–1535 (2010).
Goel, S. et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29, 255–269 (2016).
Liu, Y., Li, C., Lu, Y., Liu, C. & Yang, W. Tumor microenvironment-mediated immune tolerance in development and treatment of gastric cancer. Front. Immunol. 13, 1–17 (2022).
Terry, S. et al. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 492, 1–10 (2020).
Lee, J. et al. Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non-small cell lung cancer. Cancer 126, 2704–2712 (2020).
Filippi, I., Naldini, A. & Carraro, F. Role of the hypoxic microenvironment in the antitumor activity of tyrosine kinase inhibitors. Curr. Med. Chem. 18, 2885–2892 (2011).
Bixby, D. & Talpaz, M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematology 2009, 461–476 (2009).
Bridgeman, V. L. et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 241, 362–374 (2017).
Kuczynski, E. A. & Reynolds, A. R. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 23, 55–74 (2020).
Xie, X. et al. OSGIN1 is a novel TUBB3 regulator that promotes tumor progression and gefitinib resistance in non-small cell lung cancer. Cell Mol. Life Sci. 80 (2023).
Chen, Y.-H., Yang, Y., Xu, L.-J., Deng, Y. & Fu, J.-W. Regulation of epidermal growth factor receptor tyrosine kinase inhibitor resistance via Ambra1-mediated autophagy in non-small cell lung cancer. J. Physiol. Pharm. 74, 325–334 (2023).
Giuliano, M. et al. Upregulation of ER signaling as an adaptive mechanism of cell survival in HER2-positive breast tumors treated with anti-HER2 therapy. Clin. Cancer Res. 21, 3995–4003 (2015).
Xiao, Z., Li, M., Zhang, X., Rong, X. & Xu, H. TRIP13 overexpression promotes gefitinib resistance in non-small cell lung cancer via regulating autophagy and phosphorylation of the EGFR signaling pathway. Oncol. Rep. 49 (2023).
Watson, S. S. et al. Microenvironment-mediated mechanisms of resistance to HER2 inhibitors differ between HER2+ breast cancer subtypes. Cell Syst. 6, 329–342 (2018).
Ji, C. et al. Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer. Cancer Biol. Ther. 15, 570–577 (2014).
Dai, J. et al. LncRNA LINC00969 promotes acquired gefitinib resistance by epigenetically suppressing of NLRP3 at transcriptional and posttranscriptional levels to inhibit pyroptosis in lung cancer. Cell Death Dis. 14 (2023).
Lee, T.-G., Jeong, E.-H., Kim, S. Y., Kim, H.-R. & Kim, C. H. The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFR T790M-mutated lung cancer. Int. J. Cancer 136, 2717–2729 (2015).
Zhang, W. et al. Concomitant targeting of FLT3 and BTK overcomes FLT3 inhibitor resistance in acute myeloid leukemia through the inhibition of autophagy. Haematologica 108, 1500–1514 (2023).
Chen, R. et al. Modulation of the tumour microenvironment in hepatocellular carcinoma by tyrosine kinase inhibitors: from modulation to combination therapy targeting the microenvironment. Cancer Cell Int. 22 (2022).
Noel, B. M. et al. Multiomic profiling of tyrosine kinase inhibitor-resistant K562 cells suggests metabolic reprogramming to promote cell survival. J. Proteome Res. 18, 1842–1856 (2019).
Huang, F. et al. SLC12A8 mediates TKI resistance in EGFR-mutant lung cancer via PDK1/AKT axis. J. Cancer Res. Clin. Oncol. 149, 16729–16739 (2023).
Su, S.-F. et al. Genome-wide epigenetic landscape of lung adenocarcinoma links HOXB9 DNA methylation to intrinsic EGFR-TKI resistance and heterogeneous responses. JCO Precis. Oncol. 5, 418–431 (2021).
de Klerk, D. J., Honeywell, R. J., Jansen, G. & Peters, G. J. Transporter and lysosomal mediated (Multi)drug resistance to tyrosine kinase inhibitors and potential strategies to overcome resistance. Cancers 10 (2018).
Chhouri, H., Alexandre, D. & Grumolato, L. Mechanisms of acquired resistance and tolerance to EGFR targeted therapy in non-small cell lung cancer. Cancers 15 (2023).
Adelaiye-Ogala, R. et al. EZH2 modifies sunitinib resistance in renal cell carcinoma by kinome reprogramming. Cancer Res. 77, 6651–6666 (2017).
Ghiaur, G. & Levis, M. Mechanisms of resistance to FLT3 inhibitors and the role of the bone marrow microenvironment. Hematol. Oncol. Clin. North Am. 31, 681–692 (2017).
Ouar, Z., Lacave, R., Bens, M. & Vandewalle, A. Mechanisms of altered sequestration and efflux of chemotherapeutic drugs by multidrug-resistant cells. Cell Biol. Toxicol. 15, 91–100 (1999).
Mlejnek, P. What is the significance of lysosomal-mediated resistance to imatinib? Cells 12, 1–15 (2023).
Locher, K. P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 23, 487–493 (2016).
Lu, J. et al. Reprogramming of TAMs via the STAT3/CD47-SIRPα axis promotes acquired resistance to EGFR-TKIs in lung cancer. Cancer Lett. 564 (2023).
Lin, L., Yee, S. W., Kim, R. B. & Giacomini, K. M. SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 14, 543–560 (2015).
Diekstra, M. H. M. et al. CYP3A5 and ABCB1 polymorphisms as predictors for sunitinib outcome in metastatic renal cell carcinoma. Eur. Urol. 68, 621–629 (2015).
Chen, S. et al. Changes of tumor microenvironment in non-small cell lung cancer after TKI treatments. Front. Immunol. 14, 1094764 (2023).
Cheng, D. et al. Tumor-associated macrophages mediate resistance of EGFR-TKIs in non-small cell lung cancer: mechanisms and prospects. Front. Immunol. 14, 1–12 (2023).
Zhao, B., Zhang, Y., Lu, S. & Li, M. Macrophage renewal modes affect acquired resistance to gefitinib in EGFR-mutant lung cancer PC-9 cells. Oncol. Rep. 49 (2023).
Jeong, J. et al. Downregulation of AP1S1 causes the lysosomal degradation of EGFR in non-small cell lung cancer. J. Cell Physiol. 238, 2335–2347 (2023).
Soverini, S., Mancini, M., Bavaro, L., Cavo, M. & Martinelli, G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 17, 1–15 (2018).
Shyam Sunder, S., Sharma, U. C. & Pokharel, S. Adverse effects of tyrosine kinase inhibitors in cancer therapy: pathophysiology, mechanisms and clinical management. Signal. Transduct. Target. Ther. 8 (2023).
Dobbin, S. J. H., Petrie, M. C., Myles, R. C., Touyz, R. M. & Lang, N. N. Cardiotoxic effects of angiogenesis inhibitors. Clin. Sci. 135, 71–100 (2021).
Sundararajan, S., Kumar, A., Poongkunran, M., Kannan, A. & Vogelzang, N. J. Cardiovascular adverse effects of targeted antiangiogenic drugs: mechanisms and management. Future Oncol. 12, 1067–1080 (2016).
Du, X. L., Xia, R., Burau, K. & Liu, C.-C. Cardiac risk associated with the receipt of anthracycline and trastuzumab in a large nationwide cohort of older women with breast cancer, 1998-2005. Med. Oncol. 28, 80–90 (2011).
Curigliano, G. et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann. Oncol. 31, 171–190 (2020).
Pinkhas, D., Ho, T. & Smith, S. Assessment of pazopanib-related hypertension, cardiac dysfunction and identification of clinical risk factors for their development. Cardiooncology 3 (2017).
Chang, H.-M., Moudgil, R., Scarabelli, T., Okwuosa, T. M. & Yeh, E. T. H. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J. Am. Coll. Cardiol. 70, 2536–2551 (2017).
Díaz-Serrano, A., Gella, P., Jiménez, E., Zugazagoitia, J. & Paz-Ares Rodríguez, L. Targeting EGFR in lung cancer: current standards and developments. Drugs 78, 893–911 (2018).
Wang, L. & Wang, W. Safety and efficacy of anaplastic lymphoma kinase tyrosine kinase inhibitors in non-small cell lung cancer (Review). Oncol. Rep. 45, 13–28 (2021).
Godinas, L. et al. Tyrosine kinase inhibitors in pulmonary arterial hypertension: a double-edge sword? Semin. Respir. Crit. Care Med. 34, 714–724 (2013).
Montani, D. et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 125, 2128–2137 (2012).
Giudice, V., Vecchione, C. & Selleri, C. Cardiotoxicity of novel targeted hematological therapies. Life 10, (2020).
Mishra, A. K., Sahu, K. K., Kaul, S. & Lal, A. Dasatinib induced pleuro-pericardial effusion. Acta Biomed. 91, 142–143 (2020).
Maroun, J. A. et al. Prevention and management of chemotherapy-induced diarrhea in patients with colorectal cancer: a consensus statement by the Canadian Working Group on Chemotherapy-Induced Diarrhea. Curr. Oncol. 14, 13–20 (2007).
Wang, Z. et al. Comparison of hepatotoxicity associated with new BCR-ABL tyrosine kinase inhibitors vs imatinib among patients with chronic myeloid leukemia: a systematic review and meta-analysis. JAMA Netw. Open 4 (2021).
Nelson, R. P. et al. Desensitization to imatinib in patients with leukemia. Ann. Allergy Asthma Immunol. 97, 216–222 (2006).
Zheng, P.-P., Li, J. & Kros, J. M. Breakthroughs in modern cancer therapy and elusive cardiotoxicity: Critical research-practice gaps, challenges, and insights. Med Res. Rev. 38, 325–376 (2018).
Hamberg, P. et al. Non-islet-cell tumor induced hypoglycemia in patients with advanced gastrointestinal stromal tumor possibly worsened by imatinib. J. Clin. Oncol. 24, 30–31 (2006).
Lodish, M. B. & Stratakis, C. A. Endocrine side effects of broad-acting kinase inhibitors. Endocr. Relat. Cancer 17, 233–244 (2010).
Torino, F., Corsello, S. M., Longo, R., Barnabei, A. & Gasparini, G. Hypothyroidism related to tyrosine kinase inhibitors: an emerging toxic effect of targeted therapy. Nat. Rev. Clin. Oncol. 6, 219–228 (2009).
Pezzilli, R., Corinaldesi, R. & Morselli-Labate, A. M. Tyrosine kinase inhibitors and acute pancreatitis. JOP 11, 291–293 (2010).
Suh, C. H. et al. Pneumonitis in advanced non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitor: Meta-analysis of 153 cohorts with 15,713 patients: meta-analysis of incidence and risk factors of EGFR-TKI pneumonitis in NSCLC. Lung Cancer 123, 60–69 (2018).
Kanemura, H., Takeda, M., Shimizu, S. & Nakagawa, K. Interstitial lung disease associated with capmatinib therapy in a patient with non-small cell lung cancer harboring a skipping mutation of MET exon 14. Thorac. Cancer 12, 549–552 (2021).
Cabanillas, M. E., Hu, M. I., Durand, J.-B. & Busaidy, N. L. Challenges associated with tyrosine kinase inhibitor therapy for metastatic thyroid cancer. J. Thyroid Res. 2011, 1–9 (2011).
Kandula, P. & Agarwal, R. Proteinuria and hypertension with tyrosine kinase inhibitors. Kidney Int. 80, 1271–1277 (2011).
Fachi, M. M. et al. Haematological adverse events associated with tyrosine kinase inhibitors in chronic myeloid leukaemia: a network meta-analysis. Br. J. Clin. Pharm. 85, 2280–2291 (2019).
Jabbour, E., Deininger, M. & Hochhaus, A. Management of adverse events associated with tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia. Leukemia 25, 201–210 (2011).
Hakkola, J., Hukkanen, J., Turpeinen, M. & Pelkonen, O. Inhibition and induction of CYP enzymes in humans: an update. Arch. Toxicol. 94, 3671–3722 (2020).
Rambhatla, A., Strug, M. R., De Paredes, J. G., Cordoba Munoz, M. I. & Thakur, M. Fertility considerations in targeted biologic therapy with tyrosine kinase inhibitors: a review. J. Assist. Reprod. Genet. 38, 1897–1908 (2021).
Dou, X., Qin, Y., Huang, X. & Jiang, Q. Planned pregnancy in female patients with chronic myeloid leukemia receiving tyrosine kinase inhibitor therapy. Oncologist 24, 1141–1147 (2019).
Hua, G. et al. Real-world circulating tumor DNA analysis depicts resistance mechanism and clonal evolution in ALK inhibitor-treated lung adenocarcinoma patients. ESMO Open 7 (2022).
Choi, Y.-R. et al. Early On-treatment prediction of the mechanisms of acquired resistance to EGFR tyrosine kinase inhibitors. Cancers 14 (2022).
Gorachinov, F. et al. Nanotechnology—a robust tool for fighting the challenges of drug resistance in non-small cell lung cancer. Beilstein J. Nanotechnol. 14, 240–261 (2023).
Shaurova, T. et al. A nanotherapeutic strategy to target drug-tolerant cells and overcome EGFR tyrosine kinase inhibitor resistance in lung cancer. Cancer Commun. 43, 503–507 (2023).
Lu, X. et al. Afatinib-loaded immunoliposomes functionalized with cetuximab: a novel strategy targeting the epidermal growth factor receptor for treatment of non-small-cell lung cancer. Int. J. Pharm. 560, 126–135 (2019).
Wan, X., Zheng, X., Pang, X., Zhang, Z. & Zhang, Q. Incorporation of lapatinib into human serum albumin nanoparticles with enhanced anti-tumor effects in HER2-positive breast cancer. Colloids Surf. B Biointerfaces 136, 817–827 (2015).
Han, W. et al. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal Transduct. Target. Ther. 3 (2018).
Greish, K. et al. Micellar formulations of Crizotinib and Dasatinib in the management of glioblastoma multiforme. J. Drug Target. 26, 692–708 (2018).
Smidova, V. et al. Nanomedicine of tyrosine kinase inhibitors. Theranostics 11, 1546–1567 (2021).
Nilsson, M. B. et al. CD70 is a therapeutic target upregulated in EMT-associated EGFR tyrosine kinase inhibitor resistance. Cancer Cell 41, 340–355 (2023).
Desai, A. & Lovly, C. M. Strategies to overcome resistance to ALK inhibitors in non-small cell lung cancer: a narrative review. Transl. Lung Cancer Res. 12, 615–628 (2023).
Talwelkar, S. S. et al. PI3Kβ inhibition enhances ALK-inhibitor sensitivity in ALK-rearranged lung cancer. Mol. Oncol. 17, 747–764 (2023).
Sun, L. et al. Understanding and targeting the epigenetic regulation to overcome EGFR-TKIs resistance in human. Cancer Recent Pat. Anticancer Drug Discov. 18, 506–516 (2023).
Yochum, Z. A. et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 38, 656–670 (2019).
Qin, A. et al. Tyrosine kinase signaling-independent MET-targeting with CAR-T cells. J. Transl. Med. 21 (2023).
Camidge, D. R. et al. Phase Ib study of telisotuzumab vedotin in combination with erlotinib in patients with c-met protein–expressing non–small-cell lung cancer. J. Clin. Oncol. 41, 1105–1115 (2023).
Larcombe-Young, D., Papa, S. & Maher, J. PanErbB-targeted CAR T-cell immunotherapy of head and neck cancer. Expert Opin. Biol. Ther. 20, 965–970 (2020).
Chicas-Sett, R., Castilla Martinez, J., Hernández Blanquisett, A., Zafra, J. & Pastor-Peidro, J. Stereotactic ablative radiotherapy for acquired resistance to EGFR therapy in metastatic non-small cell lung cancer. Front. Oncol. 12 (2022).
Jiang, K. et al. Dual inhibition of CHK1/FLT3 enhances cytotoxicity and overcomes adaptive and acquired resistance in FLT3-ITD acute myeloid leukemia. Leukemia 37, 539–549 (2023).
Veiga, R. N., de Azevedo, A. L. K., de Oliveira, J. C. & Gradia, D. F. Targeting EphA2: a promising strategy to overcome chemoresistance and drug resistance in cancer. J. Mol. Med. 102, 479–493 (2024).
Loges, S. et al. The combination of AXL inhibitor bemcentinib and low dose cytarabine is well tolerated and efficacious in elderly relapsed AML patients: update from the ongoing BGBC003 phase II trial (NCT02488408). Blood 136, 14–17 (2020).
Kubasch, A. S. et al. Efficacy and safety of bemcentinib in patients with advanced myelodysplastic neoplasms or acute myeloid leukemia failing hypomethylating agents- the EMSCO phase II BERGAMO trial. Leukemia 37, 2309–2313 (2023).
Bhalla, S. et al. Phase 1 dose escalation and expansion study of bemcentinib (BGB324), a first-in-class, selective AXL inhibitor, with docetaxel in patients with previously treated advanced NSCLC. J. Clin. Oncol. 40, 1–7 (2022).
Van Tine, B. A. et al. A phase III study (APROMISS) of AL3818 (Catequentinib, Anlotinib) hydrochloride monotherapy in subjects with metastatic or advanced synovial sarcoma. J. Clin. Oncol. 39, 11505–11505 (2021).
Hecht, J. R. et al. Randomized, placebo-controlled, phase III study of first-line oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J. Clin. Oncol. 29, 1997–2003 (2011).
Li, D. et al. Anlotinib in locally advanced or metastatic medullary thyroid carcinoma: a randomized, double-blind phase IIB trial. Clin. Cancer Res. 27, 3567–3575 (2021).
Ren, S. et al. First-line treatment with camrelizumab plus famitinib in advanced or metastatic NSCLC patients with PD-L1 TPS ≥1%: results from a multicenter, open-label, phase 2 trial. J. Immunother. Cancer 12 (2024).
Han, B. et al. Effect of anlotinib as a third-line or further treatment on overall survival of patients with advanced non-small cell lung cancer: the ALTER 0303 Phase 3 Randomized Clinical Trial. JAMA Oncol. 4, 1569–1575 (2018).
Schönherr, H. et al. Discovery of lirafugratinib (RLY-4008), a highly selective irreversible small-molecule inhibitor of FGFR2. Proc Natl Acad Sci USA 121 (2024).
Cho, B. C. et al. Amivantamab plus lazertinib in osimertinib-relapsed EGFR-mutant advanced non-small cell lung cancer: a phase 1 trial. Nat. Med. 29, 2577–2585 (2023).
Shi, Y. et al. Efficacy and safety of alflutinib (AST2818) in patients with T790M mutation-positive NSCLC: a phase IIb multicenter single-arm study. J. Clin. Oncol. 38, 9602–9602 (2020).
Li, B. et al. Efficacy and safety of ASK120067 (limertinib) in patients with locally advanced or metastatic EGFR T790M-mutated non–small cell lung cancer: a multicenter, single-arm, phase IIb study. J. Clin. Oncol. 40, 9106–9106 (2022).
Shi, Y. et al. Results of the phase IIa study to evaluate the efficacy and safety of rezivertinib (BPI-7711) for the first-line treatment of locally advanced or metastatic/recurrent NSCLC patients with EGFR mutation from a phase I/IIa study. BMC Med. 21 (2023).
Zhou, Q. et al. A novel third-generation EGFR tyrosine kinase inhibitor abivertinib for EGFR T790M-mutant non–small cell lung cancer: a multicenter phase I/II study. Clin. Cancer Res. 28, 1127–1135 (2022).
Shum, E. et al. A phase 1/2 study of BLU-945 in patients with common activating EGFR -mutant non–small cell lung cancer (NSCLC): SYMPHONY trial in progress. J. Clin. Oncol. 40, 9156–9156 (2022).
Shi, Y. et al. Efficacy and safety of rezivertinib (BPI-7711) in patients with locally advanced or metastatic/recurrent EGFR T790M-mutated NSCLC: a phase 2b study. J. Thorac. Oncol. 17, 1306–1317 (2022).
Wang, M. et al. Sunvozertinib for patients in China with platinum-pretreated locally advanced or metastatic non-small-cell lung cancer and EGFR exon 20 insertion mutation (WU-KONG6): single-arm, open-label, multicentre, phase 2 trial. Lancet Respir. Med. 12, 217–224 (2024).
Cho, B. C. et al. Lazertinib versus gefitinib as first-line treatment in patients with EGFR-mutated advanced non–small-cell lung cancer: results from LASER301. J. Clin. Oncol. 41, 4208–4217 (2023).
Ma, F. et al. Pyrotinib versus placebo in combination with trastuzumab and docetaxel as first line treatment in patients with HER2 positive metastatic breast cancer (PHILA): randomised, double blind, multicentre, phase 3 trial. BMJ 383 (2023).
Drilon, A. E. et al. The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. J. Clin. Oncol. 38, 3616–3616 (2020).
Fujino, T. et al. Foretinib can overcome common on-target resistance mutations after capmatinib/tepotinib treatment in NSCLCs with MET exon 14 skipping mutation. J. Hematol. Oncol. 15, 1–14 (2022).
Xu, J. Current treatments and future potential of surufatinib in neuroendocrine tumors (NETs). Ther. Adv. Med. Oncol. 13, 1–14 (2021).
Miyazaki, I. et al. Vepafestinib is a pharmacologically advanced RET-selective inhibitor with high CNS penetration and inhibitory activity against RET solvent front mutations. Nat. Cancer 4, 1345–1361 (2023).
Lei, T. et al. Anlotinib combined with osimertinib reverses acquired osimertinib resistance in NSCLC by targeting the c-MET/MYC/AXL axis. Pharmacol. Res. 188 (2023).
Xie, L. et al. Apatinib for advanced osteosarcoma after failure of standard multimodal therapy: an open-label phase II clinical trial. Oncologist 24, 542–550 (2019).
du Bois, A. et al. Standard first-line chemotherapy with or without nintedanib for advanced ovarian cancer (AGO-OVAR 12): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 17, 78–89 (2016).
Shi, Y. et al. Safety and activity of WX-0593 (Iruplinalkib) in patients with ALK- or ROS1-rearranged advanced non-small cell lung cancer: a phase 1 dose-escalation and dose-expansion trial. Signal Transduct. Target. Ther. 7 (2022).
Parate, S., Kumar, V., Hong, J. C. & Lee, K. W. Putative dual inhibitors of mTOR and RET kinase from natural products: Pharmacophore-based hierarchical virtual screening. J. Mol. Liq. 350 (2022).
Taniguchi, H. et al. Pan-HER inhibitors overcome lorlatinib resistance caused by NRG1/HER3 activation in ALK-rearranged lung cancer. Cancer Sci. 114, 164–173 (2023).
Wu, Y.-L. et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 8, 1132–1143 (2020).
Motzer, R. J. et al. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol. 15, 286–296 (2014).
Takumi, Y. et al. MET kinase inhibitor reverses resistance to entrectinib induced by hepatocyte growth factor in tumors with NTRK1 or ROS1 rearrangements. Cancer Med. 12, 5809–5820 (2023).
Lu, S. et al. Efficacy and safety of befotertinib (D-0316) in patients With EGFR T790M-mutated NSCLC that had progressed after prior EGFR tyrosine kinase inhibitor therapy: a phase 2, multicenter, single-arm, open-label study. J. Thorac. Oncol. 17, 1192–1204 (2022).
Pottier, C. et al. Tyrosine kinase inhibitors in cancer: breakthrough and challenges of targeted therapy. Cancers 12, (2020).
Xie, L. et al. Apatinib for advanced osteosarcoma after failure of standard multimodal therapy: an open label phase 2 clinical trial. J. Clin. Oncol. 36, 11520–11520 (2018).
Blay, J.-Y. et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 21, 923–934 (2020).
Bauer, S. et al. Ripretinib versus sunitinib in patients with advanced gastrointestinal stromal tumor after treatment with imatinib (INTRIGUE): a randomized, open-label, phase III trial. J. Clin. Oncol. 40, 3918–3928 (2022).
Xu, J. et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 21, 1489–1499 (2020).
Borghaei, H. et al. SAPPHIRE: phase III study of sitravatinib plus nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. Ann. Oncol. 35, 66–76 (2024).
Chen, X. et al. First-line camrelizumab (a PD-1 inhibitor) plus apatinib (an VEGFR-2 inhibitor) and chemotherapy for advanced gastric cancer (SPACE): a phase 1 study. Signal Transduct. Target. Ther. 9 (2024).
Chen, L. et al. Famitinib with camrelizumab and nab-paclitaxel for advanced immunomodulatory triple-negative breast cancer (FUTURE-C-Plus): an open-label, single-arm, phase II trial. Clin. Cancer Res. 28, 2807–2817 (2022).
Yau, T. C. C. et al. A Phase I/II multicenter study of single-agent foretinib as first-line therapy in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 23, 2405–2413 (2017).
Nader-Marta, G. et al. Efficacy of tyrosine kinase inhibitors for the treatment of patients with HER2-positive breast cancer with brain metastases: a systematic review and meta-analysis. ESMO Open 7 (2022).
Wu, J. et al. Neoadjuvant pyrotinib, trastuzumab, and docetaxel for HER2-positive breast cancer (PHEDRA): a double-blind, randomized phase 3 trial. BMC Med. 20 (2022).
Xu, B. et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 22, 351–360 (2021).
Hartmaier, R. J. et al. Osimertinib + savolitinib to overcome acquired MET-mediated resistance in epidermal growth factor receptor-mutated, MET-amplified non-small cell lung cancer: TATTON. Cancer Discov. 13, 98–113 (2023).
Passaro, A. et al. Amivantamab plus chemotherapy with and without lazertinib in EGFR-mutant advanced NSCLC after disease progression on osimertinib: primary results from the phase III MARIPOSA-2 study. Ann. Oncol. 35, 77–90 (2024).
Zhou, Q. et al. Safety and efficacy of epitinib for EGFR-mutant non-small cell lung cancer with brain metastases: open-label multicentre dose-expansion phase Ib study. Clin. Lung Cancer 23, 353–361 (2022).
Shi, Y. et al. Furmonertinib (AST2818) versus gefitinib as first-line therapy for Chinese patients with locally advanced or metastatic EGFR mutation-positive non-small-cell lung cancer (FURLONG): a multicentre, double-blind, randomised phase 3 study. Lancet Respir. Med. 10, 1019–1028 (2022).
Choi, Y. W. et al. Differential efficacy of tyrosine kinase inhibitors (TKIs) according to the types of EGFR mutations and agents in non-small cell lung cancer (NSCLC): a real-world study. J. Clin. Oncol. 41, 21040–21040 (2023).
Choueiri, T. K. et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: the alliance A031203 CABOSUN trial. J. Clin. Oncol. 35, 591–597 (2017).
Fachi, M. M. et al. Comparative efficacy and safety of tyrosine kinase inhibitors for chronic myeloid leukaemia: a systematic review and network meta-analysis. Eur. J. Cancer 104, 9–20 (2018).
Zhang, X. et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281, 15694–15700 (2006).
Ward, C. W. & Lawrence, M. C. Ligand-induced activation of the insulin receptor: a multi-step process involving structural changes in both the ligand and the receptor. Bioessays 31, 422–434 (2009).
Olsson, A.-K., Dimberg, A., Kreuger, J. & Claesson-Welsh, L. VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 (2006).
Graveel, C. R., Tolbert, D. & Vande Woude, G. F. MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb. Perspect. Biol. 5, (2013).
Hubbard, S. R. & Gnanasambandan, K. Structure and activation of MuSK, a receptor tyrosine kinase central to neuromuscular junction formation. Biochim. Biophys. Acta 1834, 2166–2169 (2013).
De Munck, S. et al. Structural basis of cytokine-mediated activation of ALK family receptors. Nature 600, 143–147 (2021).
Hallberg, B. & Palmer, R. H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 13, 685–700 (2013).
Menck, K., Heinrichs, S., Baden, C. & Bleckmann, A. The WNT/ROR pathway in cancer: from signaling to therapeutic intervention. Cells 10 (2021).
van Amerongen, R., Mikels, A. & Nusse, R. Alternative wnt signaling is initiated by distinct receptors. Sci. Signal 1, re9 (2008).
Naitoh, A. et al. Study of lymphoscintigraphy with 99mTc rhenium colloid for diagnosis of lymphedema. Rinsho Hoshasen 28, 663–668 (1983).
Fu, Y., Chen, Y., Huang, J., Cai, Z. & Wang, Y. RYK, a receptor of noncanonical Wnt ligand Wnt5a, is positively correlated with gastric cancer tumorigenesis and potential of liver metastasis. Am. J. Physiol. Gastrointest. Liver Physiol. 318, 352–360 (2020).
Skaper, S. D. The neurotrophin family of neurotrophic factors: an overview. In: Skaper, S. (eds) Neurotrophic Factors. Methods in Molecular Biology (ed. Skaper, S.) vol. 846 1–12 (Humana Press, 2012).
Caberoy, N. B., Zhou, Y. & Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 29, 3898–3910 (2010).
Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 5, 9076–9076 (2013).
Maisonpierre, P. C. et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60 (1997).
Labrador, J. P. et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2, 446–452 (2001).
Vogel, W. F., Aszódi, A., Alves, F. & Pawson, T. Discoidin domain receptor 1 tyrosine kinase has an essential role in mammary gland development. Mol. Cell Biol. 21, 2906–2917 (2001).
Arrieta, O. et al. Updated frequency of EGFR and KRAS mutations in non small-cell lung cancer in Latin America: the Latin-American Consortium for the Investigation of Lung Cancer (CLICaP). J. Thorac. Oncol. 10, 838–843 (2015).
Banno, E. et al. Sensitivities to various epidermal growth factor receptor‐tyrosine kinase inhibitors of uncommon epidermal growth factor receptor mutations L861Q and S768I: What is the optimal epidermal growth factor receptor‐tyrosine kinase inhibitor? Cancer Sci. 107, 1134–1140 (2016).
André, F. et al. AACR project GENIE: powering precision medicine through an International Consortium. Cancer Discov. 7, 818–831 (2017).
Suda, K., Onozato, R., Yatabe, Y. & Mitsudomi, T. EGFR T790M mutation: a double role in lung cancer cell survival? J. Thorac. Oncol. 4, 1–4 (2009).
Gessi, M. et al. FGFR1 mutations in rosette-forming glioneuronal tumors of the fourth ventricle. J. Neuropathol. Exp. Neurol. 73, 580–584 (2014).
Jones, D. T. W. et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 45, 927–932 (2013).
Dulak, A. M. et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 45, 478–486 (2013).
Li, J. J. et al. FGFR genes mutation is an independent prognostic factor and associated with lymph node metastasis in squamous non-small cell lung cancer. Cancer Biol. Ther. 19, 1108–1116 (2018).
Nakamura, I. T. et al. Comprehensive functional evaluation of variants of fibroblast growth factor receptor genes in cancer. NPJ Precis. Oncol. 5 (2021).
Dienstmann, R. et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann. Oncol. 25, 552–563 (2014).
Ascione, C. M. et al. Role of FGFR3 in bladder cancer: treatment landscape and future challenges. Cancer Treat. Rev. 115 (2023).
Tomlinson, D. C., Hurst, C. D. & Knowles, M. A. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene 26, 5889–5899 (2007).
Aubertin, J., Tourpin, S., Janot, F., Ahomadegbe, J. & Radvanyi, F. Analysis of fibroblast growth factor receptor 3 G697C mutation in oral squamous cell carcinomas. Int. J. Cancer 120, 2058–2059 (2007).
Futami, T. et al. Identification of a novel oncogenic mutation of FGFR4 in gastric cancer. Sci. Rep. 9, 1–9 (2019).
Chew, N. J. et al. Evaluation of FGFR targeting in breast cancer through interrogation of patient-derived models. Breast Cancer Res. 23 (2021).
Boucher, J., Kleinridders, A. & Kahn, C. R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 6 (2014).
Cuenca, D. et al. A novel nonsense mutation in the insulin receptor gene in a patient with HAIR-AN syndrome and endometrial cancer. Mol. Genet. Metab. Rep. 35 (2023).
Esposito, D. L. et al. Novel insulin receptor substrate 1 and 2 variants in breast and colorectal cancer. Oncol. Rep. 30, 1553–1560 (2013).
Khurshid, S. et al. Splice-switching of the insulin receptor pre-mRNA alleviates tumorigenic hallmarks in rhabdomyosarcoma. NPJ Precis. Oncol. 6 (2022).
Vella, V. & Malaguarnera, R. The emerging role of insulin receptor isoforms in thyroid cancer: clinical implications and new perspectives. Int. J. Mol. Sci. 19 (2018).
Frasca, F. et al. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch. Physiol. Biochem. 114, 23–37 (2008).
Qian, H. et al. The clinical significance of platelet-derived growth factors (PDGFs) and their receptors (PDGFRs) in gastric cancer: a systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 127, 15–28 (2018).
Randomized trial of crenolanib in subjects with D842V mutated GIST. https://clinicaltrials.gov/study/NCT02847429.
Study of crenolanib for the treatment of patients with advanced GIST with the D842-related mutations and deletions in the PDGFRA gene. https://clinicaltrials.gov/study/NCT01243346.
Corless, C. L. et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 23, 5357–5364 (2005).
Joensuu, H. et al. Effect of KIT and PDGFRA mutations on survival in patients with gastrointestinal stromal tumors treated with adjuvant imatinib. JAMA Oncol. 3, 602–609 (2017).
Debiec-Rychter, M. et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer 40, 689–695 (2004).
Cassier, P. A. et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin. Cancer Res. 18, 4458–4464 (2012).
Yoo, C. et al. Efficacy of imatinib in patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors. Cancer Res. Treat. 48, 546–552 (2016).
Heinrich, M. C. et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299, 708–710 (2003).
Klempner, S. J. et al. Carving out another slice of the pie: exceptional response to single agent imatinib in an Asian female never-smoker with advanced NSCLC with a de-novo PDGFR-α N848 K mutation. Lung Cancer 124, 86–89 (2018).
Yi, E. S., Strong, C. R., Piao, Z., Perucho, M. & Weidner, N. Epithelioid Gastrointestinal Stromal Tumor With PDGFRA Activating Mutation and Immunoreactivity. Appl Immunohistochem. Mol. Morphol. 13, 157–161 (2005).
Velghe, A. I. et al. PDGFRA alterations in cancer: characterization of a gain-of-function V536E transmembrane mutant as well as loss-of-function and passenger mutations. Oncogene 33, 2568–2576 (2014).
Ozawa, T. et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 24, 2205–2218 (2010).
Dai, J. et al. Large-scale analysis of PDGFRA mutations in melanomas and evaluation of their sensitivity to tyrosine kinase inhibitors imatinib and crenolanib. Clin. Cancer Res. 19, 6935–6942 (2013).
Cheung, Y. H. et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am. J. Hum. Genet. 92, 996–1000 (2013).
Arts, F. A. et al. PDGFRB mutants found in patients with familial infantile myofibromatosis or overgrowth syndrome are oncogenic and sensitive to imatinib. Oncogene 35, 3239–3248 (2016).
Debiec-Rychter, M. et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 42, 1093–1103 (2006).
Wozniak, A. et al. Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: a European multicenter analysis based on ConticaGIST. Clin. Cancer Res. 20, 6105–6116 (2014).
Wozniak, A. et al. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann. Oncol. 23, 353–360 (2012).
Heinrich, M. C. et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 26, 5360–5367 (2008).
Szucs, Z. et al. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 13, 93–107 (2017).
Lennartsson, J., Jelacic, T., Linnekin, D. & Shivakrupa, R. Normal and oncogenic forms of the receptor tyrosine kinase Kit. Stem Cells 23, 16–43 (2005).
Carvajal, R. D. et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin. Cancer Res. 21, 2289–2296 (2015).
Pham, D. D. M., Guhan, S. & Tsao, H. KIT and Melanoma: biological Insights and Clinical Implications. Yonsei Med. J. 61, 562–571 (2020).
Funkhouser, A. T. et al. KIT Mutations correlate with higher galectin levels and brain metastasis in breast and non-small cell lung cancer. Cancers 14 (2022).
Arock, M. et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia 29, 1223–1232 (2015).
Naumann, N. et al. Adverse prognostic impact of the KIT D816V transcriptional activity in advanced systemic mastocytosis. Int. J. Mol. Sci. 22 (2021).
Abdellateif, M., Bayoumi, A. & Mohammed, M. c-Kit receptors as a therapeutic target in cancer: current insights. Onco Targets Ther. 16, 785–799 (2023).
McIntyre, A. et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res. 65, 8085–8089 (2005).
Nakai, Y. et al. KIT (c-kit oncogene product) pathway is constitutively activated in human testicular germ cell tumors. Biochem. Biophys. Res. Commun. 337, 289–296 (2005).
Sletta, K. Y., Castells, O. & Gjertsen, B. T. Colony stimulating factor 1 receptor in acute myeloid leukemia. Front. Oncol. 11 (2021).
Ridge, S. A., Worwood, M., Oscier, D., Jacobs, A. & Padua, R. A. FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc. Natl Acad. Sci. USA 87, 1377–1380 (1990).
Soares, M. J. et al. CSF1R copy number changes, point mutations, and RNA and protein overexpression in renal cell carcinomas. Mod. Pathol. 22, 744–752 (2009).
Kiyoi, H., Kawashima, N. & Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: therapeutic paradigm beyond inhibitor development. Cancer Sci. 111, 312–322 (2020).
Simons, M., Gordon, E. & Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 17, 611–625 (2016).
Mabeta, P. & Steenkamp, V. The VEGF/VEGFR axis revisited: implications for cancer therapy. Int. J. Mol. Sci. 23 (2022).
Loaiza-Bonilla, A. et al. KDR mutation as a novel predictive biomarker of exceptional response to regorafenib in metastatic colorectal cancer. Cureus 8 (2016).
Kumar, R. D. & Bose, R. Analysis of somatic mutations across the kinome reveals loss-of-function mutations in multiple cancer types. Sci. Rep. 7 (2017).
Antonescu, C. R. et al. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res. 69, 7175–7179 (2009).
Wei, L. et al. Dramatic response of CTNNB1 and VEGFR-2 mutant temporal bone squamous cell carcinoma to bevacizumab in combination with pemetrexed. Oncotarget 8, 57898–57904 (2017).
Ravi, V. et al. Antitumor response of VEGFR2- and VEGFR3-amplified angiosarcoma to pazopanib. J. Natl Compr. Cancer Netw. 14, 499–502 (2016).
Peghini, P. L. et al. Overexpression of epidermal growth factor and hepatocyte growth factor receptors in a proportion of gastrinomas correlates with aggressive growth and lower curability. Clin. Cancer Res. 8, 2273–2285 (2002).
Matsumoto, K., Umitsu, M., De Silva, D. M., Roy, A. & Bottaro, D. P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 108, 296–307 (2017).
Yano, S. et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac. Oncol. 6, 2011–2017 (2011).
De Silva, D. M. et al. Targeting the hepatocyte growth factor/Met pathway in cancer. Biochem. Soc. Trans. 45, 855–870 (2017).
Stone, A. EGFR and c-met inhibitors are effective in reducing tumorigenicity in cancer. J. Carcinog. Mutagen 5, 1–9 (2014).
Xu, J., Wang, J. & Zhang, S. Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer. Oncotarget 8, 90557–90578 (2017).
Oh, S. et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat. Biotechnol. 41, 1229–1238 (2023).
Gainor, J. F. et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis. Oncol. 1, 1–13 (2017).
Drilon, A. et al. A novel crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin. Cancer Res. 22, 2351–2358 (2016).
Mahato, A. K. & Sidorova, Y. A. RET receptor tyrosine kinase: role in neurodegeneration, obesity, and cancer. Int. J. Mol. Sci. 21 (2020).
Bardelli, A. et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science 300, 949–949 (2003).
Bonkowsky, J. L., Yoshikawa, S., O’Keefe, D. D., Scully, A. L. & Thomas, J. B. Axon routing across the midline controlled by the Drosophila Derailed receptor. Nature 402, 540–544 (1999).
Sasaki, T. et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 71, 6051–6060 (2011).
Joshi, S. K. et al. The FLT3N701K mutation causes clinical AML resistance to gilteritinib and triggers TKI sensitivity switch to quizartinib. Am. J. Hematol. 98, 364–368 (2023).
Freire, T. S., Caracelli, I., Zukerman-Schpector, J. & Friedman, R. Resistance to a tyrosine kinase inhibitor mediated by changes to the conformation space of the kinase. Phys. Chem. Chem. Phys. 25, 6175–6183 (2023).
Napolitano, A. & Vincenzi, B. Secondary KIT mutations: the GIST of drug resistance and sensitivity. Br. J. Cancer 120, 577–578 (2019).
Gramza, A. W., Corless, C. L. & Heinrich, M. C. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin. Cancer Res. 15, 7510–7518 (2009).
Chen, Y. et al. SPK1/S1P axis confers gastrointestinal stromal tumors (GISTs) resistance of imatinib. Gastric Cancer 26, 26–43 (2023).
Jiang, L. et al. Low frequency KRAS mutations in colorectal cancer patients and the presence of multiple mutations in oncogenic drivers in non-small cell lung cancer patients. Cancer Genet. 206, 330–339 (2013).
Xie, Y.-H., Chen, Y.-X. & Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 5 (2020).
Lim, M. et al. EGFR/ERBB2 Amplifications and Alterations Associated With Resistance to Lenvatinib inHepatocellular Carcinoma. Gastroenterology 164, 1006–1008 (2023).
Desbois-Mouthon, C. et al. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. 15, 5445–5456 (2009).
Gu, L., Jin, X., Liang, H., Yang, C. & Zhang, Y. Upregulation of CSNK1A1 induced by ITGB5 confers to hepatocellular carcinoma resistance to sorafenib in vivo by disrupting the EPS15/EGFR complex. Pharmacol. Res. 192 (2023).
Khan, H. Y. et al. Targeting XPO1 and PAK4 in 8505C anaplastic thyroid cancer cells: putative implications for overcoming lenvatinib therapy resistance. Int. J. Mol. Sci. 21 (2019).
Feng, H. et al. FOXK2 transcriptionally activating VEGFA induces apatinib resistance in anaplastic thyroid cancer through VEGFA/VEGFR1 pathway. Oncogene 40, 6115–6129 (2021).
Bertol, B. C. et al. Lenvatinib plus Anti-PD-1 combination therapy for advanced cancers: defining mechanisms of resistance in an inducible transgenic model of thyroid cancer. Thyroid 32, 153–163 (2022).
Patel, M. et al. Resistance to molecularly targeted therapies in melanoma. Cancers 13 (2021).
Kakadia, S. et al. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 11, 7095–7107 (2018).
Marín, A. et al. Acquired secondary HER2 mutations enhance HER2/MAPK signaling and promote resistance to HER2 kinase inhibition in breast cancer. Cancer Res. 83, 3145–3158 (2023).
Rini, B. I. & Atkins, M. B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 10, 992–1000 (2009).
Makhov, P. et al. Resistance to systemic therapies in clear cell renal cell carcinoma: mechanisms and management strategies. Mol. Cancer Ther. 17, 1355–1364 (2018).
Jeong, S. U. et al. PD-L1 upregulation by the mTOR pathway in VEGFR-TKI-resistant metastatic clear cell renal cell carcinoma. Cancer Res. Treat. 55, 231–244 (2023).
Sweeney, P. L., Suri, Y., Basu, A., Koshkin, V. S. & Desai, A. Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma. Cancer Drug Resist. 6, 858–873 (2023).
Facchinetti, F. et al. Resistance to selective FGFR Inhibitors in FGFR-driven urothelial cancer. Cancer Discov. 13, 1998–2011 (2023).
Van Cutsem, E. et al. Nintedanib for the treatment of patients with refractory metastatic colorectal cancer (LUME-Colon 1): a phase III, international, randomized, placebo-controlled study. Ann. Oncol. 29, 1955–1963 (2018).
Shi, Y.-K. et al. Safety and activity of alflutinib in patients with advanced EGFR T790M mutation non-small cell lung cancer who progressed after EGFR-TKI therapy. Ann. Oncol. 30, 1531–1531 (2019).
Lu, S. et al. AENEAS: a randomized phase III trial of aumolertinib versus gefitinib as first-line therapy for locally advanced or metastatic non–small-cell lung cancer with EGFR exon 19 deletion or L858R mutations. J. Clin. Oncol. 40, 3162–3171 (2022).
Lu, S. et al. Befotertinib (D-0316) versus icotinib as first-line therapy for patients with EGFR-mutated locally advanced or metastatic non-small-cell lung cancer: a multicentre, open-label, randomised phase 3 study. Lancet Respir. Med. 11, 905–915 (2023).
Zhao, H. et al. Apatinib plus gefitinib as first-line treatment in advanced EGFR-mutant NSCLC: the phase III ACTIVE study (CTONG1706). J. Thorac. Oncol. 16, 1533–1546 (2021).
DeAngelo, D. J. et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood 108, 3674–3681 (2006).
Shi, Y. et al. Efficacy and safety of iruplinalkib (WX-0593) in ALK-positive crizotinib-resistant advanced non-small cell lung cancer patients: a single-arm, multicenter phase II study (INTELLECT). BMC Med. 21 (2023).
Acknowledgements
D.K. is funded by a grant of the Romanian Government (bursa Henri Coandă). D.K. and C.T. are funded by a grant from the Romanian Academy of Scientists 2023-2024. C.T. is funded by an international grant of the European Hematology Association (EHA-SWG Immunotherapy Project 2024 – CAR NK cells for tumor associated macrophage immunomodulation—a new era of immunotherapy), as well as by a bilateral collaboration grant between Romania and Moldova (PN-IV-P8-8.3-ROMD-2023-0036). H.E. is funded by a national grant of the Romanian Research Ministry – PNRR 2024-2026 (PNRR/2022/C9/MCID/18, Contract No. 760278/26.03.2024). C.M.C. is supported in part by the R35 CA 197706 grant from the National Cancer Institute.
Author information
Authors and Affiliations
Contributions
Ciprian Tomuleasa, Adrian-Bogdan Tigu, Raluca Munteanu, Cristian Silviu Moldovan, David Kegyes and Anca Onaciu drafted and conceptualized the initial manuscript. Cristian-Silviu Moldovan, Anca-Onaciu and Diana Gulei designed and organized the figure and tables. Supervision and strategic directions were provided by Gabriel Ghiaur, Hermann Einsele and Carlo Maria Croce who also contributed to the key ideas that shaped the manuscript. Ciprian Tomuleasa and Carlo Maria Croce refined the manuscript. All authors have read and approved the review article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no potential conflict of interest. C.M.C. is one of the Editors-in-Chief of Signal Transduction and Targeted Therapy, but he has not been involved in the process of the manuscript handling.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tomuleasa, C., Tigu, AB., Munteanu, R. et al. Therapeutic advances of targeting receptor tyrosine kinases in cancer. Sig Transduct Target Ther 9, 201 (2024). https://doi.org/10.1038/s41392-024-01899-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01899-w
- Springer Nature Limited