
Abstract
Cancer comprises a collection of highly proliferative and heterogeneous cells growing within an adaptive and evolving tumour microenvironment. Cancer survival rates have significantly improved following decades of cancer research. However, many experimental and preclinical studies do not translate to the bedside, reflecting the challenges of modelling the complexities and multicellular basis of human disease. Organoids are novel, complex, three-dimensional ex vivo tissue cultures that are derived from embryonic stem cells, induced pluripotent stem cells or tissue-resident progenitor cells, and represent a near-physiological model for studying cancer. Organoids develop by self-organisation, and can accurately represent the diverse genetic, cellular and pathophysiological hallmarks of cancer. In addition, co-culture methods and the ability to genetically manipulate these organoids have widened their utility in cancer research. Organoids thus offer a new and exciting platform for studying cancer and directing personalised therapies. This review aims to highlight how organoids are shaping the future of cancer research.
Similar content being viewed by others
Background
Cancer mortality rates have significantly declined by ~26% over the past two decades,1 a decrease that is attributable to early diagnosis and treatment of malignancy, evidence-based clinical pathways for surveillance and management of premalignant lesions, increased awareness of health-related behaviours such as smoking and clinically focused cancer research. In spite of this success, however, cancer is the most common cause of death in the United Kingdom, and is expected to continue to remain as such, with 212,546 cancer deaths predicted for 2035.2,3
As we continue to make progress towards a ‘cure for cancer’, it is apparent that the data from many experimental and preclinical studies do not to translate from bench to bedside,4,5 an observation that is thought to reflect the challenges of modelling the complexities and multicellular basis of human disease.6 Despite these challenges, several pivotal systems, such as two-dimensional (2D) cell cultures, explants, organ-on-a-chip system and animal models, will continue to be essential to understand the biology of cancer. Nevertheless, novel and innovative model systems can improve the translational success of preclinical studies, and the methodology for tumour-derived organoid cultures has consequently emerged (Fig. 1). Organoids are complex, 3D, ex vivo tissue cultures that are derived from embryonic stem cells, induced pluripotent stem cells or tissue-resident progenitor cells. They possess spatially restricted lineage commitment and higher-order self-assembly, which makes them attractive near-physiological models.7 This review will firstly discuss the advantages and disadvantages of the experimental model systems that are currently used in cancer research, leading to a review of tumour-derived organoid model systems, including applications in cancer research, highlighting advantages, including potential utility in personalised medicine, limitations and future perspectives.

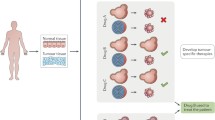
Patient-derived organoids reflect the genotype and phenotype of the original tissue, with preserved cellular heterogeneity and structural architecture. The critical steps involved in establishing a colonic organoid culture are a fresh colonic mucosa is obtained from human specimens (e.g., by biopsy or surgical resection), b colonic Lgr5+ stem cell-containing crypts are isolated and embedded in a basement membrane matrix, such as Matrigel and c colonic organoids are cultured in conditioned media containing specific growth factors and grow with a central lumen and representative apicobasal polarity. Images from Laboratory of Dr McLean, University of Aberdeen.
Current experimental model systems in cancer research
Several experimental model systems are currently applied to cancer research. Although a comprehensive overview of current laboratory models for cancer research is beyond the scope of this review, and is available elsewhere,8,9,10 the advantages and disadvantages are outlined in Fig. 2 and discussed below.

There are several model systems used to study cancer in the laboratory. Using human tumours, the most common models are two-dimensional (2D) cell lines, organ-on-a-chip technology, spheroid cultures, organoid cultures and rodent xenografts, where human tumour is implanted into live animals. The advantages and disadvantages of these models are outlined here.
2D cell culture, tissue slices and tissue explant culture
2D cell cultures—either primary cell cultures (grown directly from patient or animal tumours) or well-characterised immortalised cell lines—have been extensively used to study cancer. Although primary cell lines have a limited lifespan and are slow-growing, they are advantageous because they maintain some donor-cell characteristics and can be linked to clinicopathological data. By contrast, artificial manipulation or natural genetic mutations confer on immortalised cell lines the ability to proliferate indefinitely, making them a more convenient and well-established preclinical model, but rendering them less representative of the original tumour; furthermore, serial passages induce genotypic and phenotypic changes that might confound experimental results.11 Irrespective of their origin, 2D cell cultures cannot replicate intra-tumour cellular heterogenicity, lack the complex extracellular microenvironment, have forced apicobasal polarity and are grown as a monolayer with unnatural suspension and adherence forces. Although co-cultures and transwell assays can address some of these issues, the biological translation of 2D cell culture models can be limited.
Tissue slices and organ cultures derived from tissue explants provide the architecture, morphology and cellular composition that 2D cell cultures lack. However, these models have a short lifespan (most tissues are viable for 24 h; the liver can be viable up to 96 h) and are expensive and difficult to maintain.12
Organ-on-a-chip technology
Organ-on-a-chip technology refers to a multichannel microfluidic perfusion culture system, made from glass, plastic or a flexible polymer, that is lined with living human cells.13 This system allows more accurate modelling of organ-system physiology: for example, it facilitates the establishment of tissue–tissue interfaces, has separate vascular, extracellular and parenchymal compartments and allows for physiologically representative co-culture with microbes and immune cells.14 In cancer research, this technology has been used to study interactions between tumour cells and the extracellular milieu, cancer-associated epithelial–mesenchymal transition, angiogenesis, tumour invasion, cell migration and metastasis. Despite this impressive resume, this model does have disadvantages: for example, organ-on-a-chip platform commonly uses cell lines, and there is often significant variation and inconsistency between different chips, making experimental replication difficult.
Animal models
Preclinical animal models continue to be a key aspect of cancer research, and these have been reviewed elsewhere.15,16,17,18,19,20 Tumour growth in vivo can be induced by chemicals (e.g., azoxymethane mouse model of colorectal cancer), viruses (e.g., Friend-virus-induced erythroleukaemia in mice) or radiation (e.g., UV radiation-induced melanoma in mice). Genetically engineered animals are popular because tumours can be induced to develop in transgenic mice (e.g., mice lacking the adenomatous polyposis coli [APC] gene are used to study the adenoma–carcinoma sequence in colorectal cancer) or knockout mice (e.g., the BRCA1 conditional knockout mouse model using Cre/loxP recombination is used to study breast cancer), and key genes can be conditionally manipulated.15,21
Animal models are fundamental for translational cancer research—both for biological studies of pathogenesis and functional drug studies —and continue to be one of the cornerstone experimental approaches in the cancer research field. However, they do have limitations.16,21 Animal models are expensive, require extensive resources, and the data from many promising preclinical animal studies are often not validated in human models, or do not proceed in drug development towards clinical application,21,22 reflecting the different genetic, cellular and immunological characteristics in animals compared with humans. Steps taken to overcome these issues include transplanting human cancer tissue or cell lines into humanised rodents.23,24 These xenografts can be orthotopic (transplanted into the anatomical location from where the tumour was derived) or heterotopic (transplanted elsewhere, e.g., subcutaneously or intra-peritoneally).
3D cell models: tumour-derived organoid cultures
A critical player in organoid cultures is the stem cell, a self-renewing cell that can give rise to many different cell types within a tissue. Stem cells display unique markers such as leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5) in the intestine. In 2009, Sato et al.7 reported that single-cell-sorted Lgr5+ stem cells located at the bottom of intestinal crypts can initiate crypt–villus organoids when embedded in Matrigel (a gelatinous protein matrix that provides the structural architecture to support 3D growth), and that these intestinal organoids contained differentiated cell types that are present in the original tissue.
It is important to distinguish organoids from spheroids: both are cultured in a 3D format, but spheroids are simpler, homogeneous, 3D structures that lack the multiple cell types seen in organoids. Spheroids typically represent free-floating cell aggregates with no matrix component—they usually depend on cell–cell adhesion for viability. Spheroids can be generated from immortalised cell lines, primary cells or fragments of tissue and, as such, their viability is limited as they do not contain a progenitor phenotype. Spheroids develop a necrotic core as they grow in size, and possess no or limited tissue structure and a less representative tissue architecture (e.g., no central lumen).25 Hence, although spheroid culture is a useful 3D culture methodology, offering a bridge between traditional 2D culture and costly in vivo animal studies, organoid-based 3D culture methodology offers several advantages to spheroids owing to enhanced architectural and physiological functions.
Establishing and maintaining organoid cultures
The epithelial compartments of many tissues, including normal, premalignant tissues and tumours, have been modelled using organoids.26 Although the tissue for organoid culture is most commonly derived from surgical resection specimens,27,28 organoid cultures have been successfully established from other tissue sources, for example, endoscopic biopsy from Barrett’s oesophagus,27 needle biopsy for hepatocellular carcinoma,29 endoscopic ultrasound-scan-guided fine-needle biopsy for pancreatic ductal adenocarcinoma30 and ascitic fluid for both pancreatic and ovarian cancers.25,31 Successful tumour organoid cultures can therefore be generated from a small amount of biological material and from cancers that are difficult to access in the clinical setting. Previously, the interval between specimen collection and successful establishment of organoid culture has been dictated by the viability of fresh samples, which has limited the time, location and demographics from which a patient sample can be taken, but Tsai et al.32 published a robust method in 2018 to cryopreserve fresh human biopsy tissue and later thaw the specimen to generate gastrointestinal organoid cultures, thus overcoming this limitation. The diverse methods for tissue acquisition and the ability to cryopreserve specimens highlight the functional utility of organoid culture across a variety of cancer types and clinical situations.
Organoid cultures have been well-characterised in the literature, and this has provided a robust evidence base to validate the use of these models. For example, oesophageal adenocarcinoma organoids derived from oesophagectomy tissue specimens recapitulate the diverse genomic and transcriptomic landscape of the primary tumour,33,34 and histological assessment of these organoids demonstrated that the original tumour architecture and protein expression profile was maintained.33 This faithful representation has been reported across a variety of other tumour types, including, but not limited to, lung, ovarian, uterine, colorectal, bladder, liver, breast and biliary tract cancers.29,34,35,36,37,38 There is also evidence that epigenetic signatures in organoids appear to be reflective of those found in the primary lesion, indicating that the biology of the tumour is broadly represented.39
Once organoid cultures have been established, they require a complex and individualised combination of growth factors for survival and maintenance. It is essential to use optimised media formulations to ensure that experiments are reliable and reproducible. Organoid cultures from different tissues will have unique media requirements. Subtle changes to these cocktails can have marked consequences—for example, normal colonic organoids will outcompete colonic cancer organoids when cultured in media optimised for normal colonic organoids, potentially owing to apoptosis resulting from genomic instability in the tumour organoids.35 However, the sensitivity of organoids to growth factors can be exploited to establish many tumour organoid cultures. For example, normal colonic organoids require the ligand Wnt3a for survival, whereas the majority of colonic cancer cells demonstrate hyperactivation of the Wnt/β-catenin pathway independent of Wnt3a.40 Therefore, the selective removal of Wnt from organoid media prevents normal colonic organoids from outcompeting colonic cancer organoids.35 Not all colonic tumour cells display aberrant Wnt signalling though, and therefore, it is important to explore the implications of selecting tumour cells by their requirement(s) for specific factors.41,42 In future, it might be informative to use growth factor requirements to characterise, rather than select, tumour organoids. Nonetheless, it is important to remember that organoid function can be influenced by altering its media conditions, and it is therefore important to characterise organoid cultures before experimentation.
Long-term organoid culture is possible, with most groups reporting successful culture up to 6 months,27,33 and some groups reporting success beyond 1 year.43 Patient- and disease-specific characteristics are retained well over several passages.43 There is evidence that mutations do accumulate over time33 although this is perhaps unsurprising, given the known evolution of cancer in vivo,44,45 and this is consistent with tumour evolution in vitro.36 Tumour organoids also possess distinct organoid signatures that reflect real-life inter-patient variability.46 However, such inter-patient variability increases the sample-size requirements for robust power calculation-based experiments, which can be expensive. Ultimately, though, the expense must be weighed against the ability of this model to more accurately represent human disease.
Advantages and limitations of organoid models in cancer research
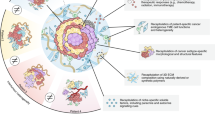
Our ability to manipulate tumour organoids further improves the utility of this culture system in cancer research. Several experimental approaches can be used to reveal novel insights into cancer pathogenesis (Fig. 3).

Tumour organoids can be manipulated to improve their functional utility for cancer research. They can be a genetically edited, for example, through CRISPR–Cas9 technology, b co-cultured with other cell types, such as immune cells, endothelial cells and stromal cells and c microinjected with microbes, antigens or chemicals. Several experimental approaches can be used to reveal novel insights into cancer pathophysiology, such as (d) immunohistochemistry (e), transwell and other 2D culture-based experiments (f) immunofluorescence, (g) organoid-on-a-chip technology and (h) xenografts.
Tumour organoids enhance the utility of 2D cell culture
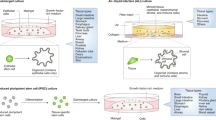
As discussed above, 2D cell cultures constitute a key experimental platform in laboratory research—a myriad of validated experiments can be performed using these simple and inexpensive cultures. Consequently, the ability to establish 2D monolayers from epithelial-derived organoids allows functional experiments to be carried out, such as wound healing and transepithelial electrical resistance assays to measure functional permeability, while maintaining the unique characteristics of ex vivo organoid cultures, such as molecular identity to the original tissue and presence of a number of different epithelial cell types, such as parietal, chief and mucous neck surface mucosal cells from the stomach (Fig. 4).47 High-throughput microscopy can be performed in 2D-organoid-derived monolayer cultures, which would be difficult to perform in the 3D equivalent.

a Organoids can be grown in monolayer culture. b Transwell cell culture systems further improve the versatility of this model by allowing access to apical (luminal) and basal surfaces. c Transwell experiments are effective at exploring how luminal antigens or cytokines impact epithelial barrier integrity in cancer (i.e., through movement of fluorescein isothiocyanate–dextran or changes in transepithelial electrical resistance). Co-culture transwell experiments may further improve this model for cancer research (i.e., by investigating the response of T cells and dendritic cells to increased barrier permeability induced by luminal antigens/cytokines in cancer). d Using organoid-derived 2D monolayers allows more complex bioimage analysis to be performed.
However, given the increased complexity of organoid models, there are some considerations for applying traditional 2D-based assays to 3D cultures. For example, organoid-derived monolayers cannot be easily passaged or propagated in 2D; instead, they achieve a homoeostatic state with balanced proliferation, differentiation and apoptosis.48 In addition, the efficiency of gene silencing using small-interfering (si)RNA is significantly reduced in the presence of serum in 2D cultures, whereas serum improves knockdown efficacy in organoid cultures by promoting the internalisation of siRNA.49
Genome editing improves the functional utility of organoids for cancer research
Genome editing has been used to improve the use of organoid models and, as such, normal/non-cancerous organoid cultures can be genetically manipulated to undergo malignant transformation. The prokaryotic clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system has revolutionised how we approach genome editing in the laboratory, allowing precise and consistent in vitro genome editing.50 As an example, in human cerebral organoids, CRISPR/Cas9 technology has been used to facilitate the expression of an oncogenic HRasG12V construct by homologous recombination into the TP53 locus, thereby simultaneously disrupting this tumour-suppressor locus.51 This approach enables putative-initiating genetic mutations to be recreated, and the natural history of tumour initiation of human gliomas to be followed. CRISPR/Cas9 has also been used to study the origin of mutational signatures in cancer by the selective deletion of critical DNA-repair genes.52
By inserting an inducible histone 2B-green fluorescent protein (GFP) reporter into patient-derived organoid cells, CRISPR/Cas9 technology has been used to track quiescent versus proliferative cells during glioblastoma recurrence.53 Due to the ability of organoids to maintain tumour cell heterogenicity, this genetic manipulation allowed researchers to compare quiescent cells from glioblastoma tumours with their proliferative counterparts, which has revealed novel insights into the pathophysiology of glioblastoma. Indeed, quiescent cells were reported to differentially express genes involved in cell-cycle control, metabolic adaptation, interaction with the extracellular matrix and mesenchymal transition, and showed higher resistance to therapy compared with proliferative cells. Both hypoxia and transforming growth factor-β were identified as potential niche factors that promoted quiescence. This use of organoid technology has therefore laid the foundations for developing new therapeutic strategies for glioblastoma by uncovering novel mechanisms of recurrence for this tumour.53
To model the well-defined progression from adenoma to carcinoma that occurs during colorectal carcinogenesis, CRISPR/Cas9 has been used to genetically engineer sequential mutations associated with stem cell niche regulation, senescence and DNA mismatch repair in colonic organoids from mice to mirror the molecular pathogenesis of serrated colorectal cancer.54 Following genetic manipulation, the resulting requirement for growth factors in the stem cell niche was exploited to select for mutant organoids, and subsequent colonoscopy-guided orthotopic injection of organoids into mice was performed to investigate the contribution of specific mutations to carcinogenesis. The resultant tumours arising in vivo were reflective of human disease. This study demonstrates how genetically modified organoids can be used to create novel orthotopic models of cancer.
Genome editing in organoids has also revealed novel insights into the pathophysiology of previously hard-to-model diseases. Barrett’s oesophagus describes distal oesophageal columnar metaplasia with malignant potential. The disease is difficult to study because available cell lines and animal models are poorly representative of the underlying biology—cell lines lack the phenotypic diversity seen in the oesophageal submucosal glands, and mouse models do not accurately model neoplastic progression.55,56 Through the use of CRISPR/Cas9 technology, patient biopsy-derived Barrett’s organoids that lack APC have been generated and used to demonstrate a fundamental role of aberrant Wnt/β-catenin signalling in the neoplastic progression of Barrett’s-associated oesophagus.57
Alternative, less costly and time-consuming methods of genomic manipulation have also been reported in the context of gene editing. Such examples include lentiviral transduction of prostate epithelial organoids to demonstrate that genetic alterations that are commonly found in human prostate cancer can be modelled in human organoid culture,58 and magnetic nanoparticle viral transduction of gastrointestinal organoids for further study in in vitro assays or in vivo functional analyses in mice.59
Organoids recreate the structural organisation of their origin tissue
Organoids can also recapitulate the spatial arrangement of their tissue of origin. When non-neoplastic bronchial mucosa failed to maintain its organoid ‘status’ during subculturing, Kim et al.60 added supplements known to promote lung development, which resulted in formation of bronchial cells; by passage 4, budding tubule-like organoids had emerged. Haematoxylin and eosin staining demonstrated that the organoids had pseudostratified epithelium, comprising basal cells and luminal cells that resembled normal bronchial mucosa. Interestingly, the non-neoplastic bronchial organoids had motile cilia found in large bundles, whereas lung cancer organoids had one single primary cilium per cell, indicating that, under the correct conditions, organoids can represent the structural organisation of their tissue of origin. In creating an organoid biobank from non-neoplastic and different subtypes of lung cancer tissue, the architectural, protein expression profile and molecular profiling was maintained from the tissue of origin.
Recreating structural morphology is especially important for cancers that arise from tissues with high structural organisation, such as the colon and rectum. Colorectal organoids self-organise to maintain apicobasal polarity with a hollow central lumen.61 This is a unique feature of organoid culture that reflects human anatomy and physiology, and it allows straightforward access to the basolateral surface via the culture media. Access to the luminal surface, however, is more difficult and, given the importance of antigens, microbiota and cell signalling receptors at the luminal surface of the epithelial barrier in colorectal carcinogenesis, there has been a focussed effort to improve luminal accessibility in organoid models. Breakdown of the epithelial barrier, with the consequent translocation of bacteria and luminal antigens through the colonic mucosa, is believed to be an important initiating event in colorectal carcinogenesis,62 and a protocol for the microinjection of fluorescently labelled dextran into patient-derived human intestinal organoids establishes a platform to study epithelial barrier dynamics in vitro.61 Breakdown of the epithelial barrier will result in increased permeability and translocation of fluorescently labelled dextran from the organoid lumen to the extracellular space. In addition to barrier integrity, the contribution of host–microbiota interactions to colorectal carcinogenesis can be studied by microinjection of live bacteria into the centre of colorectal organoids.63 Microinjection can be arduous and time consuming in the case of a large number of organoids. High-throughput microinjection of organoids is a solution to this problem, and can be achieved in the laboratory with semi-automated microinjection, microfabricated cell culture devices and computer vision systems (CVis).64 This technique could be applied to organoid cultures derived from various tumours, and could facilitate the injection not only of bacteria but also of chemical compounds, biological molecules, siRNA and other microorganisms.
One of the hallmarks of cancer is a loss of tissue organisation. Developments in microfabrication technology now enable organoids to be integrated into an extracellular matrix that contains specific biomolecules. For example, one study using normal breast organoid cultures created a defined chemoattractant gradient, using growth factors such as epidermal growth factor, to direct the formation of epithelial branches.65 This approach allows tissue-specific spatial and chemical factors to be considered in organoid cultures and, furthermore, can be adapted for use in cancer research—for example, to provide insights into factors that contribute to the loss of breast tissue organisation during carcinogenesis.
The tumour microenvironment can be represented in organoid cultures using co-culture
Many laboratory findings fail to translate to the clinic because cell cultures do not accurately recapitulate cell behaviour and function within the wider tumour microenvironment, which includes the extracellular matrix, blood vessels, signalling components and other cell types. Consequently, organoids offer a biologically relevant platform to improve translatability. Co-cultures are not a new concept in the laboratory, as they are often used to study interactions between epithelial cells and other important cell populations, such as lymphocytes, neurones and blood vessels. The successful co-culture of epithelial cancer organoids with immune cells has revealed important insights into the pathogenesis of many cancers, and the ability to genetically manipulate such organoids with or without immune cells provides a specific and relevant model for studying carcinogenesis.66,67,68 Co-culture of mouse tumour organoids with adipocytes has provided novel insights into colon cancer. For example, Wen et al.69 demonstrate that adipocytes promote the proliferation and dedifferentiation (detected by increased Lgr5 and CD44, and decreased mucin 2 and sucrase–isomaltase mRNA levels) of colon cancer organoids. The authors further suggest that adipocytes function as a metabolic regulator and energy provider to promote the growth of colon cancer cells, which offers a potential mechanism to help explain the relationship between obesity and colorectal cancer.
The extracellular matrix is not a passive bystander in cancer biology; however, the biological consequences of this are often not explored or adjusted for in traditional laboratory experiments.70 Co-culture experiments can overcome this. For example, established pancreatic ductal adenocarcinoma organoids normally develop ductal and basement membrane structures, but this organisation is lost following co-culture with pancreatic stellate cells in a collagen matrix, coincident with basement membrane destruction and increased invasion into the collagen matrix.71 Furthermore, co-culturing pancreatic cancer organoids with both stromal and immune cells leads to the activation of myofibroblast-like cancer-associated fibroblasts, an observation that was not apparent in 2D culture models.25 A model system that allows interaction between cancer cells, stromal cells and immune cells is therefore important for studying the pathogenesis of cancer.
As well as making organoid cultures more representative of the in vivo scenario, co-culture can improve the differentiation yield. For example, co-culture of human-induced pluripotent stem cells with human adipose microvascular endothelial cells leads to an increased yield of hepatocyte-like clusters and the generation of hepatocyte-like organoids that resemble mature tissue rather than cell cultures.72 There is also evidence that adding primary prostate stromal cells to 3D cultures of human prostate epithelial cells increases the formation of prostate organoids and non-random architectural organisation in the form of branching.73
Techniques to improve organoid cultures continue to emerge, such as the use of self-generating hydrogels comprising extracellular matrix derived from human tissue instead of Matrigel. For example, Mollica et al.74 describe a method for generating extracts of mammary extracellular matrix that can spontaneously gel to form hydrogels. Importantly, these hydrogels retain biological signalling responses that are different between cancer and normal epithelial organoid cultures.74
Air–liquid interface systems, in which the basal surface of stem cells is in contact with the media and the apical surface is exposed to air, have also attracted interest. This set-up can more accurately reflect the conditions of the tumour microenvironment in certain cancers, such as the luminal surface of colorectal cancer.75 Usui et al.75,76 successfully developed air–liquid interface organoid models from normal and tumour colorectal tissues of human patients, and were able to demonstrate the presence of epithelial, goblet and fibroblast cells in normal colonic tissue, and epithelial, goblet, myofibroblast and cancer stem cells in colorectal cancer tissue, as well as to show that colorectal tumour organoids were more resistant to chemotherapeutic agents than colorectal cancer cell lines. Similarly, when investigating the effect of resistance to gemcitabine treatment, co-culture of pancreatic ductal adenocarcinoma organoids with cancer-associated fibroblasts resulted in an increased IC50 when compared with organoids cultured alone,25 indicating that organoid co-culture models can also offer novel insights into treatment responses. Ensuring a representative tissue microenvironment is therefore an important consideration for organoid culture in cancer research.
Tumour organoids can enhance xenograft models
Patient-derived xenografts involve the implantation of human tissue or cells into humanised or immunodeficient rodents. This approach has provided invaluable insights into cancer invasion and metastasis; however, it can be improved further by the transplantation of organoids.77 Orthotopic transplantation is important to consider because subcutaneous xenografts often do not accurately recapitulate cancer invasion or metastasis.78 Orthotopic models of colorectal cancer have been developed from organoids and seen to produce uniform tumours that grow and metastasise reliably, depending on the metastatic potential of the cancer cells.78 One key example is the immunocompetent mouse model of colorectal cancer that recapitulates the well-defined human adenoma–carcinoma–metastasis sequence following orthotopic transplantation of colonic organoids.79 This approach can be applied to native or genetically modified human or mouse organoids: progression to adenocarcinoma occurs over 6 weeks, and spontaneous metastasis takes >20 weeks. Similar protocols use colonoscopy-guided mucosal injection and transplantation of organoids into the caecal mucosa of the mouse colon.80,81
A similar case exists in rectal cancer, for which there is a lack of anatomically relevant endoluminal rectal cancer mouse models. Ganesh et al.82 transplanted patient-derived rectal cancer organoids into mice, resulting in the generation of an invasive rectal carcinoma that metastasises to the liver and lung, as expected. Furthermore, the engrafted tumours display heterogeneous responses to chemotherapy, as also expected from clinical data.
Orthotopic transplantation can also take place after phenotypic and/or genotypic characterisation and/or manipulation of the tumour. For example, the CRISPR/Cas9 technology can be used to investigate the contribution of driver mutations in colorectal cancer.83 Such an approach could be used to generate the entire spectrum of cancer genotypes involved in carcinogenesis and metastasis,84 creating a biological library that can be used to investigate downstream phenotype changes, as has been reported.85,86 The insertion of a GFP tag by lentiviral transduction facilitates the straightforward detection of metastatic dissemination in such models.87
Cancers that develop from an orthotopically transplanted breast cancer organoid in mouse models not only reflect the morphology of the tumour of origin, but also the drug sensitivities, thereby rendering the ability to genetically modify such tumour-derived organoids invaluable in the study of drug resistance.88 Orthotopic transplantation overcomes many problems associated with other mouse models of colorectal cancer, such as a high tumour burden and tumours arising in the small intestine rather than the colorectum; as such, the use of organoids compared with cell culture clearly improves the translational ability of orthotopic transplant models.
Future perspectives
Tumour organoids can help replace, reduce and refine the use of animals in cancer research
Replacing, reducing and refining (3Rs) the use of animals in research is an international priority, and patient-derived organoid cultures represent an exciting platform to facilitate this principle. There are still limitations, such as the inability to mirror system-level interactions, multi-tissue interactions, multidirectional immune system interactions and explorations of drug pharmacokinetics and pharmacodynamics. However, these alternative organoid-based methods are evolving to study primary tumours ex vivo, and are adapting in complexity to overcome some of these limitations. As an example, metastasis adds a whole new dimension, and this process is difficult to study without the use of animal models. As discussed above, to overcome this, organ-on-a-chip technology constitutes an excellent animal-free model system for studying cancer metastasis, and can be improved by using organoid cultures. Aleman and Skardai89 have described a novel metastasis-on-a-chip system, in which colorectal cancer cells within a cancer organoid reside in a single microfluidic chamber that is connected to downstream chambers containing liver, lung and endothelial constructs in order to assess the metastatic preference of colorectal cancer cells. Other examples include the development of breast cancer-associated bone metastasis model90 and a multi-organ chip-based model of lung metastases with cell compartments representing bone, liver and brain.91 Organoids can therefore improve current disease models while helping to meet the international agenda outlined by the 3Rs.
Matrigel, which is currently widely used in the synthesis of organoids, is a basement membrane matrix with biological activity derived from Engelbreth–Holm–Swarm murine sarcomas.92 Animal-free alternatives, such as hydrogels made from alginates, do exist and have been used in novel model systems of the tissue microenvironment, such as a 3D bioprinted multicellular construct of breast tissue containing breast cancer cells and adipocytes,93 or utilising hyaluronic acid and collagen in a novel immersion bioprinting technique to allow organoid culture in 96-well plates for high-throughput drug screening, validated with patient-derived glioblastoma and sarcoma organoids.94
Could tumour organoids inform patient management?
Organoids could be a future tool to facilitate decisions regarding patient management. As an example, patient-derived tumour organoids could help determine whether a particular patient will be sensitive or resistant to specific treatments for many cancer types in a personalised medicine approach.95,96,97,98 This knowledge could be especially useful when there are a lack of robust data from large randomised control trials, which is often the case for rare and metastatic cancers. For example, this approach has been explored using organoids from appendiceal,99 neuroendocrine prostate100 and sarcoma cancers94 to test the efficacy of various chemotherapeutic agents. Researchers in the Netherlands have also established colorectal cancer organoids from ascitic fluid and peritoneal metastasis and used them to assess sensitivity to chemotherapy agents in an in vitro hyperthermic intraperitoneal chemotherapy model.101 Consistent with variable clinical outcomes following hyperthermic intraperitoneal chemotherapy, the authors reported inter-patient variability in response to commonly used chemotherapeutics, suggesting that organoids could potentially allow treatment regimens to be individualised to improve prognosis and reduce rates of recurrence.
In addition to providing insights into individualised treatment responses, organoids could also help inform on drug toxicity.102,103 For example, rimonabant, a cannabinoid receptor 1 antagonist previously used in the management of obesity, which inactivates Wnt signalling and might therefore modulate cancer stemness in colorectal cancer, was shown to be selectively toxic towards colorectal cancer organoids but not healthy colonic cells, highlighting the potential use of rimonabant as a candidate for the treatment of colorectal cancer.104 The use of organoids for assessing response and toxicity to therapy is not restricted to chemical compounds; however, patient-derived rectal cancer organoids irradiated ex vivo were seen to display heterogeneous sensitivities that correlate with the patient’s clinical response to radiotherapy.82 Furthermore, Nagle et al. used organoids to demonstrate that proton irradiation carried out in a magnetic field did not impact biological responses.105
In addition to helping to select the correct therapy for patients, organoid models could also prevent cancer patients from receiving ineffective treatments. For example, in metastatic colorectal cancer, Ooft et al. demonstrated the use of patient-derived organoids in preventing patients from receiving ineffective irinotecan-based chemotherapy; interestingly, however, these patient-derived organoids were unable to predict the outcome for treatment with 5-fluorouracil plus oxaliplatin.106
This potential approach to personalised medicine has limitations. There can be a low success rate of generating some organoid cultures, probably dependent on tissue type. For example, Li et al. report an efficiency rate of 31% for generating oesophageal adenocarcinoma organoids.33 Success rates could be improved by employing tissue-quality evaluation protocols before culturing; this approach entails preparing and histologically examining an aliquot of cell suspension to select for epithelial cell-prominent samples following tissue dissociation.60
Patient-derived organoid culture could be useful in the study of chemotherapy resistance. When a patient becomes resistant to therapy, it might be possible to select for resistant cancer cells in culture by manipulating niche factor requirements, thereby facilitating screening for drugs that are effective against the resistant cells.107 High-throughput sequencing of organoid cultures is difficult, given that these cells are not likely to proliferate fast enough to generate the cells necessary for a very large screen. However, a bespoke, clinically relevant, drug screen could be performed on the resistant organoid culture to identify effective chemotherapy agents, and this approach appears more feasible at present. In addition, new methods to overcome this issue for high-throughput applications are emerging, including the use of bioprinting with alternative support matrix combinations to allow organoid culture in small-well culture plates, for example 96- or 384-well plates.94
Individualised cancer therapy based on ex vivo experiments using a patient’s own cancer organoids represents an ambitious goal for personalised medicine, and this approach is currently not achievable and is too expensive for most healthcare systems. However, organoid biobanks could represent a realistic intermediate step towards this goal. A gastric organoid biobank has been established and comprises 64 normal, dysplastic, cancer and lymph node metastasis organoids from 34 patients.108 This biobank includes most gastric cancer subtypes, and whole-exome and transcriptome analysis data are available for these cultures. Analysis has uncovered new understanding of cancer biology and disease pathogenesis. In addition, the utility of this organoid biobank is reflected in the results of the drug-sensitivity screen as this highlights the potential impact of drugs at a very early stage of development. A breast cancer organoid biobank of >100 primary and metastatic cancers,38 a lung cancer biobank60 and an ovarian cancer biobank of 56 organoids109 have generated similar results when used for in vitro drug screening. Across these studies, the organoid tumour biobanks remarkably maintain disease-specific subtype characteristics, such as morphology, transcriptomic profile and genomic mutational analysis to the native tumour even after long-term culture.38,60,108,109 Accordingly, they offer an exciting and realistic tool for precision medicine for use after the identification of a patient’s cancer subtype through histopathological identification with validated tumour biomarkers.
Organoid cultures as novel treatment strategies
The immune response to tumours differs according to the biology of the underlying cancer. Autologous co-culture of tumour organoids derived from patients with colorectal cancer or non-small-cell lung cancer with peripheral lymphocytes leads to the expansion and enrichment of tumour-reactive T cells.110 These T cells are able to recognise and kill autologous tumour organoids but, remarkably, ignore healthy autologous organoids. This method enables us to investigate the mechanisms that underlie patient responses to immunotherapy. Furthermore, this approach could facilitate the generation of T-cell populations that could be used for autologous T-cell transfer therapy.
More direct potential therapeutic applications of organoids exist. Schwartz et al. have demonstrated that airbrush spraying of intestinal organoids onto a decellularised native extracellular matrix leads to the formation of an epithelial monolayer that resembles the intestinal surface.111 This is an exciting but underdeveloped concept that could have therapeutic benefits—for example, to help re-epithelialise areas affected by radiotherapy.
Radical surgery for breast cancer is often accompanied by radiotherapy or lymph node dissection. These adjuvant or neoadjuvant therapies are lifesaving, but increase the risk of life-long complications such as lymphoedema. In 2019, Lenti et al. investigated the transplantation of lympho-organoids into the region of dissected lymph nodes in mice.112 The lympho-organoids became fully integrated into the endogenous lymphatic system and restored lymphatic drainage. Furthermore, upon immunisation, the lympho-organoids were able to support antigen-specific endogenous immune responses. Therefore, therapeutic injection of lympho-organoids could become a novel therapeutic strategy for patients following radiotherapy or lymph node dissection for breast cancer.
Conclusions
Tumour-derived organoids are emerging as a tissue culture model that has exciting translational potential in the era of precision medicine. Tumour-derived organoids accurately represent the diverse genetic, molecular, morphological, architectural and functional pathophysiological hallmarks of cancer. Established cultures demonstrate intra-tumour and inter-patient heterogenicity, and can be further modified by genome editing, co-culture and orthotopic transplantation into rodents. In the clinic, tumour-derived organoids could be used to inform decisions on cancer treatment. In the wider setting, however, the translational impact of organoids is dependent on infrastructure, appropriate skill set and funding and, whilst the use of organoids is limited for now, there is future potential for this methodology and application to translate into clinical practice (Fig. 5). Overall, this cutting-edge method continues to evolve to provide new insights into the pathogenesis and evolution of cancer, offering the opportunity to develop new treatment strategies and enhance the impact of cancer research.

Organoids can help direct personalised medicine through a using immunohistochemical markers to subtype cancers and predicting tumour response to specific therapies via biobank data, b by predicting an individual’s response to a specific therapy by organoid culture using tumour-derived organoids. c Future applications of organoids may involve a more direct role in treatment, such as using organoids to generate autologous tumour-reactive T cells.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 66, 7–30 (2018).
Office for National Statistics. Broad causes of death by sex and single year of age. Deaths registered in England and Wales: 2017. https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/bulletins/deathsregistrationsummarytables/2017 (2018).
Smittenaar, C. R., Petersen, K. A., Stewart, K. & Moitt, N. Cancer incidence and mortality projections in the UK until 2035. Br. J. Cancer 115, 1147–1155 (2016).
Hay, M., Thomas, D. W., Craighead, J. L., Economides, C. & Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014).
Shih, H. P., Zhang, X. & Aronov, A. M. Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indications. Nat. Rev. Drug Discov. 17, 19–33 (2018).
Ireson, C. R., Alavijeh, M. S., Palmer, A. M., Fowler, E. R. & Jones, H. J. The role of mouse tumour models in the discovery and development of anticancer drugs. Br. J. Cancer 121, 101–108 (2019).
Sato, T., Vries, R. G., Snippert, H. J., van de Wetering, M., Barker, N., Stange, D. E. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Teicher, B. A. Tumor Models in Cancer Research, Second edn. (Humana Press, Totowa, NJ, 2011).
Galuschka, C., Proynova, R., Roth, B., Augustin, H. G. & Müller-Decker, K. Models in translational oncology: a public resource database for preclinical cancer research. Cancer Res. 15, 2557–2563 (2017).
Arends, M. J., White, E. S. & Whitelaw, C. B. A. Animal and cellular models of human disease. J. Pathol. 238, 137–140 (2016).
Chung, C. Y., Iida-Klein, A., Wyatt, L. E., Rudkin, G. H., Ishida, K., Yamaguchi, D. T. et al. Serial passage of MC3T3-E1 cells alters osteoblastic function and responsiveness to transforming growth factor-β1 and bone morphogenetic protein-2. Biochem. Biophys. Res. Commun. 265, 246–251 (1999).
de Graaf, I. A., Olinga, P., de Jager, M. H., Merema, M. T., de Kanter, R., van de Kerkhof, E. G. et al. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat. Protoc. 5, 1540–1551 (2010).
Sontheimer-Phelps, A., Hassell, B. A. & Ingber, D. E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 19, 65–81 (2019).
Ingber, D. E. Developmentally inspired human ‘organs on chips’. Development 145, pii:dev156125 (2018).
Phesse, T. J., Durban, V. M. & Sansom, O. J. Defining key concepts of intestinal and epithelial cancer biology through the use of mouse models. Carcinogenesis 38, 953–965 (2017).
Cheon, D. J. & Orsulic, S. Mouse models of cancer. Annu Rev. Pathol. 6, 95–119 (2011).
Wartha, K., Herting, F. & Hasmann, M. Fit-for purpose use of mouse models to improve predictivity of cancer therapeutics evaluation. Pharm. Ther. 142, 351–361 (2014).
Mendes, N., Dias Carvalho, P., Martins, F., Mendonça, S., Malheiro, A. R., Ribeiro, A. et al. Animal models to study cancer and its microenvironment. Adv. Exp. Med. Biol. 1219, 389–401 (2020).
Olson, B., Li, Y., Lin, Y., Liu, E. T. & Patnaik, A. Mouse models for cancer immunotherapy research. Cancer Discov. 8, 1358–1356 (2018).
Berman, J. N., Chiu, P. P. L. & Dellaire, G. Preclinical animal models for cancer genomics. in (eds Graham Dellaire, Jason N Berman & Robert J Arceci.) Cancer Genomics: Bench Personalized Medicine. Elsevier. Ch 8, 109–131 (Academic Press, 2014).
Gengenbacher, N., Singhal, M. & Augustin, H. G. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat. Rev. Cancer 17, 751–765 (2017).
Mak, I. W., Evaniew, N. & Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 6, 114–118 (2014).
Morton, J. J., Bird, G., Refaeli, Y. & Jimeno, A. Humanized mouse xenograft models: narrowing the tumor-microenvironment gap. Cancer Res. 76, 6153–6158 (2016).
Jin, K., Teng, L., Shen, Y., He, K., Xu, Z. & Li, G. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin. Transl. Oncol. 12, 473–480 (2010).
Tsai, S., McOlash, L., Palen, K., Johnson, B., Duris, C., Yang, Q. et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 18, 335 (2018).
Lancaster, M. A. & Huch, M. Disease modelling in human organoids. Dis. Model Mech. 12, pii:dmm039347 (2019).
Sato, T., Stange, D. E., Ferrante, M., Vries, R. G. J., Van, Es,J. H., Van den Brink, S. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Gao, M., Lin, M., Rao, M., Thompson, H., Hirai, K., Choi, M. et al. Development of patient-derived gastric cancer organoids from endoscopic biopsies and surgical tissues. Ann. Surg. Oncol. 25, 2767–2775 (2018).
Nuciforo, S., Fofana, I., Matter, M. S., Blumer, T., Calabrese, D., Boldanova, T. et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 24, 1363–1376 (2018).
Tiriac, H., Bucobo, J. C., Tzimas, D., Grewel, S., Lacomb, J. F., Rowehl, L. M. et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest. Endosc. 87, 1474–1480 (2018).
Velletri, T., Villa, E. C., Lupia, M., Riso, P. L., Luongo, R., Tobon, A. L. et al. Single cell derived organoids capture the self-renewing subpopulations of metastatic ovarian cancer. Biorxiv. Preprint at https://doi.org/10.1101/484121 (2018).
Tsai, Y. H., Czerwinski, M., Wu, A., Dame, M. K., Attili, D., Hill, E. et al. A method for cryogenic preservation of human biopsy specimens and subsequent organoid culture. Cell Mol. Gastroenterol. Hepatol. 6, 218–222.e7 (2018).
Li, X., Francies, H. E., Secrier, M., Perner, J., Miremadi, A., Galeano-Dalmau, N. et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 9, 2983 (2018).
Tamura, H., Higa, A., Hoshi, H., Hiyama, G., Takahashi, N., Ryufuku, M. et al. Evaluation of anticancer agents using patient-derived tumor organoids characteristically similar to source tissues. Oncol. Rep. 40, 635–646 (2018).
van de Wetering, M., Francies, H. E., Francis, J. M., Bounova, G., Iorio, F., Pronk, A. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015).
Lee, S. H., Hu, W., Matulay, J. T., Silva, M. V., Owczarek, T. B., Kim, K. et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell 173, 515–528.e17 (2018).
Saito, Y., Muramatsu, T., Kanai, Y., Ojima, H., Sukeda, A., Hiraoka, N. et al. Establishment of patient-derived organoids and drug screening for biliary tract carcinoma. Cell Rep. 27, 1265–1276.e4 (2019).
Sachs, N., de Ligt, J., Kopper, O., Gogola, E., Bounova, G., Weeber, F. et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386.e10 (2018).
Kraiczy, J., Nayak, K. M., Howell, K. J., Ross, A., Forbester, J., Salvestrini, C. et al. DNA methylation defines regional identity of human intestinal epithelial organoids and undergoes dynamic changes during development. Gut 68, 49–61 (2019).
Schatoff, E. M., Leach, B. I. & Dow, L. E. Wnt signaling and colorectal cancer. Curr. Colorectal Cancer Rep. 13, 101–110 (2017).
Fujii, M., Shimokawa, M., Date, S., Takano, A., Matano, M., Nanki, K. et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 18, 827–838 (2016).
Melo, F. D. S. E., Vermeulen, L., Richel, D. & Medema, J. P. Targeting Wnt signaling in colon cancer stem cells. Clin. Cancer Res. 17, 647–653 (2011).
Sachs, N., Papaspyropoulos, A., Zomer-van Ommen, D. D., Heo, I., Böttinger, L., Klay, D. et al. Long-term expanding human airway organoids for disease modelling. EMBO J. 34, pii:e100300 (2019).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Cristobal, A., van den Toorn, H. W. P., van de Wetering, M., Clevers, H., Heck, A. J. R. & Mohammed, S. Personalized proteome profiles of healthy and tumor human colon organoids reveal both individual diversity and basic features of colorectal cancer. Cell Rep. 18, 263–274 (2017).
Teal, E., Steele, N. G., Chakrabarti, J., Holokai, L. & Zavros, Y. Mouse- and human-derived primary gastric epithelial monolayer culture for the study of regeneration. J. Vis. Exp. 135, 57435 (2018).
Braverman, J. & Yilmaz, O. ̈H. From 3D organoids back to 2D enteroids. Dev. Cell 44, 533–534 (2018).
Morgan, R. G., Chambers, A. C., Legge, D. N., Coles, S. J., Greenhough, A. & Williams, A. C. Optimized delivery of siRNA into 3D tumor spheroid cultures in situ. Sci. Rep. 8, 7952 (2018).
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Ogawa, J., Pao, G. M., Shokhirev, M. N. & Verma, I. M. Glioblastoma model using human cerebral organoids. Cell Rep. 23, 1220–1229 (2018).
Drost, J., van Boxtel, R., Blokzijl, F., Mizutani, T., Sasaki, N., Sasselli, V. et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358, 234–238 (2017).
Tejero, R., Huang, Y., Katsyv, I., Kluge, M., Lin, J. Y., Tome-Garcia, J. et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 42, 252–269 (2019).
Lannagan, T. R. M., Lee, Y. K., Wang, T., Roper, J., Bettington, M. L., Fennell, L. et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 68, 684–692 (2019).
Garman, K. S., Orlando, R. C. & Chen, X. Experimental models for Barrett’s esophagus and esophageal adenocarcinoma. Am. J. Physiol. Liver Physiol. 302, 1231–1243 (2012).
McDonald, S. A. C., Graham, T. A., Lavery, D. L., Wright, N. A. & Jansen, M. The Barrett’s gland in phenotype space. Cell Mol. Gastroenterol. Hepatol. 1, 41–54 (2015).
Liu, X., Cheng, Y., Abraham, J. M., Wang, Z., Wang, Z., Ke, X. et al. Modeling Wnt signaling by CRISPR-Cas9 genome editing recapitulates neoplasia in human Barrett epithelial organoids. Cancer Lett. 436, 109–118 (2018).
Unno, K., Roh, M., Yoo, Y. A., Al-Shraideh, Y., Wang, L., Nonn, L. et al. Modelling African American prostate adenocarcinoma by inducing defined genetic alterations in organoids. Oncotarget 8, 51264–51276 (2017).
Xian, L., Chia, L., Georgess, D., Luo, L., Shuai, S., Ewald, A. J. et al. Genetic engineering of primary mouse intestinal organoids using magnetic nanoparticle transduction viral vectors for frozen sectioning. J. Vis. Exp. 147, e57040 (2019).
Kim, M., Mun, H., Sung, C. O., Cho, E. J., Jeon, H. J., Chun, S. M. et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 10, 3991 (2019).
Hill, D. R., Huang, S., Tsai, Y. H., Spence, J. R. & Young, V. B. Real-time measurement of epithelial barrier permeability in human intestinal organoids. J. Vis. Exp. 130, e56960 (2017).
Soler, A. P., Miller, R. D., Laughlin, K. V., Carp, N. Z., Klurfeld, D. M. & Mullin, J. M. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 20, 1425–1431 (1999).
Leslie, J. L., Huang, S., Opp, J. S., Nagy, M. S., Kobayashi, M., Young, V. B. et al. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect. Immun. 83, 138–145 (2015).
Williamson, I. A., Arnold, J. W., Samsa, L. A., Gaynor, L., DiSalvo, M., Cocchiaro, J. L. et al. A high-throughput organoid microinjection platform to study gastrointestinal microbiota and luminal physiology. Cell Mol. Gastroenterol. Hepatol. 6, 301–319 (2018).
Kang, T. Y., Ellison, D., Lee, S. H., Ewald, A. J. & Levchenko, A. 3D analysis of multi-cellular responses to chemoattractant gradients. J. Vis. Exp. 147, e59226 (2019).
Suarez, G., Romero-Gallo, J., Piazuelo, M. B., Sierra, J. C., Delgado, A. G., Washington, M. K. et al. Nod1 imprints inflammatory and carcinogenic responses toward the gastric pathogen Helicobacter pylori. Cancer Res. 79, 1600–1611 (2019).
Chakrabarti, J., Holokai, L., Syu, L. J., Steele, N. G., Chang, J., Wang, J. et al. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 9, 37439–37457 (2018).
Chakrabarti, J., Holokai, L., Syu, L. J., Steele, N., Chang, J., Dlugosz, A. et al. Mouse-derived gastric organoid and immune cell co-culture for the study of the tumor microenvironment. Methods Mol. Biol. 1817, 157–168 (2018).
Wen, Y. A., Xing, X., Harris, J. W., Zaytseva, Y. Y., Mitov, M. I., Napier, D. L. et al. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 8, e2593 (2017).
Conlon, G. A. & Murray, G. I. Recent advances in understanding the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 247, 629–640 (2019).
Koikawa, K., Ohuchida, K., Ando, Y., Kibe, S., Nakayama, H., Takesue, S. et al. Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer Lett. 425, 65–77 (2018).
Pettinato, G., Lehoux, S., Ramanathan, R., Salem, M. M., He, L. X., Muse, O. et al. Generation of fully functional hepatocyte-like organoids from human induced pluripotent stem cells mixed with endothelial cells. Sci. Rep. 9, 8920 (2019).
Richards, Z., McCray, T., Marsili, J., Zenner, M. L., Manlucu, J. T., Garcia, J. et al. Prostate stroma increases the viability and maintains the branching phenotype of human prostate organoids. iScience 12, 304–317 (2019).
Mollica, P. A., Booth-Creech, E. N., Reid, J. A., Zamponi, M., Sullivan, S. M., Palmer, X. L. et al. 3D bioprinted mammary organoids and tumoroids in human mammary derived ECM hydrogels. Acta Biomater. 95, 201–213 (2019).
Usui, T., Sakurai, M., Umata, K., Yamawaki, H., Ohama, T. & Sato, K. Preparation of human primary colon tissue-derived organoid using air liquid interface culture. Curr. Protoc. Toxicol. 75, 22.6.1–22.6.7 (2018).
Usui, T., Sakurai, M., Enjoji, S., Kawasaki, H., Umata, K., Ohama, T. et al. Establishment of a novel model for anticancer drug resistance in three-dimensional primary culture of tumor microenvironment. Stem Cells Int. 7053872 https://doi.org/10.1155/2016/7053872 (2016).
Lai, Y., Wei, X., Lin, S., Qin, L., Cheng, L. & Li, P. Current status and perspectives of patient- derived xenograft models in cancer research. J. Hematol. Oncol. 10, 106 (2017).
Kochall, S., Thepkaysone, M. L., García, S. A., Betzler, A. M., Weitz, J., Reissfelder, C. et al. Isolation of circulating tumor cells in an orthotopic mouse model of colorectal cancer. J. Vis. Exp. 125, 55357 (2017).
O’Rourke, K. P., Loizou, E., Livshits, G., Schatoff, E. M., Baslan, T., Manchado, E. et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 35, 577–582 (2017).
Roper, J., Tammela, T., Akkad, A., Almeqdadi, M., Santos, S. B., Jacks, T. et al. Colonoscopy-based colorectal cancer modelling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat. Protoc. 13, 217–234 (2018).
Fumagalli, A., Suijkerbuijk, S. J. E., Begthel, H., Beerling, E., Oost, K. C., Snippert, H. J. et al. A surgical orthotopic organoid transplantation approach in mice to visualize and study colorectal cancer progression. Nat. Protoc. 13, 235–247 (2018).
Ganesh, K., Wu, C., O’Rourke, K. P., Szeglin, B. C., Zheng, Y., Sauvé, C. G. et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 25, 1607–1614 (2019).
Takeda, H., Kataoka, S., Nakayama, M., Ali, M. A. E., Oshima, H., Yamamoto, D. et al. CRISPR- Cas9–mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proc. Natl Acad. Sci. USA 116, 15635–15644 (2019).
Roper, J., Tammela, T., Cetinbas, N. M., Akkad, A., Roghanian, A., Rickelt, S. et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 35, 569–576 (2017).
Nanki, K., Toshimitsu, K., Takano, A., Fujii, M., Shimokawa, M., Ohta, Y. et al. Divergent routes toward wnt and r-spondin niche independency during human gastric carcinogenesis. Cell 174, 856–869 (2018).
Fumagalli, A., Drost, J., Suijkerbuijk, S. J., van Boxtel, R., de Ligt, J., Offerhaus, G. J. et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl Acad. Sci. USA 114, E2357–E2364 (2017).
Okazawa, Y., Mizukoshi, K., Koyama, Y., Okubo, S., Komiyama, H., Kojima, Y. et al. High-sensitivity detection of micrometastases generated by GFP lentivirus-transduced organoids cultured from a patient-derived colon tumor. J. Vis. Exp. 136, e57374 (2018).
Duarte, A. A., Gogola, E., Sachs, N., Barazas, M., Annunziato, S., R de Ruiter, J. et al. BRCA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Nat. Methods 15, 134–140 (2018).
Aleman, J. & Skardal, A. A multi-site metastasis-on-a-chip microphysiological system for assessing metastatic preference of cancer cells. Biotechnol. Bioeng. 116, 936–944 (2019).
Hao, S., Ha, L., Cheng, G., Wan, Y., Xia, Y., Sosnoski, D. M. et al. A spontaneous 3D bone-on-a-chip for bone metastasis study of breast cancer cells. Small 14, e1702787 (2018).
Xu, Z., Li, E., Guo, Z., Yu, R., Hao, H., Xu, Y. et al. Design and construction of a multi-organ microfluidic chip mimicking the in vivo microenvironment of lung cancer metastasis. ACS Appl. Mater. Interfaces 8, 25840–25847 (2016).
Kibbey, M. C. Maintenance of the EHS sarcoma and Matrigel preparation. J. Tissue Cult. Meth 16, 227–230 (1994).
Chaji, S., Al-Saleh, J. & Gomillion, C. T. Bioprinted three-dimensional cell-laden hydrogels to evaluate adipocyte-breast cancer cell interactions. Gels 6, pii E10 (2020).
Maloney, E., Clark, C., Sivakumar, H., Yoo, K., Aleman, J., Rajan, S. A. P. et al. Immersion bioprinting of tumor organoids in multi-well plates for increasing chemotherapy screening throughput. Micromachines 11, pii:E208 (2020).
Bian, B., Juiz, N. A., Gayet, O., Bigonnet, M., Brandone, N., Roques, J. et al. Pancreatic cancer organoids for determining sensitivity to bromodomain and extra-terminal inhibitors (BETI). Front. Oncol. 9, 475 (2019).
Li, L., Knutsdottir, H., Hui, K., Weiss, M. J., He, J., Philosophe, B. et al. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 4, pii121490 (2019).
Saengwimol, D., Rojanaporn, D., Chaitankar, V., Chittavanich, P., Aroonroch, R., Boontawon, T. et al. A three-dimensional organoid model recapitulates tumorigenic aspects and drug responses of advanced human retinoblastoma. Sci. Rep. 8, 15664 (2018).
Mazzocchi, A. R., Rajan, S. A. P., Votanopoulos, K. I., Hall, A. R. & Skardal, A. In vitro patient-derived 3D mesothelioma tumor organoids facilitate patient-centric therapeutic screening. Sci. Rep. 8, 28886 (2018).
Votanopoulos, K. I., Mazzocchi, A., Sivakumar, H., Forsythe, S., Aleman, J., Levine, E. A. et al. Appendiceal cancer patient-specific tumor organoid model for predicting chemotherapy efficacy prior to initiation of treatment: a feasibility study. Ann. Surg. Oncol. 26, 139–147 (2019).
Puca, L., Bareja, R., Prandi, D., Shaw, R., Benelli, M., Karthaus, W. R. et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 9, 2404 (2018).
Ubink, I., Bolhaqueiro, A. C. F., Elias, S. G., Raats, D. A. E., Constantinides, A., Peters, N. A. et al. Organoids from colorectal peritoneal metastases as a platform for improving hyperthermic intraperitoneal chemotherapy. Br. J. Surg. 106, 1404–1414 (2019).
Yu, X., Fuller, A. M., Blackmon, R., Troester, M. A. & Oldenburg, A. L. Quantification of the effect of toxicants on the intracellular kinetic energy and cross-sectional area of mammary epithelial organoids by oct fluctuation spectroscopy. Toxicol. Sci. 162, 234–240 (2018).
Liu, F., Huang, J. & Liu, Z. Vincristine impairs microtubules and causes neurotoxicity in cerebral organoids. Neuroscience 404, 530–540 (2019).
Fiore, D., Ramesh, P., Proto, M. C., Piscopo, C., Franceschelli, S., Anzelmo, S. et al. Rimonabant kills colon cancer stem cells without inducing toxicity in normal colon organoids. Front. Pharm. 8, 949 (2018).
Nagle, P. W., van Goethem, M. J., Kempers, M., Kiewit, H., Knopf, A., Langendijk, J. A. et al. In vitro biological response of cancer and normal tissue cells to proton irradiation not affected by an added magnetic field. Radiother. Oncol. 137, 125–129 (2019).
Ooft, S. N., Weeber, F., Dijkstra, K. K., McLean, C. M., Kaing, S., van Werkhoven, E. et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 11, piieaay2574 (2019).
Vincan, E., Schwab, R. H. M., Flanagan, D. J., Moselen, J. M., Tran, B. M., Barker, N. et al. The central role of wnt signaling and organoid technology in personalizing anticancer therapy. Prog. Mol. Biol. Transl. 153, 299–319 (2018).
Yan, H. H. N., Siu, H. C., Law, S., Ho, S. L., Yue, S. S. K., Tsui, W. Y. et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 23, 882–897 (2018).
Kopper, O., de Witte, C. J., Lõhmussaar, K., Valle-Inclan, J. E., Hami, N., Kester, L. et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 25, 838–849 (2019).
Dijkstra, K. K., Cattaneo, C. M., Weeber, F., Chalabi, M., van de Haar, J., Fanchi, L. F. et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598 (2018).
Schwartz, D. M., Pehlivaner Kara, M. O., Goldstein, A. M., Ott, H. C. & Ekenseair, A. K. Spray delivery of intestinal organoids to reconstitute epithelium on decellularized native extracellular matrix. Tissue Eng. Part C. Methods 23, 565–573 (2017).
Lenti, E., Bianchessi, S., Proulx, S. T., Palano, M. T., Genovese, L., Raccosta, L. et al. Therapeutic regeneration of lymphatic and immune cell functions upon lympho-organoid transplantation. Stem Cell Rep. 12, 1260–1268 (2019).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to the creation of this paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
Not applicable.
Competing interests
G.I.M. is an editorial board member of the British Journal of Cancer. The remaining authors declare no competing interests.
Funding information
None.
Additional information
Note This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Porter, R.J., Murray, G.I. & McLean, M.H. Current concepts in tumour-derived organoids. Br J Cancer 123, 1209–1218 (2020). https://doi.org/10.1038/s41416-020-0993-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-0993-5
- Springer Nature Limited
This article is cited by
-
Rebuilding the microenvironment of primary tumors in humans: a focus on stroma
Experimental & Molecular Medicine (2024)
-
Azoxymethane-induced carcinogenesis-like model of mouse intestine and mouse embryonic stem cell-derived intestinal organoids
Molecular Biology Reports (2024)
-
A systematic review on the culture methods and applications of 3D tumoroids for cancer research and personalized medicine
Cellular Oncology (2024)
-
Drug screening at single-organoid resolution via bioprinting and interferometry
Nature Communications (2023)
-
Thinking in 3 dimensions: philosophies of the microenvironment in organoids and organs-on-chip
History and Philosophy of the Life Sciences (2023)