
Abstract
In acute myeloid leukemia (AML), risk stratification based on cytogenetics and mutation profiling is essential but remains insufficient to select the optimal therapy. Accurate biomarkers are needed to improve prognostic assessment. We analyzed RNA sequencing and survival data of 430 AML patients and identified HMGA2 as a novel prognostic marker. We validated a quantitative PCR test to study the association of HMGA2 expression with clinical outcomes in 358 AML samples. In this training cohort, HMGA2 was highly expressed in 22.3% of AML, mostly in patients with intermediate or adverse cytogenetics. High expression levels of HMGA2 (H + ) were associated with a lower frequency of complete remission (58.8% vs 83.4%, P < 0.001), worse 3-year overall survival (OS, 13.2% vs 43.5%, P < 0.001) and relapse-free survival (RFS, 10.8% vs 44.2%, P < 0.001). A positive HMGA2 test also identified a subgroup of patients unresponsive to standard treatments. Multivariable analyses showed that H + was independently associated with significantly worse OS and RFS, including in the intermediate cytogenetic risk category. These associations were confirmed in a validation cohort of 260 patient samples from the UK NCRI AML17 trial. The HMGA2 test could be implemented in clinical trials developing novel therapeutic strategies for high-risk AML.
Similar content being viewed by others

Introduction
In adult acute myeloid leukemia (AML), clinical outcome is predicted by age, cytogenetics and specific gene mutations.1,2,3,4,5 In the recent European LeukemiaNet (ELN) guidelines for AML genetic testing, screening for mutations in NPM1, CEBPA, RUNX1, FLT3, TP53, and ASXL1 genes in addition to chromosomal anomalies is recommended.1 It is now well accepted that the genetic and cytogenetic risk stratification guides AML consolidation therapy: patients in a favorable risk category are treated with conventional consolidation chemotherapy, whereas adverse-risk patients are usually referred for allogeneic hematopoietic stem cell transplantation (allo-HSCT), a procedure carrying an inherent mortality rate surpassing 15%.6 However, the ideal consolidation therapy remains unclear for up to 40% of AML patients classified in the intermediate-risk category, hence the need to improve prognostic assessment in this patient subgroup.1 Likewise, identification of possible long-term survivors in the adverse-risk group represents another clinical challenge.
Gene expression signatures, mostly derived from microarray studies, have been evaluated as a means to further improve AML risk stratification.7,8,9,10,11,12,13,14 Although several markers have been identified, they have not been widely adopted because of technical challenges in implementing large gene signatures in clinical settings. Global RNA-sequencing technologies, which are more accurate in estimating gene expression levels than microarray studies,15 have now been applied to a few large AML cohorts including that of The Cancer Genome Atlas (TCGA, n = 179)16 and Leucegene (n = 430).17,18,19,20,21 These data sets provide new opportunities to determine whether candidate gene expression levels can complement currently accepted prognostic tests.
In this study, we have explored the Leucegene data set using bioinformatic tools to identify genes with bimodal expression patterns that correlate with patient survival. The two best candidate genes, High Mobility Group AT-Hook 2 (HMGA2) and Pro-Apoptotic WT1 Regulator (PAWR) were evaluated in the training cohort but only HMGA2 was validated in the independent cohort. We present the development and inter-laboratory validation of a RT-qPCR HMGA2 clinical test and demonstrate its utility to refine AML risk stratification.
Patients, materials, and methods
Study design, patients, and AML sample characteristics
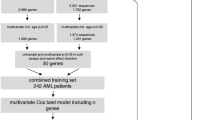
This study is part of the Leucegene project and was approved by the Research Ethics Boards of Université de Montréal and Maisonneuve-Rosemont Hospital. Diagnostic AML samples and clinical data were collected with informed consent from patients between 2002 and 2014 at nine hospitals participating in the Banque de cellules leucémiques du Québec program (BCLQ, bclq.org). The Leucegene full cohort of 430 RNA-sequenced samples (Fig. 1) was used for the discovery of new candidate prognostic markers. RNA-sequencing data are available separately,17,18,19,20,21 #GSE49642, #GSE52656, #GSE62190, #GSE66917, #GSE67039. The training cohort includes 263 de novo AML patients treated with intensive regimens sequenced in the Leucegene project and 95 additional BCLQ specimens similarly selected, which were not sequenced (Fig. 1). The median follow-up was 6.0 years. Alive patients were censored at their last follow-up (May to August 2015). Four additional patients were censored owing to loss to follow-up. Definitions of complete remission (CR), overall survival (OS), relapse-free survival (RFS) and cumulative incidence of relapse (CIR) followed ELN recommendations.1 Description of clinical characteristics and treatment protocols are provided in the Supplementary Information (Supplementary Figures S1–S2; Supplementary Tables S1–S4). AML samples (n = 70) from Australia were used to confirm the distribution of HMGA2 expression values (Fig. 1 and Supplementary Table S5). This study was approved by the Human Research Ethics Committees of the Alfred and the Box Hill Hospitals in Melbourne. The external validation cohort included 263 AML samples from intensively treated patients enrolled in the UK NCRI AML17 trial (ISRCTN55675535) approved by Wales Research Ethics Committee 3 (Fig. 1 and Table 1). Patients with intermediate- and adverse-risk cytogenetics were selected for external validation because the HMGA2 test appears useful in these risk categories. HMGA2 expression values were not available for 3 out of 263 samples.

The HMGA2 prognostic marker was identified from the RNA-sequenced samples of the Leucegene full cohort (n = 430). Criteria for marker selection were: best log-rank P values to discriminate between poor vs good survivors based on the 75th percentile of expression (in RPKM values) for each gene, high dynamic range, bimodal distribution of gene expression values, and gene expression values above one RPKM. Development, analytical, and clinical validation of the HMGA2 RT-qPCR test were performed in the training cohort (n = 358). The Australian cohort (n = 70) was used to validate the RT-qPCR expression values. The HMGA2 test was externally validated in the NCRI AML17 cohort (n = 260). AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; MDS, myelodysplastic syndromes; RPKM, reads per kilobase per million mapped reads; Tx, treatment
Cytogenetics, mutation analysis, and RNA sequencing
Cytogenetic risk was categorized according to ELN recommendations.1 Methods for leukemia cell cryopreservation and for NPM1, FLT3-ITD, and CEBPA mutation testing are described in the Supplementary Information. The workflow for RNA-sequencing and mutation analysis has been described previously.20
Quantitative PCR experiments
A RT-qPCR assay to evaluate HMGA2 expression was developed. Detailed methods including complementary DNA synthesis, primer, and probe sequences, PCR, construction of plasmid standard curves and results of analytical validation are outlined in the Supplementary Information (Methods section, Supplementary Tables S6 and S7). Normalized copy numbers (NCN) of HMGA2 were generated following Europe Against Cancer program recommendations.22
Statistical methods
Receiver operating characteristic (ROC) curves and the Youden index were used to identify a threshold between low and high HMGA2 expression values.23,24 Fisher’s exact test was used to test bivariate unadjusted associations between the marker, dichotomized as above (H + ) vs. below (H −) the threshold, and categorical variables. Probabilities of OS were estimated with Kaplan–Meier curves and compared using the log-rank test. CIR curves were estimated using competing risks analyses to account for mortality and compared with Gray’s test.25 OS was measured from the date of AML diagnosis and RFS and CIR were measured from the date of achievement of a remission. For studies in the subgroup of younger transplanted patients, time 0 was defined as the date of transplantation. Main analyses relied on multivariable regression methods to estimate the associations of the dichotomized marker with each of the clinically relevant outcomes. Multivariable models were adjusted for the following set of established prognostic variables: age, white blood cell (WBC) counts, HSCT as a time-dependent variable (except for CR prediction), cytogenetic risk and NPM1 and FLT3-ITD mutation status. TP53, RUNX1, and ASXL1 mutations were added as variables for models in the sequenced cohort and biallelic CEBPA, RUNX1, and ASXL1 mutations for models in the intermediate cytogenetic risk subgroup. The effect of age was modeled using the linear and quadratic terms, to account for its significantly non-linear relationships with most of the outcomes (Supplementary Information, Statistical Methods section). The ability of HMGA2 to enhance CR prediction was assessed with multivariable logistic regression and its independent association with the time to relapse and/or death was estimated by multivariable Cox proportional hazards regression. Flexible time-dependent model was used to test the proportional hazards assumption26 (Supplementary Information, Statistical Methods section). The Lunn-McNeil competing risks extension of the Cox model27,28 estimated associations of the marker with the hazards of either relapse or death. Statistical significance of the associations was tested using multivariable model-based Wald tests and their strength quantified by the adjusted hazard ratio (HR) or for CR, odds ratio (OR), with 95% confidence intervals. All P values were two-sided and considered statistically significant if P < 0.05. The analyses were performed with R (v3.2.2) and EZR (v3.1) softwares. Statistical methods for the NCRI AML17 cohort are described in the Supplementary Information (Statistical Methods section).
This manuscript complies with the REMARK guidelines29 (Supplementary Table S8).
Results
Identification of HMGA2 as a new prognostic marker in AML
We first investigated all annotated genes in the Leucegene full cohort (n = 430) for their potential to discriminate between patients with good vs. poor survival by analyzing survival based on the 75th percentile of expression values (Fig. 1). The best candidate prognostic markers were also selected for features that would ease their usage as clinical tests: (1) high dynamic range of expression; (2) evidence for bimodal distribution illustrative of two distinct subgroups with more than tenfold difference in reads per kilobase per million mapped reads (RPKM) values between low and high expressors, and (3) peak expression in high expressors above one RPKM. HMGA2 and PAWR were identified for test development and validation but only HMGA2 was validated in the independent NCRI AML17 validation cohort and is reported herein. Analyses of PAWR in the validation cohort are provided in Supplementary Figure S3 and Supplementary Table S9.
Notably, based on the 75th percentile of HMGA2 expression values, most genetic anomalies associated with poor survival were highly prevalent in the HMGA2 positive subgroup including samples with complex karyotype, TP53 mutations or other adverse-risk mutations such as RUNX1, ASXL1, SRSF2, and MLL (Supplementary Figure S4).
Development and validation of the HMGA2 RT-qPCR test
We developed and validated a HMGA2 RT-qPCR test in three independent AML patient cohorts (Leucegene, NCRI AML17 and Australian cohorts) and confirmed the bimodal expression pattern of HMGA2 (Fig. 1 and Supplementary Figure S5). We observed a high correlation between these results and those found by RNA sequencing or droplet digital PCR as well as a large range of expression values (Supplementary Figure S6; Fig. 2 upper panel). Using ROC curves, the cutoff for the RT-qPCR test was optimized and established at 1100 NCN in the training cohort.23 Samples with expression levels ≥ 1100 NCN are hereafter referred to as H+ and those with expression levels < 1100 NCN as H−.

Upper panel. The HMGA2 RT-qPCR test shows a large range of expression values among cytogenetic and mutation subgroups of the training cohort. HMGA2 expression levels evaluated by this test were normalized on the ABL1 control gene and expressed as NCN. The dotted line represents the assay cutoff established at 1100 NCN. Bottom panel. Frequency of patients classified in each subgroup according to the HMGA2 expression profile. Numbers in white represent HMGA2+ patients. ELN 2017 and cytogenetic risk subgroups were evaluated in the training cohort (n = 358), mutations were obtained by RNA sequencing in 263 samples of the training cohort. Abn., abnormal; biCEBPA, biallelic CEBPA mutations; ELN: European LeukemiaNet; HMGA2+ , high expression (≥1100 NCN); HMGA2−, low expression (<1100 NCN); NCN, Normalized Copy Numbers; NK, normal karyotype
In the training cohort, the HMGA2 test showed high reproducibility, robustness, and specificity (Supplementary Table S7). Inter-laboratory test validation was performed at the King’s College University of London laboratory, using 263 AML samples from patients of the NCRI AML17 trial.
HMGA2 expression profile in relation to age and genetics
In the training cohort, 38 of 123 (30.9%) older patients (≥60 years) were H + compared with 42 of 235 (17.9%) younger patients (<60 years) (P = 0.007) (Table 1). H + status was more frequent in the adverse cytogenetic risk category: 35 patients of 68 (51.5%) were H + , and in the 2017 ELN adverse-risk group: 45 patients of 87 (51.7%) were H + . Similar to RNA-sequencing results, most patients with complex and monosomal karyotype (88.9% H + , 24 of 27) or with TP53 mutations (84.2% H + , 16 of 19) were H + (Fig. 2). Among the 232 patients in the intermediate cytogenetic risk category, 41 (17.7%) were positive for HMGA2. Of those, only a low proportion were NPM1 (4.9% vs 66.0% in H−; P < 0.001) or FLT3-ITD mutated (22.0% vs 42.4% in H−; P = 0.021) (Table 1). In the favorable cytogenetic risk category (t(8;21) and inv(16)), only 3 of 54 patients (5.6%) were H + (Table 1; Fig. 2). All biallelic CEBPA mutated samples were H− (Fig. 2). In the validation cohort, high HMGA2 expression levels were detected in 72 of 214 (33.6%) intermediate cytogenetic risk patients and 24 of 35 (68.6%) adverse cytogenetic risk patients with a lower frequency in NPM1 (22.2% vs 58.0% in H−; P < 0.001) or FLT3-ITD (12.1% vs 34.4% in H−; P < 0.001) mutated samples (Table 1).
HMGA2 test is powerful to predict clinical outcomes in AML
In the training cohort, compared with H− patients, H + patients had lower CR frequency (58.8% vs 83.4%; P < 0.001) (Table 1), worse 3-year OS (13.2% vs 43.5%; P < 0.001) and RFS (10.8% vs 44.2%; P < 0.001), and a higher 3-year CIR (72.9% vs 48.1%; P = 0.004) (Fig. 3a; Supplementary Table S10). Univariate analyses showed the strong effect of H + as well as age and cytogenetics, for all these clinical outcomes (Supplementary Table S11). Among the 308 patients with ≥3 years of follow-up, only 7 of 72 H + patients were still alive at 3 years compared with 87 of 236 H− patients (9.7% vs 36.9%; P < 0.001) (Supplementary Figure S7).

From left to right for a the training cohort, b NCRI AML17 validation cohort: overall survival (OS), relapse-free survival (RFS), and cumulative incidence of relapse (CIR) curves according to high expression levels of HMGA2 (HMGA2 + , H + ) compared with low expression levels (HMGA2−, H−). c OS and RFS curves for patients ( < 60 years) of the training cohort transplanted in first complete remission (n = 60). d OS curve for patients of the NCRI AML17 cohort not classified in the high-risk category (n = 157). The NCRI high-risk category is defined in the statistical methods section in the Supplementary Information.30,31 The P values were obtained by the log-rank test for comparison of OS and RFS curves and by Gray’s test for CIR curves
Multivariable analyses, adjusted for age, WBC counts, HSCT as a time-dependent variable (except for CR prediction), cytogenetic risk and NPM1 and FLT3-ITD mutation status, revealed that H + was independently associated with a significantly higher probability of primary refractory disease (adjusted Odds ratio = 3.08, (95% confidence interval (CI), 1.44–6.59), P = 0.004), worse OS (adjusted Hazard ratio = 1.68, (95% CI, 1.17–2.43), P = 0.006) and RFS (aHR = 1.61, (95% CI, 1.02–2.55), P = 0.041) and a higher CIR (aHR = 1.67, (95% CI, 1.01–2.75), P = 0.047) (Table 2; Fig. 4). Importantly, among the 263 sequenced patients of the Leucegene prognostic cohort (Fig. 1), even after having adjusted for the 2017 ELN poor risk mutations (TP53, ASXL1, and RUNX1), HMGA2 remained a strong predictor for poor response to induction chemotherapy (aOR = 4.03, (95% CI, 1.55–10.4), P = 0.004) (Table 2) and worse OS (aHR = 1.73, (95% CI, 1.06–2.84), P = 0.030) (Supplementary Table S12). Interestingly, after adjusting for HMGA2, TP53 mutations lost their statistical significance in the OS model (Supplementary Table S12).

Forest plot for multivariable analyses of overall survival, relapse-free survival and cumulative incidence of relapse in the training cohort. aHR, adjusted hazard ratio; CI, confidence intervals; HMGA2 + , high expression ( ≥ 1100 NCN); HMGA2-, low expression (<1100 NCN); HSCT, allogeneic hematopoietic stem cell transplantation; ITD, internal tandem duplication; WBC, white blood cell counts (×109/l). As the non-linear effect of age at diagnosis is represented jointly by the two coefficients (linear and quadratic), the interpretation of each coefficient separately is not appropriate and not shown in the figure
HMGA2 test is clinically useful in intermediate genetic risk patients
The prognostic value of HMGA2 expression was also evaluated in the intermediate cytogenetic risk category. In this subgroup, a positive HMGA2 test (41 of 232 patients) also predicted poor clinical outcomes (OS: 17.3% vs 40.2% for H− patients, P = 0.024; RFS: 12.8% vs 42.0%, P = 0.010; CIR: 75.6% vs 50.6%, P = 0.028) (Fig. 5a and Supplementary Table S10). Importantly, among these H + patients, seven were negative for the six prognostically informative AML mutations (FLT3-ITD, NPM1, biallelic CEBPA, ASXL1, RUNX1, and TP53) (Fig. 5b). Moreover, in 14 additional H + patients negative for FLT3-ITD, NPM1, and biallelic CEBPA mutations, and for which mutation profiling of ASXL1, RUNX1, and TP53 genes was not available in the clinical laboratory, the HMGA2 test could have been useful to identify poor risk patients (Fig. 5b). RNA-sequencing data were available for 165 intermediate cytogenetic risk patients: mutations in ASXL1 and/or RUNX1 genes were detected in 22 patients. Only 2 of these 165 patients had TP53 mutations and were excluded from the multivariable analyses. Even after having adjusted for FLT3-ITD, NPM1, biallelic CEBPA, ASXL1 and RUNX1 mutations, the WBC count and HSCT as a time-dependent variable, the significant independent impact of H + for survival and relapse prediction was further confirmed (OS: aHR = 2.38, (95% CI, 1.26–4.50), P = 0.008; RFS: aHR = 2.67, (95% CI, 1.24–5.77), P = 0.012; CIR (aHR = 2.61, (95% CI, 1.13–6.05), P = 0.025) (Supplementary Table S13).

a From left to right for patients of the training cohort classified in the intermediate cytogenetic risk category: overall survival (OS), relapse-free survival (RFS) and cumulative incidence of relapse (CIR) curves according to high expression levels of HMGA2 (HMGA2 + , H + ) compared with low expression levels (HMGA2−, H−). The P values were obtained by the log-rank test for comparison of OS and RFS curves and by Gray’s test for CIR curves. b Results for the six prognostically informative AML mutations (FLT3-ITD, NPM1, biallelic CEBPA, ASXL1, RUNX1, and TP53) in 41 H + patients of the training cohort classified in the intermediate cytogenetic risk category are shown. Dark blue squares, presence of mutation; light blue squares, absence of mutation (WT); white squares, sample not tested or not sequenced for this mutation (NA); H + , high expression levels of HMGA2 (≥1100 NCN)
HMGA2 test in transplanted patients
Among 60 younger patients who underwent allo-HSCT in first CR, including 42 intermediate-risk patients, H + was highly predictive of poor OS (3-year OS: 13.3% vs 63.6%, P = 0.013) and RFS (3-year RFS: 15.0% vs 57.6%, P = 0.047) (Fig. 3c) and appeared to be associated with a higher CIR (3-year CIR: 65.0% vs 27.7%, P = 0.064) (Supplementary Figure S8; Supplementary Table S10). However, the number of transplanted patients was too small for multivariable analysis.
HMGA2 test also adds prognostic value in the 2017 ELN adverse-risk category
We next studied whether the HMGA2 test could improve prognostic assessment in AML patients classified according to the 2017 ELN genetic risk stratification.1 We found that 45 out of 87 ELN adverse-risk patients (51.7%) (Table 1, Supplementary Table S14) were positive for the HMGA2 test and had a significantly worse survival (Supplementary Figure S9, right panel, red curve). In this patient subgroup (ELN adverse-H + ), representing 12.6% of the entire AML training cohort, no patients were long-term survivors. In contrast, the survival of H− patients classified as adverse risk by the ELN risk stratification was similar to that of ELN intermediate-risk patients (Supplementary Figure S9, right panel, yellow and green curves). Importantly, among the 45 H + patients, eight samples harbored mutations in RUNX1 and/or ASXL1 genes (intermediate-risk cytogenetics) and 15 had mutations in TP53 (Supplementary Table S14). This finding is clinically relevant, especially if screening for these poor risk mutations is not readily available.
HMGA2 test validation in the NCRI AML17 cohort
To validate the ability of HMGA2 expression to enhance risk stratification in an independent cohort, the prognostic value of H + was assessed in the UK NCRI AML17 cohort using the same RT-qPCR assay and cutoff (Table 1). Consistent with our findings, H + was a strong predictor of a lower frequency of CR (70% vs 85.6%, P = 0.002, Table 1), poor survival (5-year OS: 21% vs 51%, P < 0.001) and a higher risk of relapse (5-year RFS: 21% vs 44%, P < 0.001 and 5-year CIR: 60% vs 46%, P = 0.003) in the validation cohort (Table 3, Fig. 3b). Multivariable logistic and Cox regression analyses were used to examine the effect of HMGA2 expression adjusted for these known prognostic variables: age, log WBC count, secondary disease, WHO/ECOG performance status, presence of adverse cytogenetics, FLT3-ITD and NPM1 mutations. These results confirmed that H + was significantly and independently associated with lower CR/CRi (CR with incomplete hematologic recovery) frequency (aOR = 3.98, (95% CI, 1.36–11.65), P = 0.010), worse OS (aHR = 2.03, (95% CI, 1.36–3.03), P < 0.001), and RFS (aHR = 2.06, (95% CI, 1.38–3.08), P < 0.001) and a higher CIR (aHR = 2.01 (95% CI, 1.28–3.14), P = 0.002) (Table 3). The utility of the HMGA2 test was also evaluated in AML patients classified using a clinical risk score to identify high-risk patients. High-risk disease was defined according to the NCRI multi-parameter risk score, based upon baseline characteristics and response to the first course of induction chemotherapy30,31 (detailed in Supplementary Information, Statistical Methods section). Importantly, among the 157 patients not classified in the NCRI high-risk category, 52 (33%) H + patients had a significantly worse survival than 105 H− patients (P = 0.002) (Fig. 3d).
Discussion
HMGA2 encodes a member of the HMGA family of proteins implicated in chromatin remodeling and transcription regulation. It is overexpressed in many human solid tumors and its upregulation was thought to be potentially associated with tumor progression and poor prognosis.32,33 This study reports the strong negative prognostic impact of HMGA2 overexpression in AML, thus justifying the development and validation of a rapid, simple and inexpensive RT-qPCR test, also optimized on the droplet digital PCR platform, which can now be implemented in clinical laboratories. Our findings reveal that high HMGA2 expression confers a significantly higher probability of primary refractory disease after an anthracycline and cytarabine based induction chemotherapy. Interestingly, in the training cohort, the HMGA2 test also reclassified 17.7% of intermediate cytogenetic risk patients into a poor risk group. These results were confirmed in the validation cohort in which 33% of patients not classified in the NCRI high-risk category were H + and had a significantly worse survival than H − patients. This new knowledge could guide clinicians to consider offering more intensive or novel consolidation therapies for these patients.
Data presented in this study also highlight the possibility that HMGA2 expression status may predict outcome following allo-HSCT, although our study does not have the power to fully address this issue.
Importantly, in a subgroup of ELN adverse genetic risk patients, a positive HMGA2 test could also predict resistance to standard treatments including allogeneic stem cell transplantation. However, these results require further validation in other AML cohorts with comprehensive mutation profiling data and classified according to the 2017 ELN genetic risk categories. Future prospective studies will determine if specific therapeutic strategies such as investigational new drugs or novel transplantation methods can improve the clinical outcome of HMGA2 positive patients.
Although age, mutations, and cytogenetic characteristics affect patient survival in AML, we demonstrate that expression of a single gene, HMGA2, is an independent prognostic factor in multivariable analyses in two independent AML cohorts. Moreover, HMGA2 appears to integrate the negative prognostic value conferred by complex karyotype and several poor risk mutations and could simplify prognostic assessment of positive cases. However, the test did not capture all poor prognosis patient subgroups. For example, MLL rearrangements and the poor prognostic NPM1 + FLT3-ITD + DNMT3A + subset3 (~ 7% and ~ 12.5% in the Leucegene cohort, respectively) were frequently associated with low expression levels of HMGA2. Based on these findings, we propose a new algorithm integrating the HMGA2 test in current strategies for AML prognostic assessment (Supplementary Figure S10). Validation of this algorithm in clinical trials is warranted.
In conclusion, this study showed that high HMGA2 expression adds significant independent prognostic value to known clinical and genetic prognostic factors in AML, and is predictive of poor clinical outcomes with standard AML therapies. The HMGA2 test could complement the current AML tests to improve treatment orientation and be integrated in ongoing and future prospective clinical trials studying innovative therapies to increase survival of HMGA2 positive AML patients.
Change history
28 February 2019
Since the publication of the original article the authors noticed the the affiliation details for Paresh Vyas are incorrect. The correct affiliation details for this author are given below:
References
Dohner, H. et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447 (2017).
Grossmann, V. et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood 120, 2963–2972 (2012).
Papaemmanuil, E. et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221 (2016).
Patel, J. P. et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366, 1079–1089 (2012).
Metzeler, K. H. et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 128, 686–698 (2016).
Saber, W. et al. Outcomes after matched unrelated donor versus identical sibling hematopoietic cell transplantation in adults with acute myelogenous leukemia. Blood 119, 3908–3916 (2012).
Bullinger, L. et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 350, 1605–1616 (2004).
Metzeler, K. H. et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood 112, 4193–4201 (2008).
Damm, F. et al. Integrative prognostic risk score in acute myeloid leukemia with normal karyotype. Blood 117, 4561–4568 (2011).
Li, Z. et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J. Clin. Oncol. 31, 1172–1181 (2013).
Rockova, V. et al. Risk stratification of intermediate-risk acute myeloid leukemia: integrative analysis of a multitude of gene mutation and gene expression markers. Blood 118, 1069–1076 (2011).
Ng, S. W. et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–437 (2016).
Chuang, M. K. et al. An mRNA expression signature for prognostication in de novo acute myeloid leukemia patients with normal karyotype. Oncotarget 6, 39098–39110 (2015).
Valk, P. J. et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 350, 1617–1628 (2004).
Zhao, S., Fung-Leung, W. P., Bittner, A., Ngo, K. & Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 9, e78644 (2014).
Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
Lavallee, V. P. et al. Identification of MYC mutations in acute myeloid leukemias with NUP98-NSD1 translocations. Leukemia 30, 1621–1624 (2016).
Lavallee, V. P. et al. RNA-sequencing analysis of core binding factor AML identifies recurrent ZBTB7A mutations and defines RUNX1-CBFA2T3 fusion signature. Blood 127, 2498–2501 (2016).
Lavallee, V. P. et al. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood 127, 3054–3061 (2016).
Lavallee, V. P. et al. EVI1-rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood 125, 140–143 (2015).
Lavallee, V. P. et al. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat. Genet. 47, 1030–1037 (2015).
Gabert, J. et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia 17, 2318–2357 (2003).
Kroll, M. H. et al. Assessment of the diagnostic accuracy of laboratory tests using receiver operating characteristic curves; approved guideline. 2nd edn, 31 (NCCLS, Wayne PA, 2011). 2011 CLSI document EP24-A2.
Schisterman, E. F., Perkins, N. J., Liu, A. & Bondell, H. Optimal cut-point and its corresponding Youden Index to discriminate individuals using pooled blood samples. Epidemiology 16, 73–81 (2005).
Scrucca, L., Santucci, A. & Aversa, F. Competing risk analysis using R: an easy guide for clinicians. Bone Marrow Transplant. 40, 381–387 (2007).
Abrahamowicz, M., MacKenzie, T. & Esdaile, J. M. Time-dependent hazard ratio: modeling and hypothesis testing with application in lupus nephritis. J. Am. Stat. Assoc. 91, 1432–1439 (1996).
Belot, A., Abrahamowicz, M., Remontet, L. & Giorgi, R. Flexible modeling of competing risks in survival analysis. Stat. Med. 29, 2453–2468 (2010).
Lunn, M. & McNeil, D. Applying Cox regression to competing risks. Biometrics 51, 524–532 (1995).
McShane, L. M. & Hayes, D. F. Publication of tumor marker research results: the necessity for complete and transparent reporting. J. Clin. Oncol. 30, 4223–4232 (2012).
Burnett, A. K., Hills, R. K., Wheatley, K., Goldstone, A. H. & Prentice, A. G. D. M. A sensitive risk score for directing treatment in younger patients with AML. Blood 108, 18 (2006).
Ling, V. et al. Utility of a clinical risk score to identify high-risk patients with de novo acute myeloid leukaemia in first remission after high-dose cytarabine (HiDAC) based induction chemotherapy. Br. J. Haematol. 160, 861–863 (2013).
Pallante, P., Sepe, R., Puca, F. & Fusco, A. High mobility group a proteins as tumor markers. Front. Med. 2, 15 (2015).
Fedele, M. & Fusco, A. HMGA and cancer. Biochim. Biophys. Acta 1799, 48–54 (2010).
Acknowledgements
This work was supported by Genome Canada and Génome Québec (G.S., J.H. and S.L.), research chairs from the Canada Research Chair program (G.S.) and Industrielle-Alliance, Université de Montréal (J.H.), and the Leukemia and Lymphoma Society (US) to J.H. M.A. is a James McGill Professor of Biostatistics at McGill University. The Banque de cellules leucémiques du Québec (BCLQ) is supported by grants from the Cancer Research Network of the Fonds de recherche du Québec-Santé. RNA-Seq read mapping and transcript quantification were performed on the supercomputer Briaree from Université de Montréal, managed by Calcul Québec and Compute Canada. The operation of this supercomputer is funded by the Canada Foundation for Innovation (CFI), NanoQuébec, Réseau de médecine génétique appliquée and the Fonds de recherche du Québec—Nature et technologies. V.P.L. was supported by a fellowship from the Cole Foundation, W.G. was supported by the Generation Trust and King’s College London. D.G. was supported by a specialist program grant from Bloodwise (13043) and King’s College London. P.V. acknowledges funding from the MRC (MHU Award G1000729, MRC Disease Team Award 4050189188), CRUK (Program Grant to PV C7893/A12796), Bloodwise (Specialist Program 13001). and the Oxford Partnership Comprehensive Biomedical Research Centre (NIHR BRC Funding scheme). We acknowledge all patients and hematologists who participate to the BCLQ program and the BCLQ team, namely Professor Giovanni D’Angelo, Sylvie Lavallée and Claude Rondeau. We acknowledge Muriel Draoui for the Leucegene project coordination, Geneviève Boucher and Patrick Gendron for RNA-Sequencing data processing (IRIC bioinformatics platform), Martine Lavoie for RT-qPCR assays and Isabel Boivin for mutation validation (training cohort), Bruno Lamontagne, Guylaine Lépine, and Julie Bergeron for NPM1, FLT3-ITD, and biallelic CEBPA mutation testing (clinical molecular laboratory, Maisonneuve-Rosemont Hospital).
Author information
Authors and Affiliations
Contributions
M.M. developed and validated the HMGA2 RT-qPCR assay and analyses for the training cohort, generated figures, tables, and co-wrote the paper, C.B. performed statistical analyses for the training cohort, generated figures and tables, and co-wrote the paper, V.P.L. contributed to the identification of the HMGA2 marker, analyzed mutations and contributed to Fig. 1 and figure S4, M.A. supervised the generation of multivariable models, C.D. performed statistical analyses, S.L. supervised the bioinformatics team, I.A. contributed to statistical analyses, A.W. and S.T. provided AML samples from Australia, S.F. and A.S. provided clinical data related to the Australian samples, W.G. performed the RT-qPCR assays for the NCRI cohort, D.G. supervised and analyzed the RT-qPCR assays for the NCRI cohort, R.K.H. performed statistical analyses for the NCRI cohort, P.V. and N.R. contributed to analyses for the NCRI cohort, G.S. contributed to project conception and identification of the HMGA2 marker and edited the manuscript, J.H. contributed to project conception and identification of the HMGA2 marker, supervised this study, analyzed cytogenetic studies, provided AML samples and clinical data of the training cohort and co-wrote the paper. All authors revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marquis, M., Beaubois, C., Lavallée, VP. et al. High expression of HMGA2 independently predicts poor clinical outcomes in acute myeloid leukemia. Blood Cancer J. 8, 68 (2018). https://doi.org/10.1038/s41408-018-0103-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-018-0103-6
- Springer Nature Limited
This article is cited by
-
Silencing LINC00987 ameliorates adriamycin resistance of acute myeloid leukemia via miR-4458/HMGA2 axis
Biology Direct (2024)
-
Heterogeneity in leukemia cells that escape drug-induced senescence-like state
Cell Death & Disease (2023)
-
ZFX-mediated upregulation of CEBPA-AS1 contributes to acute myeloid leukemia progression through miR-24-3p/CTBP2 axis
Cell Biology and Toxicology (2023)
-
A cellular hierarchy framework for understanding heterogeneity and predicting drug response in acute myeloid leukemia
Nature Medicine (2022)
-
Overexpression of Hmga2 activates Igf2bp2 and remodels transcriptional program of Tet2-deficient stem cells in myeloid transformation
Oncogene (2021)