
Abstract
The extent to which transcription factors read and respond to specific information content within short DNA sequences remains an important question that the tumor suppressor p53 is helping us answer. We discuss recent insights into how local information content at p53 binding sites might control modes of p53 target gene activation and cell fate decisions. Significant prior work has yielded data supporting two potential models of how p53 determines cell fate through its target genes: a selective target gene binding and activation model and a p53 level threshold model. Both of these models largely revolve around an analogy of whether p53 is acting in a “smart” or “dumb” manner. Here, we synthesize recent and past studies on p53 decoding of DNA sequence, chromatin context, and cellular signaling cascades to elicit variable cell fates critical in human development, homeostasis, and disease.
Similar content being viewed by others


Facts
-
Transcription factors contain DNA binding domains that enable them to recognize and bind short DNA sequences.
-
Sequence content and context in gene regulatory elements, and how that influences transcription factor occupancy, plays a crucial role in development and disease.
-
p53 is a model transcription factor, due in part to decades of investigation in the context of tumor suppression and cancer and more recently as a critical barrier to successful gene editing approaches.
-
Similar to other transcription factors, p53 binding to the genome is mediated by its DNA recognition motif, the p53 response element (p53RE), and is affected by DNA shape, chromatin state, and co-factors [1, 2].
-
p53 controls cell fate after DNA damage and other cellular insults, such as the decision to undergo apoptosis or to temporarily or permanently arrest the cell cycle, through its ability to regulate gene expression.
Open Questions
-
How do transcription factors like p53 “choose” which genomic binding sites to occupy, which genes to regulate, and ultimately, what cell fate is the best outcome at the cellular and organismal level?
-
How do DNA sequence, DNA shape, chromatin structure, p53 binding affinity and kinetics, and other non-DNA information like cell and tissue context affect p53-dependent transcription and cell fate?
-
How do other transcription factors and co-factors support or impede p53 activities on DNA and what influence do these factors have on tumor suppression and other critical p53-dependent activities?
-
How can we best modulate a cell’s apoptotic threshold to make p53 activating cancer treatment strategies more successful?
Introduction
Cell fate decisions by “smart” and “dumb” p53
The tumor suppressor p53 is best known as a transcription factor that uses its target genes to control cell fate decisions in response to cellular stress [3]. The regulation of p53 revolves around the E3-ubiquitinase MDM2, which binds p53 and leads to its proteasomal degradation in unstressed cells. Upon stress, p53 is post-translationally modified to block its interaction with MDM2, leading to elevated p53 levels [4] (Fig. 1A). Downstream of the numerous pathways that can be driven by p53, cell cycle arrest and apoptosis are arguably the most prominent fates a cell can arrive at. Thus, p53 can control the fate of a cell to survive or die in response to stress. Given the critical importance of cellular life-or-death decisions to anti-cancer strategies, therapeutic genome editing approaches, and our expanding view of aberrant p53 activity in developmental disorders, a large number of studies have addressed the question of how the p53 signaling pathway makes this ultimate decision about a cell’s life or death [5]. Similar to most of these studies, this Review focuses on the role of full-length p53 in mammals. Many p53 target genes have been associated with different pathways and cell fates [6], such as p21 (also known as CDKN1A), which promotes cell cycle arrest [7], and puma (BBC3), which stimulates apoptosis [8, 9]. The specific associations between target genes and cell fates prompted Karen Vousden, in a review article published in 2000, to ask whether p53 is “smart” and can selectively activate specific target genes to achieve a desired cell fate, or whether p53 is rather “dumb” and tries to activate all of its target genes, leaving the ultimate cell fate decisions to other signaling cues [10]. The distinction between “smart” and “dumb” p53 serves as an analogy for our understanding of p53 functionality.

A p53 is kept inactive via MDM2-mediated E3 ligase activity, which itself is inhibited by stress-dependent post-translational modifications (PTMs) to p53. In the absence of MDM2-mediated degradation, p53 protein levels rise and p53 activates transcription of a broad gene network and dictates cell fate. B The “smart” model suggests that differential binding kinetics of p53 to target gene regulatory sequences dictates cell fate decisions by p53. In this model, intrinsic sequence characteristics (DNA sequence, shape) or modulation of p53 protein activity (PTMs, co-factor binding) drive p53-mediated transcription of selective genes controlling distinct cell fates. C The “dumb” model suggests that p53 protein abundance dictates p53-dependent cell fates. In this model, p53 binds to all permissive p53REs and broadly activates all target genes. As p53 protein levels rise, interactions between p53 and cell type and condition-dependent factors ultimately determine cell fates.
“smart” p53
For many years, p53 seemed rather “smart” to most researchers in the field. For example, post-translational modifications (PTMs) [11,12,13] and co-factors [14, 15] were identified that could help p53 to select specific target genes and ultimately tip the balance of p53-dependent cell fate decisions from survival to death and vice versa. A particularly intriguing concept, at least from a genomic perspective, suggests that differences in p53 response elements (p53REs) result in differential DNA binding affinity of p53 and varying activation kinetics of the associated target genes [16], which may contribute to p53 favoring target genes linked to cell cycle arrest over those linked to apoptosis [17, 18] (Fig. 1B). Taken together, it is thought that “smart” p53 decodes different signals that a cell generates in response to various stress conditions, and then p53 scans the genome for DNA recognition sites that encode for specific target gene activation and cell fates.
“dumb” p53
What if p53 was “dumb”? The strength of the p53RE, i.e., the predicted affinity for p53 binding, has been found to have very limited correlation with biological function [19]. Consistent with these data, p53 binds to most of its canonical target genes relatively invariant across cell types and tissues [20,21,22,23]. Furthermore, p53 activates most of its target genes at the same time, and cells undergo apoptosis when a cell type- and condition-dependent threshold of p53 levels is reached [24, 25] (Fig. 1C). In summary, “dumb” p53 welcomes any activation signal that increases its abundance and subsequently binds to any accessible p53RE, where it attempts to recruit Pol II to induce transcription.
The terms “smart” and “dumb” succinctly describe the information content that is present in various upstream signaling cues as well as at different levels of the p53 signaling cascade. In the following sections, we will explore our current understanding of whether p53 behaves “smart” or “dumb”, with an initial focus on target recognition and ending with cell fate decisions.
Information content of p53REs
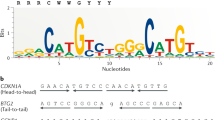
Through its DNA binding domain, p53 binds a DNA sequence motif known as the p53RE. The canonical p53RE is composed of two decameric half-sites with the consensus sequence RRRCWWGYYY (R = A/G, W = A/T, Y = C/T). Occasionally, multiple p53REs exist in the same regulatory element such as in MDM2 [26] or there exist multiple regulatory elements containing a p53RE such as in CDKN1A [27] and GDF15 [28]. Both of these arrangements may lead to combinatorial activity of p53 and higher target gene expression. The specific p53RE DNA sequence is variable and thus provides a means to encode critical information. This information is critical in enabling p53 to act “smart”. The information content of p53REs has been central to several models of p53 function. For example, it had been suggested that sequence differences in p53REs might determine whether p53 induces or represses transcription at a given locus [29, 30], but these models were later shown to be inconsistent with genome-wide data demonstrating that p53 is only associated with transcriptional activation at its binding sites [6, 31,32,33,34,35]. In general, the mechanisms that lead to the activation or repression of many genes upon p53 activation still require more in-depth study.
To make complex cell fate decisions through differential activation of target genes, the DNA sequence of a p53RE must encode information about its associated gene that p53 can effectively decode (Fig. 1B). Indeed, an early model proposed that p53REs direct p53 to regulate specific targets, such as cell cycle arrest or pro-apoptotic targets, through sequence-specific binding affinities and subsequent activation kinetics [16,17,18, 36]. Large-scale binding data from ChIP-seq experiments show a positive correlation between the occurrence of p53 binding and a p53RE’s similarity to the consensus sequence [37], which provides evidence that the DNA sequence of a p53RE can indeed influence p53 binding. Consistent with these data, machine learning models of DNA sequence features predict that p53-mediated activation is best described by its similarity to the consensus sequence of the p53RE [38, 39]. However, higher affinity binding sites do not always predict higher transcriptional output [40]. An early genome-wide analysis of p53RE DNA sequences and their predicted affinity for p53 binding revealed only a very limited correlation with the biological pathways of the associated genes [19]. However, a number of recent studies have provided new evidence that p53REs may encode high information content that can help p53 make decisions. Based on an evaluation of 250 p53REs, the authors of one study found that the variable DNA shape caused by the specific DNA sequence within p53REs may allow p53 to interpret directionality [41], i.e., p53 decodes whether to send Pol II complexes up- or downstream of the p53RE. Two other studies revived the early model in which p53REs instruct p53 to regulate cell cycle arrest or pro-apoptotic targets. While Qian et al. [17] did not identify the specific DNA information that causes p53 to favor cell cycle-associated p53REs over apoptosis-associated p53REs, the two recent studies differ markedly in the specific DNA features they identified that cause the different activation kinetics. One study reported that specific base-pair changes cause differences in the width of the minor or major DNA groove, with narrower minor grooves resulting in higher p53 binding affinity and faster target gene activation [42]. The second study found that the p53RE sequence affects DNA torsional flexibility, with high flexibility allowing for more robust p53 binding and thus faster target gene activation [43]. However, another recent study demonstrated that the DNA shape of p53REs does not generally predict its activity [39]. Although the authors have identified different mechanisms of action, they support a model in which p53 acts “smart” by decoding information from p53REs to differentially activate target genes associated with different biological functions.
Many genomic DNA fragments with p53REs have been evaluated using MPRAs (massively parallel reporter assays). Studies have used DNA fragments that bind p53 [28, 38, 44], have enhancer potential [45], or that represent essentially the entire human genome [46] and all have found that p53REs mediate significant transactivation potential. Based on these data, the authors proposed a model in which p53 binding can be sufficient to drive transcription [38, 46], implying a “dumb” p53 that attempts to activate each locus to which it binds.
In summary, the current literature contains conflicting results regarding how p53 reads and interacts with p53REs and how this translates into target gene activation. Notably, most of these observations have been made using reporter assays and do not fully recapitulate the in vivo context. These assays measure activity driven by a p53RE lacking genomic context, such as gene distance, other regulatory elements, and chromatin state. In the next section, we discuss the extent to which these properties may influence how p53 interacts with p53REs to regulate associated target genes.
Chromatin structure affects p53 binding and productivity
Chromatin, transcription, and cell fate are inextricably linked. Chromatin structure can facilitate or impede biochemical processes on DNA depending on the context, with nucleosomes generally acting as a strong barrier to transcription factor binding [47]. Gene regulatory regions occupied by nucleosomes tend to be inactive due to this inability of transcription factors to recognize and bind their cognate DNA elements. p53 is more complicated, however, as it can recognize the p53RE in both naked DNA and in certain nucleosomal contexts as part of its pioneer transcription factor activity. When p53 interacts with nucleosome-bound p53REs, it prefers p53REs oriented near nucleosome entry/exit sites [48,49,50,51]. Conversely, p53RE positions near the nucleosome dyad, i.e., the center, strongly inhibit p53 binding, providing a clear mechanism to control p53 binding and activity [48,49,50,51] (Fig. 2A). Nucleosome rotational position preferences have also been proposed to differentially regulate the activation and kinetics of cell cycle control genes such as CDKN1A and apoptotic genes such as BBC3/puma [52, 53], but this rotational positioning model has not yet been tested on a high-throughput data basis.

A p53 recognizes and binds to its RE in naked DNA and when found in certain rotational positions within nucleosomal DNA, leading to p53-dependent nucleosome remodeling and gene activation (top panel). The presence of a p53RE in other nucleosome contexts is non-permissive and prevents p53 binding and downstream gene activation (bottom panel). B Co-factors and chromatin remodelers can alter the local chromatin landscape to facilitate or impede p53 binding to its response element. Factors, like TRIM24, inhibit p53 activity (top panel), whereas other factors, like ATRX:DAXX and p63, promote p53 binding through nucleosome displacement (bottom panel). C Numerous p53 interacting proteins, e.g., p300 or iASPP, can alter p53:DNA binding kinetics and promote or repress p53-dependent transcription. D Co-binding of other transcription factors, e.g., SP1 or ATF3, to promoters and enhancers alters p53-dependent transcriptional activity.
Chromatin states may also explain observations of p53 binding and perhaps, differences in cell fate choices. The number of predicted p53 response elements (p53RE) in the human genome greatly exceeds the number of experimentally observed binding events, suggesting that DNA sequence alone is not sufficient to direct p53 binding and activity [2]. The majority of unbound p53RE are nucleosome-occupied, although the field currently lacks sufficient experimental depth and detail to determine whether these p53RE are in unfavorable rotational nucleosome positions or other restrictive chromatin states. Extensive meta-analyses of p53 genomic binding have identified two groups of p53 binding with potentially distinct activities: one invariant across cell types and another with strong cell type dependence. p53 appears to activate a common set of gene targets across cell types [21, 23, 32, 34, 54], but also activates numerous cell type-dependent targets [23, 55]. Cell type-dependent p53 binding strongly correlates with cell type-dependent differences in chromatin accessibility [22, 56], but the extent to which cell type-dependent p53 binding is causal for the observed differential activation of target genes is unclear.
Pioneer factors such as p53 recognize and bind to their DNA motifs embedded in nucleosomes in certain contexts, but also facilitate chromatin remodeling and nucleosome displacement. When does p53 binding lead to chromatin remodeling? As is often the case with chromatin structure, the answer depends on the context. In human fibroblasts, there is little evidence for p53-dependent chromatin remodeling 6 h after p53 stabilization with Nutlin-3a [57], but after 12 h of treatment with the DNA-damaging chemotherapeutic doxorubicin, p53 facilitates remodeling at a select, but still limited, number of binding sites [44]. In mouse embryonic stem cells (mESCs), p53 also led to increased chromatin accessibility after 4 h of doxorubicin treatment [58]. In addition to local chromatin regulation, p53 facilitates long-distance chromatin interactions between distal enhancers and promoters to control gene expression [59]. Still, many p53 binding locations remain “closed”, raising the question of the contexts in which p53 might remodel the local chromatin structure. Recent work in mouse embryonic stem cells provides at least one answer, but also raises additional questions. TRIM24 binds along with p53 to sites with nucleosomes containing unmethylated lysine 4 of histone H3 (H3K4). TRIM24:p53 co-binding restricts p53-dependent chromatin remodeling [58]. Conversely, transcription-associated methylation of H3K4 inhibits TRIM24 activity, thus permitting p53-dependent chromatin remodeling and activation of downstream gene targets. p53 binds to regions independent of TRIM24 that can be variably remodeled, suggesting that additional, as-of-yet unknown, cofactors also mediate p53-dependent chromatin remodeling. One such factor is the H3 chaperone ATRX:DAXX and its modulation of histone occupancy, which facilitates differential p53 binding and chromatin remodeling [60] (Fig. 2B). The breadth and impact of p53 pioneer activity, from differential binding to differential remodeling activity and target gene induction, is only beginning to be elucidated.
Although we still lack sufficiently detailed genome-wide analyses of how p53 reads and interacts with the chromatin, the current data provide the first insights into the extent to which p53 is “smart” and decodes the information itself, or whether it is “dumb” and just tries to perform its trans-activator function whenever possible. While the current data could be read to suggest that p53 is quite “smart” and can discriminate and bind p53REs in multiple nucleosomal contexts, the data may actually show that the chromatin structure, and in particular nucleosome positioning, can control p53’s ability to bind to and trans-activate a given genomic locus. In these cases, p53 does not appear to read or decode the chromatin state, but its “dumb” action is controlled by other factors that read or organize chromatin. For many sites, however, it remains unclear how p53 binding and productivity are regulated, leaving room for “smart” actions by p53.
In general, the productivity of p53 binding sites in the genome is influenced not only by the individual p53RE DNA sequence and chromatin state, but also by other factors that are located nearby. In the next section, we discuss links between such co-factors and their target-specific effects in the p53 transcriptional program.
Co-factors influence productivity and kinetics at p53 binding sites
Multiple co-factors and other transcription factors influence p53’s ability to activate transcription at a given locus. In the previous section, we discussed how TRIM24 and ATRX:DAXX can affect p53’s ability to increase DNA accessibility and to activate transcription (Fig. 2B). Many additional factors have been identified that locally affect p53 function (Fig. 2C), including traditional co-activators like p300, which directly modifies p53 and the local chromatin environment to regulate p53-dependent transcription [61]. The ASPP (ankyrin-repeat, SH3-domain, and proline-rich-region-containing protein) family consists of three members, ASPP1, ASPP2, and iASPP. ASPP1 and ASPP2 have been shown to enhance p53 binding to promoters and activation of target genes involved in apoptosis [14], while iASPP has the opposite effect, inhibiting this process [62]. While it is unclear how ASPP1 and ASPP2 direct p53 towards target genes involved in apoptosis, iASPP has been shown to bind to p53 and perturb its interaction with p53REs, resulting in reduced binding affinity and altered selectivity [63]. Another co-factor, DAZAP2, binds to a subset of p53 targets and inhibits p53 at these sites, leading to reduced activation of the targets by p53, but it is unclear how DAZAP2 selects targets [64]. Non-protein cofactors, such as long non-coding RNAs, have a role up and downstream of p53 [65], but how they may affect local p53 activity remains largely unexplored. Interestingly, it has been shown that differences in the local assembly of the preinitiation complexes that position Pol II to transcribe the gene have been shown to be caused by different core promoter arrangements that cause p53 to rapidly induce CDKN1A (p21) of short duration and slowly induce FAS of long duration [66]. Such differences could contribute to differences in the magnitude and kinetics of p53 target gene activation and ultimately influence cell fate decisions.
Transcription factors have also been shown to cooperate with p53 locally (Fig. 2D). For example, SP1 and ATF3 have been shown to cooperate with p53 in target gene activation when their DNA binding sites are in close proximity [28]. A particularly critical regulator of p53 responses is its sibling p63 (ΔNp63), which is predominantly expressed in basal epithelia. p63 can bind to most sites that p53 can bind to [37], and it enables p53 to bind to specific sites that would otherwise be inaccessible to p53 [56, 67] (Fig. 2B). Combinatorial activity of transcription factors at regulatory elements is a well-studied phenomenon. This concept is comparatively understudied in the p53 network, with opposing views on the influence of local transcription factors in driving p53-dependent activities on DNA [27, 28, 38, 56]. Recent studies combining high-throughput genomic screening and modern computational approaches like deep and machine learning confirmed a long held truth about p53: regulatory elements containing a p53RE, and thus exhibiting binding of p53, drive high levels of transcription [39, 46]. While the specific roles for other transcription factors and co-factors influencing p53RE productivity were not specifically investigated, this blend of computational and experimental power will no doubt provide new insight into p53-dependent cell fate decisions.
To summarize, p53 binding and the ability of p53 to transactivate a given locus depend on multiple layers of information, ranging from the DNA sequence of the p53RE, to the nucleosomal context in which the p53RE is located, to other factors that function at the given locus. All of these properties can influence the transcriptional program of p53. They could help a “smart” p53 to decide on target gene activation and cell fate, or they could instruct a “dumb” p53 towards preferred outcomes. In the last section, we will discuss our current understanding of the extent to which the p53 transcriptional program is affected on a genome-wide scale and how this translates into different cell fate decisions.
Gene regulation and cell fate decisions by p53
The concept that p53 can differentially activate target genes involved in different biological processes, such as cell cycle arrest and apoptosis, to achieve desired cell fates is fascinating. It is largely based on relatively low throughput analyses that have examined only a limited number of p53 target genes in cell populations. Fortunately, multiple high throughput and single cell analyses have become available and we can take an unbiased look at whether the magnitude or kinetics of p53 target gene induction varies between different modes of p53 activation and treatment durations, and how that relates to the subsequent fate of the cells.
A number of studies have evaluated the differential kinetics of p53 target gene activation. In single cells, p53 protein levels have been shown to rise in pulses in response to DNA damage, an effect that cannot be observed when cell populations are evaluated. These rapid changes in p53 protein levels have been shown to be largely driven by negative feedback regulation of p53 by MDM2 and the PPM1D phosphatase [68]. The different dynamics of p53 protein levels have been associated with different cell fates. For example, γ-irradiation resulted in p53 pulses and cell cycle arrest, and when these pulses were sustained by pharmacological treatment with the MDM2 inhibitor Nutlin-3, cells underwent apoptosis [69]. Importantly, the different p53 dynamics and cell fates were associated with different p53 target gene expression. Pulsed and sustained p53 led to the induction of cell cycle arrest genes, such as CDKN1A, GADD45A, and XPC, and negative feedback regulators, such as MDM2 and PPM1D, but sustained p53 alone led to the activation of inducers of apoptosis, such as APAF1, TP53AIP1, and BAX, and senescence, such as PML and YPEL3 [69]. Similar results were obtained with different doses and durations of treatment with the DNA-damaging chemotherapeutic agent doxorubicin [70]. Follow-up studies evaluated a much larger number of p53 target genes and used time-series gene expression analysis to determine the effect of p53 levels that vary over time, i.e., p53 pulses. These studies showed that p53 indiscriminately binds and activates its target genes in response to DNA damage, without enrichment for specific biological functions, and that subsequent differences in temporal RNA levels can be explained by differences in RNA degradation rates [20, 25, 71]. This is supported by previous data from a mouse model in which the p53-MDM2 feedback loop was disturbed by removing the p53REs from the Mdm2 gene, resulting in increased p53 activity in response to DNA damage and induction of apoptosis, while simultaneously increasing the expression of both apoptotic and cell cycle arrest-associated p53 target genes [72]. Although there are clear differences in how p53 activates individual promoters [73,74,75], genes with such promoter features do not appear to be enriched for specific biological functions. This notion is also supported by earlier transcriptome analyses that failed to identify features that discriminate between p53 target genes involved in cell cycle arrest and apoptosis [24, 33] and instead suggested a rather simple model for cell fate decisions that is based on thresholds of p53 levels [24] (Fig. 1C). Consistent with p53-dependent transcript levels, low- and high-throughput chromatin immunoprecipitation (ChIP) data show that the p53 binding signal varies between promoters with p53REs but this difference does not enrich for target genes involved in specific biological functions [20, 24, 76].
In summary, the well-characterized biophysical differences in how p53 interacts with different p53REs in different nucleosomal contexts do not appear to translate into a selective mode of target gene binding or activation by p53 that has a measurable effect on cell fate. Differences in promoter properties are clearly important for the regulation of individual promoters and their associated genes, but current data suggest that this is not overly critical for cell fate decisions after p53 induction.
Other factors, such as death receptor signaling [77] and levels of the transcription factors E2F1 [78] and MYC [79], have also been shown to have threshold mechanisms that determine whether or not apoptosis is induced. A cell’s decision to induce apoptosis is largely determined by the balance of pro- and anti-apoptotic factors, the levels of which vary between cell types and contexts. Such balances provide actionable opportunities for therapeutic intervention to achieve more successful cancer treatment strategies. Differences in cells, such as cell type and context, cause different anti-apoptotic factors to be present at levels close to a tipping point toward induction of apoptosis and thus such cells may be more or less sensitive to specific interventions (Fig. 3). Over the years, several studies have shown that changes in anti-apoptotic factors can tip a cell toward apoptosis after p53 activation. Examples include BCL-2/XL [80, 81], the inhibitors of apoptosis (IAP) family [82], and the pseudo-caspase FLIP(L) [83]. Indeed, inhibition of BCL-2 and BCL-XL has been shown to be a potent anticancer strategy when combined with p53 activation [84, 85]. Another therapeutically promising example is the pathway of the integrated stress response. The gene regulatory network of ATF4, a transcription factor that is the key mediator of the integrated stress response, overlaps with the gene regulatory network of p53, specifically sharing pro-apoptotic targets [86]. Recently, PPM1D inhibition has been shown to induce ATF4 accumulation and activation in addition to stabilizing p53. Importantly, a combination treatment with PPM1D and MDM2 inhibitors resulted in a potent induction of apoptosis [87], presumably due to increased expression of pro-apoptotic target genes.

Under low stress conditions, p53 protein expression is below the threshold for cell cycle arrest or apoptosis, allowing cells to proliferate. Cell cycle arrest occurs when p53 levels reach a specific threshold, defined by a tug-of-war between pro- and anti-proliferative factors, the levels of which depend on context such as cell type and nutrient availability. When p53 levels are high enough, cells undergo p53-dependent apoptosis. The threshold for apoptosis is determined by a tug-of-war between pro- and anti-apoptotic factors that vary in their abundance depending on the cellular context. Importantly, these factors represent therapeutically actionable opportunities to tip the balance toward apoptosis, a desired outcome in cancer treatment regimens.
Notably, the levels of anti-apoptotic proteins can be increased in response to p53 over time, thereby raising the threshold for apoptosis induction and making the rate of p53 accumulation relevant to cell fate [82, 88] (Fig. 3). p53 leads to a cell cycle arrest fate when oncogenes are hyperactive in normal human fibroblasts or when HCT116 colon cancer cells are exposed to genotoxic damage [27, 89]. When one of the p53REs of CDKN1A was removed, cells failed to arrest and continued to proliferate, showing that the p53 levels were insufficient to induce apoptosis in this context [27]. In HCT116 cells, knockout of CDKN1A changed the cellular context to favor apoptosis upon genotoxic damage. Mutation of a p53RE controlling a single pro-apoptotic p53 target, BBC3 (Puma), tipped the balance back against apoptosis and allowed the cells to proliferate [89]. In addition to the well-known cell cycle arrest and pro-apoptotic genes, p53 targets also include anti-apoptotic, pro-proliferative genes [6]. One such example is the p53 target gene KITLG. The p53RE of the KITLG gene has been shown to harbor a single nucleotide polymorphism (SNP) that is associated with reduced p53 binding and KITLG expression and increased cancer risk, presumably because the threshold for apoptosis induction is increased in individuals carrying this SNP [90].
Threshold-based mechanisms are clearly not a feature of “smart” p53, but they allow cells to combine a greater number of signals and are less fragile because they are less complex.
Conclusions and perspective
Over the past years, we have begun to understand the properties that allow p53 to bind to DNA in different chromatin contexts and the co-factors that modulate p53’s ability to transactivate a given locus and control cell fate, although much remains unknown. Yet, when do the well-supported biophysical differences between individual p53 target gene promoters translate into measurable differences in mRNA expression and cell fate? We find that current genomic and transcriptomic data suggest that information content of p53REs is rather low. This could be explained by differences in scales: small differences in p53 binding kinetics and affinity may not be overly relevant in a cell when many other factors are acting on the DNA and p53 has a relatively long time to bind and recruit Pol II. The ability to combine genome-scale assays examining the entirety of sequence space with precise genome editing approaches to validate observations in more native contexts will help resolve these questions. For now, most available evidence suggests that p53 is “dumb” rather than “smart”, trying to transactivate any locus it can bind to (Fig. 1C), instead of decoding information from DNA to selectively activate its targets. We find that it makes sense for a cell to determine its own fate based on simple and robust threshold-based mechanisms rather than on complex and thus fragile p53RE decoding strategies. From an evolutionary perspective, it is difficult to imagine a benefit for shaping and preserving nuances in p53REs to make life-or-death decisions. In the end, the tumor suppressive capacity of p53 is strong no matter whether it is “smart” or “dumb”. However, “dumb” processes might forgive errors such as minor mutations more easily and therefore may be more robust and favored during evolution – at least when it comes to critical decisions such as whether to live or die.
References
Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordân R, Rohs R. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci. 2014;39:381–99.
Sammons MA, Nguyen T-AT, McDade SS, Fischer M. Tumor suppressor p53: from engaging DNA to target gene regulation. Nucleic Acids Res. 2020;48:8848–69.
Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170:1062–78.
Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9.
Liu Y, Leslie PL, Zhang Y. Life and Death Decision-Making by p53 and Implications for Cancer Immunotherapy. Trends Cancer. 2021;7:226–39.
Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017;36:3943–56.
Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–90.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA Induces the Rapid Apoptosis of Colorectal Cancer Cells. Mol Cell. 2001;7:673–82.
Nakano K, Vousden KH. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol Cell. 2001;7:683–94.
Vousden KH. p53. Cell. 2000;103:691–4.
Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–62.
Tang Y, Luo J, Zhang W, Gu W. Tip60-Dependent Acetylation of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol Cell. 2006;24:827–39.
Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-Binding Domain Regulates Apoptosis Induction. Mol Cell. 2006;24:841–51.
Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh J-K, Zhong S, et al. ASPP Proteins Specifically Stimulate the Apoptotic Function of p53. Mol Cell. 2001;8:781–94.
Das S, Raj L, Zhao B, Kimura Y, Bernstein A, Aaronson SA, et al. Hzf Determines Cell Survival upon Genotoxic Stress by Modulating p53 Transactivation. Cell. 2007;130:624–37.
Szak ST, Mays D, Pietenpol JA. Kinetics of p53 Binding to Promoter Sites In Vivo. Mol Cell Biol. 2001;21:3375–86.
Qian H, Wang T, Naumovski L, Lopez CD, Brachmann RK. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene. 2002;21:7901–11.
Inga A, Storici F, Darden TA, Resnick MA. Differential Transactivation by the p53 Transcription Factor Is Highly Dependent on p53 Level and Promoter Target Sequence. Mol Cell Biol. 2002;22:8612–25.
Veprintsev DB, Fersht AR. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008;36:1589–98.
Hafner A, Stewart-Ornstein J, Purvis JE, Forrester WC, Bulyk ML, Lahav G. p53 pulses lead to distinct patterns of gene expression albeit similar DNA-binding dynamics. Nat Struct Mol Biol. 2017;24:840–7.
Nguyen T-AT, Grimm SA, Bushel PR, Li J, Li Y, Bennett BD, et al. Revealing a human p53 universe. Nucleic Acids Res. 2018;46:8153–67.
Hafner A, Kublo L, Tsabar M, Lahav G, Stewart-Ornstein J. Identification of universal and cell-type specific p53 DNA binding. BMC Mol Cell Biol. 2020;21:5.
Resnick-Silverman L, Zhou R, Campbell MJ, Leibling I, Parsons R, Manfredi JJ. In vivo RNA-seq and ChIP-seq analyses show an obligatory role for the C terminus of p53 in conferring tissue-specific radiation sensitivity. Cell Rep. 2023;42:112216.
Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013;20:576–88.
Jiménez A, Lu D, Kalocsay M, Berberich MJ, Balbi P, Jambhekar A, et al. Time‐series transcriptomics and proteomics reveal alternative modes to decode p53 oscillations. Mol Syst Biol. 2022;18:e10588.
Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32.
Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K, et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol. 2016;34:192–8.
Catizone AN, Uzunbas GK, Celadova P, Kuang S, Bose D, Sammons MA. Locally acting transcription factors regulate p53-dependent cis-regulatory element activity. Nucleic Acids Res. 2020;48:4195–213.
Johnson RA, Ince TA, Scotto KW. Transcriptional Repression by p53 through Direct Binding to a Novel DNA Element. J Biol Chem. 2001;276:27716–20.
Wang B, Xiao Z, Ren EC. Redefining the p53 response element. Proc Natl Acad Sci USA. 2009;106:14373–8.
Kenzelmann Broz D, Mello SS, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–31.
Fischer M, Steiner L, Engeland K. The transcription factor p53: Not a repressor, solely an activator. Cell Cycle. 2014;13:3037–58.
Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200.
Andrysik Z, Galbraith MD, Guarnieri AL, Zaccara S, Sullivan KD, Pandey A, et al. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017;27:1645–57.
Fischer M. Conservation and divergence of the p53 gene regulatory network between mice and humans. Oncogene. 2019;38:4095–109.
Jordan JJ, Menendez D, Inga A, Nourredine M, Bell D, Resnick MA. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008;4:e1000104.
Riege K, Kretzmer H, Sahm A, McDade SS, Hoffmann S, Fischer M. Dissecting the DNA binding landscape and gene regulatory network of p63 and p53. Elife. 2020;9:e63266.
Verfaillie A, Svetlichnyy D, Imrichova H, Davie K, Fiers M, Atak ZK, et al. Multiplex enhancer-reporter assays uncover unsophisticated TP53 enhancer logic. Genome Res. 2016;26:882–95.
de Almeida BP, Reiter F, Pagani M, Stark A. DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat Genet. 2022;54:613–24.
Trauernicht M, Rastogi C, Manzo SG, Bussemaker HJ, van Steensel B, van Steensel B. Optimisation of TP53 reporters by systematic dissection of synthetic TP53 response elements. Nucleic Acids Res. 2023;51:9690–702.
Senitzki A, Safieh J, Sharma V, Golovenko D, Danin-Poleg Y, Inga A, et al. The complex architecture of p53 binding sites. Nucleic Acids Res. 2021;49:1364–82.
Farkas M, Hashimoto H, Bi Y, Davuluri RV, Resnick-Silverman L, Manfredi JJ, et al. Distinct mechanisms control genome recognition by p53 at its target genes linked to different cell fates. Nat Commun. 2021;12:484.
Safieh J, Chazan A, Saleem H, Vyas P, Danin-Poleg Y, Ron D, et al. A molecular mechanism for the “digital” response of p53 to stress. Proc Natl Acad Sci. 2023;120:e2305713120.
Younger ST, Rinn JL. p53 regulates enhancer accessibility and activity in response to DNA damage. Nucleic Acids Res. 2017;45:9889–9900.
Peng T, Zhai Y, Atlasi Y, ter Huurne M, Marks H, Stunnenberg HG, et al. STARR-seq identifies active, chromatin-masked, and dormant enhancers in pluripotent mouse embryonic stem cells. Genome Biol. 2020;21:243.
Sahu B, Hartonen T, Pihlajamaa P, Wei B, Dave K, Zhu F, et al. Sequence determinants of human gene regulatory elements. Nat Genet. 2022;54:283–94.
Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82.
Sahu G, Wang D, Chen CB, Zhurkin VB, Harrington RE, Appella E, et al. p53 binding to nucleosomal DNA depends on the rotational positioning of DNA response element. J Biol Chem. 2010;285:1321–32.
Yu X, Buck MJ. Defining TP53 pioneering capabilities with competitive nucleosome binding assays. Genome Res. 2019;29:107–15.
Nishimura M, Arimura Y, Nozawa K, Kurumizaka H. Linker DNA and histone contributions in nucleosome binding by p53. J Biochem. 2020;168:669–75.
Nishimura M, Takizawa Y, Nozawa K, Kurumizaka H. Structural basis for p53 binding to its nucleosomal target DNA sequence. PNAS Nexus. 2022;1:pgac177.
Cui F, Zhurkin VB. Rotational positioning of nucleosomes facilitates selective binding of p53 to response elements associated with cell cycle arrest. Nucleic Acids Res. 2014;42:836–47.
Freewoman JM, Snape R, Cui F. Temporal gene regulation by p53 is associated with the rotational setting of its binding sites in nucleosomes. Cell Cycle. 2021;20:792–807.
Fischer M, Grossmann P, Padi M, DeCaprio JA. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44:6070–86.
Tatavosian R, Donovan MG, Galbraith MD, Duc HN, Szwarc MM, Joshi MU, et al. Cell differentiation modifies the p53 transcriptional program through a combination of gene silencing and constitutive transactivation. Cell Death Differ. 2023;30:952–65.
Karsli Uzunbas G, Ahmed F, Sammons MA. Control of p53-dependent transcription and enhancer activity by the p53 family member p63. J Biol Chem. 2019;294:10720–36.
Sammons MA, Zhu J, Drake AM, Berger SL. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015;25:179–88.
Isbel L, Iskar M, Durdu S, Weiss J, Grand RS, Hietter-Pfeiffer E, et al. Readout of histone methylation by Trim24 locally restricts chromatin opening by p53. Nat Struct Mol Biol. 2023;30:948–57.
Serra F, Nieto-Aliseda A, Fanlo-Escudero L, Rovirosa L, Cabrera-Pasadas M, Lazarenkov A, et al. p53 rapidly restructures 3D chromatin organization to trigger a transcriptional response. Nat Commun. 2024;15:2821.
Gulve N, Su C, Deng Z, Soldan SS, Vladimirova O, Wickramasinghe J, et al. DAXX-ATRX regulation of p53 chromatin binding and DNA damage response. Nat Commun. 2022;13:5033.
Tang Z, Chen W-Y, Shimada M, Nguyen UTT, Kim J, Sun X-J, et al. SET1 and p300 Act Synergistically, through Coupled Histone Modifications, in Transcriptional Activation by p53. Cell. 2013;154:297–310.
Bergamaschi D, Samuels Y, O’Neil NJ, Trigiante G, Crook T, Hsieh J-K, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33:162–7.
Chen S, Wu J, Zhong S, Li Y, Zhang P, Ma J, et al. iASPP mediates p53 selectivity through a modular mechanism fine-tuning DNA recognition. Proc Natl Acad Sci. 2019;116:17470–9.
Liebl MC, Moehlenbrink J, Becker H, Raddatz G, Abdeen SK, Aqeilan RI, et al. DAZAP2 acts as specifier of the p53 response to DNA damage. Nucleic Acids Res. 2021;49:2759–76.
Lin T, Hou P-F, Meng S, Chen F, Jiang T, Li M-L, et al. Emerging Roles of p53 Related lncRNAs in Cancer Progression: A Systematic Review. Int J Biol Sci. 2019;15:1287–98.
Morachis JM, Murawsky CM, Emerson BM. Regulation of the p53 transcriptional response by structurally diverse core promoters. Genes Dev. 2010;24:135–47.
Woodstock DL, Sammons MA, Fischer M. p63 and p53: Collaborative Partners or Dueling Rivals? Front Cell Dev Biol. 2021;9:701986.
Batchelor E, Loewer A, Mock C, Lahav G. Stimulus‐dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7:488.
Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 Dynamics Control Cell Fate. Science. 2012;336:1440–4.
Wu M, Ye H, Tang Z, Shao C, Lu G, Chen B, et al. p53 dynamics orchestrates with binding affinity to target genes for cell fate decision. Cell Death Dis. 2017;8:e3130–e3130.
Porter JR, Fisher BE, Batchelor E. p53 Pulses Diversify Target Gene Expression Dynamics in an mRNA Half-Life-Dependent Manner and Delineate Co-regulated Target Gene Subnetworks. Cell Syst. 2016;2:272–82.
Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintás-Cardama A, et al. The p53–Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013;27:1857–67.
Friedrich D, Friedel L, Finzel A, Herrmann A, Preibisch S, Loewer A. Stochastic transcription in the p53-mediated response to DNA damage is modulated by burst frequency. Mol Syst Biol. 2019;15:e9068.
Harton MD, Koh WS, Bunker AD, Singh A, Batchelor E. p53 pulse modulation differentially regulates target gene promoters to regulate cell fate decisions. Mol Syst Biol. 2019;15:e8685.
Antwi EB, Marrakchi Y, Çiçek Ö, Brox T, Di Ventura B. Requirements for mammalian promoters to decode transcription factor dynamics. Nucleic Acids Res. 2023;51:4674–90.
Kaeser MD, Iggo RD. Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proc Natl Acad Sci. 2002;99:95–100.
Spencer SL, Sorger PK. Measuring and Modeling Apoptosis in Single Cells. Cell. 2011;144:926–39.
Shats I, Deng M, Davidovich A, Zhang C, Kwon JS, Manandhar D, et al. Expression level is a key determinant of E2F1-mediated cell fate. Cell Death Differ. 2017;24:626–37.
Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, et al. Distinct Thresholds Govern Myc’s Biological Output In Vivo. Cancer Cell. 2008;14:447–57.
Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–11.
Sánchez-Rivera FJ, Ryan J, Soto-Feliciano YM, Clare Beytagh M, Xuan L, Feldser DM, et al. Mitochondrial apoptotic priming is a key determinant of cell fate upon p53 restoration. Proc Natl Acad Sci. 2021;118:e2019740118.
Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell. 2016;165:631–42.
Lees A, McIntyre AJ, Crawford NT, Falcone F, McCann C, Holohan C, et al. The pseudo-caspase FLIP(L) regulates cell fate following p53 activation. Proc Natl Acad Sci. 2020;117:17808–19.
Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, et al. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell. 2017;32:748–760.e6.
Bharti V, Watkins R, Kumar A, Shattuck-Brandt RL, Mossing A, Mittra A, et al. BCL-xL inhibition potentiates cancer therapies by redirecting the outcome of p53 activation from senescence to apoptosis. Cell Rep. 2022;41:111826.
Baniulyte G, Durham SA, Merchant LE, Sammons MA. Shared Gene Targets of the ATF4 and p53 Transcriptional Networks. Mol Cell Biol. 2023;43:426–49.
Andrysik Z, Sullivan KD, Kieft JS, Espinosa JM. PPM1D suppresses p53-dependent transactivation and cell death by inhibiting the Integrated Stress Response. Nat Commun. 2022;13:7400.
Sun T, Mu D, Cui J. Mathematical model identifies effective P53 accumulation with target gene binding affinity in DNA damage response for cell fate decision. Cell Cycle. 2018;17:2716–30.
Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci. 2007;104:4054–9.
Zeron-Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro-Giner F, et al. A Polymorphic p53 Response Element in KIT Ligand Influences Cancer Risk and Has Undergone Natural Selection. Cell. 2013;155:410–22.
Funding
The p53 research of MF is supported by the German Research Foundation (DFG) through grants FI 1993/6-1 and FI 1993/7-1. MAS was supported by NIH award GM138120. This work was made possible by generous sabbatical leave granted by the University at Albany to MAS. Funding for open access charge: Leibniz Institute on Aging – Fritz Lipmann Institute (FLI), Jena, Germany. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MF and MAS prepared the Figures and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fischer, M., Sammons, M.A. Determinants of p53 DNA binding, gene regulation, and cell fate decisions. Cell Death Differ 31, 836–843 (2024). https://doi.org/10.1038/s41418-024-01326-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-024-01326-1
- Springer Nature Limited