
Abstract
This article delves into Alzheimer’s disease (AD), a prevalent neurodegenerative condition primarily affecting the elderly. It is characterized by progressive memory and cognitive impairments, severely disrupting daily life. Recent research highlights the potential involvement of microRNAs in the pathogenesis of AD. MicroRNAs (MiRNAs), short non-coding RNAs comprising 20–24 nucleotides, significantly influence gene regulation by hindering translation or promoting degradation of target genes. This review explores the role of specific miRNAs in AD progression, focusing on their impact on β-amyloid (Aβ) peptide accumulation, intracellular aggregation of hyperphosphorylated tau proteins, mitochondrial dysfunction, neuroinflammation, oxidative stress, and the expression of the APOE4 gene. Our insights contribute to understanding AD’s pathology, offering new avenues for identifying diagnostic markers and developing novel therapeutic targets.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD): an overview
AD stands as a leading neurodegenerative disorder, predominantly seen in the elderly. Characterized by early-stage subtle memory lapses and declines in verbal fluency, AD is a major contributor to dementia, accounting for 50–75% of all cases [1]. In 2010, ~35.6 million individuals suffered from dementia globally, with projections suggesting a dramatic rise to 135 million by 2050 [1, 2]. In the United States alone, an estimated 6.7 million elderly individuals currently live with Alzheimer’s dementia, a number expected to double by 2060 in the absence of medical advancements. Notably, AD has become a significant cause of death, ranking as the sixth leading cause in the U.S. as of 2019 https://doi.org/10.1002/alz.13016. While deaths from other conditions like heart disease and HIV have declined, Alzheimer’s-related fatalities have surged by over 145% since 2000 https://doi.org/10.1002/alz.12638.
The pathological hallmark of AD is neuronal death and synaptic loss, culminating in severe cognitive dysfunction. Key neuropathological indicators include extracellular β-amyloid (Aβ) accumulation and intracellular accumulation of phosphorylated tau, leading to neurofibrillary tangles (NFTs) [3, 4]. Diagnosis and assessment of AD severity rely on memory and physical evaluations [5], with a definitive diagnosis possible only post-mortem through autopsy findings correlating Aβ deposition with clinical symptoms [6]. The challenge of diagnosing Mild Cognitive Impairment (MCI) at early stages, a potential precursor to AD, underscores the need for reliable biomarkers [7, 8]. MicroRNAs (MiRNAs) have emerged as pivotal biomarkers, playing crucial roles in AD’s progression [9,10,11,12]. Differentially expressed miRNAs have the potential to be used as biomarkers for diagnosing and monitoring AD progression. These miRNAs regulate several biological functions, with the most significant and important biological processes and pathways related mainly to the maintenance of genomic integrity, proteostasis control, regulation of apoptotic processes, and neurotrophic support [13]. This review examines the changes in miRNA expression and their consequent effects on neuronal dynamics in the AD brain, discussing their significance in advancing our understanding of the disease’s progression and potential preventative strategies.
Molecular changes and pathogenesis of AD
In AD, the brain undergoes significant structural alterations, including a drastic reduction in neuronal count, the formation of NFTs composed of hyperphosphorylated tau, and the development of Aβ or senile plaques [14, 15]. These plaques, characterized by protein deposits outside cells encircled by dystrophic neurites, predominantly consist of Aβ, stemming from aberrant metabolism of the β-amyloid precursor protein (APP). APP, a transmembrane protein, typically undergoes enzymatic cleavage to produce soluble α-APP and β-APP [16,17,18]. However, in AD patients, this metabolic pathway malfunctions, leading to the accumulation of Aβ, a process central to the pathogenesis of AD [19, 20]. These deposits incite inflammatory responses and oxidative stress, culminating in neuronal damage and death [21,22,23].
In addition to Aβ deposition, the aberrant aggregation of tau protein is a crucial aspect of AD pathogenesis. Normally, tau protein, a microtubule-associated protein, maintains neuronal structural stability [24]. In AD, however, tau undergoes abnormal phosphorylation and aggregation, resulting in the formation of NFTs [25, 26]. The exact mechanisms behind tau aggregation remain partially understood, but key hypotheses include aberrant phosphorylation and splice mutation of the tau protein. Hyperphosphorylation leads to tau proteins aggregating into fibrillary tangles, while splice mutations can alter tau protein structure, promoting aggregation [27]. These tangles accumulate inside brain neurons, forming NFTs that disrupt normal nutrient transport and signal transmission within neurons, leading to neuronal dysfunction and death [28].
AD manifests in two primary forms: early-onset Alzheimer’s disease (EOAD) and late-onset Alzheimer’s disease (LOAD) [29]. EOAD, typically occurring in individuals under 65, is a rarer genetic variant of AD. LOAD, on the other hand, is the more common form of dementia in individuals over 65 and is characterized by sporadic AD attributed to abnormal amyloid mechanisms [30]. Both EOAD and LOAD exhibit distinct memory impairments, with EOAD often presenting with nonamnestic features and LOAD patients showing increasing semantic memory disorders. These differences underscore the necessity for tailored approaches in understanding and treating each AD type [31,32,33].
Genetic research has pinpointed four genes implicated in autosomal dominant or EOAD: APP, presenilin 1 (PS1), presenilin 2 (PS2), and apolipoprotein E (APOE). The formation of amyloid plaques, primarily composed of Aβ peptides, is significantly influenced by mutations within these genes [19, 34, 35]. Specifically, mutations in APP and presenilin proteins are known to elevate the production of Aβ peptides, especially the more amyloidogenic Aβ42, which is closely linked to the development of amyloid plaques in the brain. This evidence positions the presenilin proteins as initial factors in the cascade leading to Aβ42 accumulation and plaque formation [36, 37]. APOE, a key player in lipid transport, is encoded by genes on chromosome 19 and plays a protective role against AD. Present in brain cells like oligodendrocytes, astrocytes, and microglia [35]. APOE comes in three isoforms: E2, E3, and E4, with varying frequencies in the population [38]. Interaction between APOE and Aβ peptides suggests a complex relationship where APOE binds to Aβ, potentially mitigating the formation of disease-associated fibrils [39, 40]. APOE3, for instance, is thought to shield neurons from Aβ‘s detrimental effects by facilitating its clearance [41]. In contrast, the APOE4 variant is linked to increased risks of coronary and cerebrovascular diseases, which are significant risk factors for AD [42,43,44].
Beyond genetic predispositions, environmental factors play a critical role in AD’s pathogenesis. Incidences of head trauma with loss of consciousness have been correlated with a higher prevalence of AD [45]. Aging remains the most significant risk factor, with the disease’s incidence rising as the population ages [46, 47]. Lifestyle choices, including poor diet, lack of physical activity, and exposure to pollutants [48,49,50,51], alongside cardiovascular conditions like diabetes [52,53,54], hyperlipidemia, and hypertension [55,56,57], have been associated with an increased risk of AD. Additionally, long-term exposure to toxic substances, such as pesticides and heavy metals, may elevate AD risk [58,59,60]. The aging process itself, marked by a decline in cellular function and repair capacity, contributes to AD development [61]. However, it’s crucial to note that while these environmental factors are linked to AD, they do not guarantee its onset [62, 63]. The complexity of AD’s pathogenesis, with many aspects still unknown, underscores the need for further research.
In summary, AD is a multifactorial disorder influenced by abnormal metabolism of β-APP, Aβ deposition, tau protein aggregation, genetic mutations, environmental factors, and the aging process. Understanding the molecular biology underlying AD is vital for unveiling its pathology and identifying therapeutic targets. Despite the availability of symptomatic treatments, a cure for AD remains elusive, highlighting the urgency for continued in-depth research [9,10,11]. Recent insights into the role of miRNAs in AD offer promising directions for unraveling its pathological processes and developing novel therapeutic strategies.
MiRNAs: regulatory functions and implications in AD
MiRNAs are a class of small, non-coding RNA molecules, ranging in length from 20 to 24 nucleotides, that play a pivotal role in the regulation of gene expression within eukaryotic cells. These molecules achieve regulation by binding to the messenger RNA (mRNA) of target genes, affecting both transcriptional and translational processes [64]. MiRNAs are integral to a myriad of cellular functions, including cell proliferation, differentiation, apoptosis, metabolism, and immune response. The biosynthesis of miRNAs involves several key steps. Initially, miRNA genes are transcribed in the nucleus into primary miRNA transcripts (pri-miRNAs), which can extend to hundreds of nucleotides in length [65]. These pri-miRNAs are then processed by the Drosha enzyme and its cofactor into precursor miRNA (pre-miRNA) fragments, ~70 nucleotides long [64, 66]. Subsequently, pre-miRNAs are transported to the cytoplasm [67], where they are further cleaved by the RNAse III enzyme Dicer and the TAR RNA-binding protein (TRBP) into miRNA duplexes. A helicase unwinds these duplexes, resulting in a single-stranded mature miRNA and the degradation of the complementary strand [68]. Mature miRNAs are then incorporated into the RNA-induced silencing complex (RISC), forming active RISC-miRNA complexes that modulate gene expression by interacting with target mRNAs [69] (Fig. 1). MiRNAs regulate gene expression through two primary mechanisms: by binding completely to the 3′ untranslated region (3′ UTR) of target mRNAs, leading to mRNA degradation and gene expression inhibition, or by partially binding to the 3′ UTR to repress the translation process, thereby reducing gene expression [70, 71]. Interestingly, some miRNAs can also bind to the 5′ UTR or coding sequences of mRNAs, in some cases even enhancing translation [72].

The microRNA (miRNA) biosynthesis pathway is categorized into classical and non-classical. (1) In the classical pathway, miRNA genes are transcribed to generate primary miRNAs (pri-miRNAs), which are processed by Drosha and DGCR8 to form precursor miRNAs (pre-miRNAs), and then transferred to the cytoplasm where Dicer generates double-stranded miRNAs, which ultimately bind to RISC to regulate the translation of target mRNAs; (2) the non-classical pathway directly generates double-stranded miRNA. [miRNA microRNA, Pri-miRNA primary microRNA), Pre-miRNA precursor microRNA, RISC RNA-induced silencing complex, mRNA (messenger RNA, ORF open reading frame, Ago Argonaute, DGCR8 DiGeorge Syndrome Critical Region 8, Dicer Dicer enzyme].
A collection of 3028 genes associated with the cell cycle identified in a 2016 study [73], represents a wide array of protein classes. These include nucleic acid binding proteins (594), transcription factors (305), cytoskeletal proteins (232), kinases (174), phosphatases (111), and chaperones (84). Within this set, 2125 genes have been validated as targets of 424 miRNAs, with miR-335 alone targeting 301 genes. This highlights the significant role of miRNAs in post-transcriptional regulation, where individual miRNAs can influence the expression of numerous mRNAs [74]. This capability makes miRNAs promising therapeutic targets for complex diseases, such as brain disorders, due to their ability to regulate multiple genes simultaneously.
MiRNAs have been implicated in the pathogenesis of various diseases, including cancer [75], cardiovascular diseases [76], and neurological disorders like AD [77]. In the context of cancer, aberrant miRNA expression is closely associated with tumor initiation, progression, and metastasis. In cardiovascular diseases, miRNAs regulate processes such as cardiac myocyte proliferation and apoptosis, affecting heart structure and function [78,79,80,81,82,83,84]. Specifically, in AD, alterations in miRNA expression levels have been linked to key pathological features [85], including tau protein phosphorylation and aggregation, mitochondrial dysfunction, and Aβ peptide production (Table 1). These changes in miRNA activity contribute to neuronal dysfunction and cell death, playing a critical role in the disease’s progression. The study of miRNAs in AD offers insights into the molecular mechanisms underlying the disease and highlights the potential of miRNAs as biomarkers for diagnosis and targets for therapeutic intervention [86, 87].
In conclusion, miRNAs are crucial regulators of gene expression, influencing a wide array of biological processes and disease pathologies. The aberrant expression of miRNAs is intricately linked to the development and progression of diseases, including AD. Understanding the role and mechanisms of miRNAs in the brain and their impact on disease progression is essential for developing novel diagnostic and therapeutic strategies, particularly in the context of neurodegenerative diseases like AD.
Regulation of Aβ by MiRNAs in AD
The hallmark complications of AD, such as memory impairment and cognitive decline, are closely associated with changes in synaptic plasticity [88]. One of the important characteristics of AD patients is the alteration in synaptic plasticity, which affects the brain’s ability to adapt and process information [89]. Genome-wide transcriptome studies have revealed that many key genes involved in synaptic activity are downregulated in AD [90, 91]. These miRNAs are implicated in the formation of senile plaques and NFTs, with extracellular amyloid plaques as the most notable feature. Postmortem analyses of AD patients reveal axonal and dendritic deformations and synaptic disruption in hippocampal and neocortical areas surrounding amyloid deposits [92]. The accumulation of Aβ peptides, a byproduct of the APP processed by BACE1, plays a crucial role in neuronal cytotoxicity, representing a key pathogenic factor in AD [93].
Evidence increasingly supports the role of miRNAs in modulating Aβ production. APP, a large transmembrane glycoprotein [94], undergoes cleavage by β-secretases and γ-secretases to produce Aβ, marking the amyloidogenic pathway. Alternatively, the non-amyloidogenic pathway involves α-secretase cleaving APP, preventing Aβ peptide formation [95]. Disruption in the balance between Aβ production and clearance leads to extracellular Aβ accumulation, neurofibrillary tangle formation, neuronal degeneration, and loss of function [96]. The expression levels of Aβ are intricately linked to those of APP and BACE1. Mutations in presenilin-1, a component of the γ-secretase complex involved in Aβ production, along with deficiencies in presenilin, inactive mutants, γ-secretase inhibitors, and loss of β-APP or amyloid precursor-like protein 2 (APLP2), influence p53 expression and activity, thereby affecting AD progression [97]. Specific miRNAs have been identified as either upregulated or downregulated in AD, influencing Aβ production by modulating BACE1 and/or APP expression. For instance, miRNAs such as miR-9, miR-29, miR-29a/b-1, miR-124, miR-101, miR-107, miR-298, and miR-328 have been associated with increased Aβ production [98,99,100].
MiRNAs and BACE1
In AD patients, the expression of miR-149, miR-34a-5p, miR-125b-5p, miR-15b, miR-16, miR-124, and miR-374b-5p is significantly reduced in serum and the frontal cortex, inversely correlating with BACE1 mRNA expression [101,102,103,104,105,106]. Introduction of these miRNAs into Aβ-induced AD cell models significantly decreases BACE1 mRNA levels and Aβ accumulation. Specifically, miR-149, miR-34a-5p, miR-16, miR-29c, and miR-374b-5p directly target BACE1 to exert anti-AD effects [101, 102, 105,106,107]. Animal model studies further corroborate these findings. In the hippocampus of SAMP8 mice, a common model for studying AD, researchers have observed the downregulation of miR-340 and the upregulation of BACE1 [108]. Additionally, in AD model mice, miR-22-3p and miR-340 have been shown to significantly inhibit BACE1 and reduce Aβ levels. MiR-22-3p indirectly affected MAPK14, while miR-340 has been verified to directly target BACE1 [108, 109].
Moreover, exogenous overexpression of miRNAs can critically influence Aβ production. For example, miR-29a expression in transgenic mice leads to reduced endogenous BACE1 levels and increased Aβ [110]. Conversely, miR-195 overexpression in N2a/APP695 cells results in decreased Aβ levels, while its inhibition increases Aβ levels. This suggests that reduced expression of specific miRNAs may enhance BACE1 expression and function, contributing to the aberrant Aβ production characteristic of AD [111]. Furthermore, overexpression of miR-186 in neural cells decreases Aβ levels by inhibiting BACE1 expression, highlighting the complex regulatory network of miRNAs in AD pathogenesis and offering potential therapeutic targets for modulating Aβ production [112].
MiRNAs and APP regulation in AD
Clinical research indicates that specific miRNAs play a crucial role in regulating amyloid APP levels, thereby influencing AD pathology. For instance, overexpression of miR-106a and miR-520c has been shown to significantly reduce APP levels in HEK-293 cells, suggesting a potential therapeutic pathway for AD by targeting APP synthesis [113]. Additionally, evidence points to the involvement of miR-16 in APP accumulation. Decreased miR-16 expression was observed to correlate with increased APP protein levels in the brains of spontaneously aged accelerated mouse P8 (SAMP8), a model of AD. Conversely, miR-16 overexpression led to reduced APP protein levels both in vitro and in vivo, highlighting its regulatory impact on APP [99]. Similarly, lower expressions of miR-17, miR-101, and miR-16 have been associated with elevated APP levels [114], underscoring the inhibitory role of these miRNAs on APP expression.
These findings illuminate the complex interplay between miRNAs and key AD pathogenic factors such as BACE1, APP, and Aβ, offering new perspectives on the disease’s etiology. However, the precise contribution of diminished miRNA levels to AD initiation remains to be fully elucidated. Beyond these miRNAs, others like miR-128 have been linked to increased Aβ levels and AD progression. Notably, miR-128 expression, alongside Aβ levels, was found to be significantly elevated in the cerebral cortex of 3xTg-AD mice compared to wild-type counterparts. In contrast, miR-128 knockout mice demonstrated improved cognitive functions relative to 3xTg-AD mice, suggesting a detrimental role of miR-128 in AD [115]. Moreover, miR-126 inhibition has shown neuroprotective effects against Aβ42 toxicity, indicating that both miR-128 and miR-126 may play critical roles in AD progression [116].
These insights into miRNA-mediated regulation of APP and Aβ production not only deepen our understanding of AD’s molecular basis but also hint at novel miRNA-targeted therapeutic strategies for managing or potentially altering the course of the disease.
MiRNAs, tau phosphorylation, and aggregation in AD
The pathological hallmark of AD is not only characterized by Aβ accumulation but also by the hyperphosphorylation of tau protein, leading to the formation of NFTs. This process is a critical event in the disease’s pathogenesis. The detrimental impact of altered miRNAs in AD extends beyond Aβ pathology to include significant associations with tau phosphorylation and pathological aggregation [117]. Tau, a protein encoded by the microtubule-associated protein tau (MAPT) gene, plays a vital role in stabilizing microtubules in neuron axons under normal conditions [118,119,120]. However, in AD, tau becomes hyperphosphorylated, losing its affinity for tubulin and contributing to neuronal dysfunction and degeneration [120, 121].
Landgraf et al.‘s genomic study of brain tissue miRNAs from AD patients identified significant expression changes in over 250 miRNAs across various organ systems and cell types [122]. Notably, miR-132 and miR-425-5p levels were found to be upregulated, whereas miR-124-3p and miR-512 levels were downregulated in AD brains. MiR-132, in particular, has been highlighted for its profound regulatory role within the central nervous system (CNS). Conditional deletion studies have shown that miR-132 double-knockout mice exhibit significant cognitive deficits, implicating miR-132 dysregulation in a broad spectrum of cognitive disorders [123, 124]. Furthermore, overexpression of miR-132 in neuronal models has been linked to increased neuronal apoptosis and enhanced tau phosphorylation, suggesting its pivotal role in AD pathogenesis through the modulation of tau phosphorylation pathway [125]. Similarly, miR-425-5p overexpression has been associated with increased apoptosis, activation of glycogen synthase kinase-3β (GSK-3β), and enhanced tau phosphorylation in AD models [126]. On the other hand, miR-124-3p appears to offer a neuroprotective effect by inhibiting abnormal tau hyperphosphorylation through the caveolin-1-PI3K/Akt/GSK3β pathway [127]. The reduced expression of miR-512 in late-stage AD brains, along with its association with tau protein anomalies, further underscores the complex role of miRNAs in tau pathology [128].
Additional miRNAs, such as miR-483-5p, miR-125b-5p, have been implicated in regulating signaling pathways indirectly influencing tau phosphorylation [129, 130]. For instance, miR-483-5p-mediated inhibition of ERK1/2 has been shown to reduce tau phosphorylation, linking it to tau neuropathology in AD [129]. Moreover, melatonin’s effect on tau hyperphosphorylation, potentially mediated through miR-125b-5p regulation of protein phosphatase 2A (PP2A) activity, highlights the intricate molecular interplay involved in tau regulation [130].
These findings collectively suggest that miRNAs play a critical role in AD by modulating tau phosphorylation and aggregation, offering potential therapeutic targets for intervention. The alteration in miRNA expression patterns not only affects neuronal survival but may also contribute to the accumulation of pathogenic proteins like hyperphosphorylated tau, thereby disrupting normal neuronal function and contributing to the clinical manifestations of AD.
MiRNAs and inflammatory response in AD
Inflammation significantly contributes to the pathogenesis of CNS diseases, with neuroinflammation playing a pivotal role in AD pathology [131, 132]. Glial cells, including microglia and astrocytes, are key players in the inflammatory response to Aβ toxicity [133]. Microglia, the brain’s resident macrophages, exhibit phagocytic functions that are crucial for Aβ clearance under healthy conditions. However, Aβ aggregates can induce microglial activation, leading to the release of nitric oxide (NO), reactive oxygen species (ROS), and pro-inflammatory cytokines such as IL-1β and TNF-α [134, 135]. This inflammatory response can exacerbate neuronal damage and accelerate AD pathology [136]. When activated in response to AD pathology, astrocytes transform into reactive astrocytes that produce toxic factors through the induction of their downstream target genes [137, 138]. The interaction between neurons and astrocytes, particularly through the glutamate/glutamine cycle, is essential for neurotransmission and closely linked to astrocytic energy metabolism [139, 140]. AD-induced metabolic remodeling in astrocytes significantly impacts this cycle, contributing to disease progression [141].
Recent studies have highlighted the interplay between neuroinflammation and autophagy in AD [142,143,144], with disruptions in autophagic lysosome maturation and transport leading to autophagic vacuole accumulation. This accumulation, indicative of incomplete autophagy, correlates with Aβ aggregate buildup and autophagic dysfunction [145]. The resulting imbalance in microglial and astrocytic activity, coupled with impaired immune interactions between neurons, affects synaptic plasticity, neuronal survival, and cognition [146].
Microglia’s role in neuroinflammation
Microglia serve dual roles in brain health: they are crucial for immune defense and maintaining homeostasis. Beyond acting as the brain’s immune sentinels, microglia contribute to neuronal proliferation, differentiation, and synaptic connection formation [147]. Under physiological conditions, microglia patrol the brain, providing immune surveillance and supporting neuronal survival. Aβ aggregates trigger microglia to undergo morphological and molecular changes, adopting a disease-associated microglial (DAM) phenotype characterized by the overexpression of specific receptors, chemokines, and cytokines [148]. Pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), the receptor for advanced glycation end products (RAGE), and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), play a significant role in detecting Aβ formation. For example, studies have demonstrated that TLR signaling pathways might be involved in Aβ clearance, positioning TLRs as potential therapeutic targets for AD [149, 150]. Increased RAGE expression in AD brains suggests its involvement in neuronal dysfunction and death, mediating the effects of Aβ on neurons and microglia [151]. Additionally, NLRP3 inflammasome deficiency in AD models has been shown to reduce Aβ deposition and tilt microglial responses towards an M2 phenotype, indicating the NLRP3/caspase-1 axis’s importance in AD pathogenesis [152]. The activation of PRRs leads to the activation of various transcription factors, including NF-kB, AP-1, CREB, and IRFs, which regulate a multitude of genes involved in the inflammatory response. This intricate network of microglial activation, PRR signaling, and transcription factor regulation underscores the complex role of neuroinflammation in AD and highlights the potential for targeting these pathways to modulate disease progression [153].
The role of astrocytes in neuroinflammation
Astrocytes, the most abundant glial cells in the CNS, originate from neural stem cells (NSCs) [154]. Their functions parallel those of microglia to some extent, including maintaining homeostasis and supplying neurons with metabolites and growth factors. Astrocytes play a crucial role in synaptic formation, synaptic plasticity, regulating the blood-brain barrier [155], balancing ions and fluids, and scavenging free radicals. Moreover, they are involved in bidirectional communication with neurons, essential for maintaining CNS homeostasis and neuronal survival [156]. Under pathological conditions, such as in AD, astrocytes undergo significant morphological and functional transformations, becoming reactive astrocytes. This state is characterized by cellular hypertrophy and the excessive release of neurotoxic factors. Studies have highlighted the detrimental effects of reactive astrocytes on neuronal health [157, 158]. Like microglia, astrocytes detect Aβ aggregates via TLRs and RAGE, triggering the activation of downstream genes and the production of various neurotoxic factors [159]. Aβ exposure induces astrocytes to produce ROS, nitric oxide (NO), and a range of cytokines and chemokines, exacerbating neuroinflammation and contributing to the pathology of AD [134].
MiRNAs in neuroinflammation
The inflammatory response in AD is closely linked to changes in miRNA expression. Identifying the specific miRNAs involved in the production of pro-inflammatory cytokines and proteolytic enzymes is crucial for understanding their role in AD pathology. For instance, studies have demonstrated that miR-146a overexpression in microglia can mitigate cognitive deficits, attenuate neuroinflammation, reduce Aβ levels, and prevent neuronal loss. This is achieved by promoting a shift from the M1 pro-inflammatory phenotype to the M2 anti-inflammatory phenotype [160, 161]. This shift is associated with decreased production of pro-inflammatory cytokines and enhanced phagocytosis, offering neuronal protection. Conversely, data from human AD brains indicate that increased levels of miR-146 may intensify neuroinflammation by targeting complement factor H (CFH), a key regulatory protein involved in innate immunity and inflammation, leading to a significant reduction in CFH expression [162]. Additionally, miR-132 has been implicated in the regulation of inflammation, exerting a negative regulatory effect on inflammatory factors. In models of spinal cord injury, substances like resveratrol have been shown to modulate miR-132 expression, suggesting a protective role against AD-induced inflammatory damage [163].
MiR-155, another multifunctional miRNA, has a distinct expression profile and is involved in various physiological and pathological processes, including immunity and inflammation [164, 165]. It regulates T cell function during inflammation and has been found to be highly expressed in 3xTg AD animal models. The upregulation of miR-155 leads to enhanced activation of microglia and astrocytes, triggering the release of inflammatory mediators and potentially contributing to AD progression through the modulation of T-cell functions during inflammation [166].
These insights into the roles of specific miRNAs in neuroinflammation offer potential therapeutic targets for modulating inflammatory responses in AD. Pharmacological modulation of these miRNAs could provide novel strategies for mitigating neuroinflammation and its detrimental effects on neuronal health in AD.
MiRNAs and oxidative stress in AD
Oxidative stress (OS) is a pivotal factor in the etiology of AD and other neurodegenerative disorders. It encompasses an array of detrimental events, including enhanced production of ROS, mitochondrial dysfunction, anomalies in neuronal energy metabolism, alterations in neurotrophic signaling and cellular stress responses, dysregulation of calcium handling, and impairments in autophagy [167,168,169]. Unlike DNA, RNA is more vulnerable to oxidative damage due to its predominantly single-stranded structure, exposing its bases to oxidative agents. Moreover, the proximity of cellular RNA to mitochondria, the primary sources of ROS, exacerbates its susceptibility to oxidative modifications. These modifications are not limited to protein-coding RNAs but also affect non-coding RNAs, including miRNAs, thus influencing the complex neurobiological landscape of the mammalian brain [170, 171].
Clinical and laboratory studies across neurodegenerative diseases like AD, Parkinson’s disease, and amyotrophic lateral sclerosis have consistently highlighted OS as a critical early pathogenic event [172, 173]. OS modulates the expression of various miRNAs, which in turn regulate genes integral to the OS response [174]. For instance, Jiao et al. discovered that a natural coumarin derivative, amphorin, upregulated miR-107 expression, leading to the inhibition of BACE1, a direct target of miR-107. This interaction resulted in reduced Aβ production, suggesting a therapeutic potential for miR-107 modulation in AD [175]. Similarly, miR-125b has been implicated in promoting APP and BACE1 expression, and consequently, Aβ accumulation and apoptosis, by influencing inflammatory and OS pathways via SPHK1 targeting [176].
Furthermore, miR-125b induces GSK3β activity and excessive tau phosphorylation by targeting NCAM, implicating it in AD progression [177]. MiRNA-146a, sensitive to NF-kB, modulates inflammation by targeting the 3′ UTR of CFH, a crucial inhibitor of brain inflammatory responses. Overexpression of miR-146a has been shown to induce tau phosphorylation and modulate superoxide dismutase 2 (SOD2) expression, underscoring its role in AD pathology [178]. Dysregulation of miR-146a, therefore, contributes to tau hyperphosphorylation, suggesting that inhibiting this miRNA could offer a new therapeutic approach for AD [179, 180]. MiR-200c emerges as a neuroprotective agent in the context of Aβ-induced endoplasmic reticulum stress, supporting cell survival and neurite outgrowth by downregulating PTEN and modulating insulin signaling pathways. This indicates that miR-200c could serve as both a biomarker and a therapeutic target in AD, highlighting the potential of miRNAs as tools for early diagnosis and treatment [181,182,183].
The intricate relationships between miRNAs, OS, and neurodegenerative processes in AD point to a significant role for miRNAs in cellular and biological responses to OS. Understanding these mechanisms could unveil novel therapeutic strategies centered around neuroprotective transcriptional repressors and excitatory effects associated with OS responses, offering new avenues for combating AD.
MiRNAs, mitochondrial, and synaptic dysfunction in AD
Synaptic basis of AD
The neuropathological hallmarks of AD include senile plaques composed of Aβ aggregates and NFTs formed by hyperphosphorylated tau protein within the brain [184,185,186]. Synaptic loss is closely associated with cognitive decline in AD [187, 188], prompting extensive research into the neurotoxic effects of Aβ and hyperphosphorylated tau on synaptic integrity and neuronal viability [189]. Synapses, crucial for neurotransmission and cognitive functions, become compromised in AD, marking synaptic dysfunction as a primary feature and a key driver of disease progression [190]. Aβ‘s impact on synaptic communication precedes synaptic loss, with Aβ oligomers disrupting neurotransmitter release—an early event leading to synaptic dysfunction [191]. Conversely, tau protein, under physiological conditions, interacts with multiple cellular components, influencing cell survival and function [192, 193]. Although NFTs comprise toxic tau forms, tau-induced neuronal dysfunction, and toxicity do not solely depend on these insoluble aggregates [194]. Emerging evidence suggests that soluble tau oligomers, rather than NFTs [195, 196], may exhibit pathological activity, interfering with essential intraneuronal transport [197].
Recent study utilizing single-nucleus RNA sequencing (snRNAseq) have identified distinct microglial profiles associated with AD pathology, linking them with Aβ and tau burdens. The activation of microglia by toxic proteins like Aβ and tau not only aims at clearing these pathogenic entities but can also overstimulate microglial defense mechanisms, exacerbating neuroinflammation and furthering neuropathological conditions [198]. Pathways such as Trem2, Cx3cr1, and the granulin precursor pathway play roles in modulating microglial responses and promoting the clearance of harmful stimuli [199]. However, when microglial clearance becomes compromised, chronic brain inflammation ensues, serving as both a biomarker and a contributor to AD progression [200].
Mitochondrial dysfunction and synaptic impairment are integral to AD’s pathophysiology, affecting neurotransmission and memory [201]. The role of miRNAs in regulating neuronal mitochondrial and synaptic functions has garnered increasing interest, with numerous miRNAs implicated in mitochondrial and synaptic dysfunctions identified in AD [190]. These miRNAs, many of which are upregulated in the disease, highlight the potential of miRNA modulation as a therapeutic strategy or biomarker for AD. The dysregulation of miRNAs may play a pivotal role in AD’s onset and progression, offering new insights into the molecular underpinnings of the disease and opening avenues for targeted interventions to mitigate mitochondrial and synaptic dysfunction in AD.
Integrating mitochondrial dynamics, biogenesis, and miRNAs in AD
Mitochondria, the cellular powerhouses, are critical for generating the bulk of cellular energy in the form of ATP, essential for numerous cellular functions [202]. Their evolutionary origin, theorized to stem from a symbiotic relationship between proto-eukaryotic cells and aerobic bacteria, highlights their integral role in eukaryotic cell biology [203]. This unique origin is supported by similarities between mitochondrial and bacterial DNA, including their mode of propagation through binary division. Over time, this symbiotic bacterium evolved into the mitochondria observed in today’s eukaryotic cells.
Mitochondrial dysfunction is a hallmark of AD, characterized by dynamic damage and impaired mitophagy, which are closely linked to synaptic damage [204]. The laboratory of Reddy and others has extensively explored how mitochondrial dysfunction contributes to AD progression, emphasizing the association between impaired mitochondrial dynamics and synaptic loss [201, 205,206,207]. This dysfunction includes alterations in mitochondrial DNA, protein import, and respiratory function, all contributing to the neurodegenerative process.
Recent discoveries have highlighted the presence of mitochondrial miRNAs, which regulate genes essential for mitochondrial function. These miRNAs have been implicated in the pathogenesis of AD, affecting energy production and contributing to the disease’s progression [208, 209]. For instance, miR-743a [210] and miR-23a/b [211] disrupt the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS), leading to reduced ATP production and neuronal death. Additionally, miR-210 [212], miR-338 [213], and miR-34a [214] target OXPHOS-related enzymes, further compromising neuronal energy demands. In neural cell lines, miR-16-5p has been shown to target BCL-2, an anti-apoptotic factor, inducing apoptosis and contributing to neuronal loss observed in AD [215]. The dysregulation of mitochondrial fusion protein 2 (mfn2) by miR-195 and the regulation of Nurr1 levels by miR-132 highlight the complex interplay between mitochondrial miRNAs and mitochondrial function in the context of AD [216].
Mitochondrial biogenesis is crucial for meeting the high energy demands of neurons [217], particularly in the brain, which consumes a significant portion of the body’s energy. This process is vital for sustaining continuous synaptic activity and action potential generation by neurons [218]. Kumar et al. found that miR-455-3p promotes mitochondrial biogenesis, enhancing cell survival and reducing Aβ toxicity [219]. Similarly, miR-34a [220] and miR-23a/b [221, 222] have been identified as regulators of mitochondrial biogenesis, offering neuroprotection against Aβ toxicity by modulating the SIRT1 signaling pathway.
The intricate relationship between mitochondrial dysfunction, miRNA regulation, and AD underscores the complexity of AD pathology. Mitochondrial miRNAs play a pivotal role in regulating mitochondrial dynamics, energy production, and synaptic function, directly impacting the progression of neurodegenerative diseases like AD [223]. Understanding the mechanisms by which mitochondrial dysfunction and miRNA dysregulation contribute to AD offers potential pathways for therapeutic intervention, aiming to restore mitochondrial function and neuronal health. As research continues to unravel these complex interactions, the potential for developing targeted treatments to mitigate the impact of mitochondrial dysfunction in AD grows, offering hope for future advancements in the management and treatment of this debilitating condition.
Synaptic dysfunction and mitochondrial miRNAs in AD
Synaptic activity is fundamental to brain function and neurotransmission. It underpins our ability to process information, form memories, and adapt to new experiences [224]. In AD, synaptic dysfunction is a critical pathological feature leading to cognitive decline, impaired neural processing, and ultimately, neuronal death. The intricate relationship between synaptic activity and mitochondrial function highlights the complexity of AD’s impact on neuronal health.
MiRNAs play a significant role in modulating synaptic function and neurotransmission. Specific miRNAs, such as miR-484, miR-132, and miR-212, are crucial for maintaining synaptic integrity and facilitating neurotransmitter release. miR-132 and miR-212, in particular, have been shown to enhance neurotransmitter levels, thereby supporting efficient neural signaling. However, in AD, the dysregulation of these miRNAs contributes to a decline in neurotransmitter availability, limiting neural processing capabilities and exacerbating cognitive deficits [225]. Moreover, Wingo et al. found that lower miR-484 levels were associated not only with a higher probability of having AD but also with greater depressive symptoms [226].
The role of miRNAs extends beyond the regulation of synaptic activity to encompass mitochondrial functions critical for energy production and cellular health. Mitochondrial miRNAs are of particular interest due to their potential involvement in AD pathogenesis. These miRNAs regulate genes encoding proteins essential for mitochondrial dynamics, energy metabolism, and apoptosis, directly impacting neuronal survival and function. While the significance of mitochondrial miRNAs in regulating mitochondrial health is well recognized, their specific contributions to synaptic functions remain less understood. Current knowledge on how mitochondrial miRNAs influence processes like mitochondrial transport, calcium signaling, and synaptic vesicle formation is limited. These processes are vital for effective neurotransmission and synaptic plasticity, suggesting that mitochondrial miRNAs could play a role in modulating these critical aspects of neuronal function.
The investigation into miRNAs associated with synaptic activity, neurotransmission, synaptic plasticity, and neurotoxicity has revealed only a handful of candidates. This gap in knowledge presents a significant opportunity for research aimed at identifying new miRNAs involved in these processes and understanding their regulation in the context of AD. Unraveling the specific roles of mitochondrial miRNAs in synaptic dysfunction could shed light on the molecular mechanisms driving cognitive decline in AD and offer new targets for therapeutic intervention.
Interestingly, it has been suggested that a complex interaction exists between miRNAs, mitochondrial dysfunction, and oxidative stress. On the one hand, miRNAs can regulate mitochondrial function, thus affecting the degree of oxidative stress; on the other hand, mitochondrial dysfunction and oxidative stress also affect miRNA expression and function [227, 228]. This interactive relationship plays an important role in balancing the redox state and maintaining cellular homeostasis within the cell, which is of great significance for cell survival and function [229]. The bidirectional influence between miRNAs and mitochondrial health highlights the importance of these molecules in the cellular response to oxidative stress and their potential as therapeutic targets for diseases involving mitochondrial dysfunction.
miRNAs and APOE4 in AD
The APOE4 allele is recognized as the most significant genetic risk factor for sporadic Alzheimer’s disease (sAD) [230], with its role first proposed by Strittmatter and Roses and subsequently supported by various genome-wide association studies (GWAS), cellular, and animal model research [231,232,233,234]. APOE’s primary function in the brain involves the transportation of cholesterol within high-density lipoprotein particles, but its polymorphic forms, namely APOE2, APOE3, and APOE4, show varied associations with AD risk, with APOE4 being strongly linked to increased susceptibility to the disease [235].
APOE4’s contribution to AD pathogenesis is multifaceted, affecting lipid and lipoprotein metabolism and influencing the plasma levels of APOE. Notably, the presence of APOE4 alleles is inversely related to miR-107 levels in AD patients, suggesting a genetic interaction that modulates disease risk and progression. The APOE E3/E3 genotype, conversely, is associated with higher APOE concentrations and miR-107 levels, indicating a protective or neutral effect regarding AD. Recent studies have illuminated the role of specific miRNAs in modulating the effects of APOE4 in AD [236,237,238]. For instance, the expression level of miR-195 in brain tissue and cerebrospinal fluid samples from AD patients carrying the APOE4 allele is significantly lower compared to those without the allele. This reduction in miR-195 levels correlates negatively with cognitive performance, as measured by the Mini-Mental State Examination (MMSE) score. However, upregulation of miR-195 in animal models expressing the APOE4 allele has shown promising results, including ameliorated cognitive deficits, reduced amyloid plaque burden, and decreased tau hyperphosphorylation [237]. Furthermore, the expression levels of miR-203, phosphorylated tau (p-Tau), and APOE4 significantly increase in the brain tissue of mice subjected to traumatic brain injury (TBI), linking TBI with an elevated risk of AD pathology in the context of APOE4. The use of miR-203 inhibitors has been shown to reduce the expression of APOE4 and p-Tau levels, improving hippocampal long-term potentiation (LTP) defects and ameliorating learning and memory dysfunctions induced by TBI [238].
The intricate interplay between APOE4, miRNAs, and AD pathology underscores the complexity of genetic and molecular mechanisms contributing to the disease. APOE4 not only influences lipid metabolism and amyloid-beta deposition but also interacts with specific miRNAs to modulate disease progression and severity. These findings highlight the potential of targeting miRNAs as a therapeutic strategy to mitigate the adverse effects of APOE4 in AD, offering new avenues for research and treatment development. Understanding the regulatory roles of miRNAs in the context of APOE4 can provide insights into personalized medicine approaches for managing AD, tailoring interventions based on genetic risk factors and molecular pathologies.
Conclusion and future perspective on AD research and miRNAs
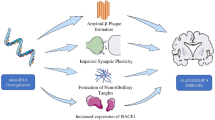
AD, traditionally characterized by the concurrent presence of Aβ plaques and tau protein tangles, is undergoing a paradigm shift in its conceptual understanding. The simplistic linear causality proposed by the original amyloid hypothesis is being reconsidered, giving way to a more nuanced view that incorporates age-related, protective, and disease-promoting factors interacting with the disease’s core mechanisms. This paper has discussed critical elements in AD pathogenesis, including Aβ deposition, intracellular hyperphosphorylated tau aggregation, synaptic loss, neuroinflammation, OS, and genetic variations, all of which are intertwined with the dysregulation of a myriad of miRNAs (Fig. 2). MiRNAs have emerged as pivotal molecules in the regulatory networks underlying AD’s complex pathology. Their ability to modulate key pathogenic processes presents a promising avenue for research and therapeutic intervention. Unlike the more complex molecular neuroimaging techniques, such as structural magnetic resonance imaging (MRI) and positron emission tomography (PET), analyzing miRNAs in bodily fluids offers a relatively straightforward and minimally invasive method for studying AD [239, 240]. This simplicity, combined with their regulatory significance, positions miRNAs as valuable biomarkers for early detection and progression monitoring of AD.

Specific microRNAs play key roles in the development of Alzheimer's disease (AD), regulating Aβ deposition, Tau hyperphosphorylation, synaptic dysfunction, neuroinflammation, oxidative stress, and genetic factors. All these factors are intertwined with the dysregulation of myriad miRNAs. These microRNAs, as biomarkers, not only help in early diagnosis and disease monitoring, but may also become new therapeutic targets and provide new ideas for intervention strategies in Alzheimer’s disease. [miR microRNA, APP amyloid precursor protein, BACE1 beta-secretase 1, PS1/2 Presenilin1/2, TNF-α tumor necrosis factor α, IL interleukin, TGF-β transforming growth factor β, NF-κB nuclear factor Kappa B, SOCS suppressor of cytokine signaling, STAT signal transducer and activator of transcription, OH hydroxyl radical, O2 oxygen, NO nitric oxide, PHF paired helical filaments, NFT neurofibrillary tangle, SOD2 superoxide dismutase 2, GSK3β glycogen synthase kinase 3β, NCAM neural cell adhesion molecule, BCL-2 B-cell lymphoma-2, MFN2 mitochondrial fusion protein 2, MDH2 malate dehydrogenase, MCL1 myeloid cell leukemia 1, ERK1/2 extracellular regulated protein kinases1/2].
To evaluate the potential of miRNAs as early non-invasive markers for AD diagnosis, a meta-analysis was conducted to screen differentially expressed miRNAs in blood exosomes of AD patients. The study revealed significantly lower levels of blood miR-212 and miR-132 in AD patients compared to healthy controls [241]. Conversely, serum levels of exosomal miR-135a, miR-193b, and miR-384 were markedly increased in AD patients [242]. Several miRNAs showing significant differential expression were selected for validation and diagnostic testing. Notably, miR-132 demonstrated a robust ability to distinguish AD from controls in blood assays [243]. Most AD patients exhibit memory cognitive decline, often linked to impaired neurogenesis. Walgrave et al. identified miR-132 as one of the most consistently downregulated miRNAs in AD. Decreased miR-132 expression was shown to impair adult hippocampal neurogenesis (AHN), a process crucial for memory and cognitive plasticity. In mouse models, miR-132 replacement in the AD hippocampus restored AHN and ameliorated associated memory deficits. This effect was influenced by Aβ pathology [244]. Deng et al. found that miR-132 expression was significantly downregulated, while MAPK1 expression was upregulated in an AD rat model. They suggested miR-132 and MAPK1 have specific binding sites, and that miR-132 can inhibit MAPK1 expression. This inhibition reduced hippocampal oxidative stress and iNOS expression, thereby improving cognitive function in AD rats [245]. El Fatimy et al. demonstrated that miR-132 exerts neuroprotective effects against AD through multiple signaling pathways. miR-132 mitigates tau protein pathology associated with tauopathy, reduces phosphorylated tau in P301S tau transgenic mice, and enhances long-term potentiation [246]. These results suggests that modulation of miR-132 expression could provide new therapeutic strategies for AD.
A comprehensive understanding of specific miRNAs’ regulatory roles in AD is essential for advancing therapeutic strategies. Future research should aim to elucidate the intricate molecular mechanisms by which miRNAs influence AD pathogenesis, with a focus on their interaction with known risk factors and pathological markers. The integration of large-scale GWAS and transcriptomics studies has significantly advanced our understanding of the genetic basis of AD [247]. By identifying associations between single nucleotide polymorphisms and AD risk, as well as differentially expressed genes associated with AD, researchers have gained insights into the molecular mechanisms underlying the disease. One promising approach involves analyzing molecular interactions within the context of disease networks, known as interactomes [248]. This method considers the collective dysfunction of disease-related genes and their interactions, providing a comprehensive view of AD pathophysiology at multiple levels [249].
Studies have revealed associations between AD and various molecular networks, including those involved in lipid metabolism and immune dysfunction. For instance, the APOE ε4 allele has been implicated in lipid metabolism, while immune dysfunction in microglia has also been linked to AD [250,251,252,253]. Recent research has identified a significant association between genetic variants in TMEM106B and APOE mRNA levels in the human brain cortex, highlighting its potential role in AD [254]. TMEM106B has also been implicated as a risk gene for frontotemporal lobar degeneration (FTLD) [255]. Further investigation has revealed specific microRNA regulators of TMEM106B, such as miR-132 and miR-212, which are attenuated in FTLD. Overexpression of TMEM106B disrupts the endosomal-lysosomal pathway and alters intracellular protein levels, suggesting its involvement in AD pathogenesis [256].
By combining genomics, proteomics, and bioinformatics methods, we can construct miRNA-target gene networks, revealing the regulatory network of miRNAs in the pathogenesis of AD. This approach will allow us to study the regulatory pathways and signaling of miRNAs, providing a comprehensive understanding of the complex mechanisms involved in AD. Ultimately, this knowledge will support the development of relevant therapeutic strategies for AD, offering significant insights into the disease’s pathogenesis and potential treatment avenues.
The potential of miRNAs as therapeutic agents in AD is vast but yet to be fully realized. Developing miRNA-based therapies will involve overcoming challenges related to delivery, targeting specificity, and off-target effects. Innovative approaches, such as nanoparticle-based delivery systems, could provide solutions to these challenges, enabling precise modulation of pathological miRNA levels within the brain. As the field moves forward, bridging the gap between research findings and clinical applications will be crucial. This transition will necessitate rigorous clinical trials to validate the efficacy and safety of miRNA-based diagnostics and therapeutics. Furthermore, personalized medicine approaches, which tailor interventions based on individual genetic and molecular profiles, could significantly enhance treatment outcomes for AD patients.
In conclusion, miRNAs represent a frontier in AD research with the potential to revolutionize diagnostics and therapeutics. As our understanding of their roles in AD deepens, the prospects for developing effective interventions to delay, halt, or reverse the progression of this devastating disease become increasingly tangible. The journey from bench to bedside is complex and fraught with challenges, but the potential rewards for patients and society are immense, driving the continued pursuit of knowledge in this promising area of neuroscience.
Data availability
Data available on request from the authors.
References
Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63–75.e2.
Sugino H, Watanabe A, Amada N, Yamamoto M, Ohgi Y, Kostic D, et al. Global trends in Alzheimer disease clinical development: increasing the probability of success. Clin Ther. 2015;37:1632–42.
Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–39.
Puzzo D, Piacentini R, Fá M, Gulisano W, Li Puma DD, Staniszewski A, et al. LTP and memory impairment caused by extracellular Aβ and Tau oligomers is APP-dependent. eLife. 2017;6:e26991.
Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and management of dementia: review. JAMA. 2019;322:1589–99.
Nordberg A. PET imaging of amyloid in Alzheimer’s disease. Lancet Neurol. 2004;3:519–27.
Nagaraj S, Zoltowska KM, Laskowska-Kaszub K, Wojda U. microRNA diagnostic panel for Alzheimer’s disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res Rev. 2019;49:125–43.
Piscopo P, Lacorte E, Feligioni M, Mayer F, Crestini A, Piccolo L, et al. MicroRNAs and mild cognitive impairment: a systematic review. Ageing Res Rev. 2019;50:131–41.
Lehmann SM, Krüger C, Park B, Derkow K, Rosenberger K, Baumgart J, et al. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 2012;15:827–35.
Liu X, Jiao B, Shen L. The epigenetics of Alzheimer’s disease: factors and therapeutic implications. Front Genet. 2018;9:579.
Wang M, Qin L, Tang B. MicroRNAs in Alzheimer’s disease. Front Genet. 2019;10:153.
Jung YY, Kim KC, Park MH, Seo Y, Park H, Park MH, et al. Atherosclerosis is exacerbated by chitinase-3-like-1 in amyloid precursor protein transgenic mice. Theranostics. 2018;8:749–66.
Mendes-Silva AP, Pereira KS, Tolentino-Araujo GT, Nicolau Ede S, Silva-Ferreira CM, Teixeira AL, et al. Shared biologic pathways between Alzheimer disease and major depression: a systematic review of MicroRNA expression studies. Am J Geriatr Psychiatry. 2016;24:903–12.
Gholami A, Dehghan G, Rashtbari S, Jouyban A. Probing the interactions of lamotrigine and phenobarbital with tau protein: experimental and molecular modeling studies. Iran J Pharm Res. 2022;21:e129599.
Ashrafian H, Zadeh EH, Khan RH. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int J Biol Macromol. 2021;167:382–94.
Funamoto S, Tagami S, Okochi M, Morishima-Kawashima M. Successive cleavage of β-amyloid precursor protein by γ-secretase. Semin Cell Dev Biol. 2020;105:64–74.
Nguyen KV. β-Amyloid precursor protein (APP) and the human diseases. AIMS Neurosci. 2019;6:273–81.
Ristori E, Donnini S, Ziche M. New insights into blood-brain barrier maintenance: the homeostatic role of β-amyloid precursor protein in cerebral vasculature. Front Physiol. 2020;11:1056.
Parihar MS, Hemnani T. Alzheimer’s disease pathogenesis and therapeutic interventions. J Clin Neurosci. 2004;11:456–67.
Mockett BG, Ryan MM. The therapeutic potential of the neuroactive peptides of soluble amyloid precursor protein-alpha in Alzheimer’s disease and related neurological disorders. Semin Cell Dev Biol. 2023;139:93–101.
Tamagno E, Guglielmotto M, Vasciaveo V, Tabaton M. Oxidative stress and beta amyloid in Alzheimer’s disease. Which comes first: the chicken or the egg? Antioxidants. 2021;10:1479.
Tsai AP, Dong C, Lin PB, Messenger EJ, Casali BT, Moutinho M, et al. PLCG2 is associated with the inflammatory response and is induced by amyloid plaques in Alzheimer’s disease. Genome Med. 2022;14:17.
Bhatia V, Sharma S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J Neurol Sci. 2021;421:117253.
Kim SH, Lim KH, Yang S, Joo JY. Boosting of tau protein aggregation by CD40 and CD48 gene expression in Alzheimer’s disease. FASEB J. 2023;37:e22702.
Ariafar S, Makhdoomi S, Mohammadi M. Arsenic and tau phosphorylation: a mechanistic review. Biol Trace Elem Res. 2023;201:5708–20.
Moore KBE, Hung TJ, Fortin JS. Hyperphosphorylated tau (p-tau) and drug discovery in the context of Alzheimer’s disease and related tauopathies. Drug Discov Today. 2023;28:103487.
Zhang W, Shen XY, Zhang WW, Chen H, Xu WP, Wei W. Corrigendum to “Di-(2-ethylhexyl) phthalate could disrupt the insulin signaling pathway in liver of SD rats and L02 cells via PPARγ“. Toxicol Appl Pharmacol. 2022;449:116091. 10.1016/j. taap.2016.12.010
Lee S, Cho HJ, Ryu JH. Innate immunity and cell death in Alzheimer’s disease. ASN Neuro. 2021;13:17590914211051908.
Tellechea P, Pujol N, Esteve-Belloch P, Echeveste B, García-Eulate MR, Arbizu J, et al. Early- and late-onset Alzheimer disease: Are they the same entity? Neurologia. 2018;33:244–53.
Dong HK, Gim JA, Yeo SH, Kim HS. Integrated late onset Alzheimer’s disease (LOAD) susceptibility genes: cholesterol metabolism and trafficking perspectives. Gene. 2017;597:10–16.
Mendez MF. Early-onset Alzheimer’s disease: nonamnestic subtypes and type 2 AD. Arch Med Res. 2012;43:677–85.
Mendez MF, Lee AS, Karve SJ, Shapira JS. Nonamnestic presentations of early-onset Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2012;27:413–20.
Joubert S, Gour N, Guedj E, Didic M, Guériot C, Koric L, et al. Early-onset and late-onset Alzheimer’s disease are associated with distinct patterns of memory impairment. Cortex. 2016;74:217–32.
Jiang H, Jayadev S, Lardelli M, Newman M. A review of the familial Alzheimer’s disease locus PRESENILIN 2 and its relationship to PRESENILIN 1. J Alzheimers Dis. 2018;66:1323–39.
Williams T, Borchelt DR, Chakrabarty P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. Mol Neurodegener. 2020;15:8.
Cash MK, Rockwood K, Fisk JD, Darvesh S. Clinicopathological correlations and cholinesterase expression in early-onset familial Alzheimer’s disease with the presenilin 1 mutation, Leu235Pro. Neurobiol Aging. 2021;103:31–41.
Yang W, Wu PF, Ma JX, Liao MJ, Xu LS, Xu MH, et al. Presenilin1 exerts antiproliferative effects by repressing the Wnt/β-catenin pathway in glioblastoma. Cell Commun Signal. 2020;18:22.
Lanfranco MF, Ng CA, Rebeck GW. ApoE lipidation as a therapeutic target in Alzheimer’s disease. Int J Mol Sci. 2020;21:6336.
Saunders TS, Jenkins N, Blennow K, Ritchie C, Muniz-Terrera G. Interactions between apolipoprotein E, sex, and amyloid-beta on cerebrospinal fluid p-tau levels in the European prevention of Alzheimer’s dementia longitudinal cohort study (EPAD LCS). EeBioMedicine. 2022;83:104241.
Troutwine BR, Hamid L, Lysaker CR, Strope TA, Wilkins HM. Apolipoprotein E and Alzheimer’s disease. Acta Pharm Sin B. 2022;12:496–510.
Nakamura T, Kawarabayashi T, Ueda T, Shimomura S, Hoshino M, Itoh K, et al. Plasma ApoE4 levels are lower than ApoE2 and ApoE3 levels, and not associated with plasma Aβ40/42 ratio as a biomarker of amyloid-β amyloidosis in Alzheimer’s disease. J Alzheimers Dis. 2023;93:333–48.
Bonaterra-Pastra A, Benítez S, Pancorbo O, Rodríguez-Luna D, Vert C, Rovira A, et al. Association of candidate genetic variants and circulating levels of ApoE/ApoJ with common neuroimaging features of cerebral amyloid angiopathy. Front Aging Neurosci. 2023;15:1134399.
Alagarsamy J, Jaeschke A, Hui DY. Apolipoprotein E in cardiometabolic and neurological health and diseases. Int J Mol Sci. 2022;23:9892.
Wu L, Zhang Y, Zhao H, Rong G, Huang P, Wang F, et al. Dissecting the association of apolipoprotein E gene polymorphisms with type 2 diabetes mellitus and coronary artery disease. Front Endocrinol. 2022;13:838547.
Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, et al. The association between head trauma and Alzheimer’s disease. Am J Epidemiol. 1990;131:491–501.
Shokhirev MN, Johnson AA. An integrative machine-learning meta-analysis of high-throughput omics data identifies age-specific hallmarks of Alzheimer’s disease. Ageing Res Rev. 2022;81:101721.
Liu RM. Aging, cellular senescence, and Alzheimer’s disease. Int J Mol Sci. 2022;23:1989.
Zhang XX, Tian Y, Wang ZT, Ma YH, Tan L, Yu JT. The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J Prev Alzheimers Dis. 2021;8:313–21.
Askarova S, Umbayev B, Masoud AR, Kaiyrlykyzy A, Safarova Y, Tsoy A, et al. The links between the gut microbiome, aging, modern lifestyle and Alzheimer’s disease. Front Cell Infect Microbiol. 2020;10:104.
Bhatti GK, Reddy AP, Reddy PH, Bhatti JS. Lifestyle modifications and nutritional interventions in aging-associated cognitive decline and Alzheimer’s disease. Front Aging Neurosci. 2019;11:369.
Xie C, Feng Y. Alcohol consumption and risk of Alzheimer’s disease: a dose-response meta-analysis. Geriatr Gerontol Int. 2022;22:278–85.
Pal I, Dey SG. The role of heme and copper in Alzheimer’s disease and type 2 diabetes mellitus. JACS Au. 2023;3:657–81.
Das S, Ramachandran AK, Halder D, Akbar S, Ahmed B, Joseph A. Mechanistic and etiological similarities in diabetes mellitus and Alzheimer’s disease: antidiabetic drugs as optimistic therapeutics in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2023;22:973–93.
Sun Y, Ma C, Sun H, Wang H, Peng W, Zhou Z, et al. Metabolism: a novel shared link between diabetes mellitus and Alzheimer’s disease. J Diabetes Res. 2020;2020:4981814.
Sáiz-Vazquez O, Puente-Martínez A, Pacheco-Bonrostro J, Ubillos-Landa S. Blood pressure and Alzheimer’s disease: a review of meta-analysis. Front Neurol. 2022;13:1065335.
Sible IJ, Nation DA. Long-term blood pressure variability across the clinical and biomarker spectrum of Alzheimer’s disease. J Alzheimers Dis. 2020;77:1655–69.
Daniela M, Grigoras C, Cuciureanu D, Constantinescu V. The circadian rhythm of arterial blood pressure in Alzheimer’s disease and vascular dementia. Acta Neurol Belg. 2023;123:129–37.
Vasefi M, Ghaboolian-Zare E, Abedelwahab H, Osu A. Environmental toxins and Alzheimer’s disease progression. Neurochem Int. 2020;141:104852.
Kumar NN, Chan YL, Chen H, Oliver BG. Editorial: effects of environmental toxins on brain health and development. Front Mol Neurosci. 2023;16:1149776.
Falandysz J, Letter to the editor: Comment on “multiannual monitoring (1974-2019) of rare earth elements in wild growing edible mushroom species in Polish forests” by Siwulski et al. 10.1016/j.chemosphere.2020.127173. A recurring question - What are the real concentrations and patterns of REE in mushrooms? Chemosphere. 2023;312:137219.
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–81.
Nguyen M, He T, An L, Alexander DC, Feng J, Yeo BTT. Predicting Alzheimer’s disease progression using deep recurrent neural networks. Neuroimage. 2020;222:117203.
Schermer MHN. Preclinical disease or risk factor? Alzheimer’s disease as a case study of changing conceptualizations of disease. J Med Philos. 2023;48:322–34.
O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. 2018;9:402.
Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta. 2010;1803:1231–43.
Starega-Roslan J, Koscianska E, Kozlowski P, Krzyzosiak WJ. The role of the precursor structure in the biogenesis of microRNA. Cell Mol Life Sci. 2011;68:2859–71.
Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10:185–91.
Ambrus AM, Frolov MV. The diverse roles of RNA helicases in RNAi. Cell Cycle. 2009;8:3500–5.
Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20:5–20.
van den Berg A, Mols J, Han J. RISC-target interaction: cleavage and translational suppression. Biochim Biophys Acta. 2008;1779:668–77.
Liu CG, Song J, Zhang YQ, Wang PC. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer’s disease. Mol Med Rep. 2014;10:2395–400.
Ørom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5’UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–71.
Bhattacharyya NP, Das E, Bucha S, Das S, Choudhury A. Regulation of cell cycle associated genes by microRNA and transcription factor. Microrna. 2016;5:180–200.
Liu B, Shyr Y, Cai J, Liu Q. Interplay between miRNAs and host genes and their role in cancer. Brief Funct Genomics. 2018;18:255–66.
Parizi PK, Yarahmadi F, Tabar HM, Hosseini Z, Sarli A, Kia N, et al. MicroRNAs and target molecules in bladder cancer. Med Oncol. 2020;37:118.
Colpaert RMW, Calore M. Epigenetics and microRNAs in cardiovascular diseases. Genomics. 2021;113:540–51.
Nikolac Perkovic M, Videtic Paska A, Konjevod M, Kouter K, Svob Strac D, Nedic Erjavec G, et al. Epigenetics of Alzheimer’s disease. Biomolecules. 2021;11:195.
Kumar S, Chawla YK, Ghosh S, Chakraborti A. Severity of hepatitis C virus (genotype-3) infection positively correlates with circulating microRNA-122 in patients sera. Dis Markers. 2014;2014:435476.
Kumar S, Batra A, Kanthaje S, Ghosh S, Chakraborti A. Crosstalk between microRNA-122 and FOX family genes in HepG2 cells. Exp Biol Med. 2017;242:436–40.
Kumar S, Vijayan M, Reddy PH. MicroRNA-455-3p as a potential peripheral biomarker for Alzheimer’s disease. Hum Mol Genet. 2017;26:3808–22.
Kumar S, Reddy PH. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim Biophys Acta. 2016;1862:1617–27.
Vijayan M, Kumar S, Yin X, Zafer D, Chanana V, Cengiz P, et al. Identification of novel circulatory microRNA signatures linked to patients with ischemic stroke. Hum Mol Genet. 2018;27:2318–29.
Zhao Y, Jaber VR, LeBeauf A, Sharfman NM, Lukiw WJ. microRNA-34a (miRNA-34a) mediated down-regulation of the post-synaptic cytoskeletal element SHANK3 in sporadic Alzheimer’s disease (AD). Front Neurol. 2019;10:28.
Lukiw WJ. microRNA-146a signaling in Alzheimer’s disease (AD) and Prion disease (PrD). Front Neurol. 2020;11:462.
Silvestro S, Bramanti P, Mazzon E. Role of miRNAs in Alzheimer’s disease and possible fields of application. Int J Mol Sci. 2019;20:3979.
Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–12.
Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. 2012;125:7–17.
Rabin LA, Smart CM, Amariglio RE. Subjective cognitive decline in preclinical Alzheimer’s disease. Annu Rev Clin Psychol. 2017;13:369–96.
Koch G, Spampinato D. Alzheimer disease and neuroplasticity. Handb Clin Neurol. 2022;184:473–9.
Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci USA. 2008;105:4441–6.
Humphries C, Kohli MA. Rare variants and transcriptomics in Alzheimer disease. Curr Genet Med Rep. 2014;2:75–84.
Kou X, Chen D, Chen N. The regulation of microRNAs in Alzheimer’s disease. Front Neurol. 2020;11:288.
Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet. 2016;388:505–17.
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32.
Kandel P, Semerci F, Mishra R, Choi W, Bajic A, Baluya D, et al. Oleic acid is an endogenous ligand of TLX/NR2E1 that triggers hippocampal neurogenesis. Proc Natl Acad Sci USA. 2022;119:e2023784119.
Sanabria-Castro A, Alvarado-Echeverría I, Monge-Bonilla C. Molecular pathogenesis of Alzheimer’s disease: an update. Ann Neurosci. 2017;24:46–54.
Alves da Costa C, Sunyach C, Pardossi-Piquard R, Sévalle J, Vincent B, Boyer N, et al. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. J Neurosci. 2006;26:6377–85.
Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci USA. 2008;105:6415–20.
Hébert SS, Horré K, Nicolaï L, Bergmans B, Papadopoulou AS, Delacourte A, et al. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol Dis. 2009;33:422–8.
Long JM, Lahiri DK. MicroRNA-101 downregulates Alzheimer’s amyloid-β precursor protein levels in human cell cultures and is differentially expressed. Biochem Biophys Res Commun. 2011;404:889–95.
Zhang J, Wang R. Deregulated lncRNA MAGI2-AS3 in Alzheimer’s disease attenuates amyloid-β induced neurotoxicity and neuroinflammation by sponging miR-374b-5p. Exp Gerontol. 2021;144:111180.
Du W, Lei C, Dong Y. MicroRNA-149 is downregulated in Alzheimer’s disease and inhibits β-amyloid accumulation and ameliorates neuronal viability through targeting BACE1. Genet Mol Biol. 2021;44:e20200064.
An F, Gong G, Wang Y, Bian M, Yu L, Wei C. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget. 2017;8:114065–71.
Gong G, An F, Wang Y, Bian M, Yu LJ, Wei C. miR-15b represses BACE1 expression in sporadic Alzheimer’s disease. Oncotarget. 2017;8:91551–7.
Zhong Z, Yuan K, Tong X, Hu J, Song Z, Zhang G, et al. MiR-16 attenuates β-amyloid-induced neurotoxicity through targeting β-site amyloid precursor protein-cleaving enzyme 1 in an Alzheimer’s disease cell model. Neuroreport. 2018;29:1365–72.
Li P, Xu Y, Wang B, Huang J, Li Q. miR-34a-5p and miR-125b-5p attenuate Aβ-induced neurotoxicity through targeting BACE1. J Neurol Sci. 2020;413:116793.
Cao Y, Tan X, Lu Q, Huang K, Tang X, He Z. MiR-29c-3p may promote the progression of Alzheimer’s disease through BACE1. J Healthc Eng. 2021;2021:2031407.
Tan X, Luo Y, Pi D, Xia L, Li Z, Tu Q. MiR-340 reduces the accumulation of Amyloid-β through targeting BACE1 (β-site amyloid precursor protein cleaving enzyme 1) in Alzheimer’s disease. Curr Neurovasc Res. 2020;17:86–92.
Ji Q, Wang X, Cai J, Du X, Sun H, Zhang N. MiR-22-3p regulates amyloid β deposit in mice model of Alzheimer’s disease by targeting mitogen-activated protein kinase 14. Curr Neurovasc Res. 2019;16:473–80.
Delay C, Mandemakers W, Hébert SS. MicroRNAs in Alzheimer’s disease. Neurobiol Dis. 2012;46:285–90.
Zhu HC, Wang LM, Wang M, Song B, Tan S, Teng JF, et al. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res Bull. 2012;88:596–601.
Kim J, Yoon H, Chung DE, Brown JL, Belmonte KC, Kim J. miR-186 is decreased in aged brain and suppresses BACE1 expression. J Neurochem. 2016;137:436–45.
Patel N, Hoang D, Miller N, Ansaloni S, Huang Q, Rogers JT, et al. MicroRNAs can regulate human APP levels. Mol Neurodegener. 2008;3:10.
Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y, et al. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol Aging. 2012;33:522–34.
Yaksh TL. TRPV1 expression regulation… A further step in defining its biology: commentary for K. Zavala et al. “The anticancer antibiotic mithramycin-A inhibits TRPV1 expression in dorsal root ganglion neurons” [Neurosci. Lett. (2014) doi:10.1016/j.neulet.2014.01.021]. Neurosci Lett. 2014;578:209–10.
Tiribuzi R, Crispoltoni L, Porcellati S, Di Lullo M, Florenzano F, Pirro M, et al. miR128 up-regulation correlates with impaired amyloid β(1-42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol Aging. 2014;35:345–56.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–26.
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44.
Aamodt EJ, Williams RC Jr. Microtubule-associated proteins connect microtubules and neurofilaments in vitro. Biochemistry. 1984;23:6023–31.
González-Billault C, Engelke M, Jiménez-Mateos EM, Wandosell F, Cáceres A, Avila J. Participation of structural microtubule-associated proteins (MAPs) in the development of neuronal polarity. J Neurosci Res. 2002;67:713–9.
Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7:656–64.
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–14.
Hansen KF, Sakamoto K, Aten S, Price KH, Loeser J, Hesse AM, et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn Mem. 2016;23:61–71.
Hernandez-Rapp J, Smith PY, Filali M, Goupil C, Planel E, Magill ST, et al. Memory formation and retention are affected in adult miR-132/212 knockout mice. Behav Brain Res. 2015;287:15–26.
Liu DY, Zhang L. MicroRNA-132 promotes neurons cell apoptosis and activates Tau phosphorylation by targeting GTDC-1 in Alzheimer’s disease. Eur Rev Med Pharmacol Sci. 2019;23:8523–32.
Yuan J, Wu Y, Li L, Liu C. MicroRNA-425-5p promotes tau phosphorylation and cell apoptosis in Alzheimer’s disease by targeting heat shock protein B8. J Neural Transm. 2020;127:339–46.
Kang Q, Xiang Y, Li D, Liang J, Zhang X, Zhou F, et al. MiR-124-3p attenuates hyperphosphorylation of Tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells. Oncotarget. 2017;8:24314–26.
Mezache L, Mikhail M, Garofalo M, Nuovo GJ. Reduced miR-512 and the elevated expression of its targets cFLIP and MCL1 localize to neurons with hyperphosphorylated tau protein in Alzheimer disease. Appl Immunohistochem Mol Morphol. 2015;23:615–23.
Nagaraj S, Want A, Laskowska-Kaszub K, Fesiuk A, Vaz S, Logarinho E, et al. Candidate Alzheimer’s disease biomarker miR-483-5p lowers TAU phosphorylation by direct ERK1/2 repression. Int J Mol Sci. 2021;22:3653.
Zhao H, Feng L, Zhong W, Zhen H, Chi Q, Wang X. Hyperphosphorylation of Tau due to the interference of protein phosphatase methylesterase-1 overexpression by MiR-125b-5p in melatonin receptor knockout mice. Int J Mol Sci. 2021;22:11850.
McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–16.
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018;4:575–90.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9:42.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421.
Kitazawa M, Yamasaki TR, LaFerla FM. Microglia as a potential bridge between the amyloid beta-peptide and tau. Ann N Y Acad Sci. 2004;1035:85–103.
Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016;37:608–20.
Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–7.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–86.
Kellner V, Menkes-Caspi N, Beker S, Stern EA. Amyloid-β alters ongoing neuronal activity and excitability in the frontal cortex. Neurobiol Aging. 2014;35:1982–91.
Andersen JV, Schousboe A, Verkhratsky A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: Integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022;217:102331.
Pomilio C, Gorojod RM, Riudavets M, Vinuesa A, Presa J, Gregosa A, et al. Microglial autophagy is impaired by prolonged exposure to β-amyloid peptides: evidence from experimental models and Alzheimer’s disease patients. Geroscience. 2020;42:613–32.
Jia R, Guardia CM, Pu J, Chen Y, Bonifacino JS. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy. 2017;13:1648–63.
Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6:24.
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91.
Chun H, Marriott I, Lee CJ, Cho H. Elucidating the interactive roles of glia in Alzheimer’s disease using established and newly developed experimental models. Front Neurol. 2018;9:797.
Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45.
Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. 2017;79:619–43.
Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2008;5:23.
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–19.
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–91.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20.
Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–84.
Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47.
Farhy-Tselnicker I, Allen NJ. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev. 2018;13:7.
Colombo E, Cordiglieri C, Melli G, Newcombe J, Krumbholz M, Parada LF, et al. Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med. 2012;209:521–35.
Singh A, Abraham WC. Astrocytes and synaptic plasticity in health and disease. Exp Brain Res. 2017;235:1645–55.
Paudel YN, Angelopoulou E, Piperi C, Othman I, Aamir K, Shaikh MF. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s disease (AD): from risk factors to therapeutic targeting. Cells. 2020;9:383.
Liang C, Zou T, Zhang M, Fan W, Zhang T, Jiang Y, et al. MicroRNA-146a switches microglial phenotypes to resist the pathological processes and cognitive degradation of Alzheimer’s disease. Theranostics. 2021;11:4103–21.
Huang Y, Lin X, Lin X. MiR-146a-5p contributes to microglial polarization transitions associated With AGEs. Mol Neurobiol. 2023;60:3020–33.
Redis RS, Calin S, Yang Y, You MJ, Calin GA. Cell-to-cell miRNA transfer: from body homeostasis to therapy. Pharmacol Ther. 2012;136:169–74.
Zhang G, Liu Y, Xu L, Sha C, Zhang H, Xu W. Resveratrol alleviates lipopolysaccharide-induced inflammation in PC-12 cells and in rat model. BMC Biotechnol. 2019;19:10.
Rastegar-Moghaddam SH, Ebrahimzadeh-Bideskan A, Shahba S, Malvandi AM, Mohammadipour A. Roles of the miR-155 in neuroinflammation and neurological disorders: a potent biological and therapeutic target. Cell Mol Neurobiol. 2023;43:455–67.
Yin Z, Herron S, Silveira S, Kleemann K, Gauthier C, Mallah D, et al. Identification of a protective microglial state mediated by miR-155 and interferon-γ signaling in a mouse model of Alzheimer’s disease. Nat Neurosci. 2023;26:1196–207.
Song J, Lee JE. miR-155 is involved in Alzheimer’s disease by regulating T lymphocyte function. Front Aging Neurosci. 2015;7:61.
Mattson MP. Late-onset dementia: a mosaic of prototypical pathologies modifiable by diet and lifestyle. NPJ Aging Mech Dis. 2015;1:15003-.
Oliver DMA, Reddy PH. Molecular basis of Alzheimer’s disease: focus on mitochondria. J Alzheimers Dis. 2019;72:S95–s116.
Wei Z, Li X, Li X, Liu Q, Cheng Y. Oxidative stress in Parkinson’s disease: a systematic review and meta-analysis. Front Mol Neurosci. 2018;11:236.
Nunomura A, Hofer T, Moreira PI, Castellani RJ, Smith MA, Perry G. RNA oxidation in Alzheimer disease and related neurodegenerative disorders. Acta Neuropathol. 2009;118:151–66.
Nunomura A, Moreira PI, Castellani RJ, Lee HG, Zhu X, Smith MA, et al. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res. 2012;22:231–48.
Singh A, Kukreti R, Saso L, Kukreti S. Oxidative stress: a key modulator in neurodegenerative diseases. Molecules. 2019;24:1583.
Teleanu DM, Niculescu AG, Lungu II, Radu CI, Vladâcenco O, Roza E, et al. An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int J Mol Sci. 2022;23:5938.
Birnbaum JH, Wanner D, Gietl AF, Saake A, Kündig TM, Hock C, et al. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018;27:121–30.
Jiao Y, Kong L, Yao Y, Li S, Tao Z, Yan Y, et al. Osthole decreases beta amyloid levels through up-regulation of miR-107 in Alzheimer’s disease. Neuropharmacology. 2016;108:332–44.
Jin Y, Tu Q, Liu M. MicroRNA‑125b regulates Alzheimer’s disease through SphK1 regulation. Mol Med Rep. 2018;18:2373–80.
Zhang L, Dong H, Si Y, Wu N, Cao H, Mei B, et al. miR-125b promotes tau phosphorylation by targeting the neural cell adhesion molecule in neuropathological progression. Neurobiol Aging. 2019;73:41–49.
Ji G, Lv K, Chen H, Wang T, Wang Y, Zhao D, et al. MiR-146a regulates SOD2 expression in H2O2 stimulated PC12 cells. PLoS ONE. 2013;8:e69351.
Lukiw WJ, Zhao Y, Cui JG. An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. 2008;283:31315–22.
Wang G, Huang Y, Wang LL, Zhang YF, Xu J, Zhou Y, et al. MicroRNA-146a suppresses ROCK1 allowing hyperphosphorylation of tau in Alzheimer’s disease. Sci Rep. 2016;6:26697.
Xu S, Zhang R, Niu J, Cui D, Xie B, Zhang B, et al. Oxidative stress mediated-alterations of the microRNA expression profile in mouse hippocampal neurons. Int J Mol Sci. 2012;13:16945–60.
Higaki S, Muramatsu M, Matsuda A, Matsumoto K, Satoh JI, Michikawa M, et al. Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS ONE. 2018;13:e0196929.
Wu Q, Ye X, Xiong Y, Zhu H, Miao J, Zhang W, et al. The protective role of microRNA-200c in Alzheimer’s disease pathologies is induced by beta amyloid-triggered endoplasmic reticulum stress. Front Mol Neurosci. 2016;9:140.
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019–31.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6.
Polanco JC, Li C, Bodea LG, Martinez-Marmol R, Meunier FA, Götz J. Amyloid-β and tau complexity - towards improved biomarkers and targeted therapies. Nat Rev Neurol. 2018;14:22–39.
Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80.
Herms J, Dorostkar MM. Dendritic spine pathology in neurodegenerative diseases. Annu Rev Pathol. 2016;11:221–50.
Kumar S, Reddy PH. The role of synaptic microRNAs in Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165937.
He Y, Wei M, Wu Y, Qin H, Li W, Ma X, et al. Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat Commun. 2019;10:1193.
Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665–704.
Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–26.
Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46.
Feuillette S, Miguel L, Frébourg T, Campion D, Lecourtois M. Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J Neurochem. 2010;113:895–903.
Bonda DJ, Castellani RJ, Zhu X, Nunomura A, Lee HG, Perry G, et al. A novel perspective on tau in Alzheimer’s disease. Curr Alzheimer Res. 2011;8:639–42.
Mroczko B, Groblewska M, Litman-Zawadzka A. The role of protein misfolding and Tau Oligomers (TauOs) in Alzheimer’s disease (AD). Int J Mol Sci. 2019;20:4661.
Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 2021;141:681–96.
Azam S, Haque ME, Kim IS, Choi DK. Microglial turnover in ageing-related neurodegeneration: therapeutic avenue to intervene in disease progression. Cells. 2021;10:150.
Park JC, Han SH, Mook-Jung I. Peripheral inflammatory biomarkers in Alzheimer’s disease: a brief review. BMB Rep. 2020;53:10–19.
Reddy PH, Oliver DM. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells. 2019;8:488.
Garcia GC, Bartol TM, Phan S, Bushong EA, Perkins G, Sejnowski TJ, et al. Mitochondrial morphology provides a mechanism for energy buffering at synapses. Sci Rep. 2019;9:18306.
Roger AJ, Muñoz-Gómez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol. 2017;27:R1177–r1192.
Cai Q, Tammineni P. Alterations in mitochondrial quality control in Alzheimer’s disease. Front Cell Neurosci. 2016;10:24.
Oliver D, Reddy PH. Dynamics of dynamin-related protein 1 in Alzheimer’s disease and other neurodegenerative diseases. Cells. 2019;8:961.
Pradeepkiran JA, Reddy PH. Defective mitophagy in Alzheimer’s disease. Ageing Res Rev. 2020;64:101191.
Bhatti JS, Tamarai K, Kandimalla R, Manczak M, Yin X, Ramasubramanian B, et al. Protective effects of a mitochondria-targeted small peptide SS31 against hyperglycemia-induced mitochondrial abnormalities in the liver tissues of diabetic mice, Tallyho/JngJ mice. Mitochondrion. 2021;58:49–58.
Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 2009;6:65–72.
Bian Z, Li LM, Tang R, Hou DX, Chen X, Zhang CY, et al. Identification of mouse liver mitochondria-associated miRNAs and their potential biological functions. Cell Res. 2010;20:1076–8.
Shi Q, Gibson GE. Up-regulation of the mitochondrial malate dehydrogenase by oxidative stress is mediated by miR-743a. J Neurochem. 2011;118:440–8.
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5.
Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29:4362–8.
Aschrafi A, Schwechter AD, Mameza MG, Natera-Naranjo O, Gioio AE, Kaplan BB. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J Neurosci. 2008;28:12581–90.
Sarkar S, Jun S, Rellick S, Quintana DD, Cavendish JZ, Simpkins JW. Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. 2016;1646:139–51.
Wang J, Tang Y, Lv X, Zhang J, Ma B, Wen X, et al. Corrigendum: “Tectoridin inhibits osteoclastogenesis and bone loss in a murine model of ovariectomy-induced osteoporosis” [Exp. Gerontol. 140: 2020; 111057. DOI:10.1016/j.exger.2020.111057]. Exp Gerontol. 2023;180:112251.
Zhang R, Zhou H, Jiang L, Mao Y, Cui X, Xie B, et al. MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res. 2016;1652:135–43.
Benaroya H. Brain energetics, mitochondria, and traumatic brain injury. Rev Neurosci. 2020;31:363–90.
Silver I, Erecińska M. Oxygen and ion concentrations in normoxic and hypoxic brain cells. Adv Exp Med Biol. 1998;454:7–16.
Kumar S, Reddy PH. A new discovery of MicroRNA-455-3p in Alzheimer’s disease. J Alzheimers Dis. 2019;72:S117–s130.
Xiong H, Chen S, Lai L, Yang H, Xu Y, Pang J, et al. Modulation of miR-34a/SIRT1 signaling protects cochlear hair cells against oxidative stress and delays age-related hearing loss through coordinated regulation of mitophagy and mitochondrial biogenesis. Neurobiol Aging. 2019;79:30–42.
Weinberg RB, Mufson EJ, Counts SE. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front Neurosci. 2015;9:430.
Tang BL. Sirt1 and the mitochondria. Mol Cells. 2016;39:87–95.
John A, Reddy PH. Synaptic basis of Alzheimer’s disease: focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res Rev. 2021;65:101208.
Gowda P, Reddy PH, Kumar S. Deregulated mitochondrial microRNAs in Alzheimer’s disease: Focus on synapse and mitochondria. Ageing Res Rev. 2022;73:101529.
Remenyi J, van den Bosch MW, Palygin O, Mistry RB, McKenzie C, Macdonald A, et al. miR-132/212 knockout mice reveal roles for these miRNAs in regulating cortical synaptic transmission and plasticity. PLoS ONE. 2013;8:e62509.
Wingo TS, Yang J, Fan W, Min Canon S, Gerasimov ES, Lori A, et al. Brain microRNAs associated with late-life depressive symptoms are also associated with cognitive trajectory and dementia. NPJ Genom Med. 2020;5:6.
Nunomura A, Perry G. RNA and oxidative stress in Alzheimer’s disease: focus on microRNAs. Oxid Med Cell Longev. 2020;2020:2638130.
de Souza/S. Nicolau E, Mendes Silva AP, Fiaux do Nascimento KK, Pereira KS, Ribeiro dos Santos G, Silva Barroso L, et al. P3-085: Interaction network of tau protein, beta-amyloid protein, and amyloid protein precursor under oxidative stress in Alzheimer’s disease. Alzheimer’s Dementia. 2015;11:P651–P652.
Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, et al. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J Neurol Sci. 2005;233:145–62.
Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1141–1154.e7.
Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, et al. Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/- mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–80.
Buttini M, Masliah E, Yu GQ, Palop JJ, Chang S, Bernardo A, et al. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am J Pathol. 2010;177:563–9.
Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24:647–57.
Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53–77.
Rasmussen KL, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Plasma apolipoprotein E levels and risk of dementia: a Mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018;14:71–80.
Prendecki M, Florczak-Wyspianska J, Kowalska M, Ilkowski J, Grzelak T, Bialas K, et al. APOE genetic variants and apoE, miR-107 and miR-650 levels in Alzheimer’s disease. Folia Neuropathol. 2019;57:106–16.
Cao J, Huang M, Guo L, Zhu L, Hou J, Zhang L, et al. MicroRNA-195 rescues ApoE4-induced cognitive deficits and lysosomal defects in Alzheimer’s disease pathogenesis. Mol Psychiatry. 2021;26:4687–701.
Zhao L, Zhang L, Zhu W, Chen H, Ding Y, Cui G. Inhibition of microRNA-203 protects against traumatic brain injury induced neural damages via suppressing neuronal apoptosis and dementia-related molecules. Physiol Behav. 2021;228:113190.
Du Y, Yu Y, Hu Y, Li XW, Wei ZX, Pan RY, et al. Genome-wide, integrative analysis implicates exosome-derived MicroRNA dysregulation in schizophrenia. Schizophr Bull. 2019;45:1257–66.
Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal fluid inflammatory cytokine aberrations in Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Front Immunol. 2018;9:2122.
Cha DJ, Mengel D, Mustapic M, Liu W, Selkoe DJ, Kapogiannis D, et al. miR-212 and miR-132 are downregulated in neurally derived plasma exosomes of Alzheimer’s patients. Front Neurosci. 2019;13:1208.
Yang TT, Liu CG, Gao SC, Zhang Y, Wang PC. The serum exosome derived MicroRNA-135a, -193b, and -384 were potential Alzheimer’s disease biomarkers. Biomed Environ Sci. 2018;31:87–96.
Liu WL, Lin HW, Lin MR, Yu Y, Liu HH, Dai YL, et al. Emerging blood exosome-based biomarkers for preclinical and clinical Alzheimer’s disease: a meta-analysis and systematic review. Neural Regen Res. 2022;17:2381–90.
Walgrave H, Balusu S, Snoeck S, Vanden Eynden E, Craessaerts K, Thrupp N, et al. Restoring miR-132 expression rescues adult hippocampal neurogenesis and memory deficits in Alzheimer’s disease. Cell Stem Cell. 2021;28:1805–1821.e8.
Deng Y, Zhang J, Sun X, Ma G, Luo G, Miao Z, et al. miR-132 improves the cognitive function of rats with Alzheimer’s disease by inhibiting the MAPK1 signal pathway. Exp Ther Med. 2020;20:159.
El Fatimy R, Li S, Chen Z, Mushannen T, Gongala S, Wei Z, et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018;136:537–55.
Ramsey SA, Liu Z, Yao Y, Weeder B. Combining eQTL and SNP annotation data to identify functional noncoding SNPs in GWAS trait-associated regions. Methods Mol Biol. 2020;2082:73–86.
Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68.
Soler-López M, Zanzoni A, Lluís R, Stelzl U, Aloy P. Interactome mapping suggests new mechanistic details underlying Alzheimer’s disease. Genome Res. 2011;21:364–76.
Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci. 2018;21:811–9.
Raj T, Shulman JM, Keenan BT, Chibnik LB, Evans DA, Bennett DA, et al. Alzheimer disease susceptibility loci: evidence for a protein network under natural selection. Am J Hum Genet. 2012;90:720–6.
Yu L, Petyuk VA, Gaiteri C, Mostafavi S, Young-Pearse T, Shah RC, et al. Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann Neurol. 2018;84:78–88.
Zhang Q, Ma C, Gearing M, Wang PG, Chin LS, Li L. Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer’s disease. Acta Neuropathol Commun. 2018;6:19.
Picard C, Miron J, Poirier J. Association of TMEM106B with cortical APOE gene expression in neurodegenerative conditions. Genes. 2024;15:416.
Piscopo P, Albani D, Castellano AE, Forloni G, Confaloni A. Frontotemporal lobar degeneration and MicroRNAs. Front Aging Neurosci. 2016;8:17.
Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. 2012;32:11213–27.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82071676), the NSFC-RGC Joint Research Scheme (32061160472).
Author information
Authors and Affiliations
Contributions
YC conceived and designed this study; YBL and QF analyzed and collated the data; MG and YD participated in the literature call and collected samples. The YBL drafted the manuscript with critical revisions from YWC and YC.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, YB., Fu, Q., Guo, M. et al. MicroRNAs: pioneering regulators in Alzheimer’s disease pathogenesis, diagnosis, and therapy. Transl Psychiatry 14, 367 (2024). https://doi.org/10.1038/s41398-024-03075-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-03075-8
- Springer Nature Limited