
Abstract
Background
LRIG1, the founding member of the LRIG (leucine-rich repeat and immunoglobulin-like domain) family of transmembrane proteins, is a negative regulator of receptor tyrosine kinases and a tumour suppressor. Decreased LRIG1 expression is consistently observed in cancer, across diverse tumour types, and is linked to poor patient prognosis. However, mechanisms by which LRIG1 is repressed are not fully understood. Silencing of LRIG1 through promoter CpG island methylation has been reported in colorectal and cervical cancer but studies in breast cancer remain limited.
Methods
In silico analysis of human breast cancer patient data were used to demonstrate a correlation between DNA methylation and LRIG1 silencing in basal/triple-negative breast cancer, and its impact on patient survival. LRIG1 gene expression, protein abundance, and methylation enrichment were examined by quantitative reverse-transcription PCR, immunoblotting, and methylation immunoprecipitation, respectively, in breast cancer cell lines in vitro. We examined the impact of global demethylation on LRIG1 expression and methylation enrichment using 5-aza-2’-deoxycytidine. We also examined the effects of targeted demethylation of the LRIG1 CpG island, and transcriptional activation of LRIG1 expression, using the RNA guided deadCas9 transactivation system.
Results
Across breast cancer subtypes, LRIG1 expression is lowest in the basal/triple-negative subtype so we investigated whether differential methylation may contribute to this. Indeed, we find that LRIG1 CpG island methylation is most prominent in basal/triple-negative cell lines and patient samples. Use of the global demethylating agent 5-aza-2’-deoxycytidine decreases methylation leading to increased LRIG1 transcript expression in basal/triple-negative cell lines, while having no effect on LRIG1 expression in luminal/ER-positive cell lines. Using a CRISPR/deadCas9 (dCas9)-based targeting approach, we demonstrate that TET1-mediated demethylation (Tet1-dCas9) along with VP64-mediated transcriptional activation (VP64-dCas9) at the CpG island, increased endogenous LRIG1 expression in basal/triple-negative breast cancer cells, without transcriptional upregulation at predicted off-target sites. Activation of LRIG1 by the dCas9 transactivation system significantly increased LRIG1 protein abundance, reduced site-specific methylation, and reduced cancer cell viability. Our findings suggest that CRISPR-mediated targeted activation may be a feasible way to restore LRIG1 expression in cancer.
Conclusions
Our study contributes novel insight into mechanisms which repress LRIG1 in triple-negative breast cancer and demonstrates for the first time that targeted de-repression of LRIG1 in cancer cells is possible. Understanding the epigenetic mechanisms associated with repression of tumour suppressor genes holds potential for the advancement of therapeutic approaches.
Similar content being viewed by others


Background
LRIG1 is a member of the “LRIG” family of single-pass transmembrane proteins, which also includes LRIG2 and LRIG3 [1, 2]. LRIG1 was identified as a negative regulator of various cell surface receptors, including ERBB [3, 4], PDGFR-A [5], MET [6], RET [7, 8] and TRKB [9] receptor tyrosine kinases. In 2012, LRIG1 was classified as a tumour suppressor, with genetic ablation leading to increased expression of ErbB receptors and the development of highly penetrant duodenal adenomas [10]. More recently, LRIG1 was reported to function as a haploinsufficient tumour suppressor in a PDGF-induced experimental glioma model, in part through negative regulation of MET [11].
Several studies have demonstrated decreased expression of LRIG1 in cancer, including lung [12, 13], breast [14, 15] and head and neck cancer [16], among others [17]. Across diverse cancer types, high expression of LRIG1 is associated with superior patient survival, in agreement with its tumour suppressor function [2, 17, 18]. This includes breast cancer, where low LRIG1 expression has been linked to decreased relapse-free, and distant metastasis-free, survival [14, 15]. As an ERα target gene, LRIG1 is most highly expressed in ER-positive luminal disease relative to other breast cancer subtypes [14], with the lowest expression observed in basal/triple-negative breast cancers (TNBC) [19, 20]. The mechanisms which contribute to LRIG1 downregulation in breast cancer are not fully understood but are known to include ERBB2-mediated repression [14] and loss of gene copy number, which conveys persistent risk of relapse in patients typically considered low-risk [15]. In the context of cancer, DNA hypermethylation at the promoter of tumour suppressor genes is a key mechanism of downregulation and is recognised as a cancer hallmark in many tumour types [21]. Interestingly, hypermethylation of the LRIG1 promoter region has been reported in colorectal cancer [22] as well as cervical cancer, where it was found to correlate with decreased progression-free survival [23]. However, studies on LRIG1 gene methylation in breast cancer remain limited.
Since persistence of LRIG1 expression is associated with superior patient survival across cancer types and because prior studies have shown that LRIG1 is epigenetically and transcriptionally regulated, we reasoned that transcriptional reactivation of LRIG1 expression in cancer cells was feasible. The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system has provided an efficient method for targeted gene activation. In the CRISPR/Cas9 system, endonuclease Cas9 is targeted to a specific DNA sequence by a single guide RNA (sgRNA) and subsequently induces double-stranded DNA cleavage [24]. A mutation of the catalytic domains of Cas9, yielding a deadCas9 (dCas9), maintains Cas9’s DNA-honing ability without inducing DNA breaks; and provides an ideal DNA targeting protein complex [25]. Recently, dCas9 fusions with the catalytic domains of a variety of effector proteins, such as ten-eleven translocation dioxygenase 1 (TET1), four tandem copies of Herpes Simplex Viral Protein 16 (VP64), and Krüppel-associated box (KRAB) have gained significance as candidate complexes that can demethylate, activate, or inactivate genes of interest, respectively [24].
We demonstrate that LRIG1 methylation is significantly increased in breast tumours compared to normal tissue, with a strong inverse correlation between LRIG1 mRNA expression and CpG island methylation. Interestingly, we find that methylation is most robust in the basal/TNBC subtype of breast cancer, both in patient samples and in cell lines representative of this subtype. Use of the global demethylating agent, 5-aza-2’-deoxycytidine (ADC), in TNBC cell lines leads to a significant increase in LRIG1 mRNA expression aligning with a significant decrease in methylation. To directly implicate DNA methylation in LRIG1 silencing, we take a targeted approach in which the catalytic domain of DNA hydroxymethyltransferase TET1 is fused to dCas9, and targeted to the LRIG1 CpG island. We show, for the first time, that targeted demethylation causes significant reactivation of LRIG1 gene expression and LRIG1 protein abundance, as well as reduces DNA methylation at the CpG island. We also demonstrate that co-expression of Tet1 and transcriptional activator, VP64, using the CRISPR/dCas9 system significantly amplifies many of these processes.
Methods
Cell culture
BT549 (Cat# HBT-122), HCC1937 (Cat# CRL-2336), MDA-MB-231 (Cat# HTB-26), MCF7 (Cat# HTB-22), T47D (Cat# HTB-133), ZR75-1 (Cat# CRL-1500) and HCT116 (Cat# CCL-247) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained as recommended. Cell lines were authenticated by short tandem repeat profiling through the University of Arizona Genetics Core and tested for mycoplasma contamination using the MycoAlert PLUS Mycoplasma Detection Kit (Cat# LT37-701, Lonza, Hayward, CA, USA). All cells tested negative for mycoplasma contamination. Cells were used for 6–8 passages, after which they were replaced with a cryopreserved stock.
ADC and panobinostat treatment
Cells were seeded in a 12-well plate at 7.5 × 105 cells per well 24 h prior to treatments. Cells were treated with 5-aza-2’-deoxycytidine (ADC) (Cat# A3656, Sigma-Aldrich, St. Louis, MO, USA) at indicated concentrations for 96 h, with culture media being replaced every 24 h. Cells were treated with 100 μM Panobinostat (Cat# S1030, SelleckChem, Houston, TX, USA) for 24 h. For the combination experiment, cells were treated with 10 μM ADC for 96 h, with 100 μM Panobinostat added in the last 24 h. On completion of treatments, media was aspirated from culture wells, cells were rinsed with 1× PBS, and frozen at −80 °C until RNA isolation was performed.
RNA extraction and reverse-transcription quantitative PCR (RT-qPCR)
RNA was collected using the PureLink RNA Mini Kit (Cat# 121183018, Thermo Fischer Scientific, Waltham, MA, USA) or RNeasy Mini Kit (Cat# 74104, QIAGEN, Germantown, MD, USA) according to the manufacturer’s protocols. High-Capacity cDNA Reverse Transcription Kit (Cat# 4368814, Thermo Fischer Scientific) or Iscript Reverse Transcription Supermix (Cat# 1708841, Bio-Rad, Hercules, CA, USA) was used to convert 0.5–1 μg of total RNA to cDNA. Quantitative PCR amplifications were conducted in a CFX96 real-time PCR system (Bio-Rad) using TaqMan probes (Life Technologies, Carlsbad, CA, USA) for LRIG1 (Hs00394267_m1), BASP1 (Hs00932356_s1), CTNND2 (Hs00181643_m1), FBXO15 (Hs00380856_m1), NPHS1 (Hs00190446_m1), KCNIP1 (Hs01557317_m1), GUSB (Hs99999908_m1), ACTB (Hs99999903_m1), GAPDH (Hs99999905_m1) and Taqman Fast Advanced Master Mix (Cat# 4444557, Thermo Fischer Scientific) or Universal SYBR Green Supermix (Cat# 1725120, Bio-Rad). Analysis was performed by the comparative Ct method under the following cycling conditions: 3 min at 95 °C, 40 cycles of 10 s at 95 °C and 30 s at 55 °C. Relative abundance was determined from the Ct values using the 2-ΔΔCt method after normalisation to GAPDH (additional reference genes, ACTB and GUSB, were examined to ensure changes in LRIG1 transcript levels were biologically accurate and not due to changes in GAPDH transcript levels, Supplemental Fig. 9).
In silico analysis of DNA methylation
The UCSC Genome Browser was used to perform a qualitative in silico analysis of methylation levels across the LRIG1 CpG island using the DNA Methylation by Reduced Representation Bisulfite Seq (RRBS) track. This track was produced as part of the ENCODE project by the Lab of Dr. Richard Myers at the HudsonAlpha Institute for Biotechnology [26] and reports the percentage of DNA molecules that exhibit cytosine methylation at more than 500,000 specific CpG dinucleotide sites in the human genome using bisulfite sequencing. For each assayed CpG, the percentage of methylated sequencing reads is reported. Methylation is represented as an 11-colour gradient with red = 100%, yellow = 50%, and green = 0% of sequenced reads methylated; respectively.
Methylated DNA immunoprecipitation (MeDIP)
Cells were seeded in 10-cm plates and grown to confluency. Genomic DNA was extracted by incubation of cells with digestion buffer (100 mM NaCl, 10 mM Tris-Cl, pH 8.0, 25 mM EDTA, pH 8.0, 0.5% SDS) and 0.1 mg/mL Proteinase K (Cat# AM2548, Thermo Fischer Scientific) for 12–18 hr at 50 °C. DNA was purified using phenol–chloroform, precipitated and dissolved in TE to a final concentration of 200 ng/μL. Genomic DNA was sonicated to an average size of 400 bp using the BioRuptor NGS (Diagenode, Denville, NJ, USA) and denatured for 10 min at 95 °C and immediately transferred on ice. In total, 2 μg of fragmented DNA and 2 μg of 5-methylcytosine antibody (Cat# 39649, Active Motif, Carlsbad, CA, USA) were added to 500 μL IP dilution buffer (16.7 mM Tris-Cl pH 8.1, 1.2 mM EDTA pH 8, 167 mM NaCl, 0.01% SDS, 1.1% Triton X-100) and immunoprecipitation of methylated DNA was performed by gentle rotation for overnight at 4 °C. After incubation for 1 hr with 2 μg Rabbit Anti-Mouse IgG (Cat# 55436, MP Biomedical, Irvine, CA, USA), immunoprecipitates were washed two times with IP Wash Buffer I (50 mM Tris-Cl pH 8.0, 150 mM NaCl, 1% NP40, 0.25% deoxycholic acid, 2 mM EDTA pH 8.0), followed by three washes with IP Wash Buffer II (100 mM Tris-Cl pH 8.0, 500 mM LiCl, 1% NP40, 1% deoxycholic acid), and one wash with IP Wash Buffer III (100 mM Tris-Cl pH 8.0, 500 mM LiCl, 1% NP40, 1% deoxycholic acid, 150 mM NaCl). DNA was eluted for 30 min with elution buffer (50 mM NaHCO3, 1% SDS, 0.1 mg/mL Proteinase K), purified using phenol/chloroform, and precipitated using ethanol according to MeDIP Protocol (Active Motif). The resulting DNA pellet was dissolved in 50 μL TE. Standard PCR reactions were performed using 1 μL of the immunoprecipitated DNA or 5% input control DNA using GoTaq polymerase (Cat# M3001, Promega, Madison, WI, USA). PCR products were separated by electrophoresis through 2% agarose gels and visualised using ethidium bromide. For qPCR analysis, 1.5 μL of immunoprecipitated DNA or 5% input control DNA was amplified with SYBR FAST mastermix (Cat# KK4600, KAPA Biosystems, Wilmington, MA, USA) or Universal SYBR Green Supermix (Cat# 1725120, Bio-Rad) using the CFX96 real-time PCR system (Bio-Rad) according to the manufacturer’s recommendations. MeDIP enrichment was calculated relative to input samples using dCq = Cq[MeDIP]-Cq[input]. MeDIP primer: LRIG1 promoter: forward 5’-GGACTGTGAGGACCCGAAC-3’, reverse 5’-GCCGCAGAGAGAACTTGG-3’, LRIG1 5’UTR: forward 5’-AAAGGGCGGCACTCACAG-3’, reverse 5’-CTGGGGACTCGCTGGACT-3’, SNRPN: forward 5’-GCAAAACAGCCAGAACGTGAA-3’, reverse 5’-GCACACGAGCAATGCCAGTAT-3’.
Western blotting
Total protein was extracted from cells using RIPA lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate]. After incubation on ice, cells were scraped from wells, lysates were vortexed and centrifuged at 13,000 × g for 15 min. Lysate supernatants were placed into new microcentrifuge tubes and protein concentrations were determined using Pierce BCA Protein Assay Kit (Cat# 23225, Thermo Fischer Scientific). A unit of 10–15 µg of cell lysate was denatured in 6× Laemmli sample buffer [50 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 0.25% b-mercaptoethanol, and bromophenol blue (1 mg/mL)] at 100 °C for 5 min. Two times Laemmli sample buffer was added to samples to bring to a final volume of ~30–40 µL. Samples were separated on an 8% SDS-polyacrylamide gel or 4–12% Novex Tris-Glycine gel (Cat# XP04120BOX, Thermo Fischer Scientific) and then transferred to the nitrocellulose membrane (Cat# 1620115, MilliporeSigma, Burlington, MA, USA). Membranes were blocked with 5% non-fat dry milk in TBST (Tris-buffered saline containing 0.05% Tween 20) and incubated with either anti-LRIG1 (Cat# 12752, Cell Signaling Technologies, Danvers, MA, USA), anti-tubulin (Cat# T5168, Sigma-Aldrich), or anti-β-Actin (AC-15, Cat# A1978, Sigma-Aldrich) primary antibodies overnight at 4 °C. After incubation, membranes were washed with TBST and then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hr at room temperature. Chemiluminescence signals were visualised using Pierce ECL (Cat# 32106, Thermo Fischer Scientific) on an Alpha Innoteceh Digital Imaging Station. Blot images are a representation of three technical replicates from at least two independent experiments. Western blot densitometry for dCas9 assay was determined using Image J. Inverted protein band value or inverted membrane background value was expressed as 255 – X, where X is the mean grey value recorded by Image J. Net band values were determined by deducting the inverted membrane background values from the inverted protein band values. Relative protein quantification was determined by taking the net band value of LRIG1 over the net band value of Actin loading control for each lane.
CRISPR-deadCas9 plasmids
The catalytic domain of mouse Tet1 (NP_001240786.1; aa 1367–2039), obtained by PCR amplification from plasmid ZFB-TET1CD. ZFB-TET1CD, was a kind gift from Marianne Rots [27]. Overhangs compatible with Gibson cloning were introduced during PCR amplification and TET1CD was then cloned into the KpnI digested dCas9 cloning vector (Addgene plasmid #100091) [28]. The resulting Tet1-dCas9 plasmid is available from Addgene (plasmid #136650). Similarly, plasmid VP64-dCas9 (Addgene plasmid #177171) was created by Gibson cloning of the VP64 activation domain into KpnI digested dCas9 cloning vector. pcDNA-dCas9-p300 Core was a gift from Charles Gersbach (Addgene plasmid #61357) [29].
sgRNA target design and off-target identification
Guide RNA (sgRNA) target sequences were designed using CHOPCHOP [30]. Only the highest scoring target sequences were chosen. Selected sgRNAs were cloned as G-N19 into AflII-digested gRNA cloning vector using Gibson assembly (Addgene plasmid #41824) [31]. sgRNA sequences are listed in Supplemental Table S2. Off-target analysis of CRISPR sgRNAs was performed using the CCTop off-target prediction tool (https://cctop.cos.uni-heidelberg.de:8043) [32]. Briefly, 20 bp spacer sequences for sgRNAs candidates 5, 6, 12, 13 and 14 (without PAM sequences) were used as the query with hg19 as the reference genome for canonical SpCas9 PAM sites (NGG). The algorithm was executed using five or less total mismatches, a maximum core length of 12, and two or less core mismatches. The list of off-target genes for sgRNA 12, 13 and 14 was overlapped to identify collective off-targets of sgRNA Combination 1 (sgC1). In a similar manner, off-targets for sgRNA 5 and 6 were overlapped to identify collective sgRNA Combination 2 (sgC2) off-targets.
Transient transfection of CRISPR/deadCas9
For CRISPR-dCas9-based transactivation of LRIG1 by RT-qPCR and targeted DNA methylation analysis, 2 × 105 cells were seeded in each well of a 12-well plate 18–24 h prior to transfection. Cells were transfected with a total mixture of 1.2 μg plasmid DNA per well (0.3 μg equimolar pooled sgRNAs, 0.7 μg equimolar pooled dCas9 complexes and 0.2 μg pBABE-puro (Addgene #1764) for Puromycin selection) using Lipofectamine 3000 and P3000 according to the manufacturer’s protocol. For western blotting, 5 × 105 cells were seeded in each well of six-well plates and a total of 3.0 μg of plasmid DNA per well was used for transfection (0.75 μg equimolar pooled sgRNAs, 1.75 μg equimolar pooled dCas9 complexes and 0.5 μg pBABE-puro). For MTS cell viability, 1 × 104 cells were seeded in each well of 96-well plates and a total of 0.12 μg plasmid DNA per well was used for transfection (0.03 μg equimolar pooled sgRNAs, 0.07 μg equimolar pooled dCas9 complexes and 0.02 μg pBABE-puro).
Transfection complexes were removed after 6–8 h and replaced with complete culture medium. 24 h post-transfection, culture medium was replaced with media containing 1–2 μg/mL Puromycin for 48 h to select for transfected cells. Untreated cells were seeded alongside transfected cells for each experiment and treated with Puromycin as a control for antibiotic selection. Following Puromycin selection, wells were gently rinsed twice with ice-cold 1× PBS to remove dead/non-transfected cell populations, and RNA extracted for RT-qPCR, lysed in RIPA for western blotting, or subjected to an MTS assay for cell viability.
Cell viability
Viability was evaluated using the CellTiter 96® AQueous One Solution Reagent (Cat# G3582, Promega), containing a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] and an electron coupling reagent (phenazine ethosulfate; PES), per the manufacturer’s instructions.
Targeted DNA methylation analysis
Genomic DNA was extracted from three independent experiments using the Quick-DNA Miniprep Kit (Cat# D3024, Zymo Research, Irvine, CA, USA) and 250–500 ng genomic DNA was bisulfite converted using the EZ DNA Methylation-Lightning Kit (Cat# D5030, Zymo Research). Bisulfite-Sequencing PCR (BSP) amplification of 25–50 ng of bisulfite converted ssDNA was carried out with ZymoTaq DNA polymerase (Cat# E2002, Zymo Research) according to the manufacturer’s instructions. MethPrimer 2.0 (http://www.urogene.org/methprimer2/) was used to design BSP primers with the degenerate primer parameter (LRIG1-BSP-F 5’-YGAGTTTTTAGYGTAAGTGTAGG-3’, LRIG1-BSP-R 5’-GTTRGAATCCTCACAATCCC-3’). Unique 6-nucleotide barcodes were added to the 5’-end of the forward primer sequence. Amplicons were then purified using the QIAquick PCR purification Kit (Cat# 28104, QIAGEN) and equal amounts were pooled. Library preparation and PE150 sequencing (CRISPR sequencing) were performed by the CCIB DNA Core Facility at Massachusetts General Hospital (Cambridge, MA). Sequence read files were demultiplexed and forward and reverse reads were merged into a single long read using FLASH2 [33]. Processed FASTQ files were aligned, and cytosine methylation states determined using Bismark [34]. All samples used for downstream analysis had a mapping efficiency of >99%. Percent methylation across 30 CG dinucleotides (CpG) sites spanning the BSP region (see Fig. 5), was determined by [% = (M/(M + U)) * 100], where M denotes methylated signal intensity and U unmethylated signal intensity, respectively. For each cell line, normalised percent methylation was determined after background subtraction of no treatment control percent methylation values.
In silico analysis of human breast cancer data
The results of computational analysis are based on data generated by The Cancer Genome Atlas (TCGA) Research Network or NCBI Gene Expression Omnibus (GEO). Beta values (β-values) are the estimate of methylation level using the ratio of methylated and unmethylated intensities. β-values range from 0 to 1, with 0 being fully unmethylated and 1 being fully methylated. At each CpG site, methylation is quantified by [β = M/(M + U + α)], where M > 0 and U > 0 denote the methylated and unmethylated signal intensities, respectively; and an offset, α, (equal to 1 by default) [35]. TCGA: β-values (Array-based DNA platform) were used to determine methylation levels of LRIG1, in adjacent normal versus breast tumour samples, and within breast tumour molecular subtypes. Welch two-sample t test, or ANOVA followed by a Tukey HSD test, was used to determine whether the means of the two groups, or three or more groups, respectively, were statistically different. The relationship between LRIG1 methylation and LRIG1 mRNA expression for breast tumour samples was determined using β-values and mRNASeq read count data. Methylation values and mRNA expression values were matched by patient sample, and correlation was computed using the Spearman rank correlation coefficient method. Molecular subtyping was based on the PAM50 (Prosigna Breast Cancer Prognostic Gene Signature Assay) profiling test. Kaplan–Meier survival plots were generated using the ‘survival’ and ‘survminer’ packages in the R statistical program. Data for Kaplan–Meier plots consisted of β-values and patient clinical information (death status and days to last contact) and used data-driven cut-offs computed by survminer. A difference in survival probability between groups was determined using the Tarone-Ware test in survminer. GSE78758: β-values (Illumina HumanMethylation450 BeadChip) were used to determine methylation levels of LRIG1 in adjacent normal, primary breast tumour, and lymph node metastases in basal/triple-negative breast cancer patients. A one-way ANOVA followed by a post ad hoc Tukey HSD test, was used to determine whether the means of the three groups were statistically different.
Statistical analysis
Statistical tests for data analysis included Welch two-sample t test, ANOVA followed by Tukey’s HSD test, log-rank test, and Student’s t test. Values are represented as mean ± standard error of the mean (SEM). For in vitro experiments, data represent at least three independent experiments (unless otherwise specified in the figure legend). In all cases, differences were considered statistically significant when P value was less than 0.05. All graphical representation and statistical analyses of data were performed using the R statistical programme or Microsoft Excel.
Results
LRIG1 methylation is increased in breast cancer
Prior studies in colorectal and cervical cancers indicate that the LRIG1 locus is hypermethylated [22, 23]. Interestingly, a recent study using canine mammary tumours as a model for human breast cancer, identified hypermethylation in the third intron of LRIG1 overlapping with a tumour suppressive PAX5 DNA binding motif. Hypermethylation at this region correlated with reduced LRIG1 gene expression, which was conserved (along with PAX5-binding motifs) in human breast cancer [36], suggesting that methylation of LRIG1 in cancer may be a species-conserved mechanism of repression. Here, we sought to explore methylation at the LRIG1 CpG island, versus intronic regions, as hypermethylation of tumour suppressor genes in the CpG islands near the promoter is a well-documented feature of cancer.
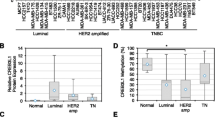
Supplemental Fig. 1 depicts the location of the promoter-proximal CpG island, relative to the transcription start site and exon 1 of the LRIG1 gene. To examine whether LRIG1 is methylated in breast cancer, we utilised the publicly available TCGA-BRCA dataset, useful for both its large sample size and matched patient methylation-mRNA expression data. (Beta values (β-values) are the estimate of methylation at a given CpG site. β-values range from 0 to 1.0, with 0 being fully unmethylated and 1.0 being fully methylated; and are quantified by β = M/(M + U), where M and U denote methylated and unmethylated signal intensities, respectively [35]). Analysis of methylation status using the HumanMethylation450 array revealed that methylation (β-value) at the LRIG1 CpG island is significantly increased in breast tumours (n = 796) compared to normal samples (n = 96) (Fig. 1a, b). Relative to the mean methylation value in normal samples, tumour samples are more highly represented at values that are 2 or ≥3 standard deviations above this normal mean (Fig. 1c). Furthermore, methylation of LRIG1 is increased in primary tumours and lymph node metastases of patients with basal breast cancer/TNBC relative to normal tissue (Fig. 1d, data from GSE78758 [37]), suggesting methylation-mediated silencing of LRIG1 may be important in breast cancer progression.

a Boxplot and (b) histograms (with overlying density plots) depicting LRIG1 β-values in normal breast tissue (n = 96) and breast tumours (n = 796) (data from TCGA BRCA). c Dot plot depicting the percent distribution of LRIG1 β-values in normal breast tissue and breast tumour samples relative to the LRIG1 β-value mean in normal samples (vertical dotted line) (data from TCGA BRCA). d Boxplot depicting LRIG1 β-values in adjacent normal breast tissue (n = 4), primary tumours (n = 23) and lymph node metastases (n = 12) from patients with triple-negative breast cancer (data from GSE78758). *P < 0.05, **P < 0.01 and ***P < 0.001. Students t test (a), Tukey’s HSD (d).
LRIG1 methylation inversely correlates with LRIG1 expression
We previously reported that LRIG1 expression is lowest in the basal-like molecular subtype of breast cancer [19]. To determine if this pattern also exists in the larger TCGA-BRCA dataset, we examined LRIG1 expression as a function of breast cancer molecular subtypes. Analysis of mRNA read count (RNASeqV2) confirmed that LRIG1 is most significantly reduced in basal breast tumours (Fig. 2a). We, therefore, queried the LRIG1 CpG island methylation status as a function of molecular subtype, as shown in Fig. 2b. Interestingly, CpG island methylation was significantly higher in basal breast tumours, as compared to the Luminal A, Luminal B and Her2+ subtypes. When tumours were segregated based on ERα status, ER-negative tumours (which encompass basal and most Her2+ breast cancers) demonstrated increased methylation (Fig. 2c), in agreement with our prior findings that LRIG1 is poorly expressed in Her2+ and basal breast tumours [19, 38], while richly expressed in ER-positive tumours [14]. We next examined the correlation between LRIG1 CpG island methylation and LRIG1 mRNA read count in breast cancer (n = 784). As shown in Fig. 2d, there is a striking inverse correlation between methylation and mRNA expression (Spearman correlation −0.63, P < 0.001), strongly suggesting that methylation decreases LRIG1 expression. (Supplemental Table 1 shows correlation by molecular subtype.) Given that ER-negative tumours show the most robust methylation, we performed Kaplan–Meier analysis on patients with ER-negative tumours to assess whether there was a correlation between LRIG1 CpG island methylation status and overall patient survival. Patients whose breast tumours expressed higher levels of LRIG1 methylation had decreased overall survival time (Fig. 2e), suggesting that LRIG1 promoter methylation is functionally important.

a Boxplot depicting LRIG1 mRNA expression (read count) in Luminal A (n = 439), Luminal B (n = 118), Her2 + (n = 37) and Basal (n = 112) breast tumours (data from TCGA). b Boxplot depicting LRIG1 β-values in Luminal A (n = 110), Luminal B (n = 46), Her2 + (n = 14) and Basal (n = 41) breast tumours (data from TCGA). c Boxplot depicting LRIG1 β-values in ER-positive (n = 565) and ER-negative (n = 167) breast tumours (data from TCGA BRCA). d Scatter plot of patient-matched LRIG1 mRNA read count and LRIG1 β-values in breast tumour samples (n = 204). Black dotted line indicates linear regression (data from TCGA BRCA). e Kaplan–Meier plot of overall survival as a function LRIG1 methylation in ER-negative tumours. Patients were segregated into groups based on ERα status and LRIG1 β-value in their primary tumour (high methylation/ER-negative: n = 135, low methylation/ER-negative: n = 32) (data from TCGA BRCA). *P < 0.05, **P < 0.01 and ***P < 0.001, respectively; Tukey’s HSD (a, b), Student’s t test (c), Spearman correlation test (d), log-rank test (e).
Recent studies have revealed that methylation of distal-regulatory regions, including enhancers, plays an important role in regulating gene expression [39, 40]. In prior work, we demonstrated that LRIG1 is an ER regulated gene [14]. Interestingly, Stone et al. reported that enhancer hypermethylation within ERα regulated genes is correlated both with decreased ERα binding and decreased gene expression [41]. Mining of supplemental data provided by Stone et al. revealed two ERα binding sites within LRIG1 enhancer regions which are hypermethylated (HumanMethylation450 CpG probes cg24150385 and cg09716921, herein labelled eCpG-1 and eCpG-2, respectively). We were curious to determine whether these elements display subtype-specific methylation, as observed for the CpG island. Indeed, as shown in Supplemental Fig. 2A, methylation of both eCpG-1 and eCpG-2 is highest in the basal subtype of breast cancer. Methylation of both elements inversely correlates with LRIG1 gene expression, in the TCGA-BRCA cohort (Supplemental Fig. 2B) and in multiple cell line models of endocrine-resistant breast cancer [41]. (Supplemental Table 1 shows correlation by molecular subtype.) Kaplan–Meier analysis of ER-negative patients revealed that patients whose breast tumours expressed higher levels of LRIG1 methylation, at these enhancer-region CpG sites, had decreased overall survival time (Supplemental Fig. 2C), suggesting that methylation at both elements is functional. Collectively, these findings suggest that loss of LRIG1 expression in basal/TNBC is multifactorial, with contributions from both promoter and enhancer methylation.
LRIG1 methylation in breast cancer cell lines
Having examined publicly available patient samples, we next examined LRIG1 CpG island methylation in breast cancer cell line models. We chose several cell lines, including MCF7 and T47D (Luminal A) and ZR75-1 (Luminal B), representative of ER-positive breast cancer, and BT549, HCC1937 (BRCA1 mutant) and MDA-MB-231, representative of basal/TNBC. LRIG1 mRNA transcript expression (Fig. 3a) and protein abundance (Fig. 3b) were comparably higher in the ER-positive breast cancer cell lines, as expected [14, 19]. To focus our analysis, we used the UCSC genome browser to query methylation levels across the LRIG1 CpG island (Fig. 3c) [26]. HCT116 colon carcinoma cells were included as a positive control, as they are reported to have high levels of LRIG1 CpG island methylation [22]. Notably, regions of the CpG island are highly methylated (red bars; 100% methylation) in HCT116 cells, while these same regions are unmethylated (green bars; 0% methylation) in MCF7 breast cancer cells. Using this information, we focused our analysis on two regions that exhibited differential methylation between HCT116 and MCF7 cell lines, designated “Exon 1” and “Promoter” in Fig. 3c. Using the methylated DNA immunoprecipitation (MeDIP) technique, which immunoprecipitates methylated DNA using an antibody specific for 5-methylcytosine [42], we assayed methylation levels across human breast cancer cell lines. A representative agarose gel (Fig. 3d) and relative methylation levels (Fig. 3e), based on qPCR, are shown for both regions. The promoter region of the small nuclear ribonucleoprotein polypeptide N gene (SNRPN/SNURF), which is heavily methylated [43], served as a control for the MeDIP technique and illustrates that differential methylation observed at the LRIG1 CpG island is not due to a global increase or decrease in methylation inherent to each cell line (Supplemental Fig. 3A, B). We found that methylation levels at the Exon 1 and Promoter regions of LRIG1 inversely correlate with LRIG1 mRNA expression levels (Fig. 3f). Interestingly, the results in the breast cancer cell lines reflected our findings in patient samples, with methylation lowest in ER-positive breast cancer lines and highest in basal/triple-negative cell lines (ER-negative). This suggests that LRIG1 silencing by methylation may be an inherent property of basal/TNBC.

a Bar plot showing relative LRIG1 transcript levels across breast cancer cell lines (n = 6). Expression was determined from the Ct values using the 2−ΔΔCt method after normalisation to GAPDH. LRIG1 transcript levels were normalised to BT549 values, which are set to 1.0. b LRIG1 protein abundance across breast cancer cell lines (n = 6), and Cos7 cells with stable overexpression of LRIG1, assessed by western blotting. Tubulin serves as a loading control. c Snapshot of LRIG1 gene and promoter-proximal CpG island in the UCSC Genome Browser hg19 build. DNA Methylation by Reduced Representation Bisulfite-Sequencing (RRBS) track depicts the location and percent methylation of CpG sites in HCT116 and MCF7 cells. Red: 100% methylation, yellow: 50% methylation, green: 0% methylation. Exon 1 and Promoter regions are highlighted. d DNA agarose gels of MeDIP-PCR show methylation levels at Exon 1 and Promoter regions across cell lines (n = 7). In all, 5% of input DNA is shown in the bottom panels. e Bar plot showing relative β-values at LRIG1 Exon 1 and Promoter regions as assayed by MeDIP-qPCR. The methylation level was determined from the Ct values using the 2−ΔΔCt method after normalisation to 5% input DNA. β-value was determined by normalisation to HCT116 Exon 1 methylation value, which is set to 1.0. f Heatmap of LRIG1 levels across replicates of transcript expression (data from (a)) compared to Exon 1 and Promoter region methylation levels (data from (e)), respectively, in breast cancer cell lines (n = 6). Values are mean ± SEM of two (d, e) or three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001. n.s. not significant (P > 0.05); Student’s t test (e).
Inhibition of DNA methylation induces LRIG1 mRNA expression
We next determined the impact of the DNA methyltransferase inhibitor, 5-aza-2’-deoxycytidine (ADC), on LRIG1 mRNA expression. As shown in Fig. 4a, ADC sequesters DNA methyltransferases resulting in the demethylation of LRIG1 DNA and subsequent transcriptional activation. ADC treatment consistently increased LRIG1 mRNA expression in the basal/TNBC cell lines (BT549, HCC1937, and MDA-MB-231), and in heavily methylated HCT116 cells at high doses (Fig. 4b). Interestingly, ADC treatment of ER-positive breast cancer cell lines (T47D, MCF7, and ZR75-1) either significantly decreased, or had little/no effect, on LRIG1 expression (Supplemental Fig. 4a). To further examine ADC-induced methylation inhibition, we used MeDIP to measure methylation levels at the LRIG1 Exon 1 and Promoter regions (from Fig. 3c), following ADC treatment. Indeed, we found that relative LRIG1 methylation levels were significantly decreased at both regions in cells treated with ADC versus vehicle control (Fig. 4c).

a Schematic representation of LRIG1 demethylation following ADC (5-Aza-2’-deoxycytidine) treatment. ADC (nucleotide structure, inset) sequesters DNA methyltransferases (DNMTs) resulting in demethylation of LRIG1 DNA and transcriptional activation. b Bar plots showing relative LRIG1 expression levels in ER-negative breast cancer cell lines (BT549, HCC1937, MDA-MB-231) and colon cancer cell line (HCT116) treated with shown concentrations of ADC for 96 h. Expression was determined from the Ct values using the 2−ΔΔCt method after normalisation to GAPDH. LRIG1 fold changes were normalised to 0 μM treatments, which are set to 1.0. c Bar plot showing relative β-values at LRIG1 Exon 1 and Promoter regions, as assayed by MeDIP-qPCR, in cells treated with vehicle control (VC) or 20 μM ADC for 96 h. Methylation level was determined from the Ct values using the 2−ΔΔCt method after normalisation to 5% input DNA. β-value was determined by normalisation to HCT116 Exon 1 VC methylation value, which is set to 1.0. Values are mean ± SEM of three (b) or two (c) independent experiments. *P < 0.05 and **P < 0.01. Student’s t test (b, c).
Since histone deacetylases (HDACs) repress gene expression by compacting chromatin structure and can be recruited to methylated DNA by methyl-binding proteins [44], we investigated whether HDACs could also be contributing to the silencing of LRIG1 in basal/TNBC. Treatment of cells with the HDAC inhibitor, Panobinostat, as a single agent did not significantly increase LRIG1 mRNA expression (Supplemental Fig. 4B). We next went on to test the combination of Panobinostat and ADC, finding this was not consistently superior to ADC alone (except in HCC1937 cells); suggesting that methylation, not deacetylation, is the dominant epigenetic mechanism of LRIG1 silencing in breast cancer.
The CRISPR-deadCas9 transactivation system reactivates LRIG1
LRIG1 mRNA and protein are consistently decreased across cancer types, including breast cancer. However, genetic alterations in LRIG1, such as copy number variation, are observed in less than 1% of breast cancers (Supplemental Fig. 5), indicating that epigenetic silencing is a dominant mechanism of LRIG1 loss. This suggests that LRIG1 may be amenable to “transcriptional reactivation”, which is now feasible using targeted approaches. Towards this end, we employed the CRISPR/deadCas9 transactivation system, which utilises a catalytically inactive Cas9 protein (dCas9) directly fused to either the: (1) ten-eleven translocation methylcytosine dioxygenase 1 (TET1) enzyme (Tet1-dCas9), (2) VP64 domain (VP64-dCas9), or (3) catalytic core of acetyltransferase p300 (dCas9-p300 Core) (Fig. 5a, b). TET enzymes accomplish demethylation by catalysing the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), and further to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). 5fC and 5caC are removed by cellular DNA repair machinery, regenerating unmethylated cytosines [45, 46]. VP64 is composed of four tandem copies of VP16 (Herpes Simplex Viral Protein 16) connected by glycine–serine linkers and acts as a strong transcriptional activator through recruitment of transcription factors (e.g., TATA-binding protein (TBP) and TBP-related factors) and recruitment of chromatin modification factors (e.g., histone-modification factors) [47]. The transcriptional co-activator p300 is a histone acetyltransferase that regulates gene expression by catalysing acetylation of histone H3 lysine 27 (H3K27) and is typically recruited to promoter and enhancer regions of target genes [29]. We designed 14 sgRNAs (Fig. 5c) spanning the LRIG1 CpG island within a ± 1 kb window on either side of the LR1G1 transcriptional start site (TSS) and evaluated the activation ability of dCas9 fusions.

a Schematic representation of construction of dCas9 plasmids. The resulting plasmids contain dCas9 in direct fusion with Tet1, VP64 or p300 (Core). b Schematic representation of the sgRNA-Tet1-dCas9 complex. dCas9 is in direct C-terminal fusion with the demethylation-initiating enzyme Tet1 or the transcriptional activators VP64 or p300 (Core). sgRNAs bind in CpG island regions of LRIG1 to activate gene expression. c Snapshot of sgRNA target sites in the LRIG1 CpG island region. Arrowheads indicate whether the sgRNA targets the forward or reverse DNA strand. Blue arrowheads indicate “+” DNA strand. Red arrowheads indicate “−” DNA strand. Bisulfite-sequencing primer (BSP) region is highlighted. Guide RNA combinations are underlined. sgC1; sgRNA combination 1 includes sgRNA 12, sgRNA 13, and sgRNA 14. sgC2; sgRNA combination 2 includes sgRNA 5 and sgRNA 6.
The dCas9 transactivation system was expressed in cell lines by lipid-based transfection followed by antibiotic selection 24 h post-transfection (HPT) to enrich for transfected cells (Supplemental Fig. 6A). As previous studies using CRISPR/dCas9 to reactivate gene expression have shown that combined delivery of multiple sgRNAs provides increased levels of endogenous gene activation [48,49,50], we tested several combinations of 2–3 guide RNAs (Supplemental Fig. 6B). We grouped guides based on their genomic locations across the LRIG1 CpG island. We performed RT-qPCR and observed significant activation of LRIG1 expression with the combination of sgRNAs 12–14 (termed sgC1) and sgRNA 5,6 (termed sgC2) paired with Tet1-dCas9, targeting regions both up- and downstream of the TSS. LRIG1 expression increased 1.3 to 3.1-fold in HCT116 cells relative to Tet1-dCas9 with no guide RNA (Fig. 6a). No significant difference was observed between cells transfected with Tet1-dCas9 alone and Tet1-dCas9 coupled with sgRNAs 9–11, 2,3,8 or 1,4,7 (Supplemental Fig. 6B). In concordance with HCT116 cells, transfection with Tet1-dCas9 and sgC1 revealed a significant upregulation of LRIG1 in BT549 (1.7-fold, Fig. 6b) and MDA-MB-231 (1.3-fold, Fig. 6c) cells when compared to cells transfected with Tet1-dCas9. (HCC1937 cells showed a 1.4-fold upregulation, but was not statistically significant, Fig. 6d). No significant upregulation was observed in basal/TNBC cell lines transfected with Tet1-dCas9 with sgC2 versus no sgRNAs. Cells treated with 10 μM ADC were included as a positive control, as we previously showed ADC treatment consistently increases LRIG1 expression. We also tested Tet1-dCas9 paired with randomised combinations of LRIG1 targeting sgRNAs and still observed significant increases in LRIG1 transcript expression (Supplemental Fig. 6C), suggesting that LRIG1 upregulation can be achieved using multiple varying combinations of guide RNAs.

a–d Bar plots showing relative LRIG1 expression levels in HCT116 (a), MDA-MB-231 (b), BT549 (c) and HCC1937 (d) cells treated with 0μM (vehicle control) or 10 μM ADC, transfected with Tet1-dCas9 alone or with guide RNA combinations (sgC1, sgC2), or transfected with a combination of Tet1-dCas9 and VP64-dCas9 alone or with guide RNA combinations (sgC1, sgC2). Expression was determined from the Ct values using the 2−ΔΔCt method after normalisation to GAPDH. LRIG1 fold changes were normalised to 0 μM ADC or no sgRNA values for each group, which are set to 1.0. e Quantification of LRIG1 protein abundance in cell lines (n = 4) transfected with Tet1-dCas9 alone, Tet1-dCas9 with sgC1 or sgC2 and Tet1-dCas9 and VP64-dCas9 with sgC1 as assessed by Western Blotting. Actin serves as a loading control. Protein abundance was determined by densitometry using Image J after normalising to Tet1-dCas9 values, which are set to 1.0. f Images of HCT116 and MDA-MB-231 blots are shown. g Line plot of change in percent methylation in HCT116 cells transfected with Tet1-dCas9, Tet1-dCas9 with guide RNA combinations (sgC1, sgC2), or a combination of Tet1-dCas9 and VP64-dCas9 with sgC1 as assessed by targeted sequencing of bisulfite converted genomic DNA across 30 CpG dinucleotides near the LRIG1 transcriptional start site. Change in percent methylation was determined after background subtracting the no treatment control percent methylation values. The X axis depicts CpG distances from LRIG1 transcriptional start site (at 0). Values are mean ± SEM of two (a–d, g) or three (e, f) independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001. Student’s t test (a–e, g).
As others have previously observed enhanced activation when co-targeting dCas9 complexes [49], we were therefore curious whether other epigenetic mechanisms, such as transcriptional activation, could also upregulate LRIG1 transcript expression. To this end, we transfected cells with VP64-dCas9 paired with sgC1 or sgC2 guide RNA combinations alone (Supplemental Fig. 6D, E) or in combination with Tet1-dCas9 (Fig. 6a–d). We observed a 3.4 to 18.9-fold upregulation of LRIG1 in basal/TNBC cells (41-fold in HCT116 cells) for sgC1, and a 1.4 to 2.3-fold increase for sgC2 (3.3-fold in HCT116 cells) when compared to cells transfected with Tet1-dCas9/VP64-dCas9 with no guides. We also tested dCas9-p300 (Core) paired with sgC1 or sgC2 guide RNA combinations alone (Supplemental Figs. 6D, E) or in combination with Tet1-dCas9 (HCT116 only, Supplemental Fig. 6E), observing no significant upregulation of LRIG1 transcript expression. Moreover, in HCT116 cells a triple combination of Tet1-dCas9, VP64-dCas9, and dCas9-p300 with sgC1 or sgC2, although significant, showed lower levels of LRIG1 expression (Supplemental Fig. 6E). As p300 regulates gene expression through acetylation, this data suggests that deacetylation does not contribute to LRIG1 silencing in line with our data from HDAC treatment in Supplemental Fig. 4B.
To investigate the possibility of off-target gene regulation, we individually analysed sgRNAs 12–14 (sgC1) and 5–6 (sgC2) sequences to identify putative genome-wide off-target sgRNA-binding sites. We then selected genes which were identified as off-targets for at least two sgRNAs in Combination 1 and for both sgRNAs in Combination 2 (Supplemental Fig. 7). RT-qPCR was performed to assess the five target genes in HCT116 cells transfected with Tet1-dCas9 or Tet1-dCas9/VP64-dCas9 with sgC1 or sgC2. We observed no significant upregulation on the expression of the putative off-target genes compared to no sgRNA control conditions (Supplemental Fig. 6F), supporting site-specific regulation of LRIG1 by dCas9 protein complexes.
The dCas9 system activates LRIG1 protein expression and inhibits cell viability
To determine whether activation of LRIG1 by Tet1-dCas9 and VP64-dCas9 extends beyond upregulation of transcript expression, we assessed LRIG1 protein levels and cell viability in HCT116 and basal/TNBC cells. For these experiments, we focused much of our attention on sgC1 as it caused higher levels of LRIG1 upregulation compared to sgC2. Only cells transfected with the combination of Tet1-dCas9 and VP64-dCas9 with sgC1 showed a significant increase (3.5 to 5.8-fold) in LRIG1 protein abundance (Fig. 6e, f and Supplemental Fig. 8A). These data may suggest that demethylation alone is not sufficient to increase LRIG1 protein abundance, and that gene-specific mRNA expression thresholds must be reached to observe concomitant protein upregulation.
We next examined the functional impact of reactivation of LRIG1 on the viability of cancer cells (Supplemental Fig. 8B). Treatment with 10 μM ADC was included as readout of global demethylation-induced changes in cell viability, and we observed significant decreases in cell viability following ADC treatment. Tet1-dCas9 and Tet1-dCas9/VP64-dCas9, both paired with sgC1, significantly decreased the viability of HCT116 cells and all three basal/TNBC cell lines when compared to no guide RNA conditions. Notably, we found that both Tet1-dCas9 with sgC1 and Tet1-dCas9/VP64-dCas9 with sgC1 were more effective at reducing cell viability compared to ADC treatment. These data suggest that targeted reactivation of LRIG1 provides superior effects compared to a global demethylation agent. This observed difference could be due to the complex crosstalk between a large number of methylated genes in the human genome, as well as their likely unequal contributions towards regulating cancer cell viability.
Tet1-dCas9 significantly reduces DNA methylation
Lastly, due to differences in reactivation of LRIG1 between the dCas9 effector domains and sgRNA combinations, we quantitatively determined the extent of demethylation in the core promoter region of LRIG1 using targeted bisulfite amplicon sequencing. We generated PCR-based amplicons which allowed us to measure percent methylation (ratio of 5-meCG/total CG) across 30 individual CpG dinucleotides in the selected region (185 base pairs, Fig. 5c) by deep sequencing. We observed significant decreases in DNA methylation across this region, starting 16–28 base pairs upstream of the LRIG1 TSS, following transfection of dCas9/sgRNA complexes. Specifically, in HCT116 cells, mean methylation decreased by 6.3%, 2.4% and 3.5% in cells transfected with Tet1-dCas9 with sgC1, Tet1-dCas9 with sgC2, and Tet1-dCas9/VP64-dCas9 with sgC1, respectively, compared to no guide RNA control (Fig. 6g, g inset). BT549 cells transfected with Tet1-dCas9 with either sgC1 or sgC2 exhibited significant decreases in methylation (Supplemental Fig. 8C, C inset). HCC1937 cells also displayed uniform decreases in methylation across experimental conditions versus Tet1-dCas9 only controls (Supplemental Fig. 8D, D inset). We observed minimal, non-significant decreases in methylation levels in MDA-MB-231 cells (Supplemental Fig. 8E). (Supplemental Fig. 8f shows methylation levels in untreated cells which were used for background subtraction calculations.) Interestingly, in HCT116, BT549, and HCC1937 cells, although the addition of VP64-dCas9 significantly reduced methylation levels compared to control, it did not further reduce percent methylation compared to Tet1-dCas9 with sgC1. In fact, in HCT116 and BT549 cells, percent methylation increased between the Tet1-dCas9 with sgC1 and Tet1-dCas9/VP64-dCas9 with sgC1 conditions. This observation may indicate that TET1 protein needs to achieve a minimum threshold of demethylation that subsequently permits transcriptional activation of LRIG1 by VP64. In addition, these data suggest in line with prior studies [49], that VP64-dCas9 does not use demethylation as a means towards transcriptional activation.
Discussion
LRIG1 is a negative regulator of oncogenic receptor tyrosine kinases and a documented tumour suppressor [51]. Indeed, LRIG1 downregulation has prognostic impact across diverse tumour types [52]. LRIG1 downregulation in cancer is widespread, with a recent study reporting “no to low” LRIG1 expression in cell lines from 22 different cancer types, and patient samples from seventeen different cancer types [53]. Analysis of a genome-wide RNAi screen [54] found that LRIG1 is one of six genes which rank in the top 1% (out of 17,080 genes) whose knockdown promotes cancer cell proliferation, across 43 different cancer cell lines [53]. Given these findings, much discussion has focused on how to “harness” LRIG1 for potential therapeutic benefit [55]. Several studies have reported that delivery of the soluble extracellular domain of LRIG1 holds promise [56, 57], although further work is needed to define the key functional domains of LRIG1. An alternative approach is to restore endogenous LRIG1 expression, although this requires greater insight into mechanisms that silence LRIG1 in cancer.
Our study focused on LRIG1 silencing in breast cancer. As LRIG1 copy number variations are rare in cancer, we hypothesised that epigenetic silencing of LRIG1 would be prevalent and explain its more significant loss in ER-negative and basal/TNBC disease. Indeed, we find that methylation of the LRIG1 CpG island and ERα-bound enhancers is significantly increased in ER-negative and basal/TNBCs and that methylation inversely correlates with LRIG1 mRNA expression. We demonstrate that subtype-specific differential methylation is observed in cell line models of ER-positive and ER-negative breast cancer and inversely correlate with LRIG1 expression in these cell lines. Furthermore, we show that robust LRIG1 methylation correlates with poor overall survival in patients with ER-negative breast tumours.
Global hypomethylating agents such as 5-aza-2’-deoxycytidine (ADC, Decitabine) are approved for the treatment of myelodysplastic syndromes and have been in use in that setting for more than a decade [58]. We used ADC, a DNA methyltransferase inhibitor, as an experimental tool to examine whether LRIG1 expression could be induced in breast cancer cells by CpG island demethylation. Indeed, ADC treatment of ER-negative/TNBC cells significantly increased LRIG1 mRNA expression. In contrast, ADC had no significant effect on LRIG1 expression in ER-positive cells, suggesting that low-level methylation observed in this setting is not functionally important. Since ADC-induced LRIG1 upregulation may occur indirectly, using methylated DNA immunoprecipitation (MeDIP) we demonstrate that demethylation occurs at the CpG island following ADC treatment. Due to its ability to globally reduce methylation levels, ADC has been implicated as a potential cancer therapeutic, with the ability to reduce cancer cell proliferation and migration and induce apoptosis [59,60,61]. In this study, we observe that ADC treatment reduces the viability of colorectal carcinoma and TNBC cells, and although high ADC concentrations (≥20 μM) likely induce apoptosis in a portion of the cell population, there is no evidence or expectation that this should impact upregulation of LRIG1.
Tumour suppressor restoration is an active area of study and holds great promise. Due to advances in CRISPR/Cas9-based technology, targeted transcriptional reactivation of tumour suppressor genes is now feasible. In fact, this has been demonstrated for PTEN and BRCA1, using dCas9 fused to the VPR effector domain (a potent transactivator consisting of VP64, p65 and Rta) and TET1; respectively [50, 62]. Genome-based CRISPR/dCas9 activation allows precise and stable editing of the epigenome with high efficiency, which could be harnessed for precision medicine and treatment of LRIG1-defincient cancers. In this study, we focused specifically on the role of the CpG island methylation in silencing LRIG1. Using Tet1-dCas9, we demonstrate for the first time, that targeted demethylation of the promoter-proximal CpG island induces LRIG1 expression in breast cancer cells. We observed that sgRNA combinations designed against binding sites furthest from the LRIG1 transcriptional starts site were most effective, suggesting that both the location of guide RNAs relative to transcriptional start and CpG sites, as well as the chosen combination, may affect the efficiency of TET1 activity. LRIG1 reactivation, both at the mRNA and protein levels, showed significant, but variable, responses across cell lines. This is consistent with previous studies using dCas9-based epigenetic editing in which target gene responses varied by cell type and genomic locus [28, 63]. However, we did observe ~1.3- to threefold upregulation of LRIG1 expression with Tet1-dCas9 which is consistent with the ~1.5 to 2.3-fold increase in target genes also reactivated via CRISPR/dCas9/TET1 demethylation [49, 62]. In addition to cell type and DNA location, variable responses may also be attributed to differences in other epigenetic modifiers of LRIG1. For example, previous studies have shown that LRIG1 is regulated by histone deacetylation [64], microRNAs [65, 66] and long noncoding RNAs [67]; all of which may contribute to the level of LRIG1 reactivation. This may result in variable levels of baseline methylation even amongst ER-negative cell lines (as observed in Fig. 3e). As the CRISPR/dCas9 system was delivered to cells via lipid-based transient transfection, we acknowledge that transfection efficiency may also contribute to observed differences in LRIG1 reactivation. However, since prior studies have shown global methylation changes and higher off-target effects with stably expressed dCas9 fusions to TET1 and DNA methyltransferase DNMT3A [49, 68, 69], we opted for transient expression, attempting to minimise these caveats. Our dCas9 transactivation system also showed specificity towards targeting LRIG1, with no observed upregulation of the five off-targeted tested.
Due to the strong effects of VP64 and p300 on gene upregulation, we sought to assess if these effectors could contribute to LRIG1 reactivation via CRISPR/dCas9. We found that the combinatorial binding of VP64-dCas9 and Tet1-dCas9 was capable of significantly upregulating LRIG1 mRNA expression and protein abundance. However, unexpectedly, these effects were not synergistic but were largely due to VP64-induced transcriptional activation as transfection with VP64-dCas9 and sgRNAs alone exhibited slightly higher LRIG1 expression compared to the Tet1-dCas9/VP64-dCas9 combination. This could be due to steric hinderance caused by large dCas9 protein complexes attempting to bind in the same genomic region [70, 71], especially regions with high binding affinity, such as CpG islands, promoters, and enhancers. However, we cannot rule out the possibilities that: (1) demethylation by the presence of dCas9 alone or TET1 alone, which has been shown in prior studies [69, 72], leads to a chromatin state more permissive of VP64-induced transcriptional activation, or (2) strong and targeted transcriptional activation by VP64 may be able to overcome or circumvent methylation-based gene silencing. Indeed, bisulfite modified sequencing shows that gene upregulation by VP64 is not dependent on demethylation.
To validate its biological relevance, we assessed the effects of Tet1/VP64-dCas9-mediated LRIG1 expression on cell viability. The significant reduction in cancer cell viability suggests that LRIG1 reactivation via CRISPR/dCas9 may have an impact on TNBC growth, however, future studies will be required to assess biological relevance in more disease-relevant models such as patient-derived organoids and in vivo mouse models. It is unclear how long demethylation and transcriptional activation by Tet1-dCas9 and VP64-dCas9, respectively, would be maintained in vivo, as well as maintenance of downstream effects. In addition, future studies, using readily available single-cell sequencing technologies, will be required to determine whether the observed reactivation of LRIG1, and other target genes, is due to uniform reactivation at the population level or strong reactivation in only a subset of the cell population. Collectively, our data provide strong evidence that LRIG1 is silenced by both CpG island and enhancer methylation in TNBC, and that the CRISPR/dCas9 system can be used to upregulate LRIG1 expression. While this study has some limitations, it provides the first evidence that a targeted approach using site-specific demethylation and transcriptional activation, is a feasible method of restoring LRIG1. As LRIG1 is known to play a crucial role in tumorigenesis and metastasis, combining CRISPR/dCas9-based reactivation with conventional therapeutic approaches could hold promise for LRIG1-silenced tumours.
Data availability
The datasets analysed in this study are available from the NCBI Gene Expression Omnibus (GEO) data repository (https://www.ncbi.nlm.nih.gov/geo) or The Cancer Genome Atlas Breast Invasive Carcinoma data repositories (https://portal.gdc.cancer.gov/projects/TCGA-BRCA).
References
Guo D, Holmlund C, Henriksson R, Hedman H. The LRIG gene family has three vertebrate paralogs widely expressed in human and mouse tissues and a homolog in Ascidiacea. Genomics. 2004;84:157–65.
Simion C, Cedano-Prieto ME, Sweeney C. The LRIG family: enigmatic regulators of growth factor receptor signaling. Endocr Relat Cancer. 2014;21:431–43.
Laederich MB, Funes-Duran M, Yen L, Ingalla E, Wu X, Carraway KL 3rd, et al. The leucine-rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. J Biol Chem. 2004;279:47050–56.
Gur G, Rubin C, Katz M, Amit I, Citri A, Nilsson J, et al. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J. 2004;23:3270–81.
Faraz M, Herdenberg C, Holmlund C, Henriksson R, Hedman H. A protein interaction network centered on leucine-rich repeats and immunoglobulin-like domains 1 (LRIG1) regulates growth factor receptors. J Biol Chem. 2018;293:3421–35.
Shattuck DL, Miller JK, Laederich M, Funes M, Petersen H, Carraway KL 3rd, et al. LRIG1 is a novel negative regulator of the Met receptor and opposes Met and Her2 synergy. Mol Cell Biol. 2007;27:1934–46.
Ledda F, Bieraugel O, Fard SS, Vilar M, Paratcha G. LRIG1 is an endogenous inhibitor of Ret receptor tyrosine kinase activation, downstream signaling, and biological responses to GDNF. J Neurosci. 2008;28:39–49.
Lindquist D, Alsina FC, Herdenberg C, Larsson C, Hoppener J, Wang N, et al. LRIG1 negatively regulates RET mutants and is downregulated in thyroid cancer. Int J Oncol. 2018;52:1189–97.
Alsina FC, Hita FJ, Fontanet PA, Irala D, Hedman H, Ledda F, et al. LRIG1 is a cell-intrinsic modulator of hippocampal dendrite complexity and BDNF signaling. EMBO Rep. 2016;17:601–16.
Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, et al. The pan-ErbB negative regulator LRIG1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 2012;149:146–58.
Mao F, Holmlund C, Faraz M, Wang W, Bergenheim T, Kvarnbrink S, et al. LRIG1 is a haploinsufficient tumor suppressor gene in malignant glioma. Oncogenesis. 2018;7:13.
Lu L, Teixeira VH, Yuan Z, Graham TA, Endesfelder D, Kolluri K, et al. LRIG1 regulates cadherin-dependent contact inhibition directing epithelial homeostasis and pre-invasive squamous cell carcinoma development. J Pathol. 2013;229:608–20.
Kvarnbrink S, Karlsson T, Edlund K, Botling J, Lindquist D, Jirstrom K, et al. LRIG1 is a prognostic biomarker in non-small cell lung cancer. Acta Oncol. 2015;54:1113–19.
Krig SR, Frietze S, Simion C, Miller JK, Fry WH, Rafidi H, et al. LRIG1 is an estrogen-regulated growth suppressor and correlates with longer relapse-free survival in ERalpha-positive breast cancer. Mol Cancer Res. 2011;9:1406–17.
Thompson PA, Ljuslinder I, Tsavachidis S, Brewster A, Sahin A, Hedman H, et al. Loss of LRIG1 locus increases risk of early and late relapse of stage I/II breast cancer. Cancer Res. 2014;74:2928–35.
Sheu JJ, Lee CC, Hua CH, Li CI, Lai MT, Lee SC, et al. LRIG1 modulates aggressiveness of head and neck cancers by regulating EGFR-MAPK-SPHK1 signaling and extracellular matrix remodeling. Oncogene. 2014;33:1375–84.
Lindquist D, Kvarnbrink S, Henriksson R, Hedman H. LRIG and cancer prognosis. Acta Oncol. 2014;53:1135–42.
Zhang Q, Shi W, Wang Q, Zhu Y, Zhai C, Wang J, et al. Clinicopathological and prognostic significance of leucine-rich repeats and immunoglobulin-like domains protein 1 (LRIG1) in malignant tumors: a meta-analysis. J Cancer. 2018;9:2895–909.
Yokdang N, Hatakeyama J, Wald JH, Simion C, Tellez JD, Chang DZ, et al. LRIG1 opposes epithelial-to-mesenchymal transition and inhibits invasion of basal-like breast cancer cells. Oncogene. 2016;35:2932–47.
Leidy J, Khan A, Kandil D. Basal-like breast cancer: update on clinicopathologic, immunohistochemical, and molecular features. Arch Pathol Lab Med. 2014;138:37–43.
Portela A, Esteller M. Epigentic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
Kou C, Zhou T, Han X, Zhuang H, Qian H. LRIG1, a 3p tumor suppressor, represses EGFR signaling and is a novel epigenetic silenced gene in colorectal cancer. Biochem Biophys Res Commun. 2015;464:519–25.
Lando M, Fjeldbo CS, Wilting SM, B CS, Aarnes EK, Forsberg MF, et al. Interplay between promoter methylation and chromosomal loss in gene silencing at 3p11-p14 in cervical cancer. Epigenetics. 2105;10:970–80.
Urbano A, Smith J, Weeks RJ, Chatterjee A. Gene-specific targeting of DNA methylation in the mammalian genome. Cancers. 2019;11:1–20.
Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–78.
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–70.
Chen H, Kazemier HG, de Groote ML, Ruiters MHJ, Xu GL, Rots MG. Induced DNA demethylation by targeting ten-eleven translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014;42:1563–74.
O’Geen H, Ren C, Nicolet CM, Perez AA, Halmai J, Le V, et al. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017;45:9901–16.
Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–17.
Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acid Res. 2019;47:171–74.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–26.
Stemmer M, Thumberger T, del Sol Keyer M, Wittbrodt J, Mateo JL. CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target predicition tool. PLoS ONE. 2015;10:e0124633.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2.
Weinhold L, Wahl S, Pechlivanis S, Hoffmann P, Schmid M. A statistical model for the analysis of beta values in DNA methylation studies. BMC Bioinforma. 2016;17:480.
Nam AR, Lee KH, Hwang HJ, Schabort JJ, An JH, Won SH. Alternative methylation of intron motifs is associated with cancer-related gene expression in both canine mammary tumor and human breast cancer. Clin Epigenetics. 2020;12:110.
Mathe A, Wong-Brown M, Locke WJ, Stirzaker C, Braye SG, Forbes JF, et al. DNA methylation profile of triple negative breast cancer-specific genes comparing lymph node positive patients to lymph node negative patients. Sci Rep. 2016;6:33435.
Miller J, Shattuck D, Ingalla E, Yen L, Borowsky A, Young L, et al. Suppression of the negative regulator LRIG1 contributes to ErbB2 overexpression in breast cancer. Cancer Res. 2008;68:8286–94.
Clermont PL, Parolia A, Liu HH, Helgason CD. DNA methylation at enhancer regions: Novel avenues for epigenetic biomarker development. Front Biosci). 2016;21:430–46.
Yao L, Berman BP, Farnham PJ. Demystifying the secret mission of enhancers: linking distal regulatory elements to target genes. Crit Rev Biochem Mol Biol. 2015;50:550–73.
Stone A, Zotenko E, Locke WJ, Korbie D, Millar EKA, Pidsley R, et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat Commun. 2015;6:7758.
Hsu HK, Weng YI, Hsu PY, Huang TH, Huang YW. Detection of DNA methylation by MeDIP and MBDCap assays: an overview of techniques. Methods Mol Biol. 2014;1105:61–70.
Glenn CC, Saitoh S, Jong MT, Filbrandt MM, Surti U, Driscoll DJ, et al. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am J Hum Genet. 1996;58:335–46.
Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25.
Tahilian M, Koh K, Shen Y, Pastor W, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5.
Ross S, Bogdanovic C. TET enzymes, DNA methylation and pluripotency. Biochem Soc Trans. 2019;47:875–85.
Beerli RR, Segal DJ, Drier B, Barbas CF 3rd. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci USA. 1998;95:14628–33.
Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–71.
Halmai JANM, Deng P, Gonzalez CE, Coggins NB, Cameron D, Carter JL, et al. Artificial escape from XCI by DNA methylation editing of the CDKL5 gene. Nucleic Acids Res. 2020;48:2372–87.
Moses C, Nugent F, Waryah CB, Garcia-Bloj B, Harvey AR, Blancafort P. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol Ther: Nucleic Acids. 2019;14:287–300.
Wang Y, Shi C, Lu Y, Poulin E, Franklin J, Coffey R. Loss of LRIG1 leads to expansion of brunner glands followed by duodenal adenomas with gastric metaplasia. Am J Pathol. 2015;185:1123–34.
Rouam S, Moreau T, Broet P. Identifying common prognostic factors in genomic cancer studies: a novel index for censored outcomes. BMC Bioinforma. 2010;11:150.
Xiong D, Wang Y, You M. Tumor intrinsic immunity related proteins may be novel tumor suppressors in some types of cancer. Sci Rep. 2019;9:10918.
Tsherniak A, Vazquez F, Montgomery P, Weir B, Kryukov G, Cowley G, et al. Defining a cancer dependency map. Cell. 2017;170:564–76.
Neirinckx V, Hedman H, Niclou S. Harnessing LRIG1-mediated inhibition of receptor tyrosine kinases for cancer therapy. Biochem Biophys Acta Rev Cancer. 2017;1868:109–16.
Johansson M, Oudin A, Tiemann K, Bernard A, Golebiewska A, Keunen O, et al. The soluble form of the tumor suppressor LRIG1 potently inhibits in vivo glioma growth irrespective of EGF receptor status. Neuro Oncol. 2013;15:1200–11.
Goldoni S, Iozzo R, Kay P, Campbell S, McQuillan A, Agnew C, et al. A soluble ectodomain of LRIG1 inhibits cancer cell growth by attenuating basal and ligand-dependent EGFR activity. Oncogene. 2007;26:368–81.
Duchmann M, Itzykson R. Clinical update on hypomethylating agents. Int J Hematol. 2019;110:161–9.
Fabrizio FP, Sparaneo A, Fontana A, Mazza T, Graziano P, Pantalone A, et al. Potential prognostic role of SPARC methylation in non-small-cell lung cancer. Cells. 2020;9:1523.
Man CH, Fung TK, Wan H, Cher CY, Fan A, Ng N, et al. Suppression of SOX7 by DNA methylation and its tumor suppressor function in acute myeloid leukemia. Blood. 2015;125:3928–36.
Patra A, Deb M, Dahiya R, Patra SK. 5-Aza-2’-deoxycytidine stress response and apoptosis in prostate cancer. Clin Epigenetics. 2011;2:339–48.
Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7:45545–56.
O’Geen H, Bates SL, Carter SS, Nisson KA, Halmai J, Fink KD, et al. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin. 2019;12:26.
Connelly JT, Mishra A, Gautrot JE, Watt FM. Shape-induced terminal differentiation of human epidermal stem cells requires p38 and is regulated by histone acetylation. PLoS ONE. 2011;6:e27259.
Qian M, Xu H, Song H, Xi H, Fang L. MiR-218-5p promotes breast cancer progression via LRIG1. Research Square. 2021, https://doi.org/10.21203/rs.3.rs-203193/v1.
Gao Y, Liu C, Zhao X, Liu C, Bi W, Jia J. hsa_circ_0000006 induces tumorigenesis through miR-361-3p targeting immunoglobulin-like domains protein 1 (LRIG1) in osteosarcoma. Ann Transl Med. 2021;9:1242.
Chen L, Fan X, Zhu J, Chen X, Liu Y, Zhou H. LncRNA MAGI2-AS3 inhibits the self-renewal of leukaemic stem cells by promoting TET2-dependent DNA demethylation of the LRIG1 promoter in acute myeloid leukaemia. RNA Biol. 2020;17:784–93.
Josipovic G, Tadic V, Klasic M, Xanki V, Beceheli I, Chung F, et al. Antagonistic and synergistic epigenetic modulation using orthologous CRISPR/dCas9-based modular system. Nucleic Acids Res. 2019;47:9637–57.
Galonska C, Charlton J, Mattei AL, Donaghey J, Clement K, Gu H, et al. Genome-wide tracking of dCas9-methyltransferase footprints. Nat Commun. 2018;9:597.
Cano-Rodriguez D, Gjaltema RAF, Jilderda LJ, Jellema P, Dokter-Fokkens J, Ruiters MHJ, et al. Writing H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat Commun. 2016;7:12284.
Verkuijl SA, Rots MG. The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr Opin Biotechnol. 2019;55:68–73.
Sapozhnikov DM, Szyf M. Unraveling the functional role of DNA demethylation at specific promoters by targeted steric blockage of DNA methyltransferase with CRISPR/dCas9. Nat Commun. 2021;12:5711.
Acknowledgements
We gratefully acknowledge the support of the NIH (MUG, MHG, DJS, CS) and The National Academies of Sciences Engineering Medicine Ford Foundation Predoctoral Fellowship (MUG).
Funding
National Academies of Science, Engineering, and Medicine—Ford Predoctoral Fellowship (Umeh Garcia), NIH T32CA009695-22 (Umeh Garcia), NIH 1K99CA256522-01 (Umeh Garcia), NIH K08NS901527 (Hayden Gephart), NIH R21HG010559-02 (Segal), NIH RO1CA118384 (Sweeney).
Author information
Authors and Affiliations
Contributions
MUG, DJS and CAS conceived and designed the study. MUG, CS, HO, DJS and CAS developed methods. All authors acquired, analysed, and interpreted data. All authors read, revised, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Umeh-Garcia, M., O’Geen, H., Simion, C. et al. Aberrant promoter methylation contributes to LRIG1 silencing in basal/triple-negative breast cancer. Br J Cancer 127, 436–448 (2022). https://doi.org/10.1038/s41416-022-01812-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-01812-8
- Springer Nature Limited
This article is cited by
-
Upregulation of p300 in paclitaxel-resistant TNBC: implications for cell proliferation via the PCK1/AMPK axis
The Pharmacogenomics Journal (2024)
-
CRISPR/dCAS9-mediated DNA demethylation screen identifies functional epigenetic determinants of colorectal cancer
Clinical Epigenetics (2023)