
Abstract
The axon guidance cue SLIT3 was identified as an osteoanabolic agent in two recent reports. However, these reports conflict in their nomination of osteoblasts versus osteoclasts as the key producers of skeletal SLIT3 and additionally offer conflicting data on the effects of SLIT3 on osteoclastogenesis. Here, aiming to address this discrepancy, we found no observable SLIT3 expression during human or mouse osteoclastogenesis and the only modest SLIT3-mediated effects on osteoclast differentiation. Conditional deletion of SLIT3 in cathepsin K (CTSK)-positive cells, including osteoclasts, had no effect on the number of osteoclast progenitors, in vitro osteoclast differentiation, overall bone mass, or bone resorption/formation parameters. Similar results were observed with the deletion of SLIT3 in LysM-positive cells, including osteoclast lineage cells. Consistent with this finding, bone marrow chimeras made from Slit3−/− donors that lacked SLIT3 expression at all stages of osteoclast development displayed normal bone mass relative to controls. Taken in context, multiple lines of evidence were unable to identify the physiologic function of osteoclast-derived SLIT3, indicating that osteoblasts are the major source of skeletal SLIT3.
Similar content being viewed by others

Introduction
Drugs for the treatment of skeletal disorders, such as osteoporosis, typically fall into one of two categories. These drugs function to either block bone resorption by osteoclasts or augment bone formation by osteoblasts. However, current treatment options still have strong limitations, due in part to both the few anabolic agents available and restrictions on the use of these agents.1,2 To continue to advance therapy for disorders of low bone mass, the identification of new strategies to promote bone formation is critical.
Axon guidance cues have emerged as agents of great interest in this regard, as they control bone formation by a variety of mechanisms, including shaping the skeletal microenvironment to support osteogenesis.3,4,5,6,7 Among them, SLIT3 was recently identified as an osteoanabolic agent that ameliorated the bone loss in mouse models in two recent reports.8,9
SLIT3 was first discovered as a repulsive axon guidance cue during neuronal migration.10 SLIT3 is now increasingly recognized to play additional physiological roles outside of the nervous system, as it is involved in immunoregulation, stem cell regulation, and cancer development.11,12,13 More recently, SLIT3 was characterized as having angiogenic functions in nonbone tissues, and SLIT3 knockout mice displayed severe developmental vascular defects in the diaphragm.14,15,16 In vitro, recombinant SLIT3 stimulation could enhance angiogenesis by increasing the proliferation, migration and tube formation of endothelial cells.15 In contrast, SLIT3 signaling inhibition significantly decreased functional blood vessel formation in human engineered tissue.17
Our prior study showed that SLIT3 is highly expressed in osteoblasts, where it acts as a proangiogenic factor in bone to increase the levels of skeletal vascular endothelium and thereby increase bone formation. Through these angiogenic effects, recombinant SLIT3 was found to have therapeutic activity in mouse models of fracture healing and postmenopausal osteoporosis.8,15 Meanwhile, a separate study reported that SLIT3 is critical for skeletal physiology and also found osteopenia and a reduction in the skeletal vascular endothelium in Slit3−/− mice.9 However, in this report, osteoclasts, as opposed to osteoblasts, were nominated as a key source of SLIT3 to control coupling between osteoblasts and osteoclasts. Given these conflicting data and the fundamental importance of SLIT3 as a promising osteoanabolic agent and physiologic signal linking bone metabolism to skeletal angiogenesis, further studies to clarify the cellular sources and targets of SLIT3 are needed.
Results
The expression of SLIT3 in osteoclastogenesis
To clarify the cellular sources of SLIT3, we first examined Slit3 expression in parallel with other osteoclast makers using real-time PCR during in vitro osteoclastogenesis. Osteoclast formation was further monitored by tartrate-resistant acid phosphatase (TRAP) staining (Fig. 1a–c). Unlike the robust Slit3 expression observed in the brain and primary osteoblasts, we were unable to detect Slit3 mRNA expression during bone marrow macrophage (BMM)-derived osteoclastogenesis at the mRNA level (Fig. 1b). To further confirm this observation, we analyzed RNA-sequencing (RNA-seq) transcriptional profiling data from macrophages, osteoclasts, and osteoblasts derived from wild-type mice. This approach also showed that Slit3 expression in osteoclasts was negligible relative to its expression in osteoblasts. Western blotting using a polyclonal SLIT3 antibody validated for specificity (Fig. 1e) also failed to detect SLIT3 expression during osteoclastogenesis (Fig. 1f). These observations are consistent with those of prior analyses of axon guidance cue expression during osteoclast and osteoblast differentiation published by other groups.5,18 Moreover, SLIT3 expression was also not detected in prior studies of human osteoclast differentiation.8,19 Thus, we were unable to find evidence that SLIT3 is expressed in osteoclast lineage cells, which contrasts with the robust expression of SLIT3 observed in osteoblasts.8

The expression of SLIT3 during osteoclastogenesis. a, b Quantitative real-time PCR analysis of mRNA expression of Ctsk, Dcstamp, Nfatc1, and Slit3 in WT BMMs treated with RANKL for 72 h using osteoblast and brain tissue as positive controls (n = 3). c Representative TRAP-staining images of WT BMMs treated with RANKL for 0, 48, and 72 h (n = 4 total images per group). d Gene expression of Slit3, Gapdh, Hprt, Nfatc1, Tm7sf4 (Dcstamp), and Sp7 in RNA-seq analysis of primary macrophages, osteoclasts and osteoblasts derived from WT mice (n = 3). e Immunoblot analysis of SLIT3 expression in whole brain lysates from Slit3+/+ and Slit3−/− mice. f Immunoblot analysis of SLIT3 expression in BMMs treated with RANKL for 48 h, with whole-brain lysates from Slit3+/+ mice and osteoblast lysates used as positive controls and whole brain lysates from Slit3−/− mice and murine bone marrow-derived endothelial cells (Neg) used as negative controls
The effect of recombinant SLIT3 on osteoclastogenesis in vitro
To assess the effects of exogenous SLIT3 on osteoclastogenesis, we treated wild-type BMMs undergoing osteoclast differentiation with recombinant SLIT3 at a level that showed bioactivity in multiple other cellular assays (1 μg·mL−1), including tube formation assays in endothelial cells.8,11,15 Although SLIT3 treatment modestly inhibited the expression of osteoclast marker genes, we were not able to observe a significant change in the number of TRAP-positive osteoclasts compared with controls (Fig. 2a–c). Similar negative results were obtained during a dose titration of SLIT3 during osteoclast differentiation (Fig. 2d). Moreover, recombinant SLIT3 treatment did not affect the osteoclast activity during an in vitro mineral resorption assay (Fig. 2e). These findings are consistent with our prior report that osteoclast numbers and serum levels of crosslinked C-telopeptide of type I collagen (CTX), a marker of osteoclast activity, were not substantially altered in Slit3−/− mice.8

The effect of recombinant SLIT3 on osteoclastogenesis. a Quantitative real-time PCR analysis of the mRNA expression of Nftac1, Itgb3, Dcstamp, Ctsk, Calcr, and Trap in BMMs treated with RANKL and recombinant SLIT3 (1 μg·mL−1) for 72 h (n = 3). b Immunoblot analysis of Nfact1 and c-fos expression in BMMs treated with RANK and recombinant SLIT3 (1 μg·mL−1) for 72 h using p38a as a loading control. c, d Osteoclast differentiation using WT BMMs stimulated with RANKL and recombinant SLIT3 for 72 h. TRAP staining (left panel) was performed, and the area of TRAP-positive cells (nuclei/cell ≥3) per well relative to the WT control was calculated (right panel) (n = 4). e Mineral resorption activity of WT BMMs cultured on calcium-coated plates with RANKL and recombinant SLIT3 (1 μg·mL−1) for 7 days. Von Kossa staining was performed to detect resorptive pit areas. Representative images of the whole well are shown in the left panel. Quantification of the resorptive area relative to the whole well area is shown in the right panel (n = 4). Values represent the mean ± s.e.m., n.s. (not statistically significant); *P < 0.05 and **P < 0.01 by unpaired two-tailed Student’s t test (c) or one-way ANOVA followed by Dunnett’s test (a, d). Scale bars: c, d, 200 μm
Deletion of Slit3 did not affect osteoclast progenitor cells in vivo
To assess the role of SLIT3 in osteoclastogenesis in vivo, we first analyzed CD117+CD11bdimCD115+ osteoclast precursors (OCPs) in male Slit3−/− mice using flow cytometry and found no apparent differences in frequency (Fig. 3a, b),20 despite the severe osteopenic phenotype observed in these mice. Next, as osteoblast-derived SLIT3 is crucial for bone mass accrual, we asked whether osteoblast-derived SLIT3 would specifically affect osteoclast precursors. To this end, we bred Slit3f/f mice to the Osx-cre deleter strain in which osteoblasts were targeted (Slit3osx). Similar to observations made following the global deletion of SLIT3, the number of osteoclast precursors in male Slit3osx mice was unaltered despite the severe osteopenia observed in these mice8 (Fig. 3c). Finally, to confirm the role of SLIT3 in osteoclasts, we bred Slit3f/f mice to a cre deleter strain in which mature osteoclasts were targeted, CTSK-cre (Slit3ctsk) mice. No apparent difference in the overall abundance of osteoclast precursors in male Slit3ctsk mice was observed, indicating that the deletion of SLIT3 in CTSK-positive cells did not disrupt osteoclast precursors (Fig. 3d). Furthermore, fluorescence-activated cell sorting (FACS)-isolated osteoclast precursors derived from Slit3ctsk mice displayed intact osteoclast differentiation capacity in vitro (Fig. 3e). Taken together, these results indicate that SLIT3 is dispensable for the generation and homeostasis of osteoclast progenitors.

SLIT3 is dispensable for the production of osteoclast progenitors. a Schematic representation of the strategy used for flow cytometric analysis of osteoclast precursors. b–d Flow cytometric analysis of osteoclast precursor populations in 5-week-old male Slit3−/− (b), Slit3osx (c) and Slit3ctsk (d) mice with the corresponding controls. Representative flow cytometry plots for each experiment are shown (left panels). The frequencies of osteoclast precursor populations in B220−- and CD11b−-gated BM cells from bilateral femurs and tibiae are presented (right panels) (n = 5). e Osteoclast differentiation using sorted osteoclast precursors from 5-week-old male Slit3ctsk mice (n = 4) and littermate controls (n = 3) stimulated with RANKL for 72 h. TRAP staining (left panel) was performed, and the area of TRAP-positive cells (≥3 nuclei/cell) per well relative to the WT control was calculated (right panel). Values represent the mean ± s.e.m., n.s. (not statistically significant) by unpaired two-tailed Student’s t test. Scale bar: 200 μm
Bone mass and skeletal vasculature are normal in Slit3 ctsk mice
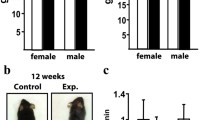
In contrast with the growth retardation and perinatal lethality observed in Slit3−/− and Slit3osx mice, Slit3ctsk mice developed normally without any gross abnormality or decrease in body weight (Fig. 4a, b). Micro-CT showed that neither male nor female Slit3ctsk mice displayed detectable alterations in bone mass in either the trabecular or cortical compartments of long bones at 5 weeks of age relative to their littermate controls (Fig. 4c, d). We further examined 4-month-old male Slit3ctsk mice versus littermate controls and also observed no detectable skeletal phenotype (Fig. 4e). Consistent with this finding, histomorphometric analysis of the vertebral bone showed that the mineralization rate, bone formation rate, and numbers of osteoblasts and osteoclasts were also not affected in Slit3ctsk mice. (Fig. 4f). In addition, in contrast to the vascular phenotype observed in Slit3−/− and Slit3osx mice, neither the visualization of endothelial cells with CD31 or EMCN immunofluorescence nor endothelial cell flow cytometry detected vascular alterations in Slit3ctsk mice (Supplementary Fig. 1a–c).8,9 Taken together, these data indicate that the deletion of SLIT3 in CTSK-positive osteoclasts was unable to alter bone remodeling and skeletal vascular endothelium, in contrast to the strong effects on both of these parameters in osteoblast lineage cells observed with the deletion of SLIT3. Finally, to further evaluate the role of SLIT3 in osteoclast lineage cells, we bred Slit3f/f mice to the cre deleter strain LysM-cre in which osteoclasts were targeted in addition to macrophages and neutrophils (Slit3lysM).21 Similar to observations in Slit3ctsk mice, female Slit3lysM mice at 8 weeks of age did not show a discernible change in bone mass accrual (Fig. 5a, b).

Conditional deletion of SLIT3 in CTSK-positive cells results in normal bone volume and normal osteoclast numbers in vivo. a, b Representative radiographic images (a) and body weights (b) of 5-week-old male Slit3ctsk mice and Slit3f/f mice (n = 6). c–e Representative micro-CT images, femoral BV/TV, cortical bone thickness, and bone morphometric analysis of trabecular bone of the distal femurs isolated from littermate Slit3ctsk mice and Slit3f/f mice. c 5-week-old female Slit3ctsk mice (n = 6) and littermate controls (n = 7); d 5-week-old male Slit3ctsk mice (n = 6) and littermate controls (n = 6); e 4-month-old male Slit3ctsk mice (n = 9) and littermate controls (n = 10). f Representative images of histological (left panel) and histomorphometric analysis (right panel) of the L3 vertebrae in male Slit3ctsk and Slit3f/f mice at 4 months of age (n = 5). Values represent the mean ± s.e.m., n.s. (not statistically significant) by an unpaired two-tailed Student’s t test. Scale bars: c–e, 1 mm; f, 500 μm

Conditional deletion of SLIT3 in LysM-positive cells results in normal bone volume. a, b Representative micro-CT images (a), femoral BV/TV, cortical bone thickness, and bone morphometric analysis of trabecular bone of the distal femurs (b) from 2-month-old female Slit3lysm mice and Slit3f/f mice (n = 8). Values represent the mean ± s.e.m., n.s. (not statistically significant) by an unpaired two-tailed Student’s t test. Scale bars, 1 mm
Ablation of SLIT3 in early osteoclast lineage cells leads to normal bone mass accrual
As CTSK-cre mediates gene deletion relatively late in osteoclast differentiation,22 we created bone marrow chimeras from Slit3−/− donors to assess the importance of SLIT3 production by early osteoclast lineage cells.23 Bone marrow was collected from 6-week-old male Slit3+/+ mice and Slit3−/− mice and then injected into lethally irradiated 6-week-old male WT mice. A CD45.1/CD45.2 congenic system was used to validate the efficiency of hematopoietic reconstitution 8 weeks after bone marrow transplantation. Flow cytometric analysis showed that B220+ cells, Gr1/CD11b+ myeloid cells and osteoclast precursors all showed nearly 100% CD45.1+ donor origin. CD3+ cells were approximately 80% CD45.1+ at this time point (Supplementary Fig. 2 a–c). Thus, high levels of donor chimerism were observed, particularly within the osteoclast precursor compartment most relevant to this study. The resulting chimeras were maintained for 12 weeks before the analysis outlined in Fig. 6a. Micro-CT analysis showed that the mice reconstituted with bone marrow cells isolated from Slit3−/− mice had normal bone mass, unlike those reconstituted with WT bone marrow cells, as illustrated by analysis of the trabecular bone volume/total volume and cortical bone thickness (Fig. 6a–c). Similarly, no alterations could be detected in the density of the bone vasculature in the recipients of Slit3−/− bone marrow (Fig. 6d). Consistent with this, the shRNA-mediated suppression of SLIT3 expression in BMMs had no effect on osteoclastogenesis in vitro (Supplementary Fig. 3a–c). Collectively, the results of both the conditional deletion of Slit3 in osteoclast lineage cells and development of bone marrow chimeras lacking SLIT3 in their hematopoietic cells, including all of the osteoclast lineage cells, indicate that osteoclasts are not a physiologically relevant source of SLIT3.

Ablation of SLIT3 in hematopoietic cells exhibits a normal bone phenotype and skeletal vasculature. a Schema showing the protocol for BM transplantation. Representative micro-CT images (b), femoral BV/TV, cortical bone thickness, and bone morphology of the distal femur trabecular bone (c) and representative confocal images of DAPI, CD31, and EMCN fluorescence in femur sections (d) isolated from bone marrow chimeras created using 5-week-old Slit3+/+ and Slit3−/− mouse donors and WT hosts (n = 6). GP growth plate. Values represent the mean ± s.e.m., n.s. (not statistically significant) by unpaired two-tailed Student’s t test. Scale bars, 500 μm
Discussion
Here, we address the discrepancies between our recent finding that osteoblasts are the major source of SLIT3 in the skeleton and another recent report nominating osteoclasts as the major source of SLIT3. In summary, multiple approaches were unable to provide evidence supporting osteoclast lineage cells as a source of SLIT3. Two separate cre lines mediating deletion in osteoclast lineage cells and bone marrow chimeras lacking SLIT3 expression in all osteoclast lineage cells displayed no detectable alteration in bone mass. These findings are in contrast to the robust phenotypes observed with either the osx-cre- or dmp1-cre-mediated deletion of SLIT3 in osteoblast lineage cells.8 Consistent with the absence of an in vivo function of SLIT3 in osteoclasts, we were also unable to observe evidence of SLIT3 expression in osteoclasts, a finding corroborated by other studies. Taken together, our results indicate that osteoclasts are not a physiologically relevant source of SLIT3 under the conditions examined, leaving osteoblasts as the major source of SLIT3 in bone.
We previously reported that SLIT3 mediates its osteoanabolic effects by promoting skeletal angiogenesis. As osteoclast lineage cells have independent mechanisms to regulate the skeletal vasculature through the secretion of PDGF-BB, the finding that the angiogenic functions of SLIT3 are restricted to osteoblasts is significant as it suggests that osteoblasts and osteoclasts each have orthogonal mechanisms to participate in the regulation of angiogenesis. This further opens the possibility of fundamental differences in osteoblast-regulated versus osteoclast-regulated vascular responses through the differential actions of SLIT3 versus PDGF-BB.23 Moreover, if SLIT3 is primarily derived from osteoblasts, it is an attractive candidate as a key mediator of the angiogenic responses induced by osteoblast-targeting anabolic agents.
In a study by Kim et al, the deletion of SLIT3 using CTSK-cre resulted in modest osteopenia at 16 weeks of age. Here, however, we did not observe a similar bone phenotype in mice of the same age and sex as those in the Kim et al. report and also did not observe phenotypes with the other approaches used here to delete SLIT3 in osteoclasts. Notably, we previously observed robust phenotypes with the cre lines used in this study.24,25 Some of these discrepancies may be due to differences in the Slit3 floxed allele mice, as the two reports utilized different mouse lines, though the deletion strategies used in each report were similar. However, the finding that bone marrow chimeras made from Slit3−/− donors failed to display skeletal phenotypes suggests that these discrepancies are not attributable solely to any idiosyncrasies of the Slit3 floxed allele utilized in the present report.
We are unable to fully exclude the possibility that osteoclasts are an additional target of SLIT3 activity based on the observation of modest effects on osteoclast gene expression after SLIT3 treatment. These effects may be mediated by the low-level expression of ROBO3 in osteoclasts.8 However, we were unable to detect an overall effect on the endpoint of osteoclast differentiation based on the formation of TRAP+ multinucleated osteoclasts in vitro or the number of either osteoclast progenitors or mature TRAP+ osteoclasts in vivo. Similarly, we were previously unable to detect alterations in markers of osteoclast resorptive activity in Slit3−/− or Slit3osx mice displaying severe osteopenia. In the context of the robust angiogenic responses evoked by SLIT3 in vivo and in vitro, this finding reinforces our model that osteoblast-derived SLIT3 acts on endothelial cells to evoke osteoanabolic responses. The differences regarding the in vitro effects of SLIT3 between the report by Kim et al. and the present report may reflect differences in the utilized source of SLIT3 or other factors. Notably, in addition to the failure of exogenous SLIT3 to exert a robust effect, in vitro genetic approaches had no effect. By serving as an endothelium-targeted osteoanabolic agent, SLIT3 may have utility when combined with traditional osteoblast- or osteoclast-targeted agents. Given that increasing evidence indicates that the optimal therapy for disorders of low bone mass may require sequential or combination therapy with multiple agents, the development of osteoanabolic agents with orthogonal action at the cellular level would be highly desired.26,27
Materials and methods
Animals
OSX-Cre, LysM-Cre, CTSK-Cre, Slit3−/−, and Slit3f/f mice were obtained or generated as described in previous studies.8,21,25 All mice were maintained under a standard 12 h dark–light cycle with chow ad libitum. All animal experiments were conducted according to the guidelines approved by the Weill Cornell Medical College subcommittee on animal care.
Osteoblast and osteoclast culture
Murine osteoclast differentiation assay was set up as previously described.28 Briefly, the whole bone marrow cells were flushed from the femora and tibiae dissected from the mutant mice and their littermates and plated on 10 cm dish for 3 days culture in α-MEM supplemented with 10% FBS in the presence of CMG14–12 supernatant as a source of M-CSF. Subsequently, we scraped the attached BMMs and seeded at a density of 4.5 × 104/cm2 for osteoclast formation with the stimulation of RANKL (40 ng·mL−1). We evaluate osteoclastogenesis using TRAP staining. Murine primary osteoblast differentiation assay was prepared as previously described.29
Osteoclast activity assay
Mineral resorption pit assay was used to evaluate the osteoclast activity as previously described.28 Briefly, BMMs were initially seeded on 96-well Corning Osteo Assay Surface Plates (Corning, Tewksbury, MA, USA) at a seeding density of 2 × 104 per well. Subsequently, osteoclast differentiation was driven by CMG14–12 supernatant and 40 ng·mL−1 RANKL. Once we observed mature osteoclasts, the cells on the plate were removed by adding 10% sodium hypochlorite solution. After twice PBS washing, von Kossa staining was performed to visualize resorptive pits. Light microscopic images were captured for each well and pit areas were quantified by Image J (National Institutes of Health, Bethesda, MD, USA).
Reverse transcription and real-time PCR
Total RNA (DNA-Free) was extracted from culture cells and tissues using an RNeasy Mini Kit (Qiagen, Germantown, MD, USA) and reverse transcription was carried on by First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). We performed real-time PCR using Fast SYBR Green Master Mix in the QuantStudio5 Real-Time PCR System (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) or hypoxanthine guanine phosphoribosyl transferase (Hprt) was used as a control for normalization. The following primers were used: Slit3-forward, 5ʹ-TCCAGTGTTCCTGAAGGCTCCT-3ʹ, and Slit3-reverse, 5ʹ-TGGCAATGCCAGGCTCCTTGTA-3ʹ; Ctsk-forward, 5ʹ-AAGATATTGGTGGCTTTGG-3ʹ, and Ctsk-reverse, 5ʹ-ATCGCTGCGTCCCTCT-3ʹ; Dcstamp-forward, 5ʹ-TTTGCCGCTGTGGACTATCTGC-3ʹ, and Dcstamp-reverse, 5ʹ-AGACGTGGTTTAGGAATGCAGCTC-3ʹ; Nfatc1-forward, 5ʹ-CCCGTCACATTCTGGTCCAT-3ʹ, and Nfatc1-reverse, 5ʹ-CAAGTAACCGTGTAGCTCCACAA-3ʹ; Itgb3-forward, 5ʹ-CCGGGGGACTTAATGAGACCACTT-3ʹ, and Itgb3-reverse, 5ʹ-ACGCCCCAAATCCCACCCATACA-3ʹ; Calcr-forward, 5ʹ-CTGAAGCTTGAGCGCCTGAGTC-3ʹ, and Calcr-reverse, 5ʹ-TGGGGTTGGGTGATTTAGAAGAAG-3ʹ; Trap-forward, 5ʹ-ACCAGCAAGGATTGCGAGGCAT-3ʹ, and Trap-reverse, 5ʹ-GGATGACAGACGGTATCAGTGG-3ʹ; Tnfrsf11b-forward, 5ʹ-CGGAAACAGAGAAGCCACGCAA-3ʹ, and Tnfrsf11b-reverse, 5ʹ-CTGTCCACCAAAACACTCAGCC-3ʹ; Gapdh-forward, 5ʹ-ATCAAGAAGGTGGTGAAGCA-3ʹ, and Gapdh-reverse, 5ʹ-GTCGCTGTTGAAGTCAGAGGA-3ʹ; and Hprt-forward, 5ʹ-CTGGTGAAAAGGACCTCTCGAAG-3ʹ, and Hprt-reverse, 5ʹ-CAAGATATCGTTGAAACGTGGA-3ʹ.
Transcriptional expression profiling by RNA-seq analysis
We purified total RNA using an RNeasy Mini Kit (Qiagen, Germantown, MD, USA) and used true-seq RNA Library preparation kits (Illumina, San Diego, CA, USA) to purify poly-A+transcripts and generate libraries with multiplexed barcode adapters according to the manufacturer’s protocols. After the quality of all samples passed control analysis using a Bioanalyzer 2100 (Agilent, Lexington, MA, USA), we constructed RNA-seq libraries per the Illumina TrueSeq RNA sample preparation kit and carried out high-throughput sequencing using the Illumina HiSeq 4000 in the Genomics Resources Core Facility of Weill Cornell Medical College. For analysis, we used STAR (version 2.3.0e)30 with the default parameters to align the reads to mm9 mouse transcripts. Then we sorted and indexed the resulting bam files using SAMtools. To obtain gene counts, we applied feature counts (version 1.4.3)31 to sorted bam files and excluded the genes without any expression counts. We normalize gene count data using the DESeq2 (version 1.4.5) R package.32
Immunoblotting
The extracts from cultured cells and total tissue were prepared using RIPA buffer or a lysis buffer [150 mmol·L−1 Tris-HCl (pH 6.8), 6% SDS, 30% glycerol, and 0.03% bromophenol blue] with 10% 2-ME. The lysates were subjected to 7.5% SDS-PAGE and transferred to Immobilon-P membranes (Millipore, Billerica, MA, USA). The transferred membranes were blocked with 5% skimmed milk and incubated with specific antibodies. The following primary antibodies were used: SLIT3 (1:1 000; R&D), Nfatc1 (1:1 000; BD Pharmagen), c-Fos (1:1 000; Santa Cruz), p38 (1:3 000; Santa Cruz), and HSP90 (1:2 000; Sigma). We detected protein bands using Western Lightning plus-ECL (PerkinElmer, Waltham, MA, USA).
Skeletal analysis
The whole-body radiographs of all mice were taken by the Faxitron X-ray system. Three dimensional skeletal images and assessments of trabecular and cortical bone from distal femur were obtained by a Scanco Medical μCT 35 system, which was calibrated weekly by scanning manufacturer-provided, resin-embedded phantoms. All sample scans were performed in 70% ethanol with an isotropic voxel size of 7 μm in the condition of an X-ray tube: 55 kVp, 0.145 mA, 600 ms integration time. The threshold of trabecular bone was 211 per mille, which corresponds to ~270 mg hydroxyapatite/ccm. The threshold of cortical bone was 350 per mille, which corresponds to ~570 mg hydroxyapatite/ccm. We reduced noise in the thresholded images using a Gaussian noise filter applied for murine bone analysis.
Histology, histomorphometry, and immunohistochemistry
The mutant mice and relative controls were subcutaneously injected with a dose of 20 mg·kg−1 on day 5 and 1 before scarification. Plastic embedding, sectioning, TRAP staining, toluidine blue staining and von Kossa staining were conducted as previously described.4 Histomorphometric analyses were carried out using the Osteomeasure Analysis System (OsteoMetrics, Atlanta, USA) as previously described29 Frozen sectioning and immunofluorescence staining were conducted according to a published protocol.33
Cell sorting and flow cytometry analysis
For OCP frequency analysis, bone marrow cells collected from Slit3+/+ and Slit3−/−mice, Slit3f/f and Slit3osx mice, Slit3f/f and Slit3ctsk mice were flushed out and removed debris using a 70 µm cell strainer. For skeletal endothelial cell analysis, the femurs and tibias from Slit3f/f and Slit3ctsk mice were isolated, and the surrounding muscles and connective tissues were cleaned. We crushed the bones in Hank’s balanced salt solution with 10 mmol·L−1 HEPES. For the enzymatically digestion, 2.5 mg·mL−1 Collagenase A and 1 U·mL−1 Dispase II were added and mixed thoroughly for 15 min incubation at 37 °C with gentle agitation. Next, we added PBS containing 0.5% FBS and 2 mmol·L−1 EDTA to stop digestion and the resulting suspensions filtered through a 40 µm cell strainer and washed twice with PBS. After washing, antimouse CD16/CD32 antibody (BD) was used to block unspecific staining for 15 min on ice. For OCP analysis, the cells were stained with BUV395-conjugated B220 antibody (BD), PerCP-Cy5.5-conjugated CD11b (BioLegend), FITC-conjugated CD115 (BioLegend) and PE/Cy7-conjugated CD117 (BioLegend) for 30 min on ice. For skeletal endothelial cell analysis, the cells were stained with FITC-conjugated CD45 (BioLegend), APC/Cy7-conjugated Ter119 (BioLegend), PE-conjugated CD31 (eBioscience) and APC-conjugated EMCN antibody (eBioscience) for 30 min on ice. DAPI solution (1 μg·mL−1) was used for live/dead exclusion. Cell sorting was performed with FACS Aria II (BD, San Jose, CA, USA) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Bone marrow transplantation
We followed a published protocol for bone marrow transplantation.34 Briefly, recipient mice (6-week-old BALB/c mice) were irradiated with a lethal dose of 875 rads 1 day before transplantation. The whole bone marrow cells from Slit3+/+ and Slit3−/−mice were harvested, and 5 million bone marrow cells per donor mouse were transplanted into each irradiated recipient via intravenous tail vein injection. The resulting bone marrow chimeras were euthanized 16 weeks after transplantation. Congenic CD45.1/CD45.2 mice were used to validate the reconstitution efficiency of bone marrow transplantation in our protocol described in Supplementary 2.
Statistical methods
All data statistical analysis in our study was performed using GraphPad Prism software (v6.0a; GraphPad, La Jolla, CA, USA). Two-tailed Student’s t test was used to determine significance for only two groups compare. One-way ANOVA with Tukey’s post-hoc tests was used to determine significance for compare between multiple groups. A P value < 0.05 indicated statistical significance. Error bars are presented as mean ± s.e.m.
References
Black, D. M. & Rosen, C. J. Postmenopausal Osteoporosis. N. Engl. J. Med. 374, 2096–2097 (2016).
Canalis, E., Giustina, A. & Bilezikian, J. P. Mechanisms of anabolic therapies for osteoporosis. N. Engl. J. Med. 357, 905–916 (2007).
Xu, R. Semaphorin 3A: a new player in bone remodeling. Cell Adh. Migr. 8, 5–10 (2014).
Fukuda, T. et al. Sema3A regulates bone-mass accrual through sensory innervations. Nature 497, 490–493 (2013).
Negishi-Koga, T. et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 17, 1473–1480 (2011).
Zhu, S. et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J. Clin. Investig. 129, 1076–1093 (2019).
Tumelty, K. E. et al. Identification of direct negative cross-talk between the SLIT2 and bone morphogenetic protein-Gremlin signaling pathways. J. Biol. Chem. 293, 3039–3055 (2018).
Xu, R. et al. Targeting skeletal endothelium to ameliorate bone loss. Nat. Med. 24, 823–833 (2018).
Kim, B. J. et al. Osteoclast-secreted SLIT3 coordinates bone resorption and formation. J. Clin. Investig. 128, 1429–1441 (2018).
Long, H. et al. Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron 42, 213–223 (2004).
Geutskens, S. B., Hordijk, P. L. & van Hennik, P. B. The chemorepellent Slit3 promotes monocyte migration. J. Immunol. 185, 7691–7698 (2010).
Geutskens, S. B. et al. Control of human hematopoietic stem/progenitor cell migration by the extracellular matrix protein Slit3. Lab Investig. 92, 1129–1139 (2012).
Dickinson, R. E. et al. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br. J. Cancer 91, 2071–2078 (2004).
Greaves, E. et al. Estrogen receptor (ER) agonists differentially regulate neuroangiogenesis in peritoneal endometriosis via the repellent factor SLIT3. Endocrinology 155, 4015–4026 (2014).
Zhang, B. et al. Repulsive axon guidance molecule Slit3 is a novel angiogenic factor. Blood 114, 4300–4309 (2009).
Yuan, W. et al. A genetic model for a central (septum transversum) congenital diaphragmatic hernia in mice lacking Slit3. Proc. Natl Acad. Sci. USA 100, 5217–5222 (2003).
Paul, J. D. et al. SLIT3-ROBO4 activation promotes vascular network formation in human engineered tissue and angiogenesis in vivo. J. Mol. Cell Cardiol. 64, 124–131 (2013).
Lattin, J. E. et al. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 4, 5 (2008).
Murata, K. et al. Hypoxia-sensitive COMMD1 integrates signaling and cellular metabolism in human macrophages and suppresses osteoclastogenesis. Immunity 47, 66–79 e65 (2017).
Tohmonda, T. et al. IRE1alpha/XBP1-mediated branch of the unfolded protein response regulates osteoclastogenesis. J. Clin. Investig. 125, 3269–3279 (2015).
Clausen, B. E., Burkhardt, C., Reith, W., Renkawitz, R. & Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999).
Lotinun, S. et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J. Clin. Investig. 123, 666–681 (2013).
Xie, H. et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 20, 1270–1278 (2014).
Greenblatt, M. B. et al. CHMP5 controls bone turnover rates by dampening NF-kappaB activity in osteoclasts. J. Exp. Med. 212, 1283–1301 (2015).
Debnath, S. et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 (2018).
Leder, B. Z. et al. Denosumab and teriparatide transitions in postmenopausal osteoporosis (the DATA-Switch study): extension of a randomised controlled trial. Lancet 386, 1147–1155 (2015).
Tsai, J. N. et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 382, 50–56 (2013).
Inoue, K. et al. Bone protection by inhibition of microRNA-182. Nat. Commun. 9, 4108 (2018).
Xu, R. et al. c-Jun N-terminal kinases (JNKs) are critical mediators of osteoblast activity in vivo. J. Bone Miner Res (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Kusumbe, A. P., Ramasamy, S. K., Starsichova, A. & Adams, R. H. Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat. Protoc. 10, 1904–1914 (2015).
Zhao, B. et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 15, 1066–1071 (2009).
Acknowledgements
R.X. holds an Award Program for the Minjiang Scholar Professorship in Fujian Province, a start-up grant from Xiamen University and support from the National Natural Science Foundation of China (81972034 to RX). M.B.G. is supported by the NIH grant DP5OD021351 and the grant NF150055 from the Department of Defense, and holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and a March of Dimes Basil O’Connor Award. B.Z. is supported by NIH grants (R01AR068970, AR071463). We thank Dr Edward Fung and Dr Douglas Ballon from Citigroup Biomedical Imaging Center for their expert assistance with micro-CT studies.
Author information
Authors and Affiliations
Contributions
R.X., B.Z., and M.B.G designed the experimental plan. N.L. and K.I. executed the experimental plan. Y.N., J.S., S.L., A.Y., X.Y., and R.S. assisted with mouse studies. R.X., B.Z., and M.B.G. wrote the manuscript and supervised the project. B.Z. is a co-senior author.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, N., Inoue, K., Sun, J. et al. Osteoclasts are not a source of SLIT3. Bone Res 8, 11 (2020). https://doi.org/10.1038/s41413-020-0086-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-020-0086-3
- Springer Nature Limited
This article is cited by
-
Schnurri-3 inhibition rescues skeletal fragility and vascular skeletal stem cell niche pathology in the OIM model of osteogenesis imperfecta
Bone Research (2024)
-
Bone controls browning of white adipose tissue and protects from diet-induced obesity through Schnurri-3-regulated SLIT2 secretion
Nature Communications (2024)
-
Messages from the Mineral: How Bone Cells Communicate with Other Tissues
Calcified Tissue International (2023)
-
Skeleton-vasculature chain reaction: a novel insight into the mystery of homeostasis
Bone Research (2021)
-
The endothelium–bone axis in development, homeostasis and bone and joint disease
Nature Reviews Rheumatology (2021)