
Abstract
Hepatocellular carcinoma (HCC) has a high mortality rate due to the lack of effective treatments and drugs. Arsenic trioxide (ATO), which has been proved to successfully treat acute promyelocytic leukemia (APL), was recently reported to show therapeutic potential in solid tumors including HCC. However, its anticancer mechanisms in HCC still need further investigation. In this study, we demonstrated that ATO inhibits tumorigenesis and distant metastasis in mouse models, corresponding with a prolonged mice survival time. Also, ATO was found to significantly decrease the cancer stem cell (CSC)-associated traits. Minichromosome maintenance protein (MCM) 7 was further identified to be a potential target suppressed dramatically by ATO, of which protein expression is increased in patients and significantly correlated with tumor size, cellular differentiation, portal venous emboli, and poor patient survival. Moreover, MCM7 knockdown recapitulates the effects of ATO on CSCs and metastasis, while ectopic expression of MCM7 abolishes them. Mechanistically, our results suggested that ATO suppresses MCM7 transcription by targeting serum response factor (SRF)/MCM7 complex, which functions as an important transcriptional regulator modulating MCM7 expression. Taken together, our findings highlight the importance of ATO in the treatment of solid tumors. The identification of SRF/MCM7 complex as a target of ATO provides new insights into ATO’s mechanism, which may benefit the appropriate use of this agent in the treatment of HCC.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC) is one of the most common solid tumors; more than 700,000 cases are diagnosed worldwide per year1. HCC has a high mortality rate due to the high metastatic potential and lack of effective treatments and drugs2. Thus, researchers have been making efforts to investigate more effective treatment options.
Arsenic trioxide (ATO), an FDA-approved drug as the first line treatment of acute promyelocytic leukemia (APL), is showing promising therapeutic potential in advanced liver cancers. As demonstrated by multiple clinical trials, HCC patients received ATO as a therapeutic agent or a chemosensitizer showed less extrahepatic metastasis and prolonged survival3,4,5. However, its anticancer mechanisms of action in HCC remain unclear. Therefore, clarifying the mechanism of ATO will help increase its clinical efficacy in HCC.
Previous researchers have reported that ATO induced growth inhibition and apoptosis in cancer cells6,7. Recently, several pieces of evidence also imply that ATO attenuates the cancer stem cells (CSCs) population with the involvement of SHH pathway8,9, Notch pathway10,11, and TGF-β pathway12. However, for most cancers, several types of inhibitors derived from normal stem cell-associated pathways such as SHH, Notch, and TGF-β have been proved to be ineffective in larger studies13. Therefore, the key functional molecules downstream of ATO which delineate its therapeutic effects in solid tumors including HCC need to be further identified.
Here, we demonstrated that ATO inhibits HCC lung metastasis and increases survival rate in a metastatic HCC xenograft model. Our results showed that ATO can inhibit the tumor-initiating capacity of HCC cells. To explore the underlying mechanism, target molecule alterations of HCC cells treated by ATO were investigated by a cDNA (complementary DNA) microarray. Minichromosome maintenance protein (MCM) 7 was further identified to be inhibited significantly after ATO treatment. Meanwhile, we found that MCM7 expression is correlated with HCC progression and prognosis in HCC patients. As we expected, MCM7 knockdown inhibits the tumor-initiating ability, and MCM7 ectopic expression abolishes the inhibitory effects of ATO. Most importantly, here for the first time, we report ATO downregulates MCM7 transcription by suppressing the transcription activity of serum response factor (SRF)/MCM7 complex. Taken together, we identify ATO as a new potential inhibitor of MCM7 protein in cancer, which will shed light on the treatment of MCM7-overexpressed HCC.
Results
ATO inhibits distant metastasis and prolongs survival of intrahepatic metastasis model mice
To minimize the toxicity of ATO on normal cells, we used various dosages of ATO to explore their effects on proliferation of normal human fetal liver L02 cells (Fig. S1A and B) and found that ATO at 3.6 μM or less had little effect on L02 cell apoptosis (Fig. S1C). Therefore, we used 3.6 μM for further assays in HCC cells and found that ATO at 3.6 μM had significant inhibitory effects on the proliferation (Fig. S1D), G0/G1 population (Fig. S1E), and the invasion of cells (Fig. S1F) but little effects on cell apoptosis (Fig. S1G). These results suggested that low dose of ATO inhibits HCC cell proliferation and invasion without inducing apoptosis.
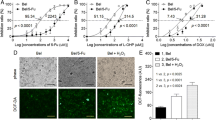
Next, we investigated whether ATO inhibits distant metastasis in a metastatic HCC xenograft model, established as we previously reported14,15,16. The mice were given 1 mg/kg ATO (conversion from human dose) in H3AsO3 solution (As2O3 + 3H2O ↔ 2H3AsO3) or PBS by intraperitoneal injection every day and were examined for lung metastasis and survival (Fig. 1a). The weight of ATO-treated mice was significantly higher than that of control mice at the indicated day (Fig. 1b). The gross observations and hematoxylin and eosin (H&E) staining of tumor foci of the liver from both groups are shown in Fig. 1c. There were significantly fewer lung metastases in the ATO group than in the control group (P = 0.0453), in which tumors were visible on the lung surface (Fig. 1d). To study the long-term effects of ATO, we evaluated the survival rates between the two groups. The survival rate was significantly higher in the ATO group than the control group (median survival = 41 days and 32 days, respectively) (Fig. 1e). The experimental cancer cachexia was significantly improved in the surviving mice of the ATO group compared with the PBS group (Fig. 1f; Supplementary Video). Taken together, these results showed ATO can inhibit HCC metastasis and prolong survival in the mouse model.

a A schematic diagram for the ATO therapy experiment. i.p. indicates intraperitoneal injection. b–d The lung metastasis assay. b The mice were weighted every other day (n = 14 for the PBS group, n = 11 for the ATO group). P values were determined by multiple t-tests. c Gross observation of nodules in livers (left) and representative images of H&E-stained liver sections (right) from ATO-treated mice and PBS-treated mice in the therapeutic experiments. Yellow circles indicate orthotopic tumors; magnification, bars, 100 μm. d shows representative examples of lungs (left) and representative images of H&E-stained lung sections (middle) from nude mice treated with ATO or PBS. Arrowheads indicate lung metastases foci. Bars, 100 μm in ×100, 20 μm in ×400. The different lung sections (5 sections per lung) were randomly chosen from each group, and the foci were quantified as the average number across the total sections per group (right). P = 0.0453, determined by two-sided Student’s t-test. e The survival curve of nude mice treated with ATO (n = 44) or PBS (n = 44) (top). P = 0.0003, by log-rank (Mantel–Cox) test. Bottom: the median survival time. f Appearance of the mice treated with ATO or PBS
ATO inhibits liver CSCs-associated traits
Increasing evidence demonstrates that CSCs contribute to tumor initiation, distal metastasis, and chemotherapy resistance17. Thus, we next investigate whether ATO affects CSCs-associated characteristics in HCC cells. Tumorsphere, an effective in vitro model for analysis of the self-renewal capability of CSCs and enrichment of these cells from bulk cancer cells, were also assessed as we previously described15,16. We found that ATO could significantly decrease the tumorsphere formation compared with the control group, whereas doxorubicin, 5-FU, or sorafenib treatment did not show significant differences (Fig. 2a). Besides, we used a combination of reported liver CSC markers18,19,20 to enrich liver CSCs. We found that ATO decreased the CD13+CD133+EpCAM+ cell population (Fig. 2b and S2A and B) and tumorsphere formation (Fig. 2c) of HCC cells in a dose-dependent manner. Quantitative realtime polymerase chain reaction (qRT-PCR) analysis showed that the expression levels of stemness-associated genes, including OCT4 and NANOG, and tumor markers, including AFP and CK19, in HCC cells were downregulated by ATO (Fig. 2d and S2C). Immunofluorescence analysis further showed that AFP expression was significantly downregulated (Fig. 2e) and the cell differentiation marker CK18 was abundant in cytoplasm after ATO treatment (Fig. S2D). To investigate whether ATO directly regulates the liver CSC population, we sorted the CD13+CD133+EpCAM+ cell population for further CSC assays. We found that the adherent growth (Fig. S2E), tumorsphere formation (Fig. 2f), expressions of OCT4, NANOG, and AFP (Fig. 2g), and chemoresistance capacity (Fig. 2h) of the sorted population were all inhibited by ATO. These results suggested that ATO inhibits liver CSCs-associated traits in vitro.

a The effects of ATO, sorafenib, 5-FU, and doxorubicin on tumorsphere formation. All the reagents were added into the tumorsphere system at day 5. Tumorsphere formation (n = 3) was quantified as the total cross-sectional areas of tumorspheres/number of tumorsphere per group (area average) at day 8. Bars, 500 μm. *P < 0.05, NS: not significant, determined by two-sided Student’s t-test. b, c The effects of ATO on CD13+/CD133+/EpCAM+ subpopulation percentage (b) and tumorsphere formation of HCC cells (c) at indicated concentrations (μM). Bars, 200 μm. Tumorsphere formation (n = 3) was quantified as the total cross-sectional areas of tumorspheres per group (area sum) (similarly hereinafter if not mentioned). “*” or “**” indicate the comparison for the “area sum” of tumorspheres with the control (0 μM) in the bar graphs. d Relative values of “stemness” gene and HCC marker gene expression in HCC cells. e Immunofluorescence analysis of AFP expressions after ATO treatment in HCC cells. Bars, 20 μm. f–h Comparison of tumorsphere formation (n = 3, f), relative values of “stemness” gene and HCC marker gene expression (by qRT-PCR, g), and chemoresistance (h) from Huh7.5.1 cells-derived CD13+/CD133+/EpCAM+ subpopulation treated with ATO or PBS. Bars, 500 μm. *P < 0.05, **P < 0.01, determined by two-sided Student’s t-test
To further investigate the effects of ATO on liver CSCs in vivo, we established an HCC xenograft model via subcutaneous injection of Hep3B cells (control group) or Hep3B cells treated with 3.6 μM ATO (ATO group) into the left or right flank of the same mouse, respectively (Fig. 3a). There were significant differences of tumor incidence in two groups (Fig. 3b–e). This effect of ATO on inhibiting tumor-initiation was also validated by using Huh7.5.1 cells (Fig. S3). We next performed serial transplantation of the SMMC-7721 cells derived from the subcutaneous tumors that received local injection of 0.72 mg/kg ATO in mice (Fig. 3f). Four mice in the PBS group (4/5) showed tumor growth, whereas only one mouse in the ATO group (1/5) showed growth at the 3 × 104 dilution in serially transplanted mice (Fig. 3g, h), suggesting that ATO inhibits liver CSCs in vivo.

a A schematic diagram for examination of the effects of ATO on tumorigenic capacity of Hep3B cells. Subcutaneous injection of the Hep3B cells into NOD/SCID mice 48 h after HCC cells pre-treated with 3.6 μM ATO in 10% H-DMEM medium (HD). After 1 month, the peeled tumors were measured for size and weight. b–d Gross observation of tumor-bearing mice (top), peeled tumors (bottom), derived from Hep3B cells at 106, 105, and 104 dilutions, respectively. e Tumor incidence of the cells from each group in diluting limitation assay. f A schematic diagram for examination of the effects of ATO on tumorigenic capacity of HCC cells via serial transplantation. ATO was given by local injection into the subcutaneous tumors of the nude mice every other day (100 μL) with the same volume of PBS as the control. After 2 weeks, the tumors were carefully removed and digested into single cells by the collagenase, which were then subcutaneously injected into the NOD/SCID mice. g Gross observation of the peeled tumors from each group. h Tumor incidence of the cells from each group in serial transplantation assay
MCM7 is downregulated in ATO-treated cancer cells
We then addressed the question of how ATO regulates liver cancer cells. We first investigated ATO-induced molecular alterations by using Affymetrix Human PrimeView mRNA microarray. A total of 201 genes were significantly changed in both ATO-treated HCC cell lines (fold change ≥2.0, P ≤ 0.05). Some of the candidate mRNAs that changed in both HCC cell lines, including HMOX1, MT1X, GADD45B, GSN, and MCM7, were validated by qRT-PCR analysis (Fig. 4a, c). Among these candidate mRNAs, MCM7, belonging to a highly conserved MCM family, attracted our attention. The MCM family, mainly consisted of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7, is essential for the initiation of eukaryotic genome replication21. Our gene ontology analysis of differentially expressed mRNAs classified by biological process demonstrated that MCM7 is involved in cellular response to drug and proliferation stimulus, in addition to DNA replication and cell cycle22 (Fig. 4b). Moreover, among the MCM family members overexpressed in multiple cancers23, only MCM7 was downregulated in both ATO-treated HCC cells (Fig. 4b, c). Therefore, we selected MCM7 for further investigation. RNA sequencing also showed that MCM7 was overexpressed in multiple cancer tissues (Fig. S4). We next examined the expression level of MCM7 protein in ATO-treated HCC cells. ATO inhibited MCM7 expression in HCC cells in a dose-dependent manner (Fig. 4d). In tumorspheres, MCM7 levels were upregulated compared with their adherent parental cells, while they were inhibited by ATO (Fig. 4e). Importantly, IHC staining of the tumors derived from mice that received local injection of ATO showed a significant reduction of MCM7 protein compared with control mice (Fig. 4f). These data suggested that ATO inhibits MCM7 expressions in vitro and in vivo.

a The microarray heat map (top) shows some of the profiles of differentially expressed mRNAs in two cells with over two-fold changes. Pseudo-colors indicate differential expression (green, transcript levels below the control; red, transcript levels greater than the control). Bottom: confirmation of some of the major candidate mRNAs by qRT-PCR analysis. *P < 0.05, **P < 0.01, determined by two-sided Student’s t-test. b GO analysis of differential expressing mRNAs classified by their biological process for MCM7 in HCC cells after ATO treatment. c qRT-PCR analysis of expressions of MCM2, MCM3, MCM4, MCM5, and MCM6 in two cells after ATO treatment. *P < 0.05, **P < 0.01, determined by two-sided Student’s t-test. d Western blot (WB) analysis of MCM7 expression in HCC cell lines for 48 h of treatment with 3.6 μM ATO (top). Bottom shows MCM7 expression in HCC cell lines treated with ATO at the indicated dosages. e qRT-PCR analysis of MCM7 expression levels in tumorspheres (n = 3) treated with ATO. WT, wild type adherent cells; Spheres-con, tumorspheres without ATO treatment; Spheres-ATO, tumorspheres with ATO treatment. *P < 0.05, **P < 0.01, determined by two-sided Student’s t-test. f H&E staining and MCM7 IHC staining of tumors derived from the mice that received local injection of ATO or PBS. Bars, 100 μm. **P < 0.01, *P < 0.05, determined by two-sided Student’s t-test
MCM7 is correlated with the tumor progression and prognosis in HCC patients
To clarify the clinical significance of MCM7, we investigated the expression profile of MCM7 in HCC tissues. We found an extensive nucleus staining of MCM7 in cancer tissues compared with a weak cytoplasmic staining in the paired nontumor tissues. A significant upregulation of MCM7 was observed in HCC tissues compared with the paired NT tissues (Fig. 5a, b). To exclude individual differences in MCM7 expression, we normalized the MCM7 expression intensities to those in the paired nontumor tissues in each pair. The total MCM7 expression was observed to be upregulated in 55.3% of HCC cases (Fig. 5b), whereas the MCM7 expression in the nuclei was strongly upregulated in 100% of HCC cases (Fig. 5c). Next, we performed a correlation analysis between the nucleic expression pattern of MCM7 and the histoprognostic factors. The elevated expression of MCM7 was highly correlated with the tumor diameter, Edmondson–Steiner grading, survival time, portal venous emboli, and AFP concentration of HCC patients (Fig. 5d, e and Supplementary Table 3). Moreover, we isolated human primary HCC cells (identified by epithelial makers including ALB and CK18, Fig. 5f). As we expected, a high MCM7 expression was observed and MCM7 expression was also inhibited by ATO treatment (Fig. 5g). Taken together, the elevated expression of MCM7 may play important roles in HCC progression.

a Representative examples of IHC staining of MCM7 (left) in HCC patients (n = 76). Quantitative analysis of the immunohistochemical expression of MCM7 (the number of MCM7 nuclear staining-positive cells/total cell number) in paired HCC tissues compared with the corresponding nontumor (NT) tissues (right). P < 0.0001, by paired t-test (two-sided). Bars, 100 μm. b A fold change (HCC/NT) plot (left) and a pie chart (right) of patient based on total MCM7 expression from HCC tissue arrays. c A log2 (HCC/NT) plot based on the ratio of nucleic MCM7 expression to total MCM7 expression. d, e Representative examples of IHC staining of MCM7 in HCC tissues at the indicated clinical grade (d). Bars, 100 μm. e The difference in percentage of MCM7 nuclear staining between the grade I-II/II, II-III, and III-IV groups of HCC patients (P = 0.0005, Fisher’s exact test). Weak nuclear staining: Integrated optical density (IOD) SUM of nuclear MCM7/IOD SUM of total MCM7 <30%; extensive nuclear staining: IOD SUM of nuclear MCM7/IOD SUM of total MCM7 >30%. f Immunofluorescence analysis of expression of ALB and CK18 in isolated primary HCC cells (left). Bars, 50 μm. g WB analysis of MCM7 expression in primary HCC cells treated with 3.6 μM of ATO
ATO inhibits liver CSCs and metastasis through targeting MCM7
To further answer the question whether ATO functions through MCM7 protein, we first validated the function of MCM7 in HCC cells. We established five stable MCM7-knockdown HCC cell lines (Fig. S5A and B), and found a decreased expression of the CSC marker CD13 (Fig. 6a). An increased expression of the cell differentiation marker CK18 is also observed in MCM7-knockdown cells (Fig. S6). Since MCM7 protein is upregulated in tumorspheres (Fig. S7), we performed tumorsphere forming assay and found that MCM7 knockdown significantly decreased the tumorsphere formation (Fig. 6b). Furthermore, we used psicoR-scramble/shMCM7 transfected Huh7.5.1 cells and HCCLM3 cells, the two cell lines with the most significant downregulation of MCM7 protein, to examine the tumor-initiating capacity of these HCC cells. The weight of tumors formed by HCCLM3-psicoR-shMCM7 cells was significantly decreased at both 1 × 106 and 1 × 105 dilutions, compared with scrambles (Fig. 6c). The tumor incidence was also decreased by MCM7 knockdown (Fig. 6d). For Huh7.5.1 cells, the scramble cells seeded at two dilutions had all formed tumors, whereas tumors were found in 7/7 and 5/7 mice in 1 × 106 and 1 × 105 knockdown group, respectively (Fig. S8A and B). The downregulation of MCM7 in formed tumors was confirmed by IHC staining (Figs. 6e and S8C). Moreover, MCM7 overexpression abolishes the inhibitory effects of ATO on tumorsphere formation (Fig. 6f–h). Our results demonstrated that downregulation of MCM7 recapitulates the inhibitory function of ATO on CSCs in vitro and in vivo.

a The flow cytometry analysis of the CD13+ percentage in psicoR-shMCM7 stable cells. b shows a fluorescence image of the tumorspheres (n = 3) from psicoR-shMCM7-GFP cells and scramble cells. Bars, 200 μm. c Gross observation of tumor-bearing NOD/SCID mice and peeled tumors derived from HCCLM3-psicoR-shMCM7-GFP cells and scramble cells at two dilutions (left). Right shows the comparison of tumor weight between the shMCM7 group and control group. d The dilutions and mouse numbers. e Representative examples (left) and quantitative analysis (right) of the IHC staining of MCM7 in the tumors derived from HCCLM3-psicoR-shMCM7 cells and scramble cells. f–h Rescue assay of CSCs by the ectopic expression of MCM7. Western blot analysis of MCM7 expression in transfected HCC cells treated with ATO or not (f). Tumorsphere formation was observed (g) and quantified (h, n = 3). Bars, 1000 μm. P value was determined by two-sided Student’s t-test
We therefore examined the effects of MCM7 on metastasis in a metastatic HCC xenograft model14,15,16 (Fig. 7a–d). Inspiringly, MCM7 knockdown inhibited more than half of the distant metastases from primary tumors in liver to lung compared with the controls (30% vs. 64.7%, P = 0.0348) (Fig. 7e). It is reported that metastatic burden is a combination of both number of metastases and the size of the metastatic tumors24. Our further analysis showed that larger metastatic foci (>100 μm) were significantly reduced in MCM7-knockdown group in contrast to the control group (Fig. 7f–h). These results strongly supported that MCM7 downregulation has a significant inhibiting effect on the distant metastasis of HCC cells.

a–d The nude mice received liver injections of MCM7-knockdown-HCCLM3 cells and scramble control cells, respectively. Representative images of gross observation of lungs (a, b) and lung metastasis foci in scanned panels of H&E-stained lung sections derived from both groups (c, d) were shown (Pannoramic MIDI, 3DHISTECH, Hungary). Blue dashed circles indicate typical macro-metastases foci (magnification: ×15) and arrowheads indicate typical micro-metastases foci in magnification inserts (×400). Bars, 1000 μm. e The metastasis rate of liver-inoculated mice with MCM7-knockdown HCC cells or control cells (P = 0.0348, by two-sided Chi-square test). f Quantitative analysis of the metastatic foci in detected lung sections based on the diameters (<100 μm and >100 μm) in each group. A total of 15 sections per lung (control group) and 17 sections per lung (MCM7-knockdown group) were randomly chosen and counted by two observers from each group. The loci diameters were measured by software (NIS-Elements F 4.30.01, Nikon). P = 0.0011, by two-sided Chi-square test. g, h A pie chart of the metastatic foci in detected lung sections based on the diameters in each group
Collectively, our data revealed that ATO inhibits liver CSCs and the metastasis capacity of HCC cells through targeting MCM7.
ATO downregulates MCM7 by suppressing the transcription activity of SRF
To explore the regulating mechanisms of ATO on MCM7 expression in HCC cells, we analyzed the possible transcription factors regulating MCM7. It is predicted that four transcription factors including peroxisome proliferator-activated receptor gamma (PPAR-γ), N-Myc, SRF, and c-Myc have the putative binding sites on MCM7 promoter (Fig. 8a). MCM7 has been reported to be a direct target of the N-Myc in neuroblastoma25. We also used PPAR-γ antagonist GW 9662 and PPAR-γ agonist Rosiglitazone to test whether PPAR-γ regulates MCM7 expression but had negative results (Fig. 8b). SRF has been identified as an oncogenic driver of HCC that had elevated expression in HCC tissues, especially in high-grade, poorly differentiated tumors26,27, consistent with the expression pattern of MCM7 in HCC. Thus, we performed chromatin immunoprecipitation (ChIP) assays to test whether SRF bind to MCM7 promoter in HCC cells. As shown in Fig. 8c, SRF binds to the MCM7 promoter covering the region between −845 bp and −625 bp.

a The transcription factor binding sites of MCM7 promoter predicted by The Champion ChiP Transcription Factor Search Portal (http://www.sabiosciences.com/chipqpcrsearch.php?app=TFBS) based on SABiosciences’ proprietary database known as DECODE. b WB analysis of MCM7 expression in HCC cell lines 7 days after treatment with 10 μM of PPAR-γ antagonist GW 9662 (M6191, Sigma-Aldrich, St. Louis, MO) and 10 μM of PPAR-γ agonist Rosiglitazone (R2408, Sigma-Aldrich). c A ChIP assay for the detection of SRF and MCM7 promoter binding specificity. M, DNA marker. d WB analysis of SRF expression at indicated time after ATO treatment in HCC cells. e WB analysis of SRF and MCM7 expression in SRF-knockdown HCC cells. f A brightfield image and quantification analysis of the tumorspheres from psicoR-shMCM7-GFP cells and scramble cells. Bars, 1000 μm. g IP with an anti-SRF antibody and analysis by immunoblotting with a MCM7 antibody and SRF antibody in HCC cells treated with ATO or PBS (con). h The dual luciferase assay for reporter gene expression in HeLa cells that were transfected with pGL4-MCM7-promoter luciferase constructs or a combination with pcDNA3.1-MCM7-expressing vectors plus pBPLV-SRF-expressing vectors, with a pGL4-basic vector as the control. Mean ± SD relative luciferase activity was calculated relative to Renilla activity. **P < 0.01, determined by Student’s t-test. i Summarized hypothesis on the ATO-SRF/MCM7 transcription axis in HCC. ATO interacts with MCM7 and inhibits the transcription activity of SRF/MCM7 complex, this in turn leads to the downregulation of MCM7, a key factor in HCC progression
Interestingly, we found that ATO could inhibit the binding of SRF to MCM7 promoter (Fig. 8c) but not constitutively downregulate the SRF expression (Fig. 8d). While in SRF-knockdown cells, MCM7 protein is significantly downregulated (Fig. 8e), the tumorsphere formation was also decreased (Fig. 8f). These results suggested that ATO downregulates MCM7 by suppressing the transcription activity of SRF in HCC cells.
SRF/MCM7 transcription complex is involved in ATO inhibiting MCM7 transcription
An interesting study suggested that MCM1 (a homolog of the mammalian SRF) in conjunction with MCM7 is involved in the regulation of MCM7 expression in Saccharomyces cerevisiae28. We then addressed the question whether ATO disrupts the transcription activity of MCM7/SRF complex in HCC cells. We used immunoprecipitation (IP) assays with anti-SRF antibody and observed an MCM7 band, of which intensity was just slightly decreased in the precipitates obtained from the ATO-treated cells (Fig. 8g). Meanwhile, dual luciferase reporter assays showed that ATO can decrease the signal intensities of pGL4-MCM7-promoter. Transduction of pcDNA3.1-MCM7 plus pBPLV-SRF resulted in an increase in MCM7 transactivation, which can be significantly inhibited by ATO (Fig. 8h). These results indicated an MCM7/SRF complex also exists in HCC cells, through which ATO disrupt the transcription activity for MCM7 without affecting the interaction between MCM7 and SRF.
Arsenic binds to various proteins containing cysteine residues29. Based on the distribution of cysteine (Cys, C) in the MCM7 protein and SRF protein, we speculated that arsenic might bind two adjacent cysteines of the MCM7 protein (Fig. S9A) by means of the As-OH, rather than the SRF protein with only one cysteine (Fig. S9B). Indeed, the dual immunofluorescence results indicated a colocalization for MCM7 and FlAsH-EDT2, a green biarsenical labeling reagent, in HCC cells (Fig. S9C). Furthermore, in vitro arsenic-protein binding assays demonstrated that FlAsH-EDT2 could bind to the full-length human recombinant MCM7 protein (Fig. S9D), while H3ASO3 significantly blocked the green fluorescence signals (Fig. S9E). We also examined whether ATO could directly degrade MCM7 protein. Even at 360 min after exposure, ATO did not induce a shift of MCM7 protein from the supernatant of cell lysates to the detergent-insoluble pellet separated from the RIPA buffer fraction as Zhang et al. previously reported30 (Fig. S9F). The enforced overexpression of MCM7 protein with pcDNA3.1-MCM7-cmyc plasmids was not reduced 48 h after ATO exposure (Fig. S9G). These results indicated that ATO may bind to the MCM7 protein but could not degrade the MCM7 protein directly.
From the data above, we supposed that ATO downregulates MCM7 expression possibly by inhibiting the transcription activity of SRF/MCM7 complex through binding MCM7 protein (Fig. 8i).
Discussion
To date, CSC-targeting agents are rare or still undergoing clinical investigations. In this study, we offered a paradigm for identifying CSCs-targeted agents from approved drugs. To demonstrate the anti-CSCs potential of ATO, we provide compelling data obtained from in vitro tumorsphere formation assay, which is widely used in selection and enrichment of normal stem cells or CSCs31, and in vivo tumorigenic assay including serial transplantation analysis, which is recommended as the gold standard for the identification of CSCs32,33, and also results from a metastatic HCC xenograft model for estimating the metastatic potential of primary liver cancer. Moreover, we revealed MCM7 as a novel and direct target of ATO in HCC. This is the first study identifying direct downstream factors of ATO in liver CSCs by screening a total of 49,395 genes. The clinical significance of the target molecule in HCC specimens further provides solid evidence for the therapeutic value of ATO in clinical practice.
MCM7 has been shown a broad-spectrum overexpression in a variety of tumors including nervous system tumors34,35, prostate36, cervical37, epidermis38, and HCC39,40. Here, downregulation of MCM7 protein is demonstrated to be involved in ATO inhibiting liver CSCs. We found that HCC patients with elevated expression of MCM7 in tumor tissues are likely to have poor prognosis. The important roles of MCM7 in HCC progression further support the therapeutic significance of ATO, which dramatically inhibits MCM7 expression. Interestingly, in the last few years, MCM proteins including MCM7 have been utilized as diagnostic and prognostic tumor markers for the involvement of DNA replication process41. However, the abundance of MCM2–MCM7 proteins in cells is far more than the replication origins, suggesting that their functions may not be limited to DNA replication, this is termed as “MCM paradox”21,22,42. Here, our study provides a possible explanation for the “MCM paradox” by uncovering a stemness-associated regulatory function of MCM7 in ATO treatment.
There are lots of intensive investigations of the clinical effects and underlying mechanisms of ATO in leukemia30,43,44; however, solid tumors including HCC are distinct from leukemia in ontogeny patterns and molecular networks6,45. SRF is a ubiquitously expressed transcription factor regulating diverse genes involved in cell growth and differentiation, migration, and apoptosis46. Here, we uncovered the important role of SRF in ATO regulating MCM7 expression for the first time. We also found ATO binds two adjacent cysteines of the MCM7 protein rather than SRF by means of the As-OH, which is consistent with recent reports29,47. We suppose this binding might induce a conformational change in this SRF/MCM7 transcription complex and attenuate its binding to the promoter of MCM7 (i.e., MCM7 autoregulation) as previously reported28, which leads to the downregulation of MCM7 transcription.
Our data suggest that ATO treatment in solid tumors, which has different regulatory mechanisms from those of standard chemotherapy, is of promising clinical applications. Given that only a few preclinical compounds inhibiting the MCM2–7 complex’s enzymatic activity and/or expression have been discovered48, and no FDA-approved drugs have been reported to target MCM complex, it is meaningful that we found ATO, approved in clinics, could target MCM7 in HCC.
Taken together, our data suggest that ATO is a promising therapeutic drug inhibiting liver CSCs and metastasis through MCM7 downregulation by suppressing the transcription activity of SRF/MCM7 complex. The identification of SRF/MCM7 complex as a direct target of ATO provides new insights into the drug’s mechanism of action, which may benefit the appropriate use of this agent in the treatment of solid tumors including HCC.
Methods
Cells and cell cultures
Human liver cancer cell lines (HCCLM3, Hep3B, Huh7.5.1, PLC, and SMMC-7721), the L02 human fetal liver cell line, and HEK-293T cell line were used. The HCCLM3 cells were from the Liver Cancer Institute of Fudan University (Shanghai, China). The Huh7.5.1 cells were maintained in our institute16. The Hep3B cells and SMMC-7721 cells were obtained from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). PLC was obtained from ATCC. The HEK-293T packaging cell line was obtained from Invitrogen (Carlsbad, CA). The L02 cells were obtained from the National Platform for Experimental Cell Resources (Beijing, China). All cells were maintained in DMEM (Sigma) containing 10% FBS (ExCell Biology, Shanghai, China) at 37 °C with 5% CO2, except the L02 cells, which were maintained in RPMI 1640 medium (Sigma). Cell lines were subjected to routine cell line quality examinations (e.g., normal morphology, mycoplasma-free) and used less than 10 passages after thawed. The fresh primary liver cancer tissues were digested into single cells (Tumor Dissociation Kit, Miltenyi Biotec, Bergisch Gladbach, Germany) and cultured in the medium, kindly provided by Prof. Li-Jian Hui (Shanghai Institute of Biochemistry and Cell Biology, CAS)49. Fresh HCC tissues were provided from Beijing Tongren Hospital of China in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from the subjects. Tumor specimens were obtained according to protocols approved by the institutional review board of the Local Ethics Committee of Beijing, China.
Tumorsphere culture
HCC cells were cultured in the tumorsphere system, as previously reported15,16.
Chromatin immunoprecipitation (ChIP)
The HCC cells were treated by 3.6 μM ATO with PBS as the control. After incubation for 48 h, we cross-linked cells with 1% formaldehyde for 10 min at 37 °C and processed them in accordance with the Chromatin Immunoprecipitation Kit (#17-295, Millipore, Burlington, MA). Briefly, after cross-linking, the DNA was sheared, 4 μL of SRF-specific antibody (#5147S Cell Signaling Technologies, Danvers, MA) was used for immunoprecipitation overnight at 4 °C with rotation, followed by incubation with 60 μL of Protein G agarose for 1 h at 4 °C with rotation. Following the reverse cross-linking step, DNA was precipitated and purified by Axyprep DNA Gel Extraction Kit (AXYGEN, Corning, NY). The purified fragmented DNA was subjected for PCR amplification to detect relative occupancy. The specific primers used for human MCM7 promoter region with the SRF binding site are listed in Supplementary Table 1. The anti-SRF antibody used is listed in Supplementary Table 2.
Animal models
Male Nu/Nu nude mice, aged 6–7 weeks, and male NOD/SCID mice, weight 18–20 g, were purchased from Vital River Laboratories (Beijing, China) or SPF (Beijing) Biotechnology Co., Ltd. (Beijing, China). Mice were housed and all experiments were performed according to international laws (EEC Council Directive 86/609, O.J. L 358. 1, December 12, 1987; Guide for the Care and Use of Laboratory Animals, United States National Research Council, 1996), relevant institutional guidelines and regulations of Beijing Medical Experimental Animal Care Commission. For diluting limitation assay of ATO-pretreated cells, 1 × 106, 1 × 105, and 1 × 104 ATO-pretreated Hep3B cells, with the control cells, were subcutaneously injected into the flanks of NOD/SCID mice, respectively. The tumor growth was monitored weekly. The subcutaneous tumors were removed and weighed 4–9 weeks later. For subcutaneous tumorigenicity of MCM7-knockdown cells, HCCLM3-psicoR-shMCM7 cells and Huh7.5.1 psicoR-shMCM7 cells at two dilutions (1 × 106 and 1 × 105), with the control cells, were subcutaneously injected into the flanks of NOD/SCID mice, respectively. The subcutaneous tumors were removed and weighed 4 weeks later. For serial transplantation of ATO-treated tumors via local injection, 5 × 106 SMMC-7721 cells were subcutaneously injected into the nude mice. Two weeks later, ATO at 0.72 mg/kg (in 100 μL) was locally injected into the subcutaneous tumors of the nude mice every other day with the same volume of PBS as the control. Approximately 2 weeks later, the tumors were carefully removed and were digested into single cells by Tumor Dissociation Kit (Miltenyi Biotec). The cells (two dilutions: 3 × 105 and 3 × 104) were subcutaneously injected into the left flank and the right flank of NOD/SCID mice (ATO-treated group vs. control group), respectively. The tumor growth was monitored weekly. The subcutaneous tumors were removed and weighed 8 weeks later. For intrahepatic metastasis model, 1.5 × 106 cells (wild type of HCCLM3 cells followed by ATO treatment; MCM7-knockdown-HCCLM3 cells and scramble control cells) were suspended in 50 μL PBS containing 30% Matrigel for each nude mouse. Each mouse was orthotopically inoculated in the left hepatic lobe under anesthesia, through a 50-μL microinjector in the upper abdomen. If the mice died after injections of HCC cells into liver within 24 h, the mice would be excluded from the analysis. The mice were sacrificed 8 weeks later. For the self-controlled mice (i.e., NOD/SCID mice received subcutaneous injection of two group cells into the left flank and the right flank, respectively), The mice were randomly grouped. Otherwise, the mice were grouped by randomized block based on body weight.
Statistical analysis
Two-sided Student’s t-tests, two-sided Chi-square test, Fisher’s exact test, and multiple t-tests were performed using GraphPad Prism 5.0 and Origin 6.1 software. Survival curves were analyzed and validated with the log-rank test. P values < 0.05 were considered significant. Results are shown as means ± S.D. The immunohistochemistry (IHC) staining intensity was analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD). The loci diameters were measured by software (NIS-Elements F 4.30.01, Nikon, Tokyo, Japan).
Accession number
The microarray data have been submitted to the GEO database (http://www.ncbi.nlm.nih.gov/geo/) and assigned the identifier GSE111935.
References
Bruix, J., Gores, G. J. & Mazzaferro, V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut 63, 844–855 (2014).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2, 16018 (2016).
Hu, H. T. et al. Arsenic trioxide intravenous infusion combined with transcatheter arterial chemoembolization for the treatment of hepatocellular carcinoma with pulmonary metastasis: long-term outcome analysis. J. Gastroenterol. Hepatol. 32, 295–300 (2017).
Liu, B. et al. Arsenic trioxide transarterial chemoembolization with and without additional intravenous administration of arsenic trioxide in unresectable hepatocellular carcinoma with lung metastasis: a single-blind, randomized trial. J. Cancer Res. Clin. Oncol. 141, 1103–1108 (2015).
Wang, H. et al. Randomized clinical control study of locoregional therapy combined with arsenic trioxide for the treatment of hepatocellular carcinoma. Cancer 121, 2917–2925 (2015).
Liu, J. X., Zhou, G. B., Chen, S. J. & Chen, Z. Arsenic compounds: revived ancient remedies in the fight against human malignancies. Curr. Opin. Chem. Biol. 16, 92–98 (2012).
Rehman, K. & Naranmandura, H. Double-edged effects of arsenic compounds: anticancer and carcinogenic effects. Curr. Drug Metab. 14, 1029–1041 (2013).
Zhang, K. Z. et al. Arsenic trioxide induces differentiation of CD133+ hepatocellular carcinoma cells and prolongs posthepatectomy survival by targeting GLI1 expression in a mouse model. J. Hematol. Oncol. 7, 28 (2014).
Han, J. B. et al. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. Onco Targets Ther. 6, 1129–1138 (2013).
Zhen, Y., Zhao, S., Li, Q., Li, Y. & Kawamoto, K. Arsenic trioxide-mediated Notch pathway inhibition depletes the cancer stem-like cell population in gliomas. Cancer Lett. 292, 64–72 (2010).
Wu, J. et al. Arsenic trioxide depletes cancer stem-like cells and inhibits repopulation of neurosphere derived from glioblastoma by downregulation of Notch pathway. Toxicol. Lett. 220, 61–69 (2013).
Li, Y. et al. Inhibition of the cancer stem cells-like properties by arsenic trioxide, involved in the attenuation of endogenous transforming growth factor beta signal. Toxicol. Sci. 143, 156–164 (2015).
Kaiser, J. The cancer stem cell gamble. Science 347, 226–229 (2015).
Jia, Y. L. et al. Epimorphin promotes human hepatocellular carcinoma invasion and metastasis through activation of focal adhesion kinase/extracellular signal-regulated kinase/matrix metalloproteinase-9 axis. Hepatology 54, 1808–1818 (2011).
Yan, X. L. et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 57, 2274–2286 (2013).
Zhou, J. N. et al. MicroRNA-125b attenuates epithelial–mesenchymal transitions and targets stem-like liver cancer cells through small mothers against decapentaplegic 2 and 4. Hepatology 62, 801–815 (2015).
Kreso, A. & Dick, J. E. Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291 (2014).
Haraguchi, N. et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Invest. 120, 3326–3339 (2010).
Ma, S. et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132, 2542–2556 (2007).
Yamashita, T. et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 136, 1012–1024 (2009).
Forsburg, S. L. Eukaryotic MCM proteins: beyond replication initiation. Microbiol. Mol. Biol. Rev. 68, 109–131 (2004).
Huang, T. H. et al. Epidermal growth factor receptor potentiates MCM7-mediated DNA replication through tyrosine phosphorylation of Lyn kinase in human cancers. Cancer Cell 23, 796–810 (2013).
Das, M., Singh, S., Pradhan, S. & Narayan, G. MCM paradox: abundance of eukaryotic replicative helicases and genomic integrity. Mol. Biol. Int. 2014, 574850 (2014).
Liu, H. et al. MYC suppresses cancer metastasis by direct transcriptional silencing of alphav and beta3 integrin subunits. Nat. Cell Biol. 14, 567–574 (2012).
Shohet, J. M. et al. Minichromosome maintenance protein MCM7 is a direct target of the MYCN transcription factor in neuroblastoma. Cancer Res. 62, 1123–1128 (2002).
Ohrnberger, S. et al. Dysregulated serum response factor triggers formation of hepatocellular carcinoma. Hepatology 61, 979–989 (2015).
Park, M. Y. et al. Expression of the serum response factor in hepatocellular carcinoma: implications for epithelial–mesenchymal transition. Int. J. Oncol. 31, 1309–1315 (2007).
Fitch, M. J., Donato, J. J. & Tye, B. K. Mcm7, a subunit of the presumptive MCM helicase, modulates its own expression in conjunction with Mcm1. J. Biol. Chem. 278, 25408–25416 (2003).
Zhao, L. et al. The reaction of arsenite with proteins relies on solution conditions. Inorg. Chem. 53, 3054–3061 (2014).
Zhang, X. W. et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 328, 240–243 (2010).
Weiswald, L. B., Bellet, D. & Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 17, 1–15 (2015).
Ailles, L. E. & Weissman, I. L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 18, 460–466 (2007).
Clarke, M. F. et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344 (2006).
Erkan, E. P. et al. Depletion of minichromosome maintenance protein 7 inhibits glioblastoma multiforme tumor growth in vivo. Oncogene 33, 4778–4785 (2014).
Lau, K. M. et al. Minichromosome maintenance proteins 2, 3 and 7 in medulloblastoma: overexpression and involvement in regulation of cell migration and invasion. Oncogene 29, 5475–5489 (2010).
Ren, B. et al. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 25, 1090–1098 (2006).
Brake, T., Connor, J. P., Petereit, D. G. & Lambert, P. F. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 63, 8173–8180 (2003).
Honeycutt, K. A. et al. Deregulated minichromosomal maintenance protein MCM7 contributes to oncogene driven tumorigenesis. Oncogene 25, 4027–4032 (2006).
Qu, K. et al. MCM7 promotes cancer progression through cyclin D1-dependent signaling and serves as a prognostic marker for patients with hepatocellular carcinoma. Cell Death Dis. 8, e2603 (2017).
Zhou, Y. M. et al. MCM7 expression predicts post-operative prognosis for hepatocellular carcinoma. Liver Int. 32, 1505–1509 (2012).
Giaginis, C., Vgenopoulou, S., Vielh, P. & Theocharis, S. MCM proteins as diagnostic and prognostic tumor markers in the clinical setting. Histol. Histopathol. 25, 351–370 (2010).
Wyrick, J. J. et al. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science 294, 2357–2360 (2001).
Hong, S. H., Yang, Z. & Privalsky, M. L. Arsenic trioxide is a potent inhibitor of the interaction of SMRT corepressor with its transcription factor partners, including the PML-retinoic acid receptor alpha oncoprotein found in human acute promyelocytic leukemia. Mol. Cell. Biol. 21, 7172–7182 (2001).
Nimmanapalli, R. et al. Arsenic trioxide inhibits translation of mRNA of bcr-abl, resulting in attenuation of Bcr-Abl levels and apoptosis of human leukemia cells. Cancer Res. 63, 7950–7958 (2003).
Rao, Y., Li, R. & Zhang, D. A drug from poison: how the therapeutic effect of arsenic trioxide on acute promyelocytic leukemia was discovered. Sci. China Life Sci. 56, 495–502 (2013).
Modak, C. & Chai, J. Serum response factor: look into the gut. World J. Gastroenterol. 16, 2195–2201 (2010).
Zhang, H. N. et al. Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proc. Natl Acad. Sci. USA 112, 15084–15089 (2015).
Simon, N. E. & Schwacha, A. The Mcm2-7 replicative helicase: a promising chemotherapeutic target. Biomed. Res. Int. 2014, 549719 (2014).
Qiu, Z. et al. Hepatocellular carcinoma cell lines retain the genomic and transcriptomic landscapes of primary human cancers. Sci. Rep. 6, 27411 (2016).
Acknowledgements
This work was supported by grants from The National Key Research and Development Program of China (2017YFA0103100, 2017YFA0103103, and 2017YFA0103104), Guangzhou Health Care and Cooperative Innovation Major Project (201508020257, 201704020224 and 201803040005), and Natural Science Foundation of China (81773137). The authors wish to thank Prof. Jian Yu (University of Pittsburgh School of Medicine) and Prof. Lin Zhang (University of Pittsburgh Cancer Institute) for critical reading and revising the manuscript, Prof. Li-Jian Hui and Dr. Zhi-Xin Qiu (Shanghai Institute of Biochemistry and Cell Biology, CAS) for kindly providing the culture medium for primary liver cancer cells, Dr. Chun-Jiang Fu (Fangshan Traditional Chinese Medicine Hospital), Ms. Huan-Huan Zhang (First Affiliated Hospital of PLA General Hospital), and Prof. Gui-Fang Dou and Mr. Peng Han in Beijing Institute of Radiation Medicine, Mrs. Jing Li and Mr. Zeng Fan in Institute of Health Service and Transfusion Medicine for help and suggestions in the study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, HY., Zhang, B., Zhou, JN. et al. Arsenic trioxide inhibits liver cancer stem cells and metastasis by targeting SRF/MCM7 complex. Cell Death Dis 10, 453 (2019). https://doi.org/10.1038/s41419-019-1676-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1676-0
- Springer Nature Limited
This article is cited by
-
Unveiling the link between arsenic toxicity and diabetes: an in silico exploration into the role of transcription factors
Toxicological Research (2024)
-
Arsenic trioxide: applications, mechanisms of action, toxicity and rescue strategies to date
Archives of Pharmacal Research (2024)
-
A combined miR-34a and arsenic trioxide nanodrug delivery system for synergistic inhibition of HCC progression after microwave ablation
Cancer Nanotechnology (2021)
-
Arsenic nano complex induced degradation of YAP sensitized ESCC cancer cells to radiation and chemotherapy
Cell & Bioscience (2020)