
Abstract
Polycythemia vera (PV) and essential thrombocythemia (ET) are Philadelphia-negative myeloproliferative neoplasms (MPNs) characterized by erythrocytosis and thrombocytosis, respectively. Approximately 95% of PV and 50–70% of ET patients harbor the V617F mutation in the exon 14 of JAK2 gene, while about 20–30% of ET patients carry CALRins5 or CALRdel52 mutations. These ET CALR-mutated subjects show higher platelet count and lower thrombotic risk compared to JAK2-mutated patients. Here, we showed that CALR-mutated and JAK2V617F-positive CD34+ cells display different gene and miRNA expression profiles. Indeed, we highlighted several pathways differentially activated between JAK2V617F- and CALR-mutated progenitors, i.e., mTOR, MAPK/PI3K, and MYC pathways. Furthermore, we unveiled that the expression of several genes involved in DNA repair, chromatin remodeling, splicing, and chromatid cohesion are decreased in CALR-mutated cells. According to the low risk of thrombosis in CALR-mutated patients, we also found the downregulation of several genes involved in thrombin signaling and platelet activation. As a whole, these data support the model that CALR-mutated ET could be considered as a distinct disease entity from JAK2V617F-positive MPNs and may provide the molecular basis supporting the different clinical features of these patients.
Similar content being viewed by others


Introduction
Philadelphia-negative myeloproliferative neoplasms (MPNs) are a heterogeneous group of clonal hematopoietic stem cell disorders with common molecular and clinical characteristics, and include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF)1,2. PV is characterized by erythrocytosis, while abnormal megakaryocytopoiesis and alterations in platelet count are distinctive features of PMF and ET3,4. Almost all PV patients harbor the JAK2 mutation (mostly the V617F mutation in exon 14 and, more rarely, deletions/insertion in exon 12), while approximately 60% of PMF and ET subjects carry the JAK2V617F mutation5,6. In addition, mutations in exon 10 of thrombopoietin receptor (MPL) gene are present in about 5% of cases with ET or PMF7.
In 2013, somatic mutations in Calreticulin (CALR) gene have been reported in 50–70% of JAK2 and MPL-negative MPNs8,9. The clinical course of CALR-mutated subjects appears to be more indolent than that of JAK2-mutated patients10,11,12. Moreover, as described by several authors, CALR-mutated ET patients show relevant differences in terms of clinical and hematologic parameters (thrombotic risk, platelet (PLT) count, white blood cell (WBC) count, hemoglobin (Hb) level) compared with JAK2V617F-positive patients10,13,14.
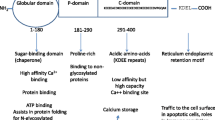
CALR is a multi-functional Ca2+-binding protein with chaperone activity mainly localized in the endoplasmic reticulum (ER). Somatic mutations of CALR frequently consist of deletions/insertions in exon 9, and generate a frameshift to a unique alternative reading frame resulting in a novel amino-acid sequence of C-terminal domain. Moreover, the mutated protein lacks the KDEL signal, which results in partial dislocation of CALR from the ER8. Recently, two different groups demonstrated that mutant CALR activates the JAK2 pathway through its association with MPL15,16 and induces thrombocytosis in a retroviral mouse model17. Unlike JAK2V617F transformed hematopoietic cells, PI3-K signaling seems to be less activated in CALR-mutated cells15, suggesting that a different activation of accessory signaling pathways could justify the differences in clinical features observed in CALR mutated patients.
In order to identify pathways deregulated by mutant CALR proteins in hematopoietic progenitors and unveil the molecular basis underlying the different clinical features of CALR-mutated ET patients, in this study we assessed gene (GEP) and miRNA expression profiles (miEP) in CD34+ cells from CALR-mutated ET patients and JAK2V617F-positive PV and ET subjects. Moreover, in order to predict deregulated mRNA–miRNA interactions involved in the disease pathogenesis and potentially affecting the clinical phenotype, we performed GEP and miEP integrative analysis.
Data analysis showed the differential activation of several signaling pathways, which could at least in part justify the distinct clinical features and outcomes of CALR-mutated and JAK2V617F-positive patients.
Subjects and methods
Patients and samples
Analysis was performed in a cohort of 50 patients diagnosed with PV (n = 26), or ET (n = 24) according to the World Health Organization (WHO)2,18. Their characteristics are reported in Table 1. PV and ET CD34+ cells were obtained from bone marrow (BM), as well as 15 controls from healthy donors (BM CTRs). All subjects provided informed written consent, and the study was performed under the local Institutional Review Board’s approved protocol. The study was conducted in accordance with the Declaration of Helsinki.
The presence of the JAK2V617F mutation and the allele burden were determined via quantitative reverse transcription polymerase chain reaction (qRT-PCR), as previously described19. ET patients were further evaluated for MPL exon 10 and CALR exon 9 mutations using the Sanger technique14.
GEP and miEP profiles and microarray data analysis
GEP and miEP were performed on the same RNA preparation using the Affymetrix technology (HG-U219 Array Strip and miRNA 2.0 array) as previously described20.
Differentially expressed genes (DEGs) and miRNAs (DEMs) were selected following a supervised approach by means of the analysis of variance module included in the Partek GS package. In particular, we considered as differentially expressed all the transcripts with a fold change contrast (FC) ≥1.5 and a false discovery rate (FDR) (q-value) < .05 in the pairwise comparisons. Downregulated genes/miRNAs in CALR-mutated samples are decreased vs. PV, JAK2V617F-positive ET and BM CTRs, as well as upregulated genes/miRNAs in CALR-mutated cells are increased vs. PV, JAK2V617F-positive ET and BM CTRs.
Functional analysis on microarray data was performed using ingenuity pathway analysis (IPA, version 01-08; Ingenuity Systems; Redwood City, CA, http://www.ingenuity.com). In order to identify regulating genes that could explain the gene expression changes in CALR-mutated ET patients, we performed IPA’s upstream regulator analysis that is able to predict the activation state of upstream transcriptional regulators based on expression of their known targets. Moreover, in order to find reliable miRNA-target interactions, we performed the integrative analysis of GEP and miEP data by using IPA’s MicroRNA Target Filter20.
In order to better compare GEP from PV and ET, gene set enrichment analysis (GSEA) was performed21. The detailed protocol for GSEA (v2.0.13; Broad Institute, Cambridge, USA) is available on the Broad Institute Gene Set Enrichment Analysis website (http://www.broad.mit.edu/gsea). The number of permutations was set to 1000.
Results
Gene and miRNA expression profiles of CD34+ cells from PV and ET patients according to CALR and JAK2 mutations
We performed gene and miRNA expression profiling in CD34+ cells from 26 PV and 24 ET patients. The clinical features of the 50 MPN patients enrolled in the study are shown in Table 1. Among the 24 ET patients, CALR mutations were detected in 7 (29%), JAK2V617F in 17 (71%), whereas all PV patients (100%) were JAK2V617F positive. As a control, 15 BM samples from healthy donors were included.
The unsupervised analysis of GEP and miEP data set through the principal component analysis (PCA, Fig. 1a) shows that that PV and ET samples cluster together and are clearly separated from BM CTRs (Supplementary Fig. 1A, B). According to PCA results, GSEA shows that the most of DEGs in PV or ET samples vs. BM CTRs is shared between the two diseases (Supplementary Fig. 1C, F). Therefore, neither GEP nor miEP could distinguish between PV and ET CD34+ samples.

Gene and miRNA expression profiles of PV and ET CD34+ cells according to JAK2 and CALR mutational status. a, b Principal component analysis (PCA) on gene and miRNA expression microarray data. The unsupervised PCA analysis on global gene expression a and miRNA expression data b were computed using Partek GS, version 6.6. BM control samples are shown as red spheres; PV samples are shown in green; JAK2V617F-positive ET samples are shown in blue; CALR-mutated ET samples are shown in orange; c, d The heat maps were computed on the gene list of DEG c and DEM d (provided in Supplementary Tables 1 and 2, respectively) using the clustering algorithm included in the Partek GS package by means of euclidean distance and average linkage. Gene coloring is based on normalized signals, as shown on the left; green indicates reduced expression, red increased expression in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET. CALR-mutated ET groups clustered separately in the dendrogram shown at the top of the heat map. e Ingenuity pathway analysis (IPA) of modulated genes in CALR-mutated ET vs. JAK2V617F-positive ET. The graph shows the most highly represented canonical pathways in the list of DEG and their overlapping connections
Noteworthy, PCA performed on both GEP and miEP data (Fig. 1a, b) shows that JAK2V617F-positive ET and PV samples are grouped together, while the CALR-mutated samples are clearly separated from PV, JAK2V617F-positive ET and BM CTR clusters.
Therefore, in order to study the differences in gene expression according to different driver mutations, we compared GEP and miEP of CD34+ cells from CALR-mutated vs. JAK2V617F-positive ET and PV patients. We found 2040 coding transcripts and 488 miRNAs differentially expressed between CALR-mutated vs. JAK2V617F-positive ET CD34+ cells, which can distinguish ET patients based on their mutational status (Fig. 1c, d, Supplementary Tables 1 and 2), while we were unable to identify any modulated genes in the pairwise comparison between PV and JAK2V617F-positive ET samples. All microarray data were submitted to the Gene Expression Omnibus repository (GEO; http://www.ncbi.nlm.nih.gov/geo, series GSE103176 and GSE53482).
ET CALR-mutated CD34+ cells show a different expression pattern of signal transducers involved in mTOR, MAPK, and PI3K pathways
The IPA functional analysis of modulated transcripts between ET CALR-mutated and JAK2V617F-positive CD34+ cells unveiled differentially activated pathways, such as “mTOR signaling”, “MAPK signaling”, “Role of BRCA1 in DNA damage response”, “ATM signaling”, “Endoplasmic Reticulum stress pathway”, “Protein ubiquitination pathway”, “RhoA signaling,” and “Thrombin signaling” (Fig. 1e). Of note, mTOR signaling appears to be inhibited in CALR-mutated ET patients, as demonstrated by the downregulation of the mTOR2 complex subunit RICTOR, and of several upstream and downstream factors (i.e., PDK1, PHIP, RHEB, PP2A, and RPS6KB1) (Fig. 2a). Similarly, we discovered a reduced expression of several signal transducers involved in MAPK and PI3K/AKT pathways (i.e., KRAS, SOS1, MAPK14, PI3KCA, PI3KR1, SHP2) in CALR-mutated ET samples (Fig. 2b).

IPA canonical pathways and upstream regulator analysis of DEG in CALR-mutated vs. JAK2V617F-positive ET. a, b IPA canonical pathways more represented in the list of DEG a mTOR signaling; b MAPK/PI3K signaling. The networks also show the predicted mRNA–miRNA interactions identified by IPA’s miRNA Target Filter. Green and red colors indicate genes/miRNA down- and upregulated, respectively, in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET c IPA upstream regulator analysis indicated the inhibition of MYC activity predicted by the decreased expression of its targets. Green colors indicate genes downregulated in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET. The blue lines display the inhibitory effect of proteins, which was confirmed by IPA Knowledge database. The gray lines indicate that the protein interactions lacked literature support to predict the activation effect. Solid lines: direct interactions; dashed lines: indirect interactions; d The histogram shows the differential expression of genes involved in cell cycle regulation and DNA replication between CALR-mutated vs. JAK2V617F-positive samples. The green block indicates a decreased expression, while the red block indicates an increased expression in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET
In order to predict potentially deregulated mRNA–miRNA interactions, we performed integrative analysis of GEP and miEP by IPA’s miRNA Target Filter. In particular, we selected DEM-DEG pairs with an anti-correlated expression pattern; among the modulated probesets in CALR-mutated vs. JAK2V617F-positive samples, we identified 34 DEMs that have at least one anti-correlated target among DEGs, whereas 720 DEGs have at least one targeting DEM showing an anti-correlated expression. Therefore, 1387 anti-correlated miRNA-target pairs were identified. Table 2 shows the miRNAs with the highest number of predicted targets.
As shown in Fig. 2a, b, the upregulation of several miRNAs (i.e., miR-326, miR-376c-3p, miR-19a-3p, miR-30e-5p, miR-21-5p, miR-152-3p, miR-503-5p) in CALR-mutated cells could cause the downregulation of mTOR/MAPK/PI3K signaling by targeting some pathway transducers. Among those, miR-152-3p is described as targeting mTOR, PI3K, and MAPK pathways in solid tumors22–24, whereas miR-503-5p, and miR-29c-3p were identified as PI3K signaling regulators in lung and bladder cancer, respectively25,26. Of note, miR-326 has been already described as regulator of MAPK pathway transducers in gliomas27. Moreover, the miR-326 upregulation could be probably ascribed to PI3K inhibition, as already reported in glioblastomas28.
Furthermore, several co-activators (i.e., ATAD2, NMI, MYCBP2) and target genes of MYC are downregulated. In fact, IPA upstream regulator analysis showed that MYC activity is lower in CALR-mutated vs. JAK2V617F-positive ET, based on the decreased expression of its known targets (Fig. 2c). Accordingly, we highlighted a decreased expression of cell cycle kinase CDK6 and of several genes involved in cell proliferation and DNA replication (i.e., MKI67, RFC1, BBX, DBF4, CDC7), as well as the upregulation of the cell cycle inhibitor CDKN1C in CALR-mutated patients (Fig. 2d).
Moreover, DNA repair process seems to be less activated in CALR-mutated cells. In fact, genes involved in BRCA1-driven DNA damage response and ATM signaling, as well as in DNA repair mechanisms (i.e., SMC, MSH, and RAD genes, SFR1, RB1, MRE11A, FAM175) are downregulated in CALR-mutated patients (Fig. 3). As showed in Fig. 3a, b, the upregulation of miR-30e-5p, miR-19a-3p, miR-224-5p, miR-503-5p, miR-301a, miR-21-5p, and miR-4310 could justify the reduced expression of DNA repair genes. Interestingly, miR-19a-3p has been already described as regulator of BARD1 expression in acute myeloid leukemia cells29.

DNA repair pathways in CALR-mutated vs. JAK2V617F-positive ET. a, b IPA canonical pathways highly represented in the list of DEG a BRCA1 in the DNA damage response; b ATM signaling. The networks also show the predicted mRNA–miRNA interactions identified by IPA’s miRNA Target Filter. Green and red colors indicate genes/miRNA down- and upregulated, respectively, in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET. c The histogram shows the differential expression between CALR-mutated vs. JAK2V617F-positive samples of genes involved in DNA repair. The green block indicates a decreased expression, while the red block indicates an increased expression in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET
CALR-mutated CD34+ cells show a different expression of CSNK1A1, chromatin remodelers, sister chromatid cohesins, and splicing factors
Among DEGs in CALR-mutated ET CD34+ cells, we underlined the downregulation of CSNK1A1 (Fig. 4a) that has been reported as favoring the clonal expansion of CD34+ progenitors30. CSNK1A1 downregulation could be ascribed to the increased expression of miR-939 and miR-30e-5p in CALR-mutated cells.

Deregulated expression of CSNK1A1 , chromatin remodeling, cohesin complex, and splicing factors in CALR-mutated ET. a Box plot shows the decreased expression level of CSNK1A1 in CALR-mutated ET. The lower part of the panel shows the overexpression of two miRNAs targeting CSNK1A1. Green and red colors indicate genes/miRNA down- and upregulated, respectively, in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET. b–d The histograms show the differential expression between CALR-mutated vs. JAK2V617F-positive samples of genes involved in b chromatin remodeling c cohesin complex d splicing factors. The green block indicates a decreased expression in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET
Interestingly, CALR-mutated cells show the decreased expression of several epigenetic regulators (i.e., EZH2, SUZ12, DNMT1, SETD2, MLL3, ARID4A, ARID4B, SETDB2), sister chromatid cohesins (i.e., SMC1A, SMC3, RAD21, STAG2), and splicing factors (i.e., SF3B1, SRSF1, ZFR), some of which were already described as mutated or inactivated in myeloid malignancies (Fig. 4b–d)31,32,33,34,35,36,37,38. As shown in Supplementary Fig. 2A, the IA identified miR-30e-5p, miR-301a-3p, miR-376c-3p, miR-152-3p, miR-19a-3p, miR-362-5p, and miR-29c-3p as key regulators of chromatin remodeling factors. The decreased expression of cohesins could be ascribed to miR-21-5p, miR-29c-3p, and miR-501-3p overexpression, whereas miR-301a-3p, miR-19a-3p, miR-30e-5p, miR-432-5p, and miR-433-3p might modulate the expression of splicing factors (Supplementary Fig. 2B, C).
Moreover, CALR-mutated cells exhibit the downregulation of several genes involved in protein ubiquitination (i.e., UBE2J1, UBE2V2, UBE3A, UCHL5, and USPs) and in Endoplasmic Reticulum stress (i.e., PERK1, GRP94, and EIF2S1) (data not shown).
As regards the expression of genes regulating cell differentiation, we found a decreased expression of several erythroid transcription factors (i.e., ZNF148, ZNF268, TWSG1, PDCD4, BCL11A, MBNL1, UFL1) in CALR-mutated cells (Fig. 5a). Of note, consistently with their role in erythropoiesis, we highlighted a direct correlation between TWSG1, ZNF148, or PDCD4 expression and Hb level (r = 0.34, r = 0.31, r = 0.32, respectively) (Supplementary Table 3).

Differential expression of erythroid and megakaryocyte regulators in CALR-mutated vs. JAK2V617F-positive ET. a The histogram shows the differential expression between CALR-mutated vs. JAK2V617F-positive samples of erythroid and megakaryocyte regulators and markers. The green block indicates a decreased expression, while the red block indicates an increased expression in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET. b Regulatory networks showing the predicted mRNA–miRNA interactions identified by IPA’s miRNA Target Filter. Green and red colors indicate genes/miRNA down- and upregulated, respectively, in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET
Moreover, the upregulation of megakaryocyte (MK) transcription factors and markers (i.e., MAF, PLK3, WIPF1)39 as well as the downregulation of inhibitors of MK commitment (i.e., IFNAR1, PTEN, SOCS6)40,41,42, could suggest that CALR-mutated CD34+ cells display the susceptibility to differentiate toward MK rather than erythroid lineage (Fig. 5a). In this regard, a positive correlation between PLK3 or WIPF1 expression levels and PLT count (r = 0.48, r = 0.40, respectively) was found. Consistently, we unveiled a negative correlation between PTEN, IFNAR1, SOCS6, or ZNF148 expression and PLT count (r = −0.41, r = −0.41, r = −0.39, r = −0.52, respectively) (Supplementary Table 3). As shown in Fig. 5b, the upregulation of miR-21-5p, miR-19a-3p, miR-301a-3p, miR-503-5p, miR-307-3p, and miR-134-5p might lead to the downregulation of erythroid positive regulators (PCDC4, MBNL1, ZNF268) and MK inhibitors (SOCS6, PTEN, IFNAR1). Of note, the role of miR-503-5p in erythrocyte lineage expansion has been already described43.
CALR-mutated cells display a reduced expression of genes related to platelet activation
Gene expression analysis highlighted the down-modulation of several proteins involved in thrombin and RhoA signaling (i.e., ROCK1, ROCK2, PPP1R12A, IQGAP2, RAPGEF6, and PIP5K), which regulate the platelet activation and aggregation (i.e., WASF2, ACTR2, RDX), in CALR-mutated patients (Fig. 6a). Moreover, we observed the reduced expression of some regulators of platelet storage and degranulation (i.e., DNML1, RAB27A, and BLOC1S6). Among the miRNAs predicted as modulators of thrombin and RhoA signaling, we underlined the upregulation of miR-376c-3p, which has been already described as involved in platelet activation44. Of note, mir-19a-3p was previously described as decreased in peripheral blood cells following acute ischemic stroke45.

Differential expression of genes involved in platelet activation in CALR-mutated vs. JAK2V617F-positive ET. a Thrombin and RhoA signaling in CALR-mutated vs. JAK2V617F-positive ET. The network also shows the predicted mRNA–miRNA interactions identified by IPA’s miRNA Target Filter. Green and red colors indicate genes/miRNA down- and upregulated, respectively, in the pairwise comparison CALR-mutated vs. JAK2V617F-positive ET. b Box plot shows the increased expression level of THBD in CALR-mutated ET samples
Finally, CALR-mutated CD34+ cells showed higher expression level of thrombomodulin (THBD), an anti-thrombotic factor expressed by endothelial and hematopoietic stem/progenitor cells (Fig. 6b)46.
Discussion
Somatic mutations in the CALR gene have been found in 50–70% of JAK2 and MPL wild-type MPN patients8,9. Recently, the molecular mechanisms underlying the pathogenetic role of CALR mutated proteins have been unveiled, demonstrating that CALR mutants activates MPL receptor, thus inducing the JAK-STAT activation as well as in JAK2- and MPL-mutated cells15,16,17.
However, as already suggested13, CALR-mutated ET show clinical features different from JAKV617F-positive ET and might be considered as a distinct disease entity from JAK2V617F-positive ET; in fact, CALR-mutated ET patients present a higher PLT count coupled with a lower thrombotic risk if compared to JAK2V617F-positive ET patients10,13,14.
According to these observations, unsupervised analysis performed on GEP and miEP data shows that CALR-mutated ET samples are clearly separated from both JAK2V617F-positive ET and PV groups, which instead cluster together. Supervised data analysis shows the differential expression of 2040 genes and 488 miRNAs distinguishing ET patients based on their mutational status; on the contrary, we were unable to identify any modulated genes or miRNAs in the pairwise comparison between PV and JAK2V617F-positive ET samples. This evidence supports the hypothesis suggested by Campbell et al.47 that ET and PV JAK2V617F-positive are distinct phases of the same disease that exhibits two different phenotypes.
The functional analysis of modulated transcripts unveiled several pathways differentially activated between JAK2V617F- and CALR-mutated progenitor cells. For instance, the expression of mTOR transducers (e.g., RICTOR) is decreased in CALR-mutated samples. RICTOR is a key component of mTORC2 complex, which plays a relevant role in leukemic cell proliferation48. Interestingly, RICTOR deletion prevents leukemogenesis in (PTEN)-deficient mouse model showing prolonged lifespan, suggesting that mTORC2 downregulation might partly explain the more indolent phenotype of CALR-mutated ET patients49.
Similarly, MAPK and PI3K pathways seem to be less activated in CALR-mutated ET: these data are consistent with the results obtained by Chachoua et al.15, which have shown that there is a strong synergy between JAK2 and PI3-K inhibitors in restraining the cytokine-independent proliferation of JAK2V617F-positive cells, unlike what occurs in the CALR-mutated cells. According to MAPK and PI3K signaling inhibition, MYC activity appears decreased as demonstrated by the huge downregulation of its transcriptional targets. In addition, we observed a strong downregulation of several proteins involved in cell cycle control and DNA replication. These evidences suggest that CALR-mutated progenitors are less proliferating compared to their JAK2V617F-positive counterparts.
Our result show that CALR mutant protein could affect signaling pathways other than JAK-STAT; for instance, the decreased expression of CSNK1A1 in CALR-mutated samples might favor the initial clonal expansion of CD34+ progenitors in ET patients30. Moreover, the downregulation of DNA repair pathways mediated by BRCA1 and ATM could suggest the impairment of DNA damage response as new pathogenetic mechanism of CALR-mutant proteins.
Similarly, the downregulation of epigenetic regulators31,32,33,34,35,36, cohesins37, and splicing factors38, which are frequently mutated or inactivated in several myeloid malignancies, could be involved in the pathogenesis of CALR-mutated disease. Furthermore, CALR mutations seem to impair essential cellular function as protein ubiquitination and ER stress response. The downregulation of genes related to “ER stress response” coupled to partial dislocation from the ER of mutant CALR lacking the KDEL signal8, suggests an inefficient response to unfolded protein accumulation in CALR-mutated cells as an additional pathogenetic mechanism (Salati S. et al., submitted).
The recent discovery of physical interaction between CALR-mutated protein and MPL receptor16 has definitely explained the link concerning CALR mutations and megakaryocytopoiesis. However, the increased of MK commitment regulator as MAF39 coupled to decreased of MK differentiation inhibitors, like IFNAR1, PTEN and SOCS6 40,41,42 could favor the higher PLT count in CALR-mutated subjects compared to JAK2V617F-positive ET patients. According to this hypothesis, our data disclosed a negative correlation between PTEN, IFNAR1, or SOCS6 expression levels and PLT count. In particular, as described by Zhang et al.41, the simultaneous downregulation of PTEN and MYC signaling in CALR-mutated MPN patients might favor the switch from granulocyte- to MK- commitment. Moreover, we highlighted the increased level of PLK3 that was already described as having a pivotal role in megakaryocyte polyploidization and differentiation and that might justify the higher number of PLT in CALR-mutated patients50. Consistently, a positive correlation between PLK3 expression level and PLT count was found.
We also unveiled a direct correlation between the hemoglobin level and the expression of some erythroid positive regulators, which are decreased in CALR-mutated patients. Therefore, CALR mutant proteins could affect the expression of several erythroid and MK differentiation-related genes, then contributing to lineage fate decision of CALR-mutated CD34+ cells.
As for reduced thrombotic risk in ET CALR-mutated patients, we uncovered the down-modulation of several proteins involved in thrombin and RhoA signaling. Moreover, several genes involved in platelet activation, aggregation, and degranulation are decreased in CALR-mutated progenitors, while the anti-thrombotic factor THBD is upregulated. These data are consistent with the results obtained by Torregrosa et al.51, showing a reduced platelet activation in CALR-mutated ET patients compared to JAK2V617F-positive ET subjects. Overall, these observations could explain the low thrombotic risk in CALR-mutated patients, even though they have a higher PLT count as compared to JAK2V617F-positive patients.
Finally, by means of integrated analysis of GEP and miEP, we identified several miRNA–mRNA interactions, which could represent novel pathogenetic mechanisms mediated by CALR mutations and could affect the patient’s clinical phenotype. In particular, we unveiled the upregulation of several miRNAs (e.g., miR-30e-5p, miR-19a-3p, miR-301a-3p, miR-152-3p, miR-29c-3p, miR-21-5p, miR-503-5p, miR-376c-3p), which are involved in the inhibition of mTOR/MAPK/PI3K signaling, DNA damage response, chromatin remodeling, alternative spicing, and PLT activation.
As a whole, this study suggests that the molecular characterization of CD34+ cells from CALR-mutated and JAK2V617F-positive ET patients, could shed light on the signaling pathways deregulated in CALR-mutated patients and elucidate their contribution to typical features of these patients, such as high platelet count and low thrombotic risk.
Overall, this study supports the pathogenetic model in which JAK2V617F-positive ET and PV could be considered as different phenotypes or phases of a single MPN characterized by JAK2 mutation47, whereas CALR-mutated ET seems to be a distinct entity both at clinical13 and at molecular level.
References
Tefferi, A. & Vardiman, J. W. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 22, 14–22 (2008).
Barbui, T. et al. The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances. Blood Rev. 30, 453–459 (2016).
Tefferi, A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am. J. Hematol. 87, 285–293 (2012).
Tefferi, A. & Barbui, T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 92, 94–108 (2017).
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005).
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Eng. J. Med. 352, 1779–1790 (2005).
Pardanani, A. D. et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 108, 3472–3476 (2006).
Klampfl, T. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Eng. J. Med. 369, 2379–2390 (2013).
Nangalia, J. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Eng. J. Med. 369, 2391–2405 (2013).
Tefferi, A. et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124, 2507–2513; quiz 615 (2014).
Tefferi, A. et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia 28, 1472–1477 (2014).
Tefferi, A. et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 28, 2300–2303 (2014).
Rumi, E. et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 123, 1544–1551 (2014).
Rotunno, G. et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 123, 1552–1555 (2014).
Chachoua, I. et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127, 1325–1335 (2016).
Araki, M. et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 127, 1307–1316 (2016).
Marty, C. et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 127, 1317–1324 (2016).
Vardiman, J. W. et al. The2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114, 937–951 (2009).
Guglielmelli, P. et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 114, 1477–1483 (2009).
Norfo, R. et al. miRNA-mRNA integrative analysis in primary myelofibrosis CD34+ cells: role of miR-155/JARID2 axis in abnormal megakaryopoiesis. Blood 124, e21–32 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. U.S.A. 102, 15545–15550 (2005).
Tsuruta, T. et al. miR-152 is a tumor suppressor microRNA that is silenced by DNA hypermethylation in endometrial cancer. Cancer Res. 71, 6450–6462 (2011).
Ge, S., Wang, D., Kong, Q., Gao, W., Sun, J. Function of MiR-152 as a tumor suppressor in human breast cancer by targeting PIK3CA. Oncol. Res 25, 1363–1371 (2017).
Xu, Q. et al. A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J. Mol. Cell Biol. 5, 3–13 (2013).
Yang, Y. et al. MiR-503 targets PI3K p85 and IKK-beta and suppresses progression of non-small cell lung cancer. Int. J. Cancer 135, 1531–1542 (2014).
Fan, Y. et al. Down-regulation of miR-29c in human bladder cancer and the inhibition of proliferation in T24 cell via PI3K-AKT pathway. Med. Oncol. 31, 65 (2014).
Liu, X., Song, B., Li, S., Wang, N. & Yang, H. Identification and functional analysis of the risk microRNAs associated with cerebral low-grade glioma prognosis. Mol. Med. Rep. 16, 1173–1179 (2017).
Nawaz, Z. et al. PI3 kinase pathway regulated miRNome in glioblastoma: identification of miR-326 as a tumour suppressor miRNA. Mol Cancer 15, 74 (2016).
Lepore, I. et al. HDAC inhibitors repress BARD1 isoform expression in acute myeloid leukemia cells via activation of miR-19a and/or b. PLoS ONE 8, e83018 (2013).
Schneider, R. K. et al. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 26, 509–520 (2014).
Khan, S. N. et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 27, 1301–1309 (2013).
Harada, T., et al. HDAC3 regulates DNMT1 expression in multiple myeloma: therapeutic implications. Leukemia (2017), https://doi.org/10.1038/leu.2017.144
Chopra, M. & Bohlander, S. K. Disturbing the histone code in leukemia: translocations and mutations affecting histone methyl transferases. Cancer Genet. 208, 192–205 (2015).
Yang, W. & Ernst, P. Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis. Curr. Opin. Hematol. 24, 322–328 (2017).
Wu, M. Y., Eldin, K. W. & Beaudet, A. L. Identification of chromatin remodeling genes Arid4a and Arid4b as leukemia suppressor genes. J. Natl Cancer Inst. 100, 1247–1259 (2008).
Parker, H. et al. 13q deletion anatomy and disease progression in patients with chronic lymphocytic leukemia. Leukemia 25, 489–497 (2011).
Mazumdar, C. & Majeti, R. The role of mutations in the cohesin complex in acute myeloid leukemia. Int. J. Hematol. 105, 31–36 (2017).
Wong, J. J., Lau, K. A., Pinello, N. & Rasko, J. E. Epigenetic modifications of splicing factor genes in myelodysplastic syndromes and acute myeloid leukemia. Cancer Sci. 105, 1457–1463 (2014).
Ruberti, S, et al. Involvement of MAF/SPP1 axis in the development of bone marrow fibrosis in PMF patients. Leukemia (2017), https://doi.org/10.1038/leu.2017.220.
Negrotto, S. et al. Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J. Thromb. Haemost. 9, 2477–2485 (2011).
Zhang, J. et al. Differential requirements for c-Myc in chronic hematopoietic hyperplasia and acute hematopoietic malignancies in Pten-null mice. Leukemia 25, 1857–1868 (2011).
Rontauroli, S. et al. miR-494-3p overexpression promotes megakaryocytopoiesis in primary myelofibrosis hematopoietic stem/progenitor cells by targeting SOCS6. Oncotarget 8, 21380–21397 (2017).
Roy, P. & Dasgupta, U. B. The microRNA hsa-miR-503 inhibits growth of K562 cell line. Blood Cells Mol. Dis. 50, 271–272 (2013).
Edelstein, L. C. et al. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat. Med. 19, 1609–1616 (2013).
Jickling, G. C. et al. MicroRNA expression in peripheral blood cells following acute ischemic stroke and their predicted gene targets. PLoS ONE 9, e99283 (2014).
Ikezoe, T. et al. Thrombomodulin enhances the antifibrinolytic and antileukemic effects of all-trans retinoic acid in acute promyelocytic leukemia cells. Exp. Hematol. 40, 457–465 (2012).
Campbell, P. J. et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 366, 1945–1953 (2005).
Hua, C. et al. Rictor/mammalian target of rapamycin 2 regulates the development of Notch1 induced murine T-cell acute lymphoblastic leukemia via forkhead box O3. Exp. Hematol. 42, 1031–1040 (2014). e1-4.
Magee, J. A. et al. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 11, 415–428 (2012).
Huang, X. et al. Physical and functional interactions between mitotic kinases during polyploidization and megakaryocytic differentiation. Cell Cycle 3, 946–951 (2004).
Torregrosa, J. M. et al. Impaired leucocyte activation is underlining the lower thrombotic risk of essential thrombocythaemia patients with CALR mutations as compared with those with the JAK2 mutation. Br. J. Haematol. 172, 813–815 (2016).
Acknowledgements
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC), IG project number #15337 and project number #10005 “Special Program Molecular Clinical Oncology 5 × 1000” to AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative, http://www.progettoagimm.it). Italian Ministry of Health (Progetti di ricerca giovani ricercatori, Ricerca Finalizzata 2011–2012), project number #GR-2011-02352109.
Authors contributions
R.Z., A.M.V., and R.M. designed the study; P.G., D.P., E.R., M.C. enrolled patients and provided clinical features; T.F. and L.C., contributed to patient sample collection and performed molecular characterization; R.Z., C.R., S.R., E.G., and S.S. performed microarray experiments; R.Z., E.B., and E.T. performed data analysis; R.Z., A.M.V., and R.M. wrote the manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zini, R., Guglielmelli, P., Pietra, D. et al. CALR mutational status identifies different disease subtypes of essential thrombocythemia showing distinct expression profiles. Blood Cancer Journal 7, 638 (2017). https://doi.org/10.1038/s41408-017-0010-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-017-0010-2
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.