
Abstract
Glutamate abnormalities in the medial prefrontal cortex (mPFC) are associated with cognitive deficits. We previously showed that homozygous deletion of CNS glutamate dehydrogenase 1 (Glud1), a metabolic enzyme critical for glutamate metabolism, leads to schizophrenia-like behavioral abnormalities and increased mPFC glutamate; mice heterozygous for CNS Glud1 deletion (C-Glud1+/− mice) showed no cognitive or molecular abnormalities. Here, we examined the protracted behavioral and molecular effects of mild injection stress on C-Glud1+/− mice. We found spatial and reversal learning deficits, as well as large-scale mPFC transcriptional changes in pathways associated with glutamate and GABA signaling, in stress-exposed C-Glud1+/− mice, but not in their stress-naïve or C-Glud1+/+ littermates. These effects were observed several weeks following stress exposure, and the expression levels of specific glutamatergic and GABAergic genes differentiated between high and low reversal learning performance. An increase in miR203-5p expression immediately following stress may provide a translational regulatory mechanism to account for the delayed effect of stress exposure on cognitive function. Our findings show that chronic glutamate abnormalities interact with acute stress to induce cognitive deficits, and resonate with gene x environment theories of schizophrenia. Stress-exposed C-Glud1+/− mice may model a schizophrenia high-risk population, which is uniquely sensitive to stress-related ‘trigger’ events.
Similar content being viewed by others
Intact glutamate transmission in the prefrontal cortex (PFC) is critical for spatial learning, cognitive flexibility, social recognition and problem solving [1,2,3,4,5,6], and is disrupted in several psychiatric disorders including schizophrenia [7, 8]. While most research pertaining to glutamate disruption and cognitive dysfunction in schizophrenia has focused on the NMDA receptor [9,10,11], recent attention has turned to manipulations that target glutamate synthesis, release or reuptake. Indeed, such manipulations significantly impact learning, memory and attention [12,13,14].
Glutamate Dehydrogenase 1 (GLUD1 in humans, GDH in rodents), encoded by the Glud1 gene, is a key regulator of glutamate metabolism in CNS. GDH is a mitochondrial enzyme, expressed mainly in astrocytes [15], where it catabolizes glutamate to α-ketoglutarate [16]. Our previous studies in mice with a homozygotic deletion of Glud1 in CNS (C-Glud1−/− mice) revealed elevated hippocampal cerebral blood volume (CBV) and glutamate levels in medial PFC (mPFC) and hippocampus, along with baseline and amphetamine-induced hyperlocomotion. These findings resonate with human schizophrenia data showing decreased Glud1 expression [14], elevated hippocampal CBV and glutamate/glutamine (Glx) levels [17, 18] and a hyperdopaminergic response to amphetamine [19]. C-Glud1−/− mice also show prominent abnormalities in tasks relevant to cognitive impairment in schizophrenia, including working, spatial and social memory tasks as well as reversal and extra-dimensional set shifting (EDSS) in the Water T-maze [14, 20]. Unlike C-Glud1−/− mice, heterozygous C-Glud1+/− mice show no substantial changes in glutamate levels in hippocampus or PFC, and control-like behavior in a range of cognitive tasks with the exception of the challenging EDSS task [14].
Stress exposure in late adolescence or early adulthood may trigger symptom eruption in schizophrenia and related disorders. Animal studies have shown that stress exposure hinders PFC-dependent working memory, social recognition and cognitive flexibility [21, 22]. Impaired glutamate signaling is implicated in stress-induced cognitive deficits [23, 24]. Furthermore, in support of an interaction between genetic and environmental factors, stress exposure alters the behavioral consequences of genetic manipulations affecting the excitatory/inhibitory (E/I) balance in tasks measuring cognitive flexibility and other PFC-dependent behaviors [25, 26]. Notably, acute stress was found to increase NMDAR- and AMPAR-mediated synaptic currents in PFC pyramidal neurons [27, 28], while chronic stress attenuates acute stress-evoked increases in extracellular glutamate [29].
Preliminary data from our lab [30] indicate that social isolation stress renders heterozygote C-Glud1+/− mice susceptible to deficits in mPFC-dependent reversal learning [31], which are absent in social stress-naïve mice. However, we and others have shown that exposure to social isolation stress alone leads to a wide range of behavioral deficits, including spatial acquisition and reversal abnormalities [32,33,34]. Chronic social isolation stress exposure also leads to abnormalities in glutamate homeostasis [34,35,36]. The impact of acute stress, in particular mild stress with negligible consequences in genetically intact mice, was not previously examined in C-Glud1+/− mutants.
In the present study, we aimed to parse out the impact of stress and Glud1 deficiency using a mild acute stress protocol [37]. We first asked whether acute stress exposure in early adulthood would ‘trigger’ cognitive abnormalities in C-Glud1+/− mice. We hypothesized that heterozygous C-Glud1+/− mice exposed to stress would exhibit deficits in capacities that require intact hippocampal and mPFC function, i.e., spatial rule acquisition, cognitive flexibility and social function, whereas Glud1 heterozygosity or stress exposure alone would not elicit behavioral abnormalities.
Second, we aimed to recognize unique gene expression patterns in the mPFC of acute stress-exposed CNS-Glud1+/− mice, using genome-wide RNA sequencing (RNA-seq). We hypothesized that acute mild stress would have little effect on its own, but when combined with Glud1 deficiency would induce long-term changes in mPFC gene expression patterns. In light of previous studies on the separate effects of Glud1 deficiency and stress [14, 20], we hypothesized that glutamate and GABA signaling pathways would be particularly affected in Glud1-deficient, stress-exposed mice.
Since behavioral and transcriptional changes were detected several weeks following stress exposure, we aimed to identify immediate stress-induced changes in mPFC. We examined immediate and long-term changes in the expression of several microRNA (miRNA) molecules, which play a key role in translational regulation [38] and were shown to be associated with stress and cognition [39,40,41,42,43,44,45]. We hypothesized that changes in the expression of these miRNAs would occur immediately following stress exposure and would persist to adulthood in stress-exposed C-Glud1+/− mice but not in their C-Cre+ or stress-naïve littermates.
Results
Behavior
Behavioral tests were run in the following order: Nesting Behavior (NB), Open Field (OF), Social Preference (SP), Social Recognition (SR) and Water T-Maze (WTM; Fig. 1a). Detailed statistical analysis is presented in Table S1.

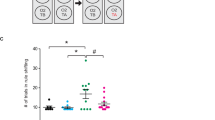
Timeline of the experiment (a); NB—Nest building, OF—Open field, SP—social preference, SR—social recognition, WTM—Water T-maze. In the Open Field, male C-Glud1+/− mice traveled less distance during the 60-min test (b, left; Time*Genotype: F(4.67,233.60) = 3.46, p = 0.005, η²p = 0.064). Female mice exhibited no genotypic differences (c, left). Male C-Glud1+/− mice exhibited an erratic pattern of time spent in the center of the open field (d, left; Time*Genotype: F(6.16,308.46) = 2.68, p = 0.013, η²p = 0.051). During the first 5 min (Bin 1), genotype and stress affected the time spent in the center (d, right; C-Glud1+/−<C-Cre+, Genotype: F(1,50) = 4.49, p = 0.038, η²p = 0.082; Stress<Control; Group: F(1,50) = 8.47, p = 0.005, η²p = 0.144). Female mice exhibited no genotypic or stress-induced differences across all bins (e, left). In Bin 1, C-Glud1+/− and stress-exposed mice spent less time in the center (e, right; Genotype: F(1,55) = 6.64, p = 0.012, η²p = 0.107; Group: F(1,55) = 8.96, p = 0.004, η²p = 0.140). In the Water T-maze, male and female data were combined. In the Acquisition phase, C-Glud1+/−/Stress mice required a larger number of trials to criterion (f, left; Genotype*Group: F(1,90) = 12.68, p < 0.001, η²p = 0.122) and exhibited higher RTv (f, right; Genotype*Group: F(1,90) = 4.94, p = 0.029, η²p = 0.052) compared to the other groups. The same pattern was observed in the Reversal phase (g, left, trials to criterion: Genotype*Group: F(1,87) = 17.79, p < 0.001, η²p = 0.170; g, right, RTv: Genotype*Group: F(1,87) = 34.05, p < 0.001, η²p = 0.281). #p < 0.055, *p < 0.05 **p < 0.01 ***p < 0.001. ‡Main effect for stress. †Group mean is statistically different from all others. NB, SP, and SR results were not significant and are provided in the SI.
In the NB test, all mice exhibited similar nesting behavior, with relatively high nesting scores regardless of Genotype, Group or Sex. No main effects or interactions were found (Fig. S1).
In the OF, males and females differed in total locomotor activity (Sex main effect; males>females) and in the time spent in center over time (Time*Sex interaction). Therefore, male and female data were analyzed separately. In male mice, all groups exhibited less locomotor activity over time, and C-Glud1+/− mice traveled less distance compared to C-Cre+ controls, regardless of stress exposure, during the first 40 min (Fig. 1bLeft) and over the entire test period (1bRight). No Group effect nor a Genotype*Group interaction was found. In female mice, all groups exhibited less locomotor activity over time (1cLeft). We found no effect for Genotype, Group or a Genotype*Group interaction (1cRight).
Examination of time spent in the center of the OF in males revealed a main effect of Time and a Time*Genotype interaction. We found a significant change over time for C-Glud1+/− mice, which exhibited linear and polynomial trends, indicative of ‘erratic’ behavior (Fig. 1bLeft). There was no change over time in C-Cre+ mice. During the first 5 min of the test (bin 1, 1dRight), C-Glud1+/− mice spent less time in the center than their C-Cre+ controls, and stress-exposed mice spent less time in the center than their stress-naïve controls. No Genotype*Group interaction was found. In females, we found a main effect for Time, but no Time*Genotype interaction (1eLeft). Similarly to male mice, C-Glud1+/− females spent less time in the center compared to their C-Cre+ littermates, and stress exposure similarly led to a decrease in time in the center (1eRight). No Genotype*Group interaction was found.
In the social behavior tests, we found no Genotype, Group or Sex effects, nor interactions between them, in the SP or SR tasks (Fig. S1).
For the WTM test, male and female data were combined since no Sex effects or interactions were found. In the acquisition phase, we found Genotype*Group interactions for both Trials to Criterion and Reaction Time variability (RTv). Post-hoc tests for both measures indicate that C-Glud1+/−/Stress mice required more trials to criterion (Fig. 1fLeft) and displayed higher RTv (1fright; Fig S2) than mice in all other groups. Data from 1 C-Glud1+/−/Stress mouse was excluded (>2 SD above mean) in the RTv analysis. RT data followed the same pattern of results: RTs were highest in the C-Glud1+/−/Stress group (not shown, see Table S1). Four mice from the C-Glud1+/−/Stress group [21.05%; significantly more than in other groups (0%)] failed to achieve criterion in the acquisition phase and received a maximal score of 60. These mice did not continue to the probe or reversal phase. In the reversal phase we found a Genotype*Group interaction for both Trials to Criterion and RTv. Post-hoc tests showed that C-Glud1+/−/Stress mice required more trials to criterion (1gLeft) and displayed higher RTv (1gRight) than mice in all other groups. Higher RTs were observed in this group as well.
RNA-seq
Differentially expressed genes (DEGs)—as can be seen in Table 1, RNA-seq analysis showed that 3107 genes (19.86% of the mPFC-expressed protein-coding genes) in the C-Glud1+/−/Stress group, 64 in the C-Glud1+/−/Control group, and 3 in the C-Cre+/Stress group were differentially expressed compared to C-Cre+/Control (FDR-corrected q value < 0.05). For a full list of DEGs in each comparison, see Table S2.
As can be seen in Fig. 2, in the C-Cre+/Stress group all 4 DEGs were downregulated (2a), in the C-Glud1+/−/Stress group the majority of the 64 DEGs were downregulated (2b), and the Glud1+/−/Stress group exhibited the highest number of DEGs, with 58.73% upregulated DEGs (2c). Examining the overlap between DEGs in our groups (2d), we found that 1 DEG in the C-Cre+/Stress group and 80% of the DEGs in the Glud1+/−/Control groups were also downregulated in the Glud1+/−/Stress group. There was only a small overlap between the C-Glud1+/−/Stress group and other groups: out of 3,107 DEGs, 3,072 (98.87%) were unique to the C-Glud1+/−/Stress group.

Volcano plots demonstrating upregulated (red) and downregulated (blue) genes in C-Cre+/Stress (a), C-Glud1+/−/Control (b) and C-Glud1+/−/Stress (c) compared to C-Cre+/Control mPFC samples. Dotted lines indicate threshold for statistical significance (−10log > 1.3, corresponding to q < 0.05). The largest number of DEGs is found in C-Glud1+/−/Stress samples, mostly (~58%) upregulated. d Venn diagram of DEG overlap between conditions. 98.8% of DEGs were unique to the C-Glud1+/−/Stress group.
To detect enriched biological categories, we performed an over-representation analysis (ORA) of unique DEGs in the C-Glud1+/−/Stress group using Enrichr [46, 47]. As can be seen in Fig. 3a, a BioPlanet 2019 ORA revealed significant changes in glutamate- and GABA-related pathways. These pathways were also prominent when other gene databases were used (e.g., GO Biological Process, Elsevier Pathway Collection; Fig S3a, b). The Elsevier Pathway Collection also pointed to the corticotropin-releasing hormone (CRH) secretion regulation pathway. Top terms also included S6K1 signaling and downregulation of ERBB4 signaling (BioPlanet), as well as chromatin organization involved in negative regulation of transcription, histone arginine methylation and postsynaptic density protein 95 clustering (GO Biological). For a list of the top 30 terms in each of the databases, see Table S3.

a Top enriched BioPlanet 2019 terms for DEGs in C-Glud1+/−/Stress mice include glutamate- and GABA-related pathways. Combined score is computed as log(p value)*Z score. b, c Relative expression of glutamatergic and GABAergic genes in C-Cre+/Stress (left; n = 7), C-Glud1+/−/Control (middle; n = 5) and C-Glud1+/−/Stress groups (right; n = 9), relative to the C-Cre+/Control reference group (n = 7). Significant expression changes in both pathways were found in the C-Glud1+/−/Stress group alone. d Functional diagram of protein-protein interactions of glutamatergic, GABAergic and stress-related DEGs in C-Glud1+/−/Stress mice, compared to C-Cre+/Control mice. Line thickness indicates interaction strength. Dotted lines: interactions between proteins from different clusters.
We then proceeded to examine expression patterns of functionally significant genes that contribute to homeostasis at the glutamate tripartite synapse, the E/I balance and cognitive function [8, 48]. C-Glud1+/−/Stress mice consistently showed expression changes in glutamate- and GABA-related genes (Fig. 3b, c). Interestingly, while Glud1 expression was not significantly altered, many of the genes that contribute to glutamate and GABA homeostasis were differentially expressed in the C-Glud1+/−/Stress group, but not in C-Cre+/Stress or C-Glud1+/−/Control mice (Fig S3a, b; see Table S4a, b for DEGs from select over-represented categories). The HPA axis function-associated genes Nr3c1 and Nr3c2 were also differentially expressed exclusively in this group.
We examined in-silico the cell-type enrichment of the DEGs in C-Glud1+/−/Stress mice using the PanglaoDB Augmented 2021 database [49] (Enrichr), and found that neurons [Combined score (CS) = 55.62], particularly pyramidal ones (CS = 22.20), were enriched. GABAergic neurons were also enriched (CS = 19.63), as were oligodendrocytes (CS = 11.09), but astrocytes were not (CS = 2.92). No cell-specific enrichment was detected in C-Glud1+/−/Control mice.
The STRING database [50] was used to examine protein-protein interactions between glutamatergic, GABAergic and stress-related DEGs (Fig. S4). This analysis revealed a functional network with 6 clusters: Glutamatergic signaling (Red; 18 genes), Postsynaptic neurotransmitter receptor internalization (Yellow; 6 genes), Glutamate biosynthetic process (Dark green; 4 genes), GABAergic signaling pathway (Pale green; 4 genes), Stress (Blue; 3 genes) and Nsf (Purple; one gene). As can be seen in Fig. 3e, proteins encoded by DEGs interact within and between clusters.
We also examined whether genes known to interact with Glud1 (STRING) were differentially expressed in C-Glud1+/−/Stress mice. Out of 63 genes known to interact with Glud1, 16 were differentially expressed in C-Glud1+/−/Stress mice (11 upregulated). Protein-protein interactions between these genes are shown in Fig. S5.
To assess the relationship between mPFC gene expression and behavior, we calculated Spearman correlations between DEG transcript levels and behavioral measures in the open field and T-maze tasks, in all mice. Thirty-eight glutamatergic, GABAergic and stress-related genes differentially expressed exclusively in the C-Glud1+/−/Stress group (see Fig. 3b, c, S4) were selected. As can be seen in Table S4, transcript levels correlated with several behavioral measures, but mostly with individual RTv in the Water T-Maze Reversal task. RTv values predicted transcript levels of 33 out of the 38 genes. Figure 4a depicts select correlations between glutamatergic, GABAergic and stress-related DEGs and individual RTv (higher RTv reflects compromised cognitive performance), derived from all 4 groups.

a RTv in the Water T-maze Reversal task correlates with expression levels of glutamatergic, GABAergic and stress-related DEGs. b K-means cluster analysis of these DEGs produces two distinct clusters. c Samples are clearly distributed between the two clusters (dots/triangles: mouse numbers). d Cluster 2 Reversal RTv scores are significantly higher than Cluster 1 scores (Z = −3.36, p < 0.001). e Glutamate, GABA and stress-related DEGs (presented in order of cluster association strength) are differentially expressed in HP (Cluster 1) and LP (Cluster 2) groups (all Z’s>3.31, FDR-corrected p’s < 0.01). ***p < 0.001.
K-means cluster analysis on these genes (jamovi (v2.2.5) ‘snowCluster’ module (v6.7.0)), excluding 2 genes due to low factor loading (Standardized;<|0.40|) with either cluster), yielded two clusters (Fig. 4b). The final model (Table S5) accounted for 74.3% of the variance, with the first PCA accounting for 66.33% of the variance. The 2 clusters separated the samples into two non-overlapping groups (4c), which differed in expression levels of clustered genes (samples in the ‘blue’ cluster have high expression levels of genes marked in blue and low expression levels genes marked in orange, and vice versa). Notably, these clusters differ in Reversal RTv scores (Mann–Whitney test; Z = −3.36, p < 0.001; 4d, Table S5). High- and low- performance mice (HP and LP, respectively) displayed differential expression of all genes in the analysis; the top 12 differentiating genes are presented in 4e (Mann–Whitney tests; all Zs>3.31, all p’s < 0.001). Notably, the majority of LP/Cluster 2 mice were C-Glud1+/−/Stress mice (orange dots; 60% of cluster 2; 33.33% of total sample).
miRNA expression
The timeline of the experiment is presented in Fig. 5a. MiRNA molecules were selected according to their relevance to stress regulation [42, 44] and cognitive function [40, 43]. For example, increased miR203 expression was found in human epileptic brains and in a mouse model of the disease, suggesting it is involved in regulating the E/I balance [41]. Increased miR203 was also induces microglial activation and production of pro-inflammatory cytokines in mouse hippocampus, as well as impaired learning [45]. We first conducted an in silico search (using the DIANA miRNA database) for targets of our selected miRNA molecules (shown in the literature to be affected by stress and associated with cognitive function) which overlap with DEGs that emerged from the RNA-seq data. We found an approximate average of 18% (SD = 0.01%) overlap between DEGs and targets of each miRNA. As can be seen in Fig. 5b, several of miRNA targets, also differentially expressed in C-Glud1+/−/Stress mice, were shared by the selected miRNAs. The miRNAs examined were found to target genes related to glutamate, GABA and postsynaptic components in pathway analysis conducted with GO biological, cellular and molecular databases; several miRNAs were found to affect the same pathways (5c).

a Timeline of miRNA experiment. b Venn diagram of target overlap between selected miRNAs. c MiRNA target shared pathways. Pathways shared by at least 2 miRNAs are presented. a MiR137-3p, b MiR137-5p, c MiR203-3p, d MiR203-5p, e MiR493-3p, f MiR493-5p, g MiR34a-5p. The combined score is calculated as the mean of the relevant miRNA target combined scores. d At T1 (immediately after stress), the expression of miR203-5p was elevated in mPFC of C-Glud1+/− /Stress mice (H(2) = 9.73, p = 0.002), and miR493-5p was upregulated in both C-Glud1+/−/Stress and C-Cre+/Stress groups (H(2) = 8.46, p = 0.008), relative to C-Cre+/Control mice. At T2 (one month after stress), miR203-5p and miR137-5p were upregulated (H(2) = 6.89, p = .024 and H(2) = 10.05, p < 0.001, respectively) whereas miR203-3p and miR34a-5p were downregulated (H(2) = 10.59, p = 0.001 and H(2) = 9.64, p = 0.001, respectively). Post hocs: #p < 0.075, *p < 0.05, **p < 0.01. C-Cre+/Control group sample size: T1 (n = 5-6), T2 (n = 4–5). e Pathway analysis of transcriptional targets of miR203-5p, which was upregulated at both T1 and T2. f Expression levels of miR203-5p and miR34a-5p, but not miR493-5p, correlate with performance in acquisition and/or reversal of the Water T-maze. Orange dots depict C-Glud1+/−/Stress mice.
Next, we asked whether the expression of these miRNAs is altered in the mPFC of C-Glud1+/−/Stress mice. Detailed statistical findings are provided in Table S6. We examined 2 time points in separate cohorts: immediately following stress exposure (T1) and approximately 1 month later (T2; the time point at which the RNA-seq experiment was performed). As can be seen in Fig. 5d, the expression of miR203-5p increased by approximately 13-fold in mPFC at T1 in C-Glud1+/−/Stress mice and remained mildly increased at T2 relative to both C-Cre+/Stress and C-Cre+/Control (dotted line) groups. The expression of miR493-5p differed significantly between groups at T1, with higher expression in C-Cre+/Stress mice and marginally higher expression in the C-Glud1+/−/Stress group; these differences did not persist at T2. Similarly, RT-PCR analysis (n:4-8/group) revealed that several of the DEG transcripts that emerged at T2 were unaltered in the mPFC at T1 (all p’s > .239; Table S6).
Differences in the expression of miR137-5p, miR203-3p and miR34a-5p emerged at T2. miR137-5p expression was upregulated in the C-Glud1+/−/Stress group compared to C-Cre+/Control mice, and marginally compared to the C-Cre+/Stress mice. miR203-3p and miR34a-5p were downregulated in C-Glud1+/−/Stress mice compared to C-Cre+/Control and C-Cre+/Stress mice. For all 3 miRNA molecules, no differences were found at T1. No differences in miR137-3p or miR493-3p were detected at either time point. The mean miRNA expression score (mean ∆∆Ct across miRNAs for each mouse) at T1 was higher in C-Glud1+/−/Stress compared to the C-Cre+/Stress (fold change: 2.60 ± 0.24) and C-Cre+/Stress (fold change: 1.21 ± 0.49) mice. No such difference was found at T2.
Analysis of individual miRNA molecules and the mean miRNA expression score at T2 in the dCA1 and vCA1 hippocampal subregions revealed no changes in the C-Glud1+/−/Stress group. In dCA1, the mean miRNA expression score and the expression of miR137-5p were upregulated in the C-Cre+/Stress group (Fig. S6).
Since mPFC miR203-5p expression increased dramatically at T1, we asked which pathways are associated with targeted transcripts of this miRNA molecule. Pathway analysis revealed targets associated with chromatin regulation, cortisol synthesis, steroid metabolism and glutamate metabolic processes (5e).
Finally, we examined the correlations between miRNA expression and behavioral measures in the water T-maze (5f; see additional behavioral measures in Table S4). Mice from all 4 groups were included. MiR203-5p expression was correlated with the number of trials to criterion in both phases, but not with variability measures. In contrast, miR34a-5p was correlated with variability measures in both phases, and with trials to criterion in reversal. MiR493-5p was not correlated with any of the measures. Overall, T-maze reversal performance was most highly correlated with GABA, glutamate and stress-related gene expression, as well as with miRNA expression levels, compared to other behavioral measures (Table S4).
Discussion
Our findings indicate that stress alters the behavioral and molecular phenotype of mice with a constitutive, CNS-specific monoallelic deletion of Glud1. Specifically, C-Glud1+/− mice exposed to mild stress, which was nearly inconsequential in C-Cre+ controls, exhibit deficits in spatial acquisition and reversal learning, alterations in prefrontal changes in glutamatergic and GABAergic gene expression, and long-lasting changes in miRNA expression. Stress-naive C-Glud1+/− mice showed only mild behavioral abnormalities in the open field and relatively limited transcriptomic alterations. Furthermore, we found that spatial discrimination and reversal learning scores correlate with mRNA and miRNA transcription levels in mPFC. These findings are particularly striking considering the mildness of the stressor and the temporal separation (i.e., 3-4 weeks) between stress exposure and behavioral/molecular testing.
Stress-exposed C-Glud1+/− mice show deficits in spatial acquisition and reversal in a water T-maze task, unlike their stress-naïve C-Glud1+/− and stress-exposed C-Cre+ littermates. We found similar deficits in stress-naïve homozygous C-Glud1−/− mice [20], and in WT mice exposed to prolonged social isolation stress in adolescence [34]. Thus, CNS Glud1 heterozygosity and mild stress have additive effects on cognitive performance in this task. Different manipulations affecting glutamate homeostasis were reported to induce compromised performance in hippocampus-dependent spatial acquisition tasks [51, 52] and mPFC-dependent reversal assays [53,54,55]. Exposure to acute stress also induces cognitive impairments [56], but its effects in mice with compromised glutamate neurotransmission have not been thoroughly studied. Notably, in our study stress preceded behavioral testing by several weeks. Apparently, the impact of early adulthood stress in our study ‘incubates’ over this time period and leads to additive effects on both the behavioral and molecular levels, in line with previous studies on stress in early life and adolescence [57].
Our findings point to expression changes in 30 genes attributable to the genetic alteration in Glud1, and no changes induced by stress exposure alone. However, the combination of Glud1 deletion and stress resulted in 3,107 DEGs. Many of these genes contribute to homeostasis at the glutamate and GABA synapses, pointing to a potentially disrupted balance between glutamate and GABA transmission in the CNS-Glud1+/− /Stress group. Notably, we found no change in Glud1 mRNA counts in C-Glud1+/− mice. However, many genes that interact with Glud1 were differentially expressed, e.g., the glutamate-glutamine cycle rate-limiting enzyme Glutaminase 1 (Gls1) [58]; the mitochondrial glutamate oxaloacetate transaminase (Got2) gene which synthesizes glutamate from aspartate and α-ketoglutarate [59], and Oxoglutarate dehydrogenase (Ogdh), a mitochondrial complex member associated with glutamate degradation [60], which regulates neurotransmitter glutamate levels and is implicated in the stress response [61]. Another gene involved in amino acid metabolism altered in C-Glud1+/− mice is the Slc6a18 gene, downregulated roughly 50% in both Glud1 groups. This gene is part of the Slc6a family, involved in transport of compounds related to regulation of inhibitory neural activity [62], and was found to be sodium and chloride dependent [63]. Interestingly, mice with knockout of this gene had higher urine glutamine and glutamate concentrations [63].
While homozygous C-Glud1−/− mice show an increase in excitatory transmission under stress-naïve conditions [14], transcriptional abnormalities implicating pyramidal neuron dysfunction in heterozygous CNS-Glud1+/− mice are triggered by stress. In CNS-Glud1+/−/Stress mice, stress may limit the ability of the mPFC to regulate glutamate homeostasis [64, 65], and hinder the competency of the mPFC to implement compensatory mechanisms critical for intact homeostasis at the glutamate tripartite and glutamate-GABA synapses. In particular, the present findings point to changes in pyramidal cells, GABA interneurons and oligodendrocytes. The function of these cell types and their interactions are disrupted in several psychiatric disorders including schizophrenia. Post-mortem PFC transcriptional data from schizophrenia patients reveals changes in pathways associated with synaptic signaling [66], glutamate signaling [66, 67], the GABAergic synapse and postsynaptic membrane [68]. Oligodendrocytic deficits could point to myelination deficits in excitatory or inhibitory neurons. In turn, dysfunctional excitatory /inhibitory neurons could impact NMDA receptors located on oligodendrocytes, thus affecting their function [69]. The association between excitatory/inhibitory neurons and oligodendrocytes was shown in human studies [70, 71] and in animal models of cognitive dysfunction [72].
Epigenetic processes may account for the unique impact of stress on the transcriptional profile of C-Glud1+/−/Stress mice. Stress was shown to impair cognitive function and alter the expression of glutamate- and GABA-related genes via epigenetic changes to DNA or RNA [73,74,75,76]. Here, we show that the expression of the stress-associated miRNA molecule miR203-5p, which targets glutamate, stress and epigenetic-associated transcriptional pathways, is upregulated immediately after stress, and remains elevated several weeks later. One possibility is that changes in miR203-5p (and/or other miRNA molecules not examined in the present study) lead to transcriptional changes, potentially through other epigenetic mechanisms such as histone methylation/acetylation and heterochromatin modifications, that escalate over time and result in impaired glutamate homeostasis and glutamate-GABA communication.
Interestingly, stress led to miRNA expression changes in dCA1 in control (Cre+), but not C-Glud1+/− mice. Spatial rule learning depends on dorsal hippocampal function, and was impaired in C- Glud1+/− mice. Taken together with the mPFC findings, these data point to a dichotomous miRNA-behavior relationship in dCA1 vs. the mPFC: in dCA1, elevated miRNA expression signifies intact spatial learning, whereas in the mPFC it signals cognitive impairment.
Performance level in the T-maze reversal task was correlated with the expression level of glutamate, GABA and several stress-related genes. Previous investigations found that performance in the Morris Water Maze, a hippocampus-dependent spatial acquisition task, is correlated with hippocampal gene expression, particularly affecting the homeostasis of glutamatergic synapses [77]. Here, we further find that genes contributing to glutamate homeostasis, e.g. the mitochondrial glutamate carrier 1 gene, Slc25a22, as well as the schizophrenia risk gene Grin1 [78], distinctly differentiate between high- and low-performers in mPFC-dependent reversal learning [31]. This further supports the importance of glutamate homeostasis, maintained by a multitude of metabolic and synaptic components, in cognitive function.
This is the first demonstration of a gene x environment interaction in mice with abnormal expression of Glud1, which is downregulated in schizophrenia [20] and predicts antipsychotic treatment efficacy [79]. Similarly to other psychiatric disorders, schizophrenia symptoms are believed to be ‘triggered’ by an external event in individuals with underlying genetic susceptibility [80]. Cognitive symptoms precede and often predict the eruption of the first psychotic episode [81]. Thus, stress-exposed C-Glud1+/− mice may provide a tool for studying high-risk populations which carry genetic susceptibility and may be particularly vulnerable to the effects of adversity.
Methods
Mice
C-Glud1−/− mice were bred at the University of Haifa mouse vivarium. Flx-Glud1 (Glud1lox/lox) mice (kind gift from Pierre Maechler, University of Geneva) were bred with mice expressing Cre recombinase under the control of the Nestin cis-regulatory sequence (Jackson labs, Sacramento CA) to generate Nestin-Cre::Glud1lox/+ (C-Glud1+/−) mice. Nestin-Cre::Glud1+/+ (C-Cre+) mice were used as controls, since they were found to display mild physiological and behavioral differences compared to Cre- mice [14]. Mice were maintained on a C57BL/6 J background. Procedures involving mice and their care were conducted in conformity with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, under approval by the University of Haifa Ethics and Animal Health Committee (587/18). Food and water were provided ad libitum. Experiments included approximately equal numbers of males and females per group. The sample size was determined based on a priori power analysis using G*Power software using a moderate effect size at an alpha error probability of 0.05 [82].
Procedure
On postnatal day (PND) 70, male and female C-Cre+ and C-Glud1+/− mice were randomly divided into Stress or Control Groups. The experiment thus consisted of 4 groups: C-Cre+/Control, C-Cre+/Stress, C-Glud1+/−/Control and C-Glud1+/−/Stress. Acute mild physical stress consisted of 3 i.p. injections of 0.9% saline within a 24-hr period: at time 0 (14:00), 21 h later and 23 h later [37]. Control (Stress-naïve) mice were held, but no injections were performed. The injection location, lighting conditions, and time were kept constant across mice, and the same experimenter administered injections to all mice. This stress protocol was previously shown to disrupt behavior and increase corticosterone levels a few hours after the stress procedure [37]. Here, we assessed behavioral and molecular phenotypes starting a week after the last injection (PND 77). The behavioral assessment consisted of the following tests: nesting, open field, social preference, social recognition, and spatial discrimination (acquisition) and reversal learning in the water T-maze [14, 34]. Mice from each group were randomly selected for mRNA/miRNA expression analysis, and were sacrificed by cervical dislocation on PND 98. A separate cohort of mice underwent the same stress procedure followed by brain removal immediately after the last i.p. injection, for examination of miRNA expression immediately following stress.
Behavioral tests
Behavioral assays are described in detail in the SI.
Briefly, Nesting assesses the ability of male and female rodents to construct a nest in their home environment. Disrupted nesting behavior is a correlate of anhedonia and self-neglect [83]. Nest quality was evaluated by 2 independent, condition-blind raters on a 1–5 scale [84]. The nesting score was calculated as the mean of the raters’ scores.
The Open field test examines spontaneous locomotion, anxiety-like behavior, and the ability to adapt to a novel location [85, 86]. Mice were put in a white unfamiliar plexiglass arena for 60 min. Trials were recorded and analyzed using Ethovision XT14.0 software (Noldus Information Technology, Leeburh, VA). Outcome measures were the distance traveled (cm; total and in each 5-min bin) and the time (sec; total and in each bin) spent in the center of the arena.
The Social preference and recognition tests assess social behavior and memory [87]. Briefly, mice were tested in a 3-chamber arena equipped with clear plexiglass cages. Mice were first tested for their preference of an unfamiliar mouse vs. a novel object. After a 60 min ITI, mice were returned to the arena and tested for recognition of novel vs. familiar social stimuli. Outcome measures were the exploration time of each stimulus, and the preference ratio (time exploring the social/total exploration time) and recognition ratio (time exploring the novel mouse/total exploration time) during the preference and recognition phases, respectively [14, 34]. Trials were recorded and analyzed using Ethovision.
Spatial rule acquisition and reversal learning were assessed in the modified Water T-maze task [88]. Mice were placed in a gray plexiglass T-shaped maze filled with water (25 °C ± 1). Following an arm preference test (3 trials), mice were trained to swim to an underwater platform located in the less-preferred arm, until a criterion of 5 consecutive correct choices was achieved (Acquisition phase). After a 3-min ITI, a probe test was conducted, and the platform was placed in the opposite arm. Mice were tested until a 5 consecutive correct trial criterion was reached (Reversal). Outcome measures for each phase were: (i) the number of trials to criterion, (ii) the reaction time (RT: time (sec) to reach the end of any arm), and (iii) RT variability (RTv: the standard deviation of the differences in RTs between each pair of successive trials, per mouse. A higher number of trials to criterion, longer RTs and higher RTv reflect compromised performance.
mRNA and miRNA expression analysis
Mice were sacrificed 14 days after the end of the behavioral battery (PND 98) by cervical dislocation. The mPFC, consisting of the prelimbic and infralimbic subregions, and the dorsal and ventral CA1 subregions of the hippocampus (dCA1 and vCA1, respectively), were removed bilaterally using 0.5 mm punches [89].
Genome wide RNA-Seq
Full procedural details are provided in the SI. RNA was extracted from 28 mPFC samples (n = 5–9 per group, approximately equal numbers of males and females) as previously described [20, 90], and sent to the Technion Genome Center for genome-wide RNA sequencing (RNA-seq) and bioinformatical analysis. The experiment was comprised of two batches; the first included samples from three of the four groups (C-Cre+/Control, C-Cre+/Stress and C-Glud1+/−/Stress), and the second included new samples from the three aforementioned groups, and added the forth group (C-Glud1+/−/Control). Findings from both batches were combined, taking into account batch and sex effects. RNA was prepared using the SMARTer Stranded Total RNA-Seq Kit v2 – Pico preparation kit according to the manufacturer’s instructions. RNA-seq library preparation, sequencing (using the Illumina HiSeq 2500 sequencer for first batch; Illumina NextSeq 550 for the second), and data analysis was performed by the Technion Genome Center. The quality of the libraries was evaluated using FASTQC (v 0.11.5), quality and adapter trimming was conducted via trim galore (uses cutadapt v 1.10), and mapping was conducted via Tophat2 v 2.1.0, (uses short read aligner Bowtie2 v 2.2.6). At the end of this process, the total reads after trimming ranged between 35–46 million reads per sample. Gene counting was conducted via HTseq-count (v0.6.1). Due to the ribosomal depletion process in the library preparation protocol and the expected ribosomal sequence reads, counting was performed with a modified annotation file which includes the 45 s ribosome annotation for better accuracy. Only counted reads without 45 s were used for the subsequent analysis. Uniquely mapped reads, aligned with high confidence to a single genomic location, ranged between 25–38 million reads per sample. Differential gene expression was performed by DESeq2 (v1.28.1).
MiRNA /mRNA expression analysis
The expression of MiR137-3p, MiR137-5p, MiR34a-5p, MiR203-3p, MiR203-5p, MiR493-3p and MiR493-5p and mRNA of 4 select DEGs were assessed by quantitative reverse transcription PCR (qRT-PCR). Reactions were carried out using a StepOne qRT-PCR system (Applied Biosystems). Fold-change values were calculated using the ∆∆Ct method [91] relative to the housekeeping genes RNU6 and RNU66 (miRNA) or HPRT (mRNA). A mean expression score for all 7 miRNAs was calculated as the mean ∆∆Ct value across miRNAs for each mouse.
Statistical analysis
Full description of statistical tests is provided in the SI. Statistical analyses were performed using SPSS 27 software (IBM SPSS Statistics, New York, United States), Prism 8 (GraphPad software, Inc.) or jamovi v 2.2.5, following normality and homogeneity of variances assumptions verification. Factorial analysis (ANOVA) was applied to behavioral data. Male and female data were analyzed separately in cases of a Sex main effect or interaction with the Sex variable. Significant interactions were followed by LSD post hoc comparisons.
In the RNA-seq experiment, normalization and differential expression analysis of uniquely mapped reads were conducted using the DESeq2 R package (v1.28.1). Three paired comparisons were conducted (C-Cre+/Stress, C-Glud1+/−/Control and C-Glud1+/−/Stress were each compared to C-Cre+/Controls) using negative binomial GLM. An FDR correction [92] was computed (q < 0.05): only genes with an FDR-corrected q value lower than 0.05 were considered statistically significant. Over-representation analysis (ORA), analysis of functional networks of protein-protein interactions, transcript cluster analysis, and correlations were performed (see SI and Results for details).
miRNA and mRNA data were analyzed using non-parametric Kruskal–Wallis tests, with Monte Carlo simulation for exact p values. Dunn tests were used to test post-hoc comparisons. Correlations between transcriptional and behavioral data were FDR-corrected (q < 0.05).
Data availability
The RNA-seq data from this study are available at https://doi.org/10.5061/dryad.6q573n63k.
References
Labrie V, Lipina T, Roder JC. Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Psychopharmacol (Berl). 2008;200:217–230.
Monaco SA, Gulchina Y, Gao W-J. NR2B subunit in the prefrontal cortex: a double-edged sword for working memory function and psychiatric disorders. Neurosci Biobehav Rev. 2015;56:127–38.
Granseth B, Andersson FK, Lindström SH. The initial stage of reversal learning is impaired in mice hemizygous for the vesicular glutamate transporter (VGluT1). Genes Brain Behav. 2015;14:477–85.
Karlsson R-M, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology. 2009;34:1578–89.
Sanderson DJ, Gray A, Simon A, Taylor AM, Deacon RMJ, Seeburg PH, et al. Deletion of glutamate receptor-A (GluR-A) AMPA receptor subunits impairs one-trial spatial memory. Behav Neurosci. 2007;121:559.
Lim J, Kim E, Noh HJ, Kang S, Phillips BU, Kim DG, et al. Assessment of mGluR5 KO mice under conditions of low stress using a rodent touchscreen apparatus reveals impaired behavioural flexibility driven by perseverative responses. Mol Brain. 2019;12:37.
Dienel SJ, Schoonover KE, Lewis DA. Cognitive dysfunction and prefrontal cortical circuit alterations in schizophrenia: developmental trajectories. Biol Psychiatry. 2022;92:450–9.
Bauminger H, Gaisler-Salomon I. Beyond NMDA receptors: homeostasis at the glutamate tripartite synapse and its contributions to cognitive dysfunction in schizophrenia. Int J Mol Sci. 2022;23:8617.
Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–27.
Krystal JH, Abi-Saab W, Perry E, D’Souza DC, Liu N, Gueorguieva R, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl). 2005;179:303–9.
Gandal MJ, Sisti J, Klook K, Ortinski PI, Leitman V, Liang Y, et al. GABAB-mediated rescue of altered excitatory–inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl Psychiatry. 2012;2:e142–e142.
Balschun D, Moechars D, Callaerts-Vegh Z, Vermaercke B, van Acker N, Andries L, et al. Vesicular glutamate transporter VGLUT1 has a role in hippocampal long-term potentiation and spatial reversal learning. Cereb Cortex. 2010;20:684–693.
Chadman KK, Watson DJ, Stanton ME. NMDA receptor antagonism impairs reversal learning in developing rats. Behav Neurosci. 2006;120:1071–83.
Lander SS, Khan U, Lewandowski N, Chakraborty D, Provenzano FA, Mingote S, et al. Glutamate dehydrogenase-deficient mice display schizophrenia-like behavioral abnormalities and CA1-specific hippocampal dysfunction. Schizophr Bull. 2019;45:127–37.
Aoki C, Milner TA, Sheu KF, Blass JP, Pickel VM. Regional distribution of astrocytes with intense immunoreactivity for glutamate dehydrogenase in rat brain: implications for neuron-glia interactions in glutamate transmission. J Neurosci. 1987;7:2214–31.
Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology (Basel). 2017;6:11.
Nakahara T, Tsugawa S, Noda Y, Ueno F, Honda S, Kinjo M, et al. Glutamatergic and GABAergic metabolite levels in schizophrenia-spectrum disorders: a meta-analysis of 1H-magnetic resonance spectroscopy studies. Mol Psychiatry. 2022;27:744–57.
Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938.
Schulz J, Zimmermann J, Sorg C, Menegaux A, Brandl F. Magnetic resonance imaging of the dopamine system in schizophrenia—a scoping review. Front Psychiatry. 2022;13, https://doi.org/10.3389/fpsyt.2022.925476.
Lander SS, Chornyy S, Safory H, Gross A, Wolosker H, Gaisler-Salomon I. Glutamate dehydrogenase deficiency disrupts glutamate homeostasis in hippocampus and prefrontal cortex and impairs recognition memory. Genes Brain Behav. 2020;e12636.
Schoofs D, Preuß D, Wolf OT. Psychosocial stress induces working memory impairments in an n-back paradigm. Psychoneuroendocrinology. 2008;33:643–53.
Zhang F, Yuan S, Shao F, Wang W. Adolescent social defeat induced alterations in social behavior and cognitive flexibility in adult mice: effects of developmental stage and social condition. Front Behav Neurosci. 2016;10, https://doi.org/10.3389/fnbeh.2016.00149.
Reznikov LR, Grillo CA, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J. Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci. 2007;25:3109–14.
Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience. 1997;77:65–73.
Matsumoto Y, Niwa M, Mouri A, Noda Y, Fukushima T, Ozaki N, et al. Adolescent stress leads to glutamatergic disturbance through dopaminergic abnormalities in the prefrontal cortex of genetically vulnerable mice. Psychopharmacology (Berl). 2017;234:3055–74.
Page CE, Alexander J, Shepard R, Coutellier L. Npas4 deficiency interacts with adolescent stress to disrupt prefrontal GABAergic maturation and adult cognitive flexibility. Genes Brain Behav. 2018;17:e12459.
Gulyaeva NV. Glucocorticoid regulation of the glutamatergic synapse: mechanisms of stress-dependent neuroplasticity. J Evol Biochem Physiol. 2021;57:564–576.
Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13:22–37.
Jett JD, Bulin SE, Hatherall LC, McCartney CM, Morilak DA. Deficits in cognitive flexibility induced by chronic unpredictable stress are associated with impaired glutamate neurotransmission in the rat medial prefrontal cortex. Neuroscience. 2017;346:284–97.
Gaisler-Salomon I, Lander SS, Chakraborty D, Asraf K, Safory H, Wolosker H, et al. 21.3 Neuronal-astrocytic regulation of glutamate homeostasis: relevance to cognitive dysfunction in schizophrenia. Schizophr Bull. 2019;45:S124–S124.
Salazar RF, White W, Lacroix L, Feldon J, White IM. NMDA lesions in the medial prefrontal cortex impair the ability to inhibit responses during reversal of a simple spatial discrimination. Behav Brain Res. 2004;152:413–24.
Niu L, Luo SS, Xu Y, Wang Z, Luo D, Yang H, et al. The critical role of the hippocampal NLRP3 inflammasome in social isolation-induced cognitive impairment in male mice. Neurobiol Learn Mem. 2020;175:107301.
Chen W, An D, Xu H, Cheng X, Wang S, Yu W, et al. Effects of social isolation and re-socialization on cognition and ADAR1 (p110) expression in mice. PeerJ. 2016;4:e2306.
Lander SS, Linder-Shacham D, Gaisler-Salomon I. Differential effects of social isolation in adolescent and adult mice on behavior and cortical gene expression. Behav Brain Res. 2017;316:245–54.
Shao Y, Yan G, Xuan Y, Peng H, Huang Q-J, Wu R, et al. Chronic social isolation decreases glutamate and glutamine levels and induces oxidative stress in the rat hippocampus. Behav Brain Res. 2015;282:201–8.
Almeida-Santos AF, Carvalho VR, Jaimes LF, de Castro CM, Pinto HP, Oliveira TPD, et al. Social isolation impairs the persistence of social recognition memory by disturbing the glutamatergic tonus and the olfactory bulb-dorsal hippocampus coupling. Sci Rep. 2019;9:473.
Advani T, Koek W, Hensler JG. Gender differences in the enhanced vulnerability of BDNF+/− mice to mild stress. Int J Neuropsychopharmacol. 2009;12:583–8.
Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93–103.
Cummings E, Donohoe G, Hargreaves A, Moore S, Fahey C, Dinan TG, et al. Mood congruent psychotic symptoms and specific cognitive deficits in carriers of the novel schizophrenia risk variant at MIR-137. Neurosci Lett. 2013;532:33–38.
Cosgrove D, Harold D, Mothersill O, Anney R, Hill MJ, Bray NJ, et al. MiR-137-derived polygenic risk: effects on cognitive performance in patients with schizophrenia and controls. Transl Psychiatry. 2017;7:e1012–e1012.
Lee S-T, Jeon D, Chu K, Jung K-H, Moon J, Sunwoo J, et al. Inhibition of miR-203 reduces spontaneous recurrent seizures in mice. Mol Neurobiol. 2017;54:3300–8.
Zhou M, Wang M, Wang X, Liu K, Wan Y, Li M, et al. Abnormal expression of MicroRNAs induced by chronic unpredictable mild stress in rat hippocampal tissues. Mol Neurobiol. 2018;55:917–35.
Gullett JM, Chen Z, O’Shea A, Akbar M, Jiang B, Rani A, et al. Micro RNA predicts cognitive performance in healthy older adults. Neurobiol Aging. 2020, https://doi.org/10.1016/j.neurobiolaging.2020.07.023.
Haramati S, Navon I, Issler O, Ezra-Nevo G, Gil S, Zwang R, et al. MicroRNA as repressors of stress-induced anxiety: the case of amygdalar miR-34. J Neurosci. 2011;31:14191–203.
Li S, Li L, Li J, Liang X, Song C, Zou Y. miR-203, fine-tunning neuroinflammation by juggling different components of NF‐κB signaling. J Neuroinflammation. 2022;19:84.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013;14:128.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7.
Cherlyn SYT, Woon PS, Liu JJ, Ong WY, Tsai GC, Sim K. Genetic association studies of glutamate, GABA and related genes in schizophrenia and bipolar disorder: a decade of advance. Neurosci Biobehav Rev. 2010;34:958–77.
Franzén O, Gan L-M, Björkegren JLM PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019, https://doi.org/10.1093/database/baz046.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613.
Dzirasa K, Ramsey AJ, Takahashi DY, Stapleton J, Potes JM, Williams JK, et al. Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J Neurosci. 2009;29:8215–24.
Ramsey AJ. NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog Brain Res. 2009;179:51–8.
Liu X, Li J, Guo C, Wang H, Sun Y, Wang H, et al. Olanzapine reverses MK-801-induced cognitive deficits and region-specific alterations of NMDA receptor subunits. Front Behav Neurosci. 2018;11, https://doi.org/10.3389/fnbeh.2017.00260.
Watson DJ, Stanton ME. Medial prefrontal administration of MK-801 impairs T-maze discrimination reversal learning in weanling rats. Behavioural Brain Res. 2009;205:57–66.
Li J-T, Su Y-A, Wang H-L, Zhao Y-Y, Liao X-M, Wang X-D, et al. Repeated blockade of NMDA receptors during adolescence impairs reversal learning and disrupts GABAergic interneurons in rat medial prefrontal cortex. Front Mol Neurosci. 2016;9, https://doi.org/10.3389/fnmol.2016.00017.
Joffe ME, Santiago CI, Stansley BJ, Maksymetz J, Gogliotti RG, Engers JL, et al. Mechanisms underlying prelimbic prefrontal cortex mGlu3/mGlu5-dependent plasticity and reversal learning deficits following acute stress. Neuropharmacology. 2019;144:19–28.
Andersen SL, Teicher MH. Stress, sensitive periods and maturational events in adolescent depression. Trends Neurosci. 2008;31:183–91.
Gaisler-Salomon I, Miller GM, Chuhma N, Lee S, Zhang H, Ghoddoussi F, et al. Glutaminase-deficient mice display hippocampal hypoactivity, insensitivity to pro-psychotic drugs and potentiated latent inhibition: relevance to schizophrenia. Neuropsychopharmacology. 2009;34:2305–22.
Mellis A-T, Misko AL, Arjune S, Liang Y, Erdélyi K, Ditrói T, et al. The role of glutamate oxaloacetate transaminases in sulfite biosynthesis and H2S metabolism. Redox Biol. 2021;38:101800.
Bunik V, Kaehne T, Degtyarev D, Shcherbakova T, Reiser G. Novel isoenzyme of 2-oxoglutarate dehydrogenase is identified in brain, but not in heart. FEBS J. 2008;275:4990–5006.
Graf A, Trofimova L, Loshinskaja A, Mkrtchyan G, Strokina A, Lovat M, et al. Up-regulation of 2-oxoglutarate dehydrogenase as a stress response. Int J Biochem Cell Biol. 2013;45:175–89.
Babenko VN, Smagin DA, Kovalenko IL, Galyamina AG, Kudryavtseva NN. Differentially expressed genes of the Slc6a family as markers of altered brain neurotransmitter system function in pathological states in mice. Neurosci Behav Physiol. 2020;50:199–209.
Singer D, Camargo SMR, Huggel K, Romeo E, Danilczyk U, Kuba K, et al. Orphan transporter SLC6A18 is renal neutral amino acid transporter B0AT3. J Biol Chem. 2009;284:19953–60.
Kinlein SA, Wallace NK, Savenkova MI, Karatsoreos IN. Chronic hypothalamic-pituitary-adrenal axis disruption alters glutamate homeostasis and neural responses to stress in male C57Bl6/N mice. Neurobiol Stress. 2022;19:100466.
Egerton A, Grace AA, Stone J, Bossong MG, Sand M, McGuire P. Glutamate in schizophrenia: Neurodevelopmental perspectives and drug development. Schizophr Res. 2020;223:59–70.
Zeppillo T, Schulmann A, Macciardi F, Hjelm BE, Föcking M, Sequeira PA, et al. Functional impairment of cortical AMPA receptors in schizophrenia. Schizophr Res. 2022;249:25–37.
Maycox PR, Kelly F, Taylor A, Bates S, Reid J, Logendra R, et al. Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol Psychiatry. 2009;14:1083–94.
Yu Z, Ueno K, Funayama R, Sakai M, Nariai N, Kojima K, et al. Sex-specific differences in the transcriptome of the human dorsolateral prefrontal cortex in schizophrenia. Mol Neurobiol. 2023;60:1083–98.
Fessel J. Abnormal oligodendrocyte function in schizophrenia explains the long latent interval in some patients. Transl Psychiatry. 2022;12:120.
Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–80.
Takahashi N, Sakurai T, Davis KL, Buxbaum JD. Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog Neurobiol. 2011;93:13–24.
Maas DA, Eijsink VD, Spoelder M, van Hulten JA, de Weerd P, Homberg JR, et al. Interneuron hypomyelination is associated with cognitive inflexibility in a rat model of schizophrenia. Nat Commun. 2020;11:2329.
Zhang T-Y, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 2010;30:13130–37.
Bagot RC, Tse YC, Nguyen H-B, Wong AS, Meaney MJ, Wong TP. Maternal care influences hippocampal N-methyl-D-aspartate receptor function and dynamic regulation by corticosterone in adulthood. Biol Psychiatry. 2012;72:491–8.
Mitchell AC, Javidfar B, Bicks LK, Neve R, Garbett K, Lander SS, et al. Longitudinal assessment of neuronal 3D genomes in mouse prefrontal cortex. Nat Commun. 2016;7:12743.
Nasca C, Zelli D, Bigio B, Piccinin S, Scaccianoce S, Nisticò R, et al. Stress dynamically regulates behavior and glutamatergic gene expression in hippocampus by opening a window of epigenetic plasticity. Proc Natl Acad Sci. 2015;112:14960–65.
Reshetnikov VV, Kisaretova PE, Ershov NI, Shulyupova AS, Oshchepkov DYU, Klimova NV, et al. Genes associated with cognitive performance in the Morris water maze: an RNA-seq study. Sci Rep. 2020;10:22078.
Liu Y, Ding M, Zhang X, Liu Y, Xuan J, Xing J, et al. Association between polymorphisms in the GRIN1 gene 5′ regulatory region and schizophrenia in a northern Han Chinese population and haplotype effects on protein expression in vitro. BMC Med Genet. 2019;20:26.
Savushkina OK, Tereshkina EB, Prokhorova TA, Boksha IS, Burminskii DS, Vorobyeva EA, et al. Platelet glutamate dehydrogenase activity and efficacy of antipsychotic therapy in patients with schizophrenia. J Med Biochem. 2020;39:54–9.
Karl T, Arnold JC. Schizophrenia: a consequence of gene-environment interactions? Front Behav Neurosci. 2014;8:435.
Gunnell D, Harrison G, Rasmussen F, Fouskakis D, Tynelius P. Associations between premorbid intellectual performance, early-life exposures and early-onset schizophrenia: cohort study. Br J Psychiatry. 2002;181:298–305.
Faul F, Erdfelder E, Buchner A, Lang A-G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav Res Methods. 2009;41:1149–60.
Pedersen CS, Sørensen DB, Parachikova AI, Plath N. PCP-induced deficits in murine nest building activity: employment of an ethological rodent behavior to mimic negative-like symptoms of schizophrenia. Behav. Brain Res. 2014;273:63–72.
Deacon RMJ. Assessing nest building in mice. Nat Protoc. 2006;1:1117–9.
Choleris E, Thomas AW, Kavaliers M, Prato FS. A detailed ethological analysis of the mouse open field test: effects of diazepam, chlordiazepoxide and an extremely low frequency pulsed magnetic field. Neurosci Biobehav Rev. 2001;25:235–60.
Archer J. Tests for emotionality in rats and mice: a review. Anim Behav. 1973;21:205–35.
Lee E, Rhim I, Lee JW, Ghim J-W, Lee S, Kim E, et al. Enhanced neuronal activity in the medial prefrontal cortex during social approach behavior. J Neurosci. 2016;36:6926–36.
Ayalon L, Doron R, Weiner I, Joel D. Amelioration of behavioral deficits in a rat model of Huntington’s disease by an excitotoxic lesion to the globus pallidus. Exp Neurol. 2004;186:46–58.
Keith BJ, Franklin MA, Paxinos G. The mouse brain in stereotaxic coordinates, compact: the coronal plates and diagrams. Amsterdam: Elsevier Science; 2008.
Zaidan H, Galiani D, Gaisler-Salomon I. Pre-reproductive stress in adolescent female rats alters oocyte microRNA expression and offspring phenotypes: pharmacological interventions and putative mechanisms. Transl Psychiatry. 2021;11:113.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc: Ser B (Methodol). 1995;57:289–300.
Acknowledgements
The authors thank Hagar Bauminger and Dr. Maya Lalzar for helpful comments on the manuscript and data analysis, respectively. This work was made possible by grant support from the Israel Science Foundation (ISF; 1481/20) and from the ISF-NSFC (2401/18) to IGS.
Author information
Authors and Affiliations
Contributions
KA and IGS made a significant contribution to conceptualization, study design, data collection, statistical analysis and interpretation of results, as well as to drafting, writing, and critically reviewing the article. HZ made a significant contribution to data collection and interpretation, as well as manuscript editing. BN made a significant contribution to data collection.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Asraf, K., Zaidan, H., Natoor, B. et al. Synergistic, long-term effects of glutamate dehydrogenase 1 deficiency and mild stress on cognitive function and mPFC gene and miRNA expression. Transl Psychiatry 13, 248 (2023). https://doi.org/10.1038/s41398-023-02534-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-023-02534-y
- Springer Nature Limited