
Abstract
The most remarkable finding in synthetic lethality (SL) is the hypersensitivity to PARP inhibitors (PARPis) of the tumors harboring defects in genes involved in homologous repair (HR) such as BRCA1/2. Despite initial responsiveness to PARPi, the penetrance of the synthetic lethal interactions between BRCA1/2 genes and PARPi is incomplete. Thus, a significant proportion of HR-defective tumors experience intrinsic or acquired resistance, representing a key challenge of clinical research. An expanded concept of SL is opening new ways and includes novel forms of genetic interactions, investigating not only traditional SL of pairs genes but also SL between biological pathways that regulate the same essential survival cell function. In this context, recent research showed that HR and theta-mediated end-joining (TMEJ) pathways exhibit SL. DNA polymerase theta (Polθ) is encoded by the POLQ gene and is a key component of the TMEJ, an essential backup pathway, intrinsically mutagenic, to repair resected double-strand breaks (DSBs) when the non-homologous end joining (NHEJ) and HR are impaired. Polθ is broadly expressed in normal tissues, overexpressed in several cancers, and typically associated with poor outcomes and shorter relapse-free survival. Notably, HR-deficient tumor cells present the characteristic mutational signatures of the error-prone TMEJ pathway. According to this observation, the loss of HR proteins, such as BRCA1 or BRCA2, contributes to increasing the TMEJ-specific genomic profile, suggesting synthetic lethal interactions between loss of the POLQ and HR genes, and resulting in the emerging interest for Polθ as a potential therapeutic target in BRCA1/2-associated tumors.
This review summarizes the converging roles of the POLQ and HR genes in DNA DSB repair, the early-stage clinical trials using Polθ inhibitor to treat HR-defective tumors and to overcome BRCA-reversion mutations responsible for therapeutic resistance, and the novel pleiotropic effects of Polθ, paving the way for the development of unexplored synthetic lethality strategies.
Similar content being viewed by others

Introduction
The concept of synthetic lethality (SL) opened novel therapeutic avenues for targeting homologous recombination (HR) deficient tumors [1]. Several SL-based targeted therapies have been approved in clinical practice, starting from the simple genetic concept of the interaction between two genes, whose simultaneous perturbation causes cell death [2].
The therapeutic approach of synthetic lethality primarily focused on targeting DNA damage response (DDR) pathways. The synthetic lethal action of PARP-inhibitors (PARPis) in Breast cancer susceptibility gene 1 (BRCA1) and breast cancer susceptibility gene 2 (BRCA2)-deficient tumors provided key improvement in the clinical outcomes of patients with homologous recombination deficiency (HRD), mainly ovarian, breast and prostate cancer patients [3]. Currently, the biggest challenge of the research in these tumors is the SL-targeted drug resistance.
Building upon the crucial role of DDR pathway in SL, several potential synthetic lethal genes have been identified, such as Ataxia-Telangiectasia Mutated (ATM), Enhancer of Zeste Homolog-2 (EZH2), Ataxia Telangiectasia and Rad3-related (ATR), and Checkpoint Kinase 1 (CHK1) genes [4]. An expanded concept of SL is opening new ways and includes novel forms of synthetic lethal interactions, investigating not only traditional SL of pairs genes but also SL between two biological pathways that regulate the same essential survival cell function [4]. In this context, recent research showed that Homologous Recombination Repair (HRR) and theta-mediated end joining (TMEJ) pathways exhibit SL [5]. TMEJ, also known as alternative non-homologous end joining (alt-NHEJ) or microhomology-mediated end joining (MMEJ), is an essential backup pathway to repair resected double-strand breaks (DSBs) when the canonical repair pathways non-homologous end joining (NHEJ) and HR repair (HRR) are impaired [6].
Activation of TMEJ involves several enzymes, including PARP-1, and the repair of DSBs is facilitated by specific DNA polymerase, primarily DNA polymerase theta (Polθ) [7]. The depletion of the POLQ gene, coding for Polθ, seems to be synthetically lethal when combined with BRCA1/2 pathogenic variants (PVs). Thus, Polθ recently emerged as a novel and potential therapeutic target in HRD tumors [8].
Although the genetic concept of LS seems to be simple, the DDR pathway is composed of intricate mechanisms and genes, making the identification of suitable targets challenging [9]. At the same time, SL interactions that work in one tumor type are sometimes ineffective in another. The findings in preclinical research only provide a potential mechanism to be explored, and whether the Polθ-inhibitors may have clinical relevance in the BRCA-mutated tumors still needs confirmation in the ongoing clinical trials.
In this review, we focused on the expanding landscape of the SL-driven discovery framework, with a focus on Polθ as a novel, targetable, vulnerability in HRD cancers.
From DNA damage to unfunctional DNA repair systems: an itinerary towards synthetic lethality
Cell survival relies on genome integrity and accurate cell function. DNA repair pathways represent an intricate intersection of processes, where specific perturbations can potentially lead to genetic instability. Among DDR pathways, four key mechanisms of DSBs play a central role: HR, the NHEJ, the alt-NHEJ or MMEJ, and the single-strand annealing (SSA) [10] (Table 1). Cancer cells can exploit these DNA repair mechanisms to their advantage, predominantly upregulating alternative repair pathways to maintain the integrity of their DNA: this interchange of pathways can ultimately boost the cancer cells’ survival [11].
This survival strategy is evident in the context of BRCA1/2 mutated cancers, where the loss of one repair pathway is compensated by another, leading to tumoral resilience and progression [12].
However, this compensatory reliance also presents a therapeutic vulnerability, placing the key concept that underlies synthetic lethality. The clinical success of small-molecule inhibitors of PARP in BRCA-mutated cancers demonstrated the success of this approach [13, 14]. In 2005, the identification of synthetic lethal interaction between the loss of BRCA1/2 and the PARP-inhibition, emerged as a new paradigm for the identification of synthetic lethal targets in cancer, going beyond the traditional concept of oncogene addiction, where targeted alterations are druggable oncogenes [14, 15].
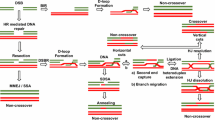
BRCA1/2 function in HR, the repair pathway that utilizes the replicated sister chromatid as a template for precise DSB repair. PARPi effectively impedes the base excision repair (BER) mechanism of DNA single-strand break (SSB), with the consequent transition from SSB to DSB. In patients with BRCA1/2 PV, cancer cells are incapable of completing DSB repair through the high-fidelity HRR, and the use of alternative repair mechanisms results in genomic instability, leading to hypersensitivity to PARP inhibition [16] (Fig. 1).

a In the BRCA-proficient cells the HR repair system is physiologically involved in DNA repair, with DNA damage correctly repaired; b In the BRCA-deficient cells, the absence of efficient homologous recombination, along with the PARP-inhibition, leads to the use of base excision repair (BER) as compensatory pathway, resulting in genomic instability and cell death.
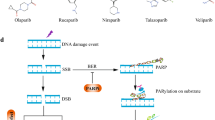
PARPis, including olaparib, niraparib, rucaparib, and talazoparib, have been approved by the FDA as maintenance therapy in ovarian, primary peritoneum, fallopian tube, breast, pancreatic, and prostate cancers that have BRCA1/2 PVs or the HRD status, with differences in therapy indications across the tumor type [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. Unfortunately, the clinical benefit of PARPi is limited by the emergence of acquired or inherent resistance mechanisms in approximately 40–70% of patients [17, 18] (Table 2). Furthermore, the recent expansion of approved PARPis, not restricted to patients carrying BRCA1/2-PVs, produced a strong clinical need to explore a new wave of genetic cancer targets for synthetic lethal interactions [19, 20] (Created with BioRender.com).
Expanding the synthetic lethality paradigm: beyond PARP inhibition in BRCA and p53-deficient tumors
Clinically, the penetrance of the synthetic lethal interactions between BRCA1/2 genes and PARPi is incomplete. Thus, some HRD patients, especially those with advanced tumors, despite the presence of a BRCA PV, showed either de novo or acquired PARPi-resistance [32]. Acquired resistance occurs through multiple mechanisms: from reverse BRCA1/2 mutations, in which the molecules’ function is reacquired and the HR repair system restored, to non-reverse mutations occurring in other aside pathways, like epigenetic modifications and restorations of ADPribosylations, better and efficient drug efflux, ultimately causing the resistance to PARPis [32]. This reversion and non-reversion resistance, not only underscores the extreme adaptability of cancer cells but also highlights the urgency for novel synthetic lethal interaction identification [21]. Furthermore, as our understanding of the complexity of cancer-cell DNA repair networks continues to evolve, numerous gene pairs are being identified as further potential candidates for synthetic lethal therapy.
In this context, several molecules involved in different shelter systems, such as Ataxia-Telangiectasia Mutated (ATM), Enhancer of Zeste Homolog-2 (EZH2), Ataxia Telangiectasia and Rad3-related (ATR), and Checkpoint Kinase 1 (CHK1), have been considered to overcome PARP resistance in BRCA-mutated cancers as well as in other tumors [22].
Among these molecules, EZH2 and ATM, still involved in the HR repair system, emerged. EZH2, is an enzymatic catalytic subunit of polycomb repressive complex 2 (PRC2) that modulates target gene expression via trimethylation of Lys-27 in histone 3 (H3K27me3) [23]. In BRCA-deficient cancers, EZH2 is often found to be overexpressed, which makes it a potential tumor target [23]. Research by Ratz et al. has explored the benefits of combining EZH2 inhibitors, like the GSK126, with chemotherapy agents such as cisplatin, to improve cancer cells’ death, using cell lines derived from BRCA1-deficient and -proficient mouse mammary tumors [23]. The effect of EZH2 was also maximized by ATM, a crucial molecule for DSB repairs, whose inhibition was already found effective in BRCA1-deficient cells [23, 24]. ATM is also involved in the MRN complex, a key HR player. ATM recruitment and activation lead to its target phosphorylation, blocking the cell cycle and initiating DNA repair [25]. Studies have shown that simultaneous inhibition of EZH2 and ATM in BRCA-deficient cells amplifies DNA damage, leading to increased apoptosis and significantly reduced tumor growth. This effect is particularly notable when using GSK126 (EZH2-inhibitor) in combination with AZD1390 (ATM inhibitor), compared to their use alone. Interestingly, cells with functional BRCA genes do not exhibit the same level of cytotoxicity, suggesting an effectiveness of EZH2/ATM inhibition specific to the presence of BRCA1 mutation context [26].
Despite these advances, the lack of clear and significant data on these molecules is still predominant and affects their use in clinical practice to overcome PARPi resistance.
Several studies have highlighted another potential synthetic lethal interaction between ATR and CHK1 within the broader context of p53-mutated cancers. Tumors harboring mutant p53 (mutp53) often are resistant to conventional cancer therapies, tend to progress more rapidly, and generally, have poorer prognosis. Given that mutations in p53 enhance cancer metastasis and proliferation, targeting this signaling pathway might represent a new strategic approach [27]. Tumor cells with p53 mutations are particularly vulnerable to cell cycle arrest in the intra-S and G2 phases, a vulnerability also attributed to checkpoint regulators like ATR. By phosphorylating CHK1, ATR manages the cell cycle and DNA damage response by recognizing specific single-stranded DNA locations [28]. ATR/CHK1 inhibitors, able to block such response, could potentiate the effects of cytotoxic drugs or radiation therapy, increasing tumor cells’ death in p53-deficient scenarios. ATR inhibitors, for instance, stall replication forks and cause chromosomal breakage, sensitizing tumors to DNA-damaging therapies [29]. This is especially effective in cancers experiencing replication stress or with deficient DNA repair mechanisms, such as triple-negative breast cancer, castration-resistant prostate cancer, and other cancers with BRCA1/BRCA2 mutations [30, 31, 33,34,35]. Furthermore, they can resensitize tumor cells to PARP inhibitors when resistance develops, such as in the presence of mutations that overcome BRCA1-mediated DNA repair pathways [32, 36, 37]. Similarly, CHK1 inhibitors target cells under replication stress, amplifying the effects of chemotherapeutic agents like gemcitabine, cisplatin, and camptothecin, enhancing damage from PARP inhibitors [38, 39]. When used with immune checkpoint inhibitors, such as the anti-PD-L1 therapy, they showed synergistic anti-tumor responses, as demonstrated in mouse models of small-cell lung cancer [40]. Currently, clinical trials involving these inhibitors are investigating the optimal timing and dosage for using these inhibitors in combination with both conventional chemotherapies and new therapeutic agents to minimize side effects and maximize treatment efficacy.
Other synthetic lethal interactors of BRCA genes are explored, represented by proteins with physiological functions in DDR pathways, including Fanconi anemia complementation group D2 (FANDC2), radiation sensitive 52 (RAD52), apurinic/apyrimidinic endodeoxyribonuclease 2 (APE2), and Flap structure-specific endonuclease 1 (FEN1) [41]. Among these molecules, Polθ represents an attractive target.
Polθ and BRCA: converging roles in DNA DSB repair
Deficiency in one DNA repair pathway in cancer cells leads to genetic instability and hyper-dependence on compensatory pathways. This dependency represents a key tumor vulnerability that can be exploited to target cancer cells [22].
Polθ is a DNA polymerase, encoded by the POLQ gene, involved in numerous DNA repair pathways, including TMEJ, a backup pathway for DNA repair when NHEJ, HR, or BER are compromised [42]. TMEJ is intrinsically mutagenic. Resected DNA DSBs, which cannot be repaired through HR in BRCA1/2-mutant cancer cells, enter the Polθ-dependent TMEJ, introducing typical sequence alterations represented by the microhomology-flanked deletions and the templated insertions.
Notably, HRR-deficient tumor cells present the characteristic mutational signatures caused by the high mutagenicity of the driving force of the error-prone TMEJ pathway [43]. According to this observation, the loss of HRR proteins, such as BRCA1 or BRCA2, contributes to increasing the TMEJ-specific genomic profile, suggesting synthetic lethal interactions between loss of the POLQ gene and HRR genes like BRCA1/2, ATM, and FANCD2, and resulting in the emerging interest for Polθ as a potential therapeutic target in BRCA1/2-associated tumors.
Interestingly, the similarity between genomic scar signatures of HRD in BRCA-associated tumors and TMEJ genomic scar pattern indicates the hyperactive TMEJ as a mechanism to investigate a potential driver of genomic scar in many cancer types, maybe involved in the lack of full correlation between HRD genomic signatures and functional HRR assays [44, 45].
Moreover, using a CRISPR genetic screen, a recent study identified 140 synthetic lethal genes with Polθ. In this study, 29.7% of breast cancers in the TCGA cohort having genetic alterations in at least one of these 140 Polθ synthetic lethal genes and genomic scar pattern of TMEJ hyperactivity, underlining the potential benefit to target Polθ to improve cancer patients clinical outcomes [41, 44]. Consistent with these results, Polθ-inhibition enhances cell death in HRD cells exposed to cisplatin or PARPi [46]. Therefore, depletion of Polθ using Polθ inhibitor in combination with or after a PARPi or platinum salts has been proposed as a new strategy to treat HRD tumors and to overcome BRCA-gene reversion mutations responsible for therapeutic resistance [22].
Polθ: the synthetic lethal interactions with other DDR-related genes and the effect on innate immunity activation
Although Polθ inhibition is emerging as a strategy to improve the synthetic lethality and cell death in BRCA-mutated tumors, Polθ showed synthetic lethal interactions also with other DNA repair-related genes that control DSB repair and HR.
The first example is the Tp53BP1. BRCA1 functions in HR promoting DNA end resection through inhibition of proteins that inhibit this pathway, such as the Tp53BP1 [47]. Thus, Tp53BP1 loss-of-function enhances DNA end resection, induces end-joining pathway hyperactivation, and Polθ-dependent survival [48]. Defects in the 53BP1/Shieldin DNA repair complex are known sources of PARPi resistance [22]. Therefore, Polθi not only has clinical potential as a therapeutic strategy in BRCA-associated tumors but could also be used to target PARPi resistance [22].
A recent study showed that the type of HR gene mutation had an impact on DNA end resection, and represents a key determinant of TMEJ pathway addiction and the specific response to Polθi. In this study, human isogenic PALB2- and BRCA2-deficient cells, were more sensitive to Polθi compared to BRCA1-deficient cells, with clinical implications for future therapy of mutation-dependent Polθ vulnerabilities [48].
Another study demonstrated that synthetic lethality with Polθ loss required mammalian RAD52 [49]. RAD52 is involved in the HR repair pathway by promoting numerous recombination-mediated repair and replication mechanisms and showed a critical role in cells lacking BRCA1/2 or PALB2 genes. Interestingly, in 53BP1-mutated and BRCA2-deficient cells, RAD52 influenced Polθ inhibition. RAD52 provided toxicity by suppressing DNA synthesis and promoting DNA resections, chromosomal breaks, and micronucleus formation, with subsequent cell death. Instead, a BRCA1-mutated context did not produce similar results. These observations underline the importance of the patient’s genetic background during the treatments for the resistance onset [49, 50]. The mechanistic basis associated with a genotype-dependent influence of Polθ inhibitor mechanisms remains to be clarified.
Some recent studies explored the potential innate immunity activation mediated by Polθ depletion through the cyclic GMP-AMP synthase–stimulator of interferon genes (cGAS-STING) signaling pathway [51]. Polθ inhibition causes the accumulation of cytosolic DNA and activates the cGAS-STING signaling pathway in BRCA2-mutated pancreatic cancers. This enhances the expression of Type I interferon (IFN), the transcription and secretion of several inflammatory cytokines and chemokines, thus increasing the intratumoral T cell infiltration [52]. The preliminary evidence on Polθi and STING agonists synergy represents a potential therapeutic strategy to combine DDR inhibitors and immunotherapy in the population of HR-deficient pancreatic adenocarcinoma [51, 52].
Another two molecules showing synthetic lethal interactions with Polθ are FANCD2 and APE2. FANCD2 acts in the HR, but also in the Fanconi anemia pathway, which repairs the inter-strand DNA crosslinks [53]. FANCD2 also facilitates the Polθ recruitment at the DNA damage sites during TMEJ activation. Preclinical and clinical studies demonstrated a synthetic lethality relationship between Polθ and FANCD2 in several cancers, such as esophageal squamous cell carcinoma [51]. Double knockdown of POLQ and FANCD2 resulted in hypersensitivity to cisplatin in lung cancer cell lines [54], and hypersensitivity to PARP inhibitors in FANCD2-deficient ovarian carcinoma cells while, in vivo, the double knockdown reduced the tumor volumes of xenotransplants of human ovarian tumors [46].
Recent discoveries have highlighted how APE2 nuclease is a key effector of MMEJ repair and it is partly related to the Polθ-mediated repair pathway. The role of APE2 in MMEJ contributes to the addiction of HR-deficient cells to APE2, and this feature could be explored for the treatment of HRD tumors [55].
Another interesting scenario includes Polθ as an adjuvant of radiotherapy. ART558 and, even better, the more recent and stable, ART899 made different tumor cells more susceptible to radiotherapy. The radiosensitivity is favored in S-phase and hypoxic conditions, characteristics predominant in tumor cells, often limiting chemotherapy response and generating resistance development. Evaluation of Polθ in combination with radiotherapy is now under investigation in a clinical trial (NCT04991480). A clinical confirmation would open a new promising therapeutic front, more precise and with less off-target effects [56].
Data provided so far only proved Polθ‘s potential. Despite its intricate nature and the research at a preliminary level, this enzyme could represent a promising target for innovative therapeutic strategy (Fig. 2).

Beyond BRCA-mutated tumors, Polθ showed synthetic lethal interactions also with other DNA repair-related genes that control DSB repair and HR, such as 53BP1, RAD52, PALB2, and FANCD2. Furthermore, the effect of the cGAS-STING immunological and inflammatory pathway, on the MMEJ repair system through APE2, and the synergy with radiotherapy, was recently highlighted (Created with BioRender.com).
Polθ: the role in cancer prognosis and clinical outcomes
Polθ is emerging as a prognostic marker, as well as how potential therapeutic target in BRCA-associated tumors. Polθ begins to show broader implications in tumor biology and cancer patient clinical outcomes [57].
Overexpression of Polθ has been consistently reported across a wide spectrum of cancers, including breast, ovarian, lung, gastric, colorectal, and prostate cancers (Table 3) [57,58,59,60,61]. Conversely, in non-tumor cells, it is expressed at a low level or completely not expressed [62,63,64]. The overexpression was associated with aggressive tumor behavior and adverse clinical prognosis, highlighted by shorter relapse-free survival and overall survival [65].
As previously highlighted, the expression of Polθ is characteristically linked to distinct mutational signatures, predominantly in cancers with HRD, suggesting Polθ upregulation as a compensatory survival mechanism for HR-deficient cancer cells. Intriguingly, Polθ expression is not solely beneficial to HR-deficient cancer cells but appears to offer HR-proficient cells an enhanced ability to survive DNA replication stress, thereby contributing to genomic stability. This multifaceted role of Polθ underlines its prognostic relevance, with overexpression being a marker of poor outcomes in diverse cancer types [66]. In breast cancer, high levels of Polθ are linked with estrogen receptor (ER) negative tumors and higher tumor grades, which are known markers of poor prognosis [58, 67]. Recent studies suggest that Polθ overexpression is associated with an intrinsic mechanism of resistance to genotoxic treatments and radiotherapy. Higgins GS et al. showed that the depletion of Polθ resulted in a marked radio-sensitization in tumor cells contrasting with minimal effects on normal tissue cells, indicating a potential for tumor-specific therapeutic exploitation [58]. Further inquiry into the therapeutic potential of Polθ-inhibition revealed its role in the response to chemotherapeutic agents that generate replicative stress. Studies in ovarian cancer revealed a strong association between Polθ expression and platinum chemotherapy resistance. Lower expression levels of Polθ have been associated with increased chemo-sensitivity, indicating its role as a negative predictor of chemotherapy response [59]. In prostate cancer, Polθ overexpression has been associated with the development of castration-resistant prostate cancer (CRPC) and resistance to olaparib. This resistance identifies Polθ as a critical determinant of treatment outcomes and highlights the enzyme as a candidate for targeted therapy. Targeting Polθ could enhance the effectiveness of treatments like docetaxel and olaparib, making prostate cancer cells more responsive to standard therapy [57, 68].
Recently, several studies have explored the mutational status of other functionally related HR genes, beyond BRCA1/2 and the established HR-associated genes such as ATM, CHEK2, and PALB2. Particularly, the study of D. R. Principe et al. in a large pan-cancer cohort, showed high rates of mutations in many HR-associated genes unexplored in the predictive setting of PARP inhibition, including POLQ, and frequently associated with poor clinical outcomes in tumors like cutaneous melanomas adenocarcinoma and squamous lung carcinoma, gastric, esophageal and head and neck cancers [69]. Therefore, Polθ‘s overexpression and/or POLQ mutation across various malignancies and its correlation with poor clinical outcomes position it as a significant prognostic marker. It plays a pivotal role in cancer cell survival under stress, particularly in HR-deficient contexts, making it an attractive target for therapeutic intervention. The current focus on Polθ inhibitors and their capacity to resensitize cancer cells to standard treatments opens new avenues for cancer therapy, warranting further exploration into their clinical applicability across both HR-proficient and HR-deficient tumors. The therapeutic manipulation of Polθ expression and function, therefore, holds substantial promise for improving the prognosis of cancer patients and overcoming resistance to conventional therapies.
Polθ-inhibitors: a chemical overview
The potential to exploit the synthetic lethal effects of the POLQ/HR genes has been investigated through novel small-molecule inhibitors.
From a structural point of view, Polθ is a multifunctional protein divided into two domains linked by an undefined central region: a DNA polymerase domain (Polθ-pol, residues 1819–2590, Fig. 3a) and a helicase-like domain (Polθ-hel, residues 32–899; Fig. 3a). These domains are essential in the TMEJ process, while the central part plays a regulatory role [70, 71]. In this light, selective inhibition of Polθ-pol and/or Polθ-hel represents a promising efficient strategy in anticancer selective treatment development [22, 72, 73]. In the last decade, several studies led to the identification of different Polθ-hel and/or Polθ-pol inhibitors, and, given the increasing interest at a clinical level, new more powerful, and selective Polθ inhibitors are still being investigated and here highlighted.

a 3D structure of the Polθ-helicase and polymerase domains (PDB code 5AGA; PDB code 6XBU); b 2D-Chemical structures of Polθ-hel inhibitors: size-expanded nucleotides [1]; heteroarylmethylene and acetamido derivatives [2]; ART558, ART812 and RP-6685 [3]; thiazoleurea and heterocyclic substituted urea derivatives [4]. 2D-Chemical structures of Polθ-pol inhibitors: thiadiazole derivatives and novobiocin [5].
Polθ-polymerase Inhibitors
Among the Polθ inhibitors landscape, DNA nucleotide derivatives, first analyzed by Temple University in 2017, are considered the ancient ones. These size-expanded nucleotides recognized by Polθ as their substrates inhibit the polymerase activity. Differently from the physiological ones, these synthetic derivatives present a 4, 5, or 6-membered additional ring modified with different substituents. The incorporation of these two consecutive deoxyribonucleoside monophosphates led to an effective Polθ inhibition, probably due to a polymerase active site distortion [74, 75]. Figure 3b-1 shows the generalized 2D-chemical structure of such size-expanded nucleotides.
Other classes of Polθ-pol inhibitors, presenting a completely different chemical structure, were heteroarylmethylene and acetamido derivatives, both consisted of two (hetero)aromatic groups (Ar1 and Ar2) separated by four atoms (N/C-C-C-X). In some derivatives, the atoms N/C-C are part of a heteroaromatic ring (A3), while in the acetamide derivative, the atoms N-C are part of a tertiary amide. The distance between the two aromatic groups (Ar1 and Ar2) plays a pivotal role in their activity and effectiveness [76, 77]. Figure 3b-2 highlights the 2D-chemical compound structures, which exhibit high and significant affinity, with an IC50 in the range of 0.2–10 μM.
The evolution of these inhibitors arrived in 2021 when Ideaya and collaborators synthesized cyclic acetamido derivatives, in which a nitrogen heterocycle, blocking the stereochemistry of the molecule, was added. Among this new category, compound ART558 (Fig. 3b-3) was extensively investigated, showing the best potency with an IC50 of 7.9 nM, 3 orders of magnitude lower [22, 78]. At the same level ART812 (Fig. 3b-3), in which additional hydroxyl and carbonyl groups increased the metabolic stability. ART812 exhibited good oral bioavailability in both mouse and rat models, as well as moderate-to-high exposure after a single oral dose, a relevant feature for a clinical application. In vivo pharmacodynamic studies determined ART812 target engagement and a twofold increase induction of micronuclei in reticulocytes, a marker of DNA damage, after 4-day treatment at the maximum tolerated dose of 150 mg/kg BID, a result analogous to Polθ loss in knockout mice.
Concurrently, a new series of Polθ-pol inhibitors are reported, such as the RP-6685 (Fig. 3b-3), with an IC50 of 5.8 nM. Structurally, RP-6685 still presents two linked aromatics groups, fundamental for inhibitors’ activity. In this compound, the usual linker N-C( = O)-C-X was shortened by deleting the X atom.
A very high affinity was also reported with thiazoleurea and heterocyclic substituted urea derivatives. These derivatives, structurally different, are characterized by the presence of a central urea moiety connected to thiazole/pyrazine [79, 80]. Such compounds (Fig. 3b-4) with a Polθ IC50 of 7 nM and 2 nM, respectively, lead to a strong and effective affinity.
Polθ-helicase inhibitors
To enhance Polθ effective inhibition, many efforts, even though not comparable to the Polθ-pol subunit, have been made also in Polθ-hel inhibitors research. Among them, thiadiazolyl derivatives, characterized by the presence of a central N-thiadiazolyl acetoamide group with a methoxyl side, stand out. Here, two aromatic groups are linked to the acetoamide methoxy moieties. Figure 3b-5 shows an example of such compounds, which possess an IC50 < 200 nM [81, 82].
Still, in this scenario, the 2-oxo-2H-chromene, naphthalene, and quinoline derivatives are of particular interest [83]. Among the synthesized compounds, Novobiocin (NVB) (Fig. 3b-5), an antibiotic derived from streptomyces, is the only one already studied. In vivo studies have shown that the use of NVB reduces tumor growth in genetically modified mice with a BRCA1 deficiency, and also increases the survival of tumor-bearing mice [72]. Currently, a clinical trial with NVB is now ongoing.
Over the last decade, chemical research on Polθ-inhibitors has progressed beyond the initial discovery phase, identifying always new promising candidates as potential anticancer drugs.
Polθ-Inhibitors in the clinical setting: an overview of the ongoing phase I/II trials
Breakthroughs on Polθ, already led to the same phase I/II clinical trials. These trials are ongoing and will play the role of critical milestones in bringing Polθ inhibitors from preclinical promises to clinical applications, addressing essential aspects such as efficacy, safety, and potential benefits for cancer patients (Table 4).
The NCT05898399 is a Phase I/II trial enrolling up to 250 patients with advanced or metastatic cancer. It explores the combination of ART6043 with olaparib or talazoparib, both oral PARPi. The study was designed in four different parts: part A1 (ART6043 as monotherapy), part A2 and A3 (ART6043 in combination with olaparib or talazoparib, respectively), and part B (ART6043 in combination with a PARPi or a PARPi alone). Participants are treated with these drugs in 21-day cycles. Primary outcome measures included the number of participants with dose-limiting toxicities (DLTs) and progression-free survival (PFS). Secondary outcome measures focus on the number of participants with adverse events, best overall response (BOR), objective response rate (ORR), disease control rate (DCR), duration of response (DOR), change in tumor size, and change in cancer antigen 125 (CA-125) levels.
The NCT04991480 is a Phase I/II trial evaluating the efficacy of drug ART4215 in 390 estimated patients with advanced or metastatic solid tumors, specifically focusing on safe and recommended doses, either alone and in combination with talazoparib or niraparib. This study aims to understand the side effects and effectiveness of ART4215 in these settings. The trial includes various experimental parts, such as part B2 for participants with solid cancers showing sensitivity to Polθ inhibition and part B3 for participants with HER2-negative BRCA breast cancer patients. Primary outcome measures include the number of participants with DLTs and PFS, while secondary outcomes focus on BOR, ORR, DCR, DOR, and changes in tumor size.
The NCT06077877 was a Phase I/II study designed to evaluate GSK4524101 administration, alone or in combination with niraparib, for the treatment of 135 patients with advanced solid tumors. It is structured in two parts: part 1 focuses on the food effect of GSK4524101 and part 2 combines GSK4524101 with niraparib. The primary outcome measures for part 1 include the number of participants with DLTs, treatment-emergent adverse events (AEs), serious adverse events (SAEs), and the percentage of participants who receive all planned doses. In part 2, the confirmed ORR is the key outcome. Secondary outcomes involve pharmacokinetic parameters like Area Under Curve (AUC) and maximum Concentration (Cmax) of GSK4364973, a metabolite of GSK4524101, and plasma concentration of niraparib.
Finally, the NCT05687110 (Phase I, National Cancer Institute, NCI) trial, was designed to evaluate the safe and optimal novobiocin dose in up to 30 estimated patients with solid tumors characterized by DNA repair gene alteration. Participants are receiving novobiocin sodium and are undergoing procedures like tumor biopsy, biospecimen collection, and medical imaging scans. The primary outcome measure is the maximum tolerated dose (MTD) and recommended phase 2 dose for continuous Novobiocin administration. Secondary outcome measures include plasma concentrations of Novobiocin and biological effectiveness, defined as an increase in RAD51-foci positive cells. Other outcome measures focus on Polθ mRNA levels and ATM immunohistochemistry (IHC).
Each of the trials encounters typical challenges of early-phase research, such as difficulties in patient recruitment and the necessity for accurate patient selection. Additionally, potential side effects and the determination of the optimal dosage for clinical use are still in need of careful evaluation. These challenges underscore the complexities of translating preclinical promising molecular targets into effective clinical therapies, emphasizing the need for cautious optimism.
Emerging data from preclinical studies are providing hopeful signs that these ongoing trials may confirm. The current trials on Polθ inhibitors, particularly in combination with PARPi such as olaparib, niraparib, and talazoparib, highlight an emerging trend in cancer treatment. This strategy aims to discover synergistic effects that could improve the overall effectiveness of these therapies. Notably, the use of Polθ inhibitors has the potential to be effective, in a clinical setting, not just in treating cancers with BRCA-gene defects but also in overcoming resistance to PARPi, which often occurs due to reversion mutations [22, 72].
Looking forward, the outcomes of these trials are anticipated to significantly shape the future of Polθ inhibitor research. If successful, they could pave the way for expanded investigations into Polθi across various cancer types, validating their clinical utility. Furthermore, positive results may encourage the exploration of combination therapies, refining patient selection criteria, and potentially positioning Polθi as a standard component in cancer treatment protocols.
Conclusion
The most remarkable finding in synthetic lethality is the hypersensitivity to PARPis and the benefit in clinical outcomes of HRD-associated cancer patients, mainly ovarian, breast, and prostate cancer patients. Approximately 50% of HRD tumors show innate or acquired drug resistance to PARPis, and overcoming the resistance mechanism is one of the major challenges of clinical research in the BRCA and other non-BRCA, HR-associated, tumors.
Polθ is largely explored as a drug target and potential synthetic lethal partner of a novel form of SL. Exploiting the central role of Polθ in TMEJ, and the emerging link between TMEJ, Polθ, and BRCA-gene reversion mutations, Polθ inhibitors seem to have clinical potential not only in targeting HRD-defective tumors but also to prevent or delay the onset of PARPi resistance.
Encouragingly, although the clinical trials exploring these Polθ/HR-genes interaction are only at an early stage, preliminary reports demonstrated that the inhibition of Polθ is a promising cancer treatment strategy, and identify Polθ as emerging target for rational combinations with PARP inhibitors.
In addition, Polθ showed synthetically lethal interactions with other DDR genes, such as RAD52 controlling in SSA, the ATM, ATR, and FANCD2 genes working in the HR pathway, and TP53BP1 involved NHEJ, expanding the landscape for future clinical applications. Other pleiotropic effects, like the improved radio-sensitization of p53-mutated tumor cells, and the new role in the tumor immune environment through the cGAS-STING signaling pathway activation, pave the way for the development of unexplored synthetic lethality strategies. Further work is needed to optimize the therapeutic window, bridging the genetic and molecular insights to oncological clinical practice.
References
Russo A, Incorvaia L, Malapelle U, Del Re M, Capoluongo E, Vincenzi B, et al. The tumor-agnostic treatment for patients with solid tumors: a position paper on behalf of the AIOM- SIAPEC/IAP-SIBioC-SIF Italian Scientific Societies. Crit Rev Oncol Hematol. 2021;165:103436.
O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18:613–23.
Gori S, Barberis M, Bella MA, Buttitta F, Capoluongo E, Carrera P, et al. Recommendations for the implementation of BRCA testing in ovarian cancer patients and their relatives. Crit Rev Oncol Hematol. 2019;140:67–72.
Li S, Topatana W, Juengpanich S, Cao J, Hu J, Zhang B, et al. Development of synthetic lethality in cancer: molecular and cellular classification. Signal Transduct Target Ther. 2020;5:241.
Bhamidipati D, Haro-Silerio JI, Yap TA, Ngoi N. PARP inhibitors: enhancing efficacy through rational combinations. Br J Cancer. 2023;129:904–16.
Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, et al. Essential roles for polymerase θ-mediated end joining in the repair of chromosome breaks. Mol Cell. 2016;63:662–73.
Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–7.
Schrempf A, Bernardo S, Arasa Verge EA, Ramirez Otero MA, Wilson J, Kirchhofer D, et al. POLθ processes ssDNA gaps and promotes replication fork progression in BRCA1-deficient cells. Cell Rep 2022;41:111716.
Russo A, Incorvaia L, Capoluongo E, Tagliaferri P, Galvano A, Del Re M, et al. The challenge of the molecular tumor board empowerment in clinical oncology practice: a position paper on behalf of the AIOM- SIAPEC/IAP-SIBioC-SIC-SIF-SIGU-SIRM Italian Scientific Societies. Crit Rev Oncol Hematol. 2022;169:103567.
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204.
Lee JK, Choi YL, Kwon M, Park PJ. Mechanisms and consequences of cancer genome instability: lessons from genome sequencing studies. Annu Rev Pathol. 2016;11:283–312.
Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64.
Curtin NJ, Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov. 2020;19:711–36.
Russo A, Incorvaia L, Capoluongo E, Tagliaferri P, Gori S, Cortesi L, et al. Implementation of preventive and predictive BRCA testing in patients with breast, ovarian, pancreatic, and prostate cancer: a position paper of Italian Scientific Societies. ESMO Open. 2022;7:100459.
Ryan CJ, Mehta I, Kebabci N, Adams DJ. Targeting synthetic lethal paralogs in cancer. Trends Cancer. 2023;9:397–409.
Li H, Liu ZY, Wu N, Chen YC, Cheng Q, Wang J. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer. 2020;19:107.
Kim D, Nam HJ. PARP inhibitors: clinical limitations and recent attempts to overcome them. Int J Mol Sci. 2022;23:8412.
Incorvaia L, Passiglia F, Rizzo S, Galvano A, Listì A, Barraco N, et al. “Back to a false normality”: new intriguing mechanisms of resistance to PARP inhibitors. Oncotarget. 2017;8:23891–904.
Bono M, Fanale D, Incorvaia L, Cancelliere D, Fiorino A, Calò V, et al. Impact of deleterious variants in other genes beyond BRCA1/2 detected in breast/ovarian and pancreatic cancer patients by NGS-based multi-gene panel testing: looking over the hedge. ESMO Open. 2021;6:100235.
Fanale D, Incorvaia L, Filorizzo C, Bono M, Fiorino A, Calò V, et al. Detection of germline mutations in a cohort of 139 patients with bilateral breast cancer by multi-gene panel testing: impact of pathogenic variants in other genes beyond BRCA1/2. Cancers. 2020;12:2415.
Groelly FJ, Fawkes M, Dagg RA, Blackford AN, Tarsounas M. Targeting DNA damage response pathways in cancer. Nat Rev Cancer. 2023;23:78–94.
Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun. 2021;12:3636.
Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–9.
Puppe J, Opdam M, Schouten PC, Jóźwiak K, Lips E, Severson T, et al. EZH2 is overexpressed in BRCA1-like breast tumors and predictive for sensitivity to high-dose platinum-based chemotherapy. Clin Cancer Res. 2019;25:4351–62.
Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J Clin Invest. 2021;131:e139333.
Ratz L, Brambillasca C, Bartke L, Huetzen MA, Goergens J, Leidecker O, et al. Combined inhibition of EZH2 and ATM is synthetic lethal in BRCA1-deficient breast cancer. Breast Cancer Res. 2022;24:41.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008.
Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27.
Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–7.
Tu X, Kahila MM, Zhou Q, Yu J, Kalari KR, Wang L, et al. ATR inhibition is a promising radiosensitizing strategy for triple-negative breast cancer. Mol Cancer Ther. 2018;17:2462–72.
Tang Z, Pilié PG, Geng C, Manyam GC, Yang G, Park S, et al. ATR inhibition induces CDK1-SPOP signaling and enhances anti-PD-L1 cytotoxicity in prostate cancer. Clin Cancer Res. 2021;27:4898–909.
Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31:318–32.
Thomas A, Takahashi N, Rajapakse VN, Zhang X, Sun Y, Ceribelli M, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell. 2021;39:566–79.e7.
El Touny LH, Hose C, Connelly J, Harris E, Monks A, Dull AB, et al. ATR inhibition reverses the resistance of homologous recombination deficient MGMT. Oncotarget. 2021;12:2114–30.
Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, et al. The ATR inhibitor AZD6738 synergizes with gemcitabine. Mol Cancer Ther. 2018;17:1670–82.
Nam AR, Yoon J, Jin MH, Bang JH, Oh KS, Seo HR, et al. ATR inhibition amplifies antitumor effects of olaparib in biliary tract cancer. Cancer Lett. 2021;516:38–47.
Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7:76534–50.
Massey AJ, Stephens P, Rawlinson R, McGurk L, Plummer R, Curtin NJ. mTORC1 and DNA-PKcs as novel molecular determinants of sensitivity to Chk1 inhibition. Mol Oncol. 2016;10:101–12.
Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest. 2012;122:1541–52.
Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–61.
Patel PS, Algouneh A, Hakem R. Exploiting synthetic lethality to target BRCA1/2-deficient tumors: where we stand. Oncogene. 2021;40:3001–14.
Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci USA. 2013;110:7720–5.
Schrempf A, Slyskova J, Loizou JI. Targeting the DNA repair enzyme polymerase θ in cancer therapy. Trends Cancer. 2021;7:98–111.
Feng W, Simpson DA, Carvajal-Garcia J, Price BA, Kumar RJ, Mose LE, et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat Commun. 2019;10:4286.
Incorvaia L, Perez A, Marchetti C, Brando C, Gristina V, Cancelliere D, et al. Theranostic biomarkers and PARP-inhibitors effectiveness in patients with non-BRCA associated homologous recombination deficient tumors: still looking through a dirty glass window? Cancer Treat Rev. 2023;121:102650.
Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518:258–62.
Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54.
Krais JJ, Glass DJ, Chudoba I, Wang Y, Feng W, Simpson D, et al. Genetic separation of Brca1 functions reveal mutation-dependent Polθ vulnerabilities. Nat Commun. 2023;14:7714.
Ronson GE, Starowicz K, Anthony EJ, Piberger AL, Clarke LC, Garvin AJ, et al. Mechanisms of synthetic lethality between BRCA1/2 and 53BP1 deficiencies and DNA polymerase theta targeting. Nat Commun. 2023;14:7834.
Belan O, Sebald M, Adamowicz M, Anand R, Vancevska A, Neves J, et al. POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Mol Cell. 2022;82:4664–80.e9.
Li J, Ko JM, Dai W, Yu VZ, Ng HY, Hoffmann JS, et al. Depletion of DNA polymerase theta inhibits tumor growth and promotes genome instability through the cGAS-STING-ISG pathway in esophageal squamous cell carcinoma. Cancers. 2021;13:3204.
Oh G, Wang A, Wang L, Li J, Werba G, Weissinger D, et al. POLQ inhibition elicits an immune response in homologous recombination-deficient pancreatic adenocarcinoma via cGAS/STING signaling. J Clin Invest. 2023;133:e165934.
Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49.
Dai CH, Chen P, Li J, Lan T, Chen YC, Qian H, et al. Co-inhibition of pol θ and HR genes efficiently synergize with cisplatin to suppress cisplatin-resistant lung cancer cells survival. Oncotarget. 2016;7:65157–70.
Fleury H, MacEachern MK, Stiefel CM, Anand R, Sempeck C, Nebenfuehr B, et al. The APE2 nuclease is essential for DNA double-strand break repair by microhomology-mediated end joining. Mol Cell. 2023;83:1429–45.e8.
Rodriguez-Berriguete G, Ranzani M, Prevo R, Puliyadi R, Machado N, Bolland HR, et al. Small-molecule Polθ inhibitors provide safe and effective tumor radiosensitization in preclinical models. Clin Cancer Res. 2023;29:1631–42.
Kuei CH, Lin HY, Lin MH, Lee HH, Lin CH, Lee WJ, et al. DNA polymerase theta repression enhances the docetaxel responsiveness in metastatic castration-resistant prostate cancer. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165954.
Higgins GS, Harris AL, Prevo R, Helleday T, McKenna WG, Buffa FM. Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget. 2010;1:175–84.
Long J, Zhu JY, Liu YB, Fu K, Tian Y, Li PY, et al. Helicase POLQ-like (HELQ) as a novel indicator of platinum-based chemoresistance for epithelial ovarian cancer. Gynecol Oncol. 2018;149:341–9.
Smolinska A, Singer K, Golchert J, Smyczynska U, Fendler W, Sendler M, et al. DNA polymerase theta plays a critical role in pancreatic cancer development and metastasis. Cancers. 2022;14:4077.
Allera-Moreau C, Rouquette I, Lepage B, Oumouhou N, Walschaerts M, Leconte E, et al. DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis. 2012;1:e30.
Kawamura K, Bahar R, Seimiya M, Chiyo M, Wada A, Okada S, et al. DNA polymerase theta is preferentially expressed in lymphoid tissues and upregulated in human cancers. Int J Cancer. 2004;109:9–16.
Pillaire MJ, Selves J, Gordien K, Gourraud PA, Gentil C, Danjoux M, et al. A ‘DNA replication’ signature of progression and negative outcome in colorectal cancer. Oncogene. 2010;29:876–87.
Pismataro MC, Astolfi A, Barreca ML, Pacetti M, Schenone S, Bandiera T, et al. Small molecules targeting DNA polymerase theta (POLθ) as promising synthetic lethal agents for precision cancer therapy. J Med Chem. 2023;66:6498–522.
Wood RD, Doublié S. DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair. 2016;44:22–32.
Goullet de Rugy T, Bashkurov M, Datti A, Betous R, Guitton-Sert L, Cazaux C, et al. Excess Polθ functions in response to replicative stress in homologous recombination-proficient cancer cells. Biol Open. 2016;5:1485–92.
Lemée F, Bergoglio V, Fernandez-Vidal A, Machado-Silva A, Pillaire MJ, Bieth A, et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc Natl Acad Sci USA. 2010;107:13390–5.
Nappi L, Mazurek S, Khazamipour N, Janfaza S, Jia A, Ozcan G, et al. 21P polymerase theta inhibition in homologous recombination-deficient prostate cancer. ESMO Open. 2024;9.
Principe DR, Narbutis M, Koch R, Rana A. Frequency and prognostic value of mutations associated with the homologous recombination DNA repair pathway in a large pan cancer cohort. Sci Rep. 2020;10:20223.
Seki M, Marini F. Wood RDPOLQ (Pol theta), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 2003;31:6117–26.
Black SJ, Ozdemir AY, Kashkina E, Kent T, Rusanov T, Ristic D, et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat Commun. 2019;10:4423.
Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat Cancer. 2021;2:598–610.
Bubenik M, Mader P, Mochirian P, Vallée F, Clark J, Truchon JF, et al. Identification of RP-6685, an orally bioavailable compound that inhibits the DNA polymerase activity of Polθ. J Med Chem. 2022;65:13198–215.
Kent T, Rusanov TD, Hoang TM, Velema WA, Krueger AT, Copeland WC, et al. DNA polymerase θ specializes in incorporating synthetic expanded-size (xDNA) nucleotides. Nucleic Acids Res. 2016;44:9381–92.
Pomerantz, RT, Kool ET. Compositions and methods of treatment using expanded-size DNA analogs. World Patent WO2018035410. 2018.
Beck HP, Dillon M, Jones B, Martinez LP. Heteroarylmethylene derivatives as DNA polymerase theta inhibitors. World Patent WO2020160213. 2020.
Beck HP, Jones BT, Martinez LP. Acetamido-amino and acetamido-sulfur derivatives as DNA polymerase theta inhibitors. World Patent WO2022026548. 2022.
Stockley ML, Ferdinand A, Benedetti G, Blencowe P, Boyd SM, Calder M, et al. Discovery, characterization, and structure-based optimization of small-molecule in vitro and in vivo probes for human DNA polymerase theta. J Med Chem. 2022;65:13879–91.
Blencowe P, Charles M, Ekwuru T, MacDonald E, McCarron H, Rigoreau L. Thiazoleureas as anticancer agents. World Patent WO2020030924. 2020.
Blencowe P, Charles M, Cridland A, Ekwuru T, Heald R, MacDonald E, et al. Heterocyclic substituted ureas, for use against cancer. World Patent WO2020030925. 2020.
Barsanti PA, Beck HP, Fleury M, Jones BT, McSpadden ED, Pei Z, et al. Substituted thiadiazolyl derivatives as DNA polymerase theta inhibitors. World Patent WO2022118210. 2022.
Barsanti PA, Beck HP, Fleury M, Knox JE, McSpadden ED, Jones BT, et al. Thiadiazolyl derivatives as DNA polymerase theta inhibitors. World Patent WO2020243459. 2020.
D’Andrea A, Blagg BSJ, Davis RE, Zhou J. Compounds and methods for treating cancer. World Patent WO2021046220. 2021.
Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–31.
Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583.
Caldecott KW. DNA single-strand break repair. Exp Cell Res. 2014;329:2–8.
Jiricny J. Postreplicative mismatch repair. Cold Spring Harb Perspect Biol. 2013;5:a012633.
Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46.
Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–70.
Jeggo PA, Löbrich M. DNA double-strand breaks: their cellular and clinical impact? Oncogene. 2007;26:7717–9.
Mladenov E, Mladenova V, Stuschke M, Iliakis G. New facets of DNA double strand break repair: radiation dose as key determinant of HR versus c-NHEJ engagement. Int J Mol Sci. 2023;24:14956.
Perrault R, Wang H, Wang M, Rosidi B, Iliakis G. Backup pathways of NHEJ are suppressed by DNA-PK. J Cell Biochem. 2004;92:781–94.
Wang H, Perrault AR, Takeda Y, Qin W, Iliakis G. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2003;31:5377–88.
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl J Med. 2019;381:317–27.
Kindler HL, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Overall survival results from the POLO trial: a phase III study of active maintenance olaparib versus placebo for germline BRCA-mutated metastatic pancreatic cancer. J Clin Oncol. 2022;40:3929–39.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-Resistant Prostate Cancer. N. Engl J Med. 2020;382:2091–102.
Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N. Engl J Med. 2021;384:2394–405.
Geyer CE, Garber JE, Gelber RD, Yothers G, Taboada M, Ross L, et al. Overall survival in the OlympiA phase III trial of adjuvant olaparib in patients with germline pathogenic variants in BRCA1/2 and high-risk, early breast cancer. Ann Oncol. 2022;33:1250–68.
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl J Med. 2017;377:523–33.
Robson ME, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. OlympiAD extended follow-up for overall survival and safety: olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Eur J Cancer. 2023;184:39–47.
Litton JK, Hurvitz SA, Mina LA, Rugo HS, Lee KH, Gonçalves A, et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: final overall survival results from the EMBRACA trial. Ann Oncol. 2020;31:1526–35.
González-Martín A, Pothuri B, Vergote I, Graybill W, Lorusso D, McCormick CC, et al. Progression-free survival and safety at 3.5years of follow-up: results from the randomised phase 3 PRIMA/ENGOT-OV26/GOG-3012 trial of niraparib maintenance treatment in patients with newly diagnosed ovarian cancer. Eur J Cancer. 2023;189:112908.
Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381:2416–28.
Ray-Coquard I, Leary A, Pignata S, Cropet C, González-Martín A, Marth C, et al. Olaparib plus bevacizumab first-line maintenance in ovarian cancer: final overall survival results from the PAOLA-1/ENGOT-ov25 trial. Ann Oncol. 2023;34:681–92.
Del Campo JM, Matulonis UA, Malander S, Provencher D, Mahner S, Follana P, et al. Niraparib maintenance therapy in patients with recurrent ovarian cancer after a partial response to the last platinum-based chemotherapy in the ENGOT-OV16/NOVA trial. J Clin Oncol. 2019;37:2968–73.
Mirza MR, Benigno B, Dørum A, Mahner S, Bessette P, Barceló IB, et al. Long-term safety in patients with recurrent ovarian cancer treated with niraparib versus placebo: results from the phase III ENGOT-OV16/NOVA trial. Gynecol Oncol. 2020;159:442–8.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–61.
Banerjee S, Moore KN, Colombo N, Scambia G, Kim BG, Oaknin A, et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22:1721–31.
DiSilvestro P, Banerjee S, Colombo N, Scambia G, Kim BG, Oaknin A, et al. Overall survival with maintenance olaparib at a 7-year follow-up in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation: the SOLO1/GOG 3004 trial. J Clin Oncol. 2023;41:609–17.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87.
Rao X, Xing B, Wu Z, Bin Y, Chen Y, Xu Y, et al. Targeting polymerase θ impairs tumorigenesis and enhances radiosensitivity in lung adenocarcinoma. Cancer Sci. 2023;114:1943–57.
Shinmura K, Kato H, Kawanishi Y, Yoshimura K, Tsuchiya K, Takahara Y, et al. POLQ overexpression is associated with an increased somatic mutation load and PLK4 overexpression in lung adenocarcinoma. Cancers. 2019;11:722.
Acknowledgements
L.I., G.B., and A.R. were supported by the Piano Nazionale di Ripresa e Resilienza (PNRR) project - Italian Network of Excellence for advanced diagnosis (INNOVA); PNC-E3-2022-23683266; PNC-HLS-DA (C43C22001630001).
Author information
Authors and Affiliations
Contributions
TD Bazan Russo, C Mujacic, E Di Giovanni, MC Vitale, C Ferrante Bannera, U Randazzo, S Contino, M Bono, V Gristina, A Galvano, A Perez, writing – original draft; G Badalamenti, A Russo and V Bazan, conceptualization, and supervision; L Incorvaia, conceptualization, writing – original draft, and supervision. The manuscript has been read and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bazan Russo, T.D., Mujacic, C., Di Giovanni, E. et al. Polθ: emerging synthetic lethal partner in homologous recombination-deficient tumors. Cancer Gene Ther (2024). https://doi.org/10.1038/s41417-024-00815-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41417-024-00815-2
- Springer Nature America, Inc.