
Abstract
Older adults with major depressive disorder (MDD) or early cognitive decline during the subjective cognitive decline (SCD) stage may exhibit neuropsychiatric symptoms such as anxiety, depression, and subtle cognitive impairment. The clinicopathological features and biological mechanisms of MDD differ from those of SCD among older adults; these conditions thus require different treatment strategies. This study enrolled 82 participants above 50 years old with normal cognitive levels from the communities to examine biomarker–behavior correlations between MDD (n = 23) and SCD (n = 23) relative to a normal control (NC) group (n = 36). Multidomain assessments were performed for all participants, including immunomagnetic reduction tests to detect plasma beta-amyloid (Aβ), total tau (Tau), phosphorylated tau-181 (p-Tau181), neurofilament light chain, and glial fibrillary acidic protein (GFAP). This study observed that depressive symptoms in MDD were associated with amyloid pathology (plasma Aβ40 vs. HADS-D: R = 0.45, p = 0.031; Aβ42/Aβ40 vs. HADS-D: R = −0.47, p = 0.024), which was not observed in the NC (group difference p < 0.05). Moreover, cognitive decline in MDD was distinguished by a mixed neurodegenerative process involving amyloid (plasma Aβ42 vs. facial memory test: R = 0.48, p = 0.025), tau (Tau/Aβ42 vs. digit symbol substitution test (DSST): R = −0.53, p = 0.01), and astrocytic injury (plasma GFAP vs. Montreal cognitive assessment score: R = −0.44, p = 0.038; plasma GFAP vs. DSST: R = −0.52, p = 0.014), findings that did not apply to the NC (group difference p < 0.05). Moreover, this study revealed different biomarker–behavior correlations between individuals with SCD and the NC. Compared with the NC, cognitive decline in the SCD group might be unrelated to amyloid pathology and instead might be early manifestations of tau pathology. This study underscores the difference in clinicopathological features between MDD and SCD among older adults, which differ from those of the NC. These findings enhance our understanding of the mechanisms underlying MDD and SCD in older individuals.
Similar content being viewed by others

Introduction
Overlap of depression and cognitive decline in older individuals
Depression, anxiety, and cognitive decline are frequently experienced by older individuals, both in cases of late-life depression and preclinical dementia [1, 2] Cognitive complaints and depressive symptoms frequently occur together [3], and anxiety and depression increase with the degree of subjective cognitive problems [4]. Thus, distinguishing between late-life depression and early dementia is challenging.
For older individuals, cognitive problems can be the first sign of depressive disorders [5] and resemble dementia, a phenomenon known as “pseudodementia” [6, 7]. Older individuals experiencing depression may encounter difficulties with attention, executive function, and working memory [8]. Moreover, they exhibit a lack of motivation to engage with cognitive dysfunction, experience distress due to impaired cognitive activity, and frequently respond with “I don’t know” on cognitive assessments [9]. These cognitive problems can assist in identifying depressive disorders in older individuals [10]. However, depressive symptoms may also precede dementia [11,12,13]. Approximately 20% of patients with mild cognitive impairment (MCI) have depressive symptoms [14], and those patients with MCI who experience depression are at a greater risk of developing Alzheimer’s disease (AD) [15].
Distinguishing late-onset depression from early cognitive impairment in older individuals based on clinical symptoms is challenging. In the preclinical stage of dementia, also referred to as subjective cognitive decline (SCD), individuals may experience cognitive problems but maintain normal performance on standard cognitive assessments [16, 17]. Increased depression and subtle cognitive decline characterize individuals with SCD compared with individuals who do not experience SCD [1, 4]. Moreover, cognitive problems and depression symptoms are also evident in older individuals with major depressive disorder (MDD) [18]. Although cognitive and depressive symptoms often co-occur, studies on SCD have typically concentrated on either cognition or depression; few have examined them in conjunction [19]. Furthermore, biomarker research has rarely explored the early preclinical stage of AD. Although positron emission tomography has been used to study cognitively normal older individuals with depression [20,21,22,23] or subjective cognitive problems [24,25,26], a parallel design has not been used to differentiate between these clinically similar conditions. Finally, although several studies on the molecular pathogenesis of these conditions have employed biofluid neurodegenerative biomarkers to examine MDD [27,28,29,30,31] or SCD [32,33,34], the distinct mechanisms underlying the conditions remain poorly understood. Therefore, the present study examined groups of individuals with MDD, SCD, or no symptoms (healthy controls) to investigate blood biomarkers and behavioral data to improve understanding of the clinicopathological features of these two disorders, which often present with similar symptoms.
Panels of blood biomarkers in neuropsychiatric diseases
Beta-amyloid (Aβ), tau protein, neurofilament light chain (NfL), and glial fibrillary acidic protein (GFAP) are neurodegenerative proteins associated with neuropsychiatric disorders. These proteins are biomarkers for neuron injury, glial cell dysfunction, pathological protein deposition, and brain waste clearance processes. They enter the blood following release from brain tissues into the cerebrospinal fluid (CSF) and are transported to the peripheral blood [35,36,37]. Owing to the trace amounts of these proteins in the blood, specialized techniques such as single molecule array and immunomagnetic reduction (IMR) are required to measure them accurately [38]. In addition, studying of neuropsychiatric diseases involve combinations of biomarkers instead of a single marker to offer a more comprehensive understanding of disease mechanisms [39,40,41].
Most neurodegenerative biomarkers are essential proteins of nervous systems that are released or aggregated during neurodegeneration. For example, amyloid precursor protein family members are crucial for neural development, synaptogenesis, synaptic function, and axonal growth [42], and Aβ protein is vital to neuronal growth and survival [43]. However, brain amyloid deposition and abnormal blood Aβ levels are associated with the pathogenesis of AD [44, 45], cerebral amyloid angiopathy [46], normal aging [44, 47], and major depression [29].
Tau is a protein associated with microtubules; its aggregation and deposition can cause neurodegenerative diseases. Tauopathies are categorized into two types: primary and secondary [48]. Primary tauopathies comprise progressive supranuclear palsy, corticobasal degeneration, and frontotemporal dementia. Secondary tauopathies develop as AD progresses when tau proteins aggregate in response to amyloid deposition, forming neurofibrillary tangles that spread from the entorhinal cortex throughout the brain [49].
Neurofilaments are fundamental proteins for axon construction, and NfL is their most abundant and soluble component. Upon axonal injury, NfL is released into both the CSF and blood in stable forms, rendering it a dependable biomarker for detecting axonal injury in various neurological diseases, such as AD, Parkinson’s disease, traumatic brain injury, and multiple sclerosis [50].
GFAP is a specific structural protein of astrocytes and has served as the classical marker of astrocytes in both in vitro and in vivo studies [51]. Blood levels of GFAP increase in response to the development of reactive astrogliosis in the brain. For example, an elevated plasma GFAP level is associated with brain Aβ deposition along the AD continuum [52].
These biomarkers have varied molecular bases. In the present study, combinations of biomarkers were employed to address pathological changes in the brains of older individuals with MDD and SCD and to distinguish these two conditions from similar clinical presentations. This study employed ultrasensitive nanotechnology, specifically IMR, to produce plasma biomarker panels [38, 53]. The study employed multidomain assessments encompassing mental, cognitive, physical, and fatigue evaluations. By examining biomarker–behavior correlations in individuals with MDD and SCD relative to healthy controls, this study identified unique clinicopathological features of the neuropsychiatric symptoms of MDD and SCD in older individuals.
Materials and methods
Participants
The participants were enrolled in the Northeastern Taiwan Community Medicine Research Cohort (NTCMRC; identifier NCT04839796 on ClinicalTrials.gov) from 2020 to 2022, which was managed by The Community Medicine Research Center of the Chang Gung Memorial Hospital in Keelung, Taiwan. The Institutional Review Board of Chang Gung Memorial Hospital approved this study (approval nos. 201901350B0, 201901353B0, 201901352B0, and 201800289A3). All the participants were well informed of the study protocol and completed a signed informed consent before entering the study. All methods were performed in accordance with the Declaration of Helsinki.
All the participants were aged over 50 years. Those with major organ failure, including severe heart, kidney, and liver diseases and thyroid diseases, were not recruited. Those with abnormal objective cognitive levels on standard cognitive tests [54], falling within the criteria for MCI [55, 56], dementia [57], or having a history of stroke, epilepsy, brain tumor, traumatic brain injury, cranial surgery, or neurodevelopmental disorders were excluded.
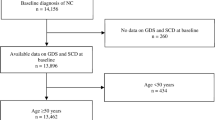
All the participants underwent the Mini-International Neuropsychiatric Interview [58], which allowed for the identification of those with MDD. The participants without psychiatric diseases were queried for subjective cognitive complaints using an AD8 questionnaire [59, 60]. An AD8 cutoff score of 2 points was further utilized to divide the psychiatric-normal participants into SCD and normal control (NC) (Fig. 1). This SCD group fulfilled the criteria of SCD definition, which included (1) self-perceiving of persistent cognitive decline when compared with previous cognitive level, (2) unimpaired objective cognitive performance, (3) being free from psychiatric diseases, and (4) subjective cognitive complaints not relevant to acute events of brain insults [17, 61].

Participants over 50 were included in the study, but only those without major organ failure (such as heart, renal, or liver failure) and unimpaired cognitive function were included. Using the Mini-International Neuropsychiatric Interview (MINI), participants with MDDs were segmented into an MDD group. Those without psychiatric disorders but with subjective cognitive problems (AD8 score ≥ 2) were segmented into an SCD group, whereas those without such problems were segmented into an NC group.
Multidomain assessments
Multidomain assessments, including blood biomarkers, mental states (depression and anxiety), cognition, physical activity, and fatigability, were utilized to conduct associative studies.
The mental assessments were performed using self-reported depression and anxiety in the hospital anxiety and depression scale (HADS); it consisted of seven questions for depression and another seven for anxiety, which were subtotaled into two subscales: the anxiety subscale (HADS-A) and the depression subscale (HADS-D). The HADS questionnaire rates depression and anxiety based solely on mental symptoms, avoiding including physical factors that may affect these conditions. The scores for each subscale ranged from 0 to 21 points, with a higher score indicating more severe anxiety or depression [62]. In addition, a proficient researcher evaluated the Hamilton depression rating scale (Ham-D) [63] and the Hamilton anxiety rating scale (Ham-A) [64]. The assessment measured both physical and psychological symptoms of depression and anxiety. A higher score indicated a greater likelihood of experiencing these conditions.
The cognitive assessments first used the Montreal Cognitive Assessment (MoCA) to determine the normality of objective cognition level. A MoCA score lower than one standard deviation below the mean of age- and education-adjusted norms was considered an impairment in objective cognitive performance [54]. Additionally, a comprehensive cognitive evaluation was conducted by a set of tests, including the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-cog) for global cognition [65, 66]; other tests for specific cognitive domains were the facial memory test (FMT), digit symbol substitution test (DSST), digit span test (DST), letter-number sequencing (LNS), and category fluency (CF) [67, 68].
To assess physical activity levels, we used the International Physical Activity Questionnaire Short Form (IPAQ-SF). This questionnaire asked participants to report their physical activity in four categories: vigorous-intensity activity, moderate-intensity activity, walking, and sitting. We then calculated the metabolic equivalent of task (MET) in minutes per week based on the duration and intensity of these activities. Finally, we classified participants’ daily physical activity levels as high, moderate, or low based on their IPAQ-SF responses [69].
The Pittsburgh Fatigability Scale (PFS) assessed fatigue levels in older adults at risk of mobility decline. This scale was established in 2015 and translated and validated into traditional Chinese in Taiwan. The PFS measures mental and physical fatigue through a 10-item performance-based questionnaire covering social, sedentary, lifestyle, light-intensity, and moderate to high-intensity activities [70, 71].
Immunomagnetic reduction (IMR) to detect plasma biomarkers
The IMR technology uses magnetic nanobeads to detect trace amounts of neurodegenerative proteins at the picogram level in plasma [72]. It leverages the magnetic susceptibility reduction upon antigen (the target protein) and antibody (precoated on magnetic nanobeads) reaction. A superconducting quantum interference device (SQUID) detects the changes in an alternating current field. The level of magnetic susceptibility reduction is linked to the number of bound proteins and can be converted into a concentration measurement using a standard curve for reference [53, 73].
To conduct the IMR experiment, plasma was first separated from peripheral blood and stored at −80 °C. During the IMR reaction, 60 μl of plasma was mixed with compatible IMR reagents that contained magnetic nanobeads pre-coated with antibodies for Aβ42, Aβ40, total tau (Tau), phosphorylated tau-181 (p-Tau181), NfL, and GFAP, respectively. The assay was performed using an IMR analyzer (XacPro-S, MagQu, Taiwan), and each measurement was duplicated to obtain an averaged quantitation of protein concentration in the samples.
Statistical analysis
Pearson’s correlation analysis evaluated the associations between biomarkers and behavior data, and the correlation matrix was corrected with a false discovery rate (FDR). To analyze group differences in biomarker and behavior data, qi-square statistics were used to compare categorical parameters, and independent t-tests were used to process continuous variables. In addition, to compare the biomarker-behavior correlations between groups, the data were processed by z-score normalization and used in a general linear model (GLM). A p-value < 0.05 was considered statistical significance. The analysis was performed on R (Bell Laboratories, Lucent Technologies, NJ, USA).
Results
Participant demographics
The final enrollment was 82 participants, including 23 MDD, 23 SCD, and 36 NC (Fig. 1). Compared with NC, the MDD and SCD groups had no difference in demographics, including age (66.91 ± 3.10 in MDD, 68.78 ± 6.07 in SCD, 67.17 ± 3.03 in NC; MDD vs. NC p = 0.757 and SCD vs. NC p = 0.246), sex (female ratio 82.6%, 43.5%, 61.1%; p = 0.080 and 0.185), and education (10.87 ± 2.17, 10.91 ± 4.21, 10.33 ± 3.44 years; p = 0.554 and 0.565) (Table 1A). The blood biomarker panel including Aβ42, Aβ40, Tau, p-Tau181, NfL, GFAP, Aβ42/Aβ40 ratio, Tau/Aβ42 ratio, and p-Tau181/Aβ42 ratio did not differ between MDD vs. NC (all p > 0.05) and SCD vs. NC (all p > 0.05) (Table 1B).
In comparison to NC, individuals with MDD and SCD exhibited higher anxiety (HADS-A 9.00 ± 4.05 in MDD, 5.65 ± 4.31 in SCD, 2.81 ± 2.70 in NC; MDD vs. NC p < 0.001 and SCD vs. NC p = 0.003) and depression tendency (HADS-D 7.91 ± 4.03, 5.22 ± 4.26, 2.81 ± 2.74; p < 0.001 and 0.021; Table 1C), more subjective cognitive complaints (AD8 score 3.13 ± 0.97, 3.91 ± 1.95, 0.17 ± 0.38; p < 0.001 and <0.001), and lower cognitive performance in categorical fluency (CF-color 11.95 ± 2.87, 12.35 ± 4.18, 15.14 ± 3.77; p = 0.001 and 0.010; Table 1D). Physical activity was not different between groups (Table 1E). Besides, the MDD showed higher fatigue levels in mental and physical fatigability than the NC (PFS physical total score 22.59 ± 8.38 and 15.26 ± 10.10, p = 0.011; mental total score 21.05 ± 8.56 and 11.64 ± 8.96, p < 0.001); no significant difference was observed between SCD and NC (Table 1F).
Correlation matrix of biomarker–behavior data
For a general trend of the biomarker–behavior relationship, a correlation matrix displayed the correlations of multidomain assessments and blood biomarker panels for all enrolled participants (Fig. 2). Significant biomarker–behavior correlations were found in plasma Aβ42 and CF-fruit, plasma Aβ40 and MoCA and forward DST, Aβ42/Aβ40 ratio and MoCA and forward DST (FDR-corrected p < 0.05). The individual correlation matrices of each group can be found in Supplementary Fig. S1.

Lower plasma Aβ42 level is associated with more impaired cognitive performance in category fluency (CF-fruit). Higher plasma Aβ40 level and lower Aβ42/Aβ40 ratio are associated with lower global cognition (MoCA score) and worse working memory in the forward digit span test (DST-f). Additionally, cognitive tests have strong and consistent correlations with each other. Furthermore, anxiety and depression are positively associated with physical and mental fatigue. Higher mental fatigue is associated with lower cognitive performance in cognitive processing speed (DSST), and semantic fluency (CF-animal, color, and city), in addition to lower physical activity (IPAQ-SF MET). The matrix presents the correlation coefficients exceeding the false discovery rate (FDR) correction. Supplementary Fig. S1 depicts the correlation matrices of individual MDD, SCD, and NC groups.
Biomarker–behavior correlations between MDD and NC
Further comparison of the biomarker–behavior correlations between MDD and NC showed significant differences in mental, cognitive, and physical dimensions.
The relationship of depression tendency (HADS-D) with plasma Aβ40 (R = 0.45, p = 0.031 in MDD vs. R = 0.072, p = 0.67 in NC) and Aβ42/Aβ40 ratio (R = −0.47, p = 0.024 in MDD vs. R = −0.16, p = 0.34 in NC) was significant in MDD but insignificant in NC (group difference p = 0.005 and 0.005; Fig. 3A).

Compared with the NC, participants in the MDD group exhibited a more pronounced association between blood amyloid levels and depression tendency, as indicated by the positive association of plasma Aβ40 with HADS-D (group difference p = 0.005; A left) and the negative association of the Aβ42/Aβ40 ratio with HADS-D (group difference p = 0.005; A right). Furthermore, the associations between biomarkers and cognition in the MDD group differed from those in the NC, showing a stronger positive association between plasma Aβ42 and facial recognition (FMT, p = 0.034; B left-upper) and a stronger negative association between the Tau/Aβ42 ratio and cognitive processing in coding compared with NC individuals (DSST, p = 0.030; B right-upper). Additionally, higher plasma GFAP levels were associated with poorer cognitive performance in MoCA (p = 0.039; B left-lower) and DSST (p = 0.012; B right-lower), an association not observed in NC participants. Notably, MDD participants engaging in more physical activity, indicated by a higher metabolic equivalent, exhibited higher plasma p-Tau181 levels (p = 0.014; C left) and a higher p-Tau181/Aβ42 ratio (p = 0.011; C right). Moreover, plasma biomarkers did not exhibit differential associations with total PFS mental and physical fatigue scores between the MDD and NC groups.
There were significant differences in the correlations between biomarkers and cognition. MDD patients with a lower plasma Aβ42 level had impaired facial recognition ability (plasma Aβ42 vs. FMT: R = 0.48, p = 0.025); this correlation is insignificant in NC individuals (p = 0.095; group difference p = 0.034). In MDD, a higher Tau/Aβ42 ratio was associated with worse cognitive performance in the DSST (Tau/Aβ42 vs. DSST: R = −0.53, p = 0.01), while there was no such correlation in NC (p = 0.94; group difference p = 0.030). Additionally, the MDD group exhibited stronger negative correlations between plasma GFAP and MoCA score (R = −0.44, p = 0.038) and DSST score (R = −0.52, p = 0.014) than the insignificant correlations in the NC group (p = 0.62 and 0.19) (group difference p = 0.039 and 0.012; Fig. 3B).
In MDD, higher physical activity in metabolic equivalent was related to higher plasma p-Tau181 (R = 0.49, p = 0.020) and p-Tau181/Aβ42 ratio (R = 0.50, p = 0.017). However, these correlations were insignificant in NC (p = 0.89 and 0.92) and differed from those in MDD (group difference p = 0.014 and 0.011; Fig. 3C).
When studying the correlation between fatigue levels and blood biomarkers, no significant differences in the correlations of biomarkers and PFS sum scores of mental and physical fatigability were noticed between MDD and NC (all p > 0.05).
Biomarker–behavior correlations between SCD and NC
The results exhibited a negative correlation between anxiety and plasma p-Tau181 level in NC (plasma p-Tau181 vs. Ham-A: R = −0.37, p = 0.024); however, this correlation was not observed in SCD (R = −0.19, p = 0.37) (group difference p = 0.044; Fig. 4A).

The biomarker–mental health correlation analysis comparing individuals with SCD and NC revealed a negative correlation between anxiety and plasma p-Tau181 in NC but not in those with SCD (group difference p = 0.044; A). In biomarker–cognition correlation studies, a trend of higher plasma Tau being associated with superior semantic fluency (CF-animal and fruit) in NC had the opposite direction among those with SCD; however, the correlations for those with SCD were not statistically significant (group differences all p < 0.05; B). Moreover, the negative correlation of plasma Aβ40 and the positive correlation of Aβ42/Aβ40 ratio with semantic fluency (CF-animal and color) and working memory (DST-f) were present in the NC group but not in the SCD group (group differences all p < 0.05; C). Additionally, no differences were observed between the SCD and NC groups in terms of correlations between physical biomarkers and fatigability (all group differences p > 0.05).
Biomarker-cognition analysis revealed that NC exhibited a trend of higher plasma Tau correlating with better cognitive performance in category fluency (plasma Tau vs. CF-animal: R = 0.34, p = 0.040; plasma Tau vs. CF-fruit: R = 0.35, p = 0.037). In contrast, the tau-cognition trend is reversed but insignificant in SCD (CF-animal: R = −0.37, p = 0.082; CF-fruit: R = −0.079, p = 0.72) (group difference all p < 0.05; Fig. 4B).
In addition, those in the NC group with an elevated plasma Aβ40 level and lower Aβ42/Aβ40 ratio had poorer semantic fluency (CF-animal and color) and worse working memory performance in DST-f (all p < 0.05). This trend was not observed in the SCD participants (all p > 0.05) (group differences all p < 0.05; Fig. 4C).
Besides, there was no noticeable difference regarding the biomarker–physical and biomarker–fatigability relationship between SCD and NC (group difference all p > 0.05).
Discussion
In summary, this study provides insights into the biological mechanisms behind the similar behaviors observed in older individuals experiencing depression and cognitive decline. The use of biological approaches to delineate neuropsychiatric diseases is now standard practice. The National Institute on Aging-Alzheimer’s Association (NIA-AA) published an updated diagnostic framework for AD in 2018, ushering in a new era of biological definitions for neurodegenerative diseases [74], from diagnosis based on symptoms [57, 75] to precise identification using biomarkers [74]. The mechanisms underlying psychiatric disorders have been uncovered in a growing body of biomarker research [76, 77]. In the present study, the authors utilized ultrasensitive IMR tests to assess plasma levels of neurodegenerative proteins in middle-aged or older individuals diagnosed with MDD and SCD who exhibited depressive and cognitive symptoms. Symptoms of depression in individuals with MDD may be associated with amyloid pathology. Similarly, declines in cognitive function may be associated with the combined neurodegeneration caused by tau, amyloid, and astrocyte injury. By contrast, amyloid pathology does not have a significant effect on the cognitive decline observed in SCD. Instead, the decline in cognitive function in SCD may be associated with tau pathology.
Correlations of biomarker and depression symptoms in MDD
Depressive symptoms in older individuals may indicate neurodegenerative processes in the brain. Studies have uncovered elevated tau deposition in the brain of older adults with depression [78] and revealed correlations between the degree of depression with tau levels in the inferior temporal lobe and entorhinal area [79]. Other studies have revealed an increase in the global amyloid burden associated with escalating depression symptoms in cognitively normal older adults [22, 23]. In patients with MDD, amyloid deposition escalates more in the precuneus and parietal lobe than in healthy controls [20]. These critical regions of tau and amyloid accumulation in MDD are similar to those in AD [80]. Nevertheless, the symptoms associated with amyloid deposition differ between MDD and AD; it is linked to depression symptoms in MDD but related to cognitive symptoms in AD.
Beyond mere variations in the presentations of amyloid pathology associated with cognitive and depressive symptoms in AD and MDD, longitudinal studies have suggested two explanations for the intricate relationship between AD and MDD. One theory suggests that depressive symptoms are secondary manifestations of brain Aβ deposition and are indicative of prodromal AD [81, 82]. The other theory suggests that primary depression and depressive symptoms are associated with amyloid pathology [20, 31, 83, 84] and may increase the risk of developing AD [85]. In the second theory, the potential pathways linking depression as a dementia risk factor involve increases in glucocorticoid stimulating hippocampal atrophy, the provocation of amyloid plaque formation, proinflammatory activation, changes in nerve growth factors, or shared vascular risk factors that promote the development of dementia [85].
Wu et al. used IMR and F-florbetapir PET18 in older adults with MDD without dementia to establish the correlations of plasma Aβ42, Aβ40, and Aβ42/Aβ40 ratios with cerebral amyloid deposition in the precuneus, parietal lobe, and posterior cingulate cortex [29]. Older individuals with MDD typically exhibit a higher level of plasma Aβ40 and a lower Aβ42/Aβ40 ratio than those without MDD [28, 30, 31]. However, the present study’s plasma biomarker results of IMR indicated no significant difference in plasma amyloid biomarkers between the MDD and NC groups. Several factors may account for this nonsignificance. First, small effect sizes may be difficult to detect, given the small sample size. Second, the correlation between depression and Aβ pathology was state-dependent, and the degree of depression was mild among the MDD participants in this study. The state-dependent relationship between depression and Aβ was evident in that elevated brain Aβ deposition was correlated with the progressive emergence of anxious-depressive symptoms over time among cognitively normal older individuals [22]. Plasma Aβ markers also exhibit a severity-dependent trend of depressive symptoms in older individuals, regardless of their cognitive function [86]. The severity of depression increases over time with decreases in the plasma Aβ42/Aβ40 ratio [31]. Therefore, owing to the severity-dependent nature and the low depression severity in the present study’s sample, the comparison between MDD and NC groups was constrained, limiting the ability to detect differences in plasma Aβ markers, producing results that were not statistically significant.
Although not indicative of a difference in plasma Aβ markers between MDD and NC, this study’s biomarker–behavior regression analysis revealed that plasma Aβ40 is positively associated and Aβ42/Aβ40 is negatively associated with HADS-D score in those with MDD (Fig. 3A). In another biomarker study on individuals with MDD without dementia, Wu and colleagues identified elevated plasma Aβ40 levels detected using IMR as being positively associated with brain PET amyloid burden, and the Aβ42/40 ratio as negatively associated with brain PET amyloid burden [29]. Consistently, a decreased plasma Aβ42/40 ratio and increased brain PET Aβ has been observed along the AD continuum from cognitively normal [87], SCD [33, 88], MCI [40], to AD dementia [89, 90]. Consequently, the inverse association between plasma Aβ40 levels and the Aβ42/40 ratio with depression and cognitive impairment suggests a correlation between increased amyloid deposition in the brain and higher clinical severity in both MDD and AD.
Correlations of biomarker and cognitive symptoms in MDD
The biomarker–cognition associations in older individuals with MDD are complex. Previous studies have demonstrated that elevated amyloid deposition [22, 23] and changes in blood amyloid markers [30, 31] are present in late-life depression. Sun et al. described amyloid-associated depression in 2008, noting its association with increased cognitive impairment, including memory, visuospatial ability, and executive function [81]. Moreover, tau deposition has been observed in patients with late-life depression [78, 79, 91]. A study on the CSF biomarker panel detected tau changes in late-life depression associated with AD pathology, but these changes were not observed in late-life depression without AD pathology [92]. The present study’s observation of worse cognitive performance associated with lower plasma Aβ42 levels, a higher Tau/Aβ42 ratio, and higher plasma GFAP in individuals with MDD than in NC highlights the presence of mixed neurodegenerative pathologies in the cognitive problems associated with late-life depression.
Blood GFAP is a reliable indicator of reactive astrocytes associated with brain injuries because it specifically originates from brain astrocytes [51]. Increased plasma GFAP indicates astrocytic injury and is more strongly inversely related to MoCA and DSST scores in individuals with MDD than in NC. Re-evaluations of the DSST have revealed that it can measure cognitive processing speed and attention, demonstrating high sensitivity for general cognitive decline in the aging population [93, 94]. Therefore, these results suggest that astrocytic injury impairs global cognition in older individuals with depression. Studies have demonstrated an increase in GFAP-positive astrocytes in the dorsolateral prefrontal cortex of individuals with late-onset MDD compared with age-matched individuals without the condition. Additionally, individuals with young-onset MDD exhibit fewer GFAP-positive astrocytes than those with late-onset MDD, indicating an age-dependent astrocytic pathology in MDD [95, 96].
The role of circulating amyloid peptides in MDD and AD
In individuals with MDD, elevated peripheral Aβ levels are believed to originate from the brain crossing the blood–brain barrier but are also derived from platelets [97]. These circulating Aβ peptides can heighten platelet activation [98,99,100] and disrupt endothelial cell function [101]. increasing the susceptibility to AD in affected individuals. Additionally, platelet activation is associated with depression. A previous study proposed that heightened platelet activation and enhanced release of Aβ40 are potential mechanisms contributing to brain abnormalities, amyloidosis, and an elevated risk of AD in individuals with depression [99].
Circulating Aβ has been proposed to precipitate AD. The proposed mechanisms involve increased mitochondrial oxidative stress and neurotoxicity, induced inflammatory response, and induced hyperphosphorylation of tau protein [102]. Peripheral blood Aβ is also implicated in the pathogenesis of AD by influencing peripheral innate immune cells [103]. The present study did not identify differences in plasma Aβ markers between groups; larger sample sizes may reveal more obvious differences. The contribution of peripheral Aβ to the onset of depressive symptoms and cognitive decline may be substantial, requiring additional exploration into its role in the progression of AD in older individuals with depression.
Biomarker–behavior associations in SCD
Dementia is believed to be a progressive process. The accumulation of neurodegenerative proteins begins at the preclinical stage [104, 105]. In the early stages, subjective cognitive problems precede the onset of dementia [106, 107]. Subjective cognitive problems in cognitively normal older individuals may be identified through molecular imaging as part of the neurodegenerative process. A positive association exists between the global Aβ burden, as determined by a Pittsburgh Compound B (PiB) PET scan, and subjective memory complaints [24]. Furthermore, subjective cognitive problems were observed to be increased when Aβ deposition was combined with neurodegeneration biomarkers in a study employing concurrent PiB-PET, FDG-PET, and structural MRI [25]. Still, Aβ deposition demonstrated regional variations, where the medial and lateral frontal areas were more closely associated with subjective cognitive complaints [26]. Unlike the effects of the global Aβ burden, tau has a region-specific influence on subjective cognition, particularly in the entorhinal cortex, the initial site of tauopathy in the development of AD [26].
The use of blood biomarkers can help to uncover the complex pathogenesis of SCD. As noted in a blood biomarker study that covered the complete dementia continuum, plasma p-Tau181, Aβ42/Aβ40, and pTau181/Aβ42 ratios differentiate MCI from SCD (area under the curve [AUC] 0.64–0.72, p < 0.05). The combination of plasma NfL and the aforementioned biomarker panel further differentiates AD from SCD (AUC 0.67–0.85, p < 0.05) [108]. Furthermore, heterogeneous combinations of ATN (amyloid, tau, neurodegeneration) components indicate clinical progression in SCD [109]. Therefore, a comprehensive view of multiple biomarkers facilitates identifying the biological status of SCD.
Although SCD is considered a possible form of preclinical AD, the present study failed to demonstrate differences in AD-related plasma biomarkers between SCD and NC groups. The heterogeneous nature of SCD explains this absence of difference. Not every case of SCD follows the same clinicopathological progression because SCD can precede both AD and non-AD dementia, with varied rates of progression to dementia [110]. Moreover, participant recruitment from communities may have resulted in the selection of individuals with relatively milder SCD than those from memory clinics. The dementia converting rate of SCD from various sources varies; SCD identified in memory clinics is associated with a higher risk of developing dementia than SCD identified in community cohorts [110]. Neuroimaging studies have also demonstrated that SCD identified in memory clinics exhibits more complex brain atrophy patterns than the purely temporal atrophy observed in SCD from community sources [111]. Therefore, studying the full spectrum of SCD from various sources might further delineate the biological nature of SCD.
Although the levels of plasma biomarkers did not differ between the SCD and NC groups in the present study, interactions of SCD in biomarker–behavior correlations were observed. In the NC group, individuals with better cognitive performance had higher plasma Tau levels. By contrast, this trend was inverted in the SCD group, although it did not reach statistical significance. This pattern may indicate an early effect of tauopathy on the onset of cognitive decline in preclinical dementia. Moreover, plasma Aβ levels were not associated with cognitive performance in the SCD group, although they were in the NC group. However, the lack of a significant correlation between Aβ and cognition in SCD does not indicate the absence of amyloid pathology, as evidenced by studies demonstrating Aβ-related cognitive decline in SCD. Specifically, a reduction in CSF Aβ42 levels has been associated with self-reported memory and language declines [112]. The other study observed that brain amyloid deposition is associated with cognitive impairment in individuals with SCD [113]. Using amyloid PET and cognitive assessment, the interaction between SCD and amyloid deposition was observed to be predictive of declines in episodic memory and overall cognitive function throughout an extended follow-up period [114]. However, studies on the mechanisms driving cognitive decline in SCD are inconclusive, and further investigation is required due to the conflicting nature of the data.
The authors previously observed that individuals reporting subjective cognitive problems frequently exhibit symptoms of anxiety and depression [4]. Nevertheless, the biomarker and behavior analysis did not corroborate the hypothesis that neurodegeneration accounts for the heightened predisposition to anxiety and depression in SCD. Whether the affective alterations in SCD observed in functional MRI studies stem from heightened self-awareness or functional compensation remains unclear [115]. Further research is required to determine the mechanisms underlying the cognitive alterations in SCD.
Limitations
This study has several limitations. First, the sample sizes for the MDD, SCD, and NC groups were relatively small, making the results less robust and generalizable. Second, this study exclusively included participants from community settings, excluding those with cognitive deficits. Consequently, the unique biomarker–behavior characteristics identified in this study may not be generalizable to older adults with cognitive impairment. Third, these biomarker–behavior features remain unchanged as older MDD patients manifest cognitive impairments or as SCD advances to MCI and dementia remains uncertain. Moreover, whether these characteristics suggest an increased risk of progression to MCI or dementia is unclear. Longitudinal studies are crucial for exploring the connection between these characteristics and cognitive decline progression. Fourth, although at least one study suggested that amyloid pathology is responsive to treatment resistance in MDD [116], this study did not evaluate the relationship between biomarkers and treatment response in the participants. Fifth, because the study did not investigate the duration of depression, the association of disease duration with biomarkers could not be determined. Sixth, APOE genotypes were not available in this study. A previous study showed that individuals with SCD recruited from memory clinics exhibit a higher prevalence of APOE4 genes than those from the community [110]. Because the APOE genotype is crucial as a phenotypic conversion factor in the transition from SCD to MCI [117] and from MCI to AD [118], future studies should explore including APOE genotyping and the expansion of sample size to address the heterogeneous nature of SCD. Finally, although this study utilized blood-based biomarkers, validation using CSF biomarkers and brain molecular imaging correlation could not be achieved in this study setting.
Conclusions
This study employed a shared control group to investigate the neuropsychiatric implications of blood biomarkers in older adults with MDD and SCD within the same community cohort. Standard cognitive assessments were used to verify the normal cognitive function of each participant. IMR was used to measure plasma biomarkers, illuminating the biological mechanisms behind neuropsychiatric symptoms. Observations of distinct biomarker–behavior characteristics in each group, compared with the NC, clarified the unique pathogenesis of these two conditions, which may present with similar neuropsychiatric symptoms in older individuals. In older adults with MDD, depressive symptoms are associated with amyloid pathology, whereas cognitive challenges emerge from a combination of amyloid, tau, and astrocytic pathologies. By contrast, patients with SCD do not have a robust connection to neurodegenerative biomarkers. However, the distinct biomarker–cognition correlations in the SCD, when compared with the NC suggests that cognitive decline in SCD may not be related to amyloid pathology but may indicate early stages of tauopathy. Acknowledging that not every individual with SCD progresses to MCI or dementia is crucial because SCD’s transitional nature may conceal the connection between biomarkers and cognitive impairments at this stage. Nevertheless, this study underscores the distinct cognitive difficulties of older adults with MDD and SCD. Utilizing blood-based neurodegenerative biomarkers may assist in differentiating the underlying causes of cognitive decline in older individuals, leading to precise diagnoses and tailored treatments.
Data availability
The datasets generated and/or analyzed during the current study are are available from the corresponding author on reasonable request.
References
Hill NL, Mogle J, Wion R, Munoz E, DePasquale N, Yevchak AM, et al. Subjective cognitive impairment and affective symptoms: a systematic review. Gerontologist. 2016;56:e109–e27.
Ma L. Depression, anxiety, and apathy in mild cognitive impairment: current perspectives. Front Aging Neurosci. 2020;12:9.
Hohman TJ, Beason-Held LL, Resnick SM. Cognitive complaints, depressive symptoms, and cognitive impairment: are they related? J Am Geriatr Soc. 2011;59:1908–12.
Wei YC, Huang LY, Chen CK, Lin C, Shyu YC, Chen YL, et al. Subjective cognitive decline in the community is affected at multiple aspects of mental health and life quality: a cross-sectional study of the community medicine of Keelung Chang Gung Memorial Hospital. Dement Geriatr Cogn Dis Extra. 2019;9:152–62.
Zlatar ZZ, Moore RC, Palmer BW, Thompson WK, Jeste DV. Cognitive complaints correlate with depression rather than concurrent objective cognitive impairment in the successful aging evaluation baseline sample. J Geriatr Psychiatry Neurol. 2014;27:181–7.
Kiloh LG. Pseudo-dementia. Acta Psychiatr Scand. 1961;37:336–51.
Kang H, Zhao F, You L, Giorgetta CDV, Sarkhel S, et al. Pseudo-dementia: a neuropsychological review. Ann Indian Acad Neurol. 2014;17:147–54.
Dillon C, Tartaglini MF, Stefani D, Salgado P, Taragano FE, Allegri RF. Geriatric depression and its relation with cognitive impairment and dementia. Arch Gerontol Geriatr. 2014;59:450–6.
Brodaty H, Connors MH. Pseudodementia, pseudo-pseudodementia, and pseudodepression. Alzheimers Dement (Amst). 2020;12:e12027
Wei Y-C, Huang L-Y, Lin C, Shyu Y-C, Chen C-K. Taiwanese depression questionnaire and AD8 questionnaire for screening late-life depression in communities. Neuropsychiatr Disease Treatment. 2021;17:747–55.
Wilson RS, Barnes LL, Mendes de Leon CF, Aggarwal NT, Schneider JS, Bach J, et al. Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology. 2002;59:364–70.
Han FF, Wang HX, Wu JJ, Yao W, Hao CF, Pei JJ. Depressive symptoms and cognitive impairment: a 10-year follow-up study from the Survey of Health, Ageing and Retirement in Europe. Eur Psychiatry. 2021;64:e55.
Yaffe K, Blackwell T, Gore R, Sands L, Reus V, Browner WS. Depressive symptoms and cognitive decline in nondemented elderly women: a prospective study. Arch Gen Psychiatry. 1999;56:425–30.
Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. JAMA. 2002;288:1475–83.
Modrego PJ, Ferrandez J. Depression in patients with mild cognitive impairment increases the risk of developing dementia of Alzheimer type: a prospective cohort study. Arch Neurol. 2004;61:1290–3.
Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292–323.
Jessen F, Amariglio RE, van Boxtel M, Breteler M, Ceccaldi M, Chetelat G, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement. 2014;10:844–52.
Mukku SSR, Dahale AB, Muniswamy NR, Muliyala KP, Sivakumar PT, Varghese M. Geriatric depression and cognitive impairment—an update. Indian J Psychol Med. 2021;43:286–93.
Gatchel JR, Rabin JS, Buckley RF, Locascio JJ, Quiroz YT, Yang HS, et al. Longitudinal association of depression symptoms with cognition and cortical amyloid among community-dwelling older adults. JAMA Netw Open. 2019;2:e198964.
Wu KY, Hsiao IT, Chen CS, Chen CH, Hsieh CJ, Wai YY, et al. Increased brain amyloid deposition in patients with a lifetime history of major depression: evidenced on 18F-florbetapir (AV-45/Amyvid) positron emission tomography. Eur J Nucl Med Mol Imaging. 2014;41:714–22.
Yasuno F, Kazui H, Morita N, Kajimoto K, Ihara M, Taguchi A, et al. High amyloid-beta deposition related to depressive symptoms in older individuals with normal cognition: a pilot study. Int J Geriatr Psychiatry. 2016;31:920–8.
Donovan NJ, Locascio JJ, Marshall GA, Gatchel J, Hanseeuw BJ, Rentz DM, et al. Longitudinal association of amyloid beta and anxious-depressive symptoms in cognitively normal older adults. Am J Psychiatry. 2018;175:530–7.
Krell-Roesch J, Lowe VJ, Neureiter J, Pink A, Roberts RO, Mielke MM, et al. Depressive and anxiety symptoms and cortical amyloid deposition among cognitively normal elderly persons: the Mayo Clinic Study of Aging. Int Psychogeriatr. 2018;30:245–51.
Amariglio RE, Becker JA, Carmasin J, Wadsworth LP, Lorius N, Sullivan C, et al. Subjective cognitive complaints and amyloid burden in cognitively normal older individuals. Neuropsychologia. 2012;50:2880–6.
Amariglio RE, Mormino EC, Pietras AC, Marshall GA, Vannini P, Johnson KA, et al. Subjective cognitive concerns, amyloid-beta, and neurodegeneration in clinically normal elderly. Neurology. 2015;85:56–62.
Buckley RF, Hanseeuw B, Schultz AP, Vannini P, Aghjayan SL, Properzi MJ, et al. Region-specific association of subjective cognitive decline with tauopathy independent of global beta-amyloid burden. JAMA Neurol. 2017;74:1455–63.
Blasko I, Kemmler G, Jungwirth S, Wichart I, Krampla W, Weissgram S, et al. Plasma amyloid beta-42 independently predicts both late-onset depression and Alzheimer disease. Am J Geriatr Psychiatry. 2010;18:973–82.
Nascimento KK, Silva KP, Malloy-Diniz LF, Butters MA, Diniz BS. Plasma and cerebrospinal fluid amyloid-beta levels in late-life depression: a systematic review and meta-analysis. J Psychiatr Res. 2015;69:35–41.
Wu KY, Hsiao IT, Chen CH, Liu CY, Hsu JL, Huang SY, et al. Plasma Abeta analysis using magnetically-labeled immunoassays and PET (18)F-florbetapir binding in non-demented patients with major depressive disorder. Sci Rep. 2018;8:2739.
Natsume S, Baba H, Maeshima H, Saida T, Yoshinari N, Shimizu K, et al. Clinical course and serum amyloid beta levels in elderly patients with major depressive disorder. J Affect Disord. 2022;315:156–61.
Pomara N, Bruno D, Plaska CR, Ramos-Cejudo J, Osorio RS, Pillai A, et al. Plasma amyloid-beta dynamics in late-life major depression: a longitudinal study. Transl Psychiatry. 2022;12:301.
Muller S, Preische O, Gopfert JC, Yanez VAC, Joos TO, Boecker H, et al. Tau plasma levels in subjective cognitive decline: results from the DELCODE study. Sci Rep. 2017;7:9529.
Verberk IMW, Slot RE, Verfaillie SCJ, Heijst H, Prins ND, van Berckel BNM, et al. Plasma amyloid as prescreener for the earliest Alzheimer pathological changes. Ann Neurol. 2018;84:648–58.
Kim KY, Park J, Jeong YH, Kim HJ, Lee E, Park JY, et al. Plasma amyloid-beta oligomer is related to subjective cognitive decline and brain amyloid status. Alzheimers Res Ther. 2022;14:162.
Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180–93.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra11.
Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci. 2015;35:518–26.
Lue LF, Guerra A, Walker DG. Amyloid beta and tau as Alzheimer’s disease blood biomarkers: promise from new technologies. Neurol Ther. 2017;6:25–36.
Stocker H, Beyer L, Perna L, Rujescu D, Holleczek B, Beyreuther K, et al. Association of plasma biomarkers, p-tau181, glial fibrillary acidic protein, and neurofilament light, with intermediate and long-term clinical Alzheimer’s disease risk: results from a prospective cohort followed over 17 years. Alzheimers Dement. 2023;19:25–35.
Chatterjee P, Pedrini S, Doecke JD, Thota R, Villemagne VL, Dore V, et al. Plasma Abeta42/40 ratio, p-tau181, GFAP, and NfL across the Alzheimer’s disease continuum: a cross-sectional and longitudinal study in the AIBL cohort. Alzheimers Dement. 2023;19:1117–1134.
de Wolf F, Ghanbari M, Licher S, McRae-McKee K, Gras L, Weverling GJ, et al. Plasma tau, neurofilament light chain and amyloid-beta levels and risk of dementia; a population-based cohort study. Brain. 2020;143:1220–32.
Müller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. 2017;18:281–98.
Bishop GM, Robinson SR. Physiological roles of amyloid-beta and implications for its removal in Alzheimer’s disease. Drugs Aging. 2004;21:621–30.
Mayeux R, Honig LS, Tang M-X, Manly J, Stern Y, Schupf N, et al. Plasma Aβ40 and Aβ42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–90.
Teunissen CE, Verberk IMW, Thijssen EH, Vermunt L, Hansson O, Zetterberg H, et al. Blood-based biomarkers for Alzheimer’s disease: towards clinical implementation. Lancet Neurol. 2022;21:66–77.
Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008;18:253–66.
Rodrigue KM, Kennedy KM, Devous MD Sr., Rieck JR, Hebrank AC, Diaz-Arrastia R, et al. β-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012;78:387–95.
Zhang Y, Wu KM, Yang L, Dong Q, Yu JT. Tauopathies: new perspectives and challenges. Mol Neurodegener. 2022;17:28.
Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22.
Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90:870–81.
Messing A, Brenner M. GFAP at 50. ASN Neuro. 2020;12:1759091420949680.
Benedet AL, Mila-Aloma M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78:1471–83.
Chiu MJ, Yang SY, Horng HE, Yang CC, Chen TF, Chieh JJ, et al. Combined plasma biomarkers for diagnosing mild cognition impairment and Alzheimer’s disease. ACS Chem Neurosci. 2013;4:1530–6.
Rossetti HC, Lacritz LH, Cullum CM, Weiner MF. Normative data for the Montreal Cognitive Assessment (MoCA) in a population-based sample. Neurology. 2011;77:1272–5.
Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–8.
Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, et al. Mild cognitive impairment—beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 2004;256:240–6.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Lecrubier Y, Sheehan D, Weiller E, Amorim P, Bonora I, Harnett, et al. The Mini International Neuropsychiatric Interview (MINI). A short diagnostic structured interview: reliability and validity according to the CIDI. Eur Psychiatry. 1997;12:224–31.
Yang YH, Galvin JE, Morris JC, Lai CL, Chou MC, Liu CK. Application of AD8 questionnaire to screen very mild dementia in Taiwanese. Am J Alzheimers Dis Other Demen. 2011;26:134–8.
Galvin JE, Roe CM, Powlishta KK, Coats MA, Muich SJ, Grant E, et al. The AD8: a brief informant interview to detect dementia. Neurology. 2005;65:559–64.
Molinuevo JL, Rabin LA, Amariglio R, Buckley R, Dubois B, Ellis KA, et al. Implementation of subjective cognitive decline criteria in research studies. Alzheimers Dement. 2017;13:296–311.
Bjelland I, Dahl AA, Haug TT, Neckelmann D. The validity of the hospital anxiety and depression scale. An updated literature review. J Psychosom Res. 2002;52:69–77.
Williams JB. A structured interview guide for the Hamilton depression rating scale. Arch Gen Psychiatry. 1988;45:742–7.
Maier W, Buller R, Philipp M, Heuser I. The Hamilton Anxiety Scale: reliability, validity and sensitivity to change in anxiety and depressive disorders. J Affect Disord. 1988;14:61–8.
Wei YC, Chen CK, Lin C, Chen PY, Hsu PC, Lin CP, et al. Normative data of mini-mental state examination, Montreal cognitive assessment, and Alzheimer’s disease assessment scale-cognitive subscale of community-dwelling older adults in Taiwan. Dement Geriatr Cogn Disord. 2022;51:365–76.
Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141:1356–64.
Chen JH, Chen HY. WAIS-III (Chinese Version): administration and scoring manual: Wechsler Adult Intelligence Scale. Taipei: Chinese Behavioral Science Corporation; 2002.
Wechsler DPC. WAIS-III: administration and scoring manual: Wechsler Adult Intelligence Scale. San Antonio, TX: Psychological Corporation; 1997.
Hagstromer M, Oja P, Sjostrom M. The International Physical Activity Questionnaire (IPAQ): a study of concurrent and construct validity. Public Health Nutr. 2006;9:755–62.
Glynn NW, Santanasto AJ, Simonsick EM, Boudreau RM, Beach SR, Schulz R, et al. The Pittsburgh fatigability scale for older adults: development and validation. J Am Geriatr Soc. 2015;63:130–5.
Lin C, Glynn NW, Gmelin T, Wei Y-C, Chen Y-L, Huang C-M, et al. Validation of the traditional Chinese version of the Pittsburgh fatigability scale for older adults. Clin Gerontol. 2022;45:606–18.
Chong JR, Ashton NJ, Karikari TK, Tanaka T, Scholl M, Zetterberg H, et al. Blood-based high sensitivity measurements of beta-amyloid and phosphorylated tau as biomarkers of Alzheimer’s disease: a focused review on recent advances. J Neurol Neurosurg Psychiatry. 2021;92:1231–41.
Chieh JJ, Yang SY, Jian ZF, Wang WC, Horng HE, Yang HC, et al. Hyper-high-sensitivity wash-free magnetoreduction assay on biomolecules using high-Tc superconducting quantum interference devices. J Appl Phys. 2008;103:014703.
Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–44.
Kalia M, Costa ESJ. Biomarkers of psychiatric diseases: current status and future prospects. Metabolism. 2015;64:S11–5.
Garcia-Gutierrez MS, Navarrete F, Sala F, Gasparyan A, Austrich-Olivares A, Manzanares J. Biomarkers in psychiatry: concept, definition, types and relevance to the clinical reality. Front Psychiatry. 2020;11:432.
Babulal GM, Roe CM, Stout SH, Rajasekar G, Wisch JK, Benzinger TLS, et al. Depression is associated with tau and not amyloid positron emission tomography in cognitively normal adults. J Alzheimers Dis. 2020;74:1045–55.
Gatchel JR, Donovan NJ, Locascio JJ, Schultz AP, Becker JA, Chhatwal J, et al. Depressive symptoms and tau accumulation in the inferior temporal lobe and entorhinal cortex in cognitively normal older adults: a pilot study. J Alzheimers Dis. 2017;59:975–85.
van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21:21–35.
Sun X, Steffens DC, Au R, Folstein M, Summergrad P, Yee J, et al. Amyloid-associated depression: a prodromal depression of Alzheimer disease? Arch Gen Psychiatry. 2008;65:542–50.
Qiu WQ, Zhu H, Dean M, Liu Z, Vu L, Fan G, et al. Amyloid-associated depression and ApoE4 allele: longitudinal follow-up for the development of Alzheimer’s disease. Int J Geriatr Psychiatry. 2016;31:316–22.
Direk N, Schrijvers EM, de Bruijn RF, Mirza S, Hofman A, Ikram MA, et al. Plasma amyloid beta, depression, and dementia in community-dwelling elderly. J Psychiatr Res. 2013;47:479–85.
Harrington KD, Gould E, Lim YY, Ames D, Pietrzak RH, Rembach A, et al. Amyloid burden and incident depressive symptoms in cognitively normal older adults. Int J Geriatr Psychiatry. 2017;32:455–63.
Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol. 2011;7:323.
Twait EL, Wu JH, Kamarioti M, Basten M, van der Flier WM, Gerritsen L, et al. Association of amyloid-beta with depression or depressive symptoms in older adults without dementia: a systematic review and meta-analysis. Transl Psychiatry. 2024;14:25.
Fandos N, Perez-Grijalba V, Pesini P, Olmos S, Bossa M, Villemagne VL, et al. Plasma amyloid beta 42/40 ratios as biomarkers for amyloid beta cerebral deposition in cognitively normal individuals. Alzheimers Dement (Amst). 2017;8:179–87.
de Rojas I, Romero J, Rodriguez-Gomez O, Pesini P, Sanabria A, Perez-Cordon A, et al. Correlations between plasma and PET beta-amyloid levels in individuals with subjective cognitive decline: the Fundacio ACE Healthy Brain Initiative (FACEHBI). Alzheimers Res Ther. 2018;10:119.
Doecke JD, Perez-Grijalba V, Fandos N, Fowler C, Villemagne VL, Masters CL, et al. Total Abeta42/Abeta40 ratio in plasma predicts amyloid-PET status, independent of clinical AD diagnosis. Neurology. 2020;94:e1580–e91.
Janelidze S, Teunissen CE, Zetterberg H, Allue JA, Sarasa L, Eichenlaub U, et al. Head-to-head comparison of 8 plasma amyloid-beta 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78:1375–82.
Schultz SA, Gordon BA, Mishra S, Su Y, Morris JC, Ances BM, et al. Association between personality and tau-PET binding in cognitively normal older adults. Brain Imaging Behav. 2020;14:2122–31.
Siafarikas N, Kirsebom BE, Srivastava DP, Eriksson CM, Auning E, Hessen E, et al. Cerebrospinal fluid markers for synaptic function and Alzheimer type changes in late life depression. Sci Rep. 2021;11:20375.
Jaeger J. Digit symbol substitution test: the case for sensitivity over specificity in neuropsychological testing. J Clin Psychopharmacol. 2018;38:513–9.
Hoyer WJ, Stawski RS, Wasylyshyn C, Verhaeghen P. Adult age and digit symbol substitution performance: a meta-analysis. Psychol Aging. 2004;19:211–4.
Si X, Miguel-Hidalgo JJ, O’Dwyer G, Stockmeier CA, Rajkowska G. Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology. 2004;29:2088–96.
Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY, Stockmeier CA, et al. Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biol Psychiatry. 2000;48:861–73.
Chen M, Inestrosa NC, Ross GS, Fernandez HL. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem Biophys Res Commun. 1995;213:96–103.
Canobbio I, Abubaker AA, Visconte C, Torti M, Pula G. Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front Cell Neurosci. 2015;9:65.
Pomara N, Murali Doraiswamy P. Does increased platelet release of Abeta peptide contribute to brain abnormalities in individuals with depression? Med Hypotheses. 2003;60:640–3.
Pomara N, Doraiswamy PM, Willoughby LM, Roth AE, Mulsant BH, Sidtis JJ, et al. Elevation in plasma Abeta42 in geriatric depression: a pilot study. Neurochem Res. 2006;31:341–9.
Park L, Zhou P, Koizumi K, El Jamal S, Previti ML, Van Nostrand WE, et al. Brain and circulating levels of Abeta1-40 differentially contribute to vasomotor dysfunction in the mouse brain. Stroke. 2013;44:198–204.
Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017;38:1205–35.
Shi M, Chu F, Zhu F, Zhu J. Peripheral blood amyloid-beta involved in the pathogenesis of Alzheimer’s disease via impacting on peripheral innate immune cells. J Neuroinflamm. 2024;21:5.
van Oostveen WM, de Lange ECM. Imaging techniques in Alzheimer’s disease: a review of applications in early diagnosis and longitudinal monitoring. Int J Mol Sci. 2021;22:2110.
Therriault J, Zimmer ER, Benedet AL, Pascoal TA, Gauthier S, Rosa-Neto P. Staging of Alzheimer’s disease: past, present, and future perspectives. Trends Mol Med. 2022;28:726–41.
Kaup AR, Nettiksimmons J, LeBlanc ES, Yaffe K. Memory complaints and risk of cognitive impairment after nearly 2 decades among older women. Neurology. 2015;85:1852–8.
Mitchell AJ, Beaumont H, Ferguson D, Yadegarfar M, Stubbs B. Risk of dementia and mild cognitive impairment in older people with subjective memory complaints: meta-analysis. Acta Psychiatr Scand. 2014;130:439–51.
Gerards M, Schild AK, Meiberth D, Rostamzadeh A, Vehreschild JJ, Wingen-Heimann S, et al. Alzheimer’s disease plasma biomarkers distinguish clinical diagnostic groups in memory clinic patients. Dement Geriatr Cogn Disord. 2022;51:182–92.
Ebenau JL, Timmers T, Wesselman LMP, Verberk IMW, Verfaillie SCJ, Slot RER, et al. ATN classification and clinical progression in subjective cognitive decline: the SCIENCe project. Neurology. 2020;95:e46–58.
Slot RER, Sikkes SAM, Berkhof J, Brodaty H, Buckley R, Cavedo E, et al. Subjective cognitive decline and rates of incident Alzheimer’s disease and non-Alzheimer’s disease dementia. Alzheimers Dement. 2019;15:465–76.
Pini L, Wennberg AM. Structural imaging outcomes in subjective cognitive decline: community vs. clinical-based samples. Exp Gerontol. 2021;145:111216.
Miebach L, Wolfsgruber S, Polcher A, Peters O, Menne F, Luther K, et al. Which features of subjective cognitive decline are related to amyloid pathology? Findings from the DELCODE study. Alzheimers Res Ther. 2019;11:66.
Ryu SY, Hong YJ, Ho S, Jeong JH, Park KH, Kim S, et al. Subtle cognitive deficits are associated with amyloid-beta positivity, but not severity of self-reported decline: results from the CoSCo study. Dement Geriatr Cogn Disord. 2022;51:159–67.
Vogel JW, Varga Dolezalova M, La Joie R, Marks SM, Schwimmer HD, Landau SM, et al. Subjective cognitive decline and beta-amyloid burden predict cognitive change in healthy elderly. Neurology. 2017;89:2002–9.
Wei Y-C, Kung Y-C, Huang W-Y, Lin C, Chen Y-L, Chen C-K, et al. Functional connectivity dynamics altered of the resting brain in subjective cognitive decline. Front Aging Neurosci. 2022;14:817137.
Li P, Hsiao IT, Liu CY, Chen CH, Huang SY, Yen TC, et al. Beta-amyloid deposition in patients with major depressive disorder with differing levels of treatment resistance: a pilot study. EJNMMI Res. 2017;7:24.
Caselli RJ, Chen K, Locke DE, Lee W, Roontiva A, Bandy D, et al. Subjective cognitive decline: self and informant comparisons. Alzheimers Dement. 2014;10:93–8.
Moreno-Grau S, Ruiz A. Genome research in pre-dementia stages of Alzheimer’s disease. Expert Rev Mol Med. 2016;18:e11.
Acknowledgements
This research was partly supported by grants from the Chang Gung Research Project to Y.-C. Wei (CRRPG2K0033), C. Lin (CRRPG2K0023), C.-K. Chen (CRRPG2K0013), P.-Y. Chen (CRRPG2K0043), the Community Medicine Research Center of Keelung Chang Gung Memorial Hospital (CLRPG2L0053), the Ministry of Science and Technology, Taiwan to C.-P. Lin (NSTC 112-2321-B-A49-008-), and Advanced National Defense Technology & Research Program. In addition, the authors thank the research assistants Jui-Yi Lee, Chun-Min Chang, and Hsin-Ju Hu. This manuscript was edited by Wallace Academic Editing.
Author information
Authors and Affiliations
Contributions
Y-CW, Y-CK, CL, and C-PL conception and design of the work. Y-CW, C-KC, CL, W-YH, P-YC, and Y-CS did data acquisition. Y-CW and Y-CK analyzed and interpreted data. Y-CW, Y-CK, and C-HY drafted the work. C-KC, C-PL, and C-HY substantively revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, YC., Kung, YC., Lin, C. et al. Differential neuropsychiatric associations of plasma biomarkers in older adults with major depression and subjective cognitive decline. Transl Psychiatry 14, 333 (2024). https://doi.org/10.1038/s41398-024-03049-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-03049-w
- Springer Nature Limited